

Abstract
The amphotericin B-perforated whole-cell patch clamp technique was used to determine the modulation of L-type Ca2+ channels by protein kinase C (PKC)-mediated pathways in adult rat ventricular myocytes.
Application of 10 nM endothelin-1 (ET-1) increased peak Ca2+ current (ICa) by 28·2 ± 2·5 % (n = 13) and slowed current decay. These effects were prevented by the endothelin receptor antagonist PD145065 (10 μM) and by the PKC inhibitor chelerythrine (8 μM).
To establish if direct activation of PKC mimicked the ET-1 effect, the active and inactive phorbol esters (phorbol-12-myristate-13-acetate and 4α-phorbol-12, 13-didecanoate) were tested. Both phorbol esters (100 nM) resulted in a small (∼10 %) increase in ICa, suggesting PKC-independent effects.
Bath application of dioctanoylglycerol (diC8), a diacylglycerol (DAG) analogue which is capable of directly activating PKC, caused a gradual decline in peak ICa (50·4 ± 6·2 %, n = 5) and increased the rate of current decay. These effects were unaffected by the PKC inhibitor chelerythrine (8 μM).
Intracellular photorelease of caged diC8 with 3 or 10 s exposure to UV light produced a concentration-dependent increase in peak ICa (20·7 ± 8·5 % (n = 8) for 3 s UV and 60·8 ± 11·4 % (n = 13) for 10 s UV), which could be inhibited by chelerythrine.
Our results demonstrate that both ET-1 and intracellularly photoreleased diC8 increase ICa by a PKC-mediated pathway, which is in direct contrast to the PKC-independent inhibition of ICa produced by bath-applied diC8. We conclude that specific cellular pools of DAG are crucially important in the regulation of ICa by PKC.
Many extracellular hormones and neurotransmitters regulate the electrical and contractile properties of cardiac muscle. These neurohormones can bind to a variety of transmembrane receptors, initiating multiple signalling cascades leading to regulatory changes in the myocyte. A subset of G-protein-coupled receptors act via Gq to stimulate phospholipase C (PLC), which hydrolyses phosphatidylinositol 4,5-bisphosphate, generating inositol trisphosphate (IP3) and DAG (Berridge, 1997). The liberated DAG can then activate PKC, which phosphorylates a wide spectrum of cardiac proteins responsible for myocardial excitability and contraction (Puceat & Vassort, 1996; Dorn & Brown, 1999). In cardiac muscle, multiple receptors, including endothelin (Endoh et al. 1998; Dorn & Brown, 1999), α1-adrenergic (Stiles, 1996) and angiotensin II receptors (Van Heugten et al. 1996), have been demonstrated to couple with Gq and ultimately lead to activation of PKC.
The L-type Ca2+ channel plays a critical role in cardiac excitability and in excitation-contraction coupling, and thus it represents an important potential target for PKC modulation. However, the effect of activation of PKC on cardiac L-type Ca2+ channels remains unclear. For example, some studies of ET-1 have shown clear increases in ICa (Bkaily et al. 1995), while others have shown a decrease in ICa (Cheng et al. 1995) or no effect (Habuchi et al. 1992). Similar apparently conflicting findings have resulted from studies of direct activators of PKC, such as diC8, 1-oleoyl-2-acetyl-sn-glycerol (OAG) as well as phorbol esters (Dosemeci et al. 1988; Walsh & Kass, 1988; Lacerda et al. 1988; Tseng & Boyden, 1991; Schreur & Liu, 1996; Zhang et al. 1997). These contradictory data may be caused by the wide differences in experimental conditions, including different species, tissues, preparations and temperatures. In addition, the method employed to measure ICa could be of critical importance in maintaining the physiological response to ET-1 and other neurohormones. Most of the initial studies were performed using the ruptured whole-cell patch clamp technique. In contrast, the perforated whole-cell technique allows voltage-clamp characterization of ICa without the dialysis of large molecules and proteins that occurs with the ruptured patch technique. This has recently been revealed to be of critical importance in characterizing the effect of α1-adrenergic and arginine vasopressin regulation of L-type Ca2+ channels in isolated rat and guinea-pig ventricular myocytes (Liu & Kennedy, 1998; Kurata et al. 1999).
Walker and colleagues have developed a novel tool to investigate the role of intracellular DAG and related activation of PKC by synthesizing a caged DAG analogue, caged diC8, which can be released in a controlled fashion by near-UV light (Huang et al. 1996). In isolated adult rat ventricular myocytes, intracellular photorelease of diC8 induced a strong positive inotropic effect, demonstrated by enhanced cell shortening, and this effect was stereospecific, concentration-dependent, and blocked by a PKC inhibitor, chelerythrine (Pi et al. 1997). Further investigation demonstrated that the positive inotropic effect was due primarily to a large increase in the intracellular Ca2+ transient in response to photorelease of diC8 (Pi & Walker, 1998). The increased Ca2+ transient did not reflect increased sarcoplasmic reticulum Ca2+ load as assessed by the caffeine releasable pool, nor did it reflect changes in sarcoplasmic reticulum Ca2+ uptake (Pi & Walker, 1998). Therefore, we hypothesized that influx of Ca2+ through L-type Ca2+ channels may increase following photorelease of caged diC8 via activation of PKC pathways.
The purpose of the present study was to determine the modulation of rat ventricular L-type Ca2+ channels by PKC activation using the perforated patch technique. We used ET-1 as a representative agonist to activate Gq-PKC pathways and compared those results to putative direct activation of PKC using phorbol esters, bath-applied diC8, and photoreleased diC8. Our results demonstrate that ET-1 increases ICa in a PKC-dependent fashion that can be mimicked by intracellular photoreleased diC8 but not by bath-applied diC8 or phorbol esters.
Some of the preliminary results from this study have been presented to the Biophysical Society (He et al. 1999).
METHODS
Ventricular myocyte isolation
Single ventricular myocytes were enzymatically isolated from the hearts of adult male Sprague-Dawley rats (200–250 g) killed with metofane (inhalation for 3 min) following a protocol approved by the University of Wisconsin Animal Care and Use Committee as previously described (Pi et al. 1997). Briefly, hearts were rapidly excised, cannulated, and subjected to retrograde perfusion on a Langendorff apparatus at 37°C via the aorta with oxygenated Ringer solution of the following composition (mM): 125 NaCl, 2 NaH2PO4, 5 KCl, 1·2 MgSO4, 25 Hepes, 5 sodium pyruvate, 11 glucose, and 1 CaCl2 (pH adjusted to 7·4 with NaOH). The hearts were briefly perfused with Ca2+-free Ringer solution followed by Ca2+-free Ringer solution containing 0·6 mg ml−1 collagenase and 0·36 mg ml−1 hyaluronidase. The left ventricle was cut away from the rest of the tissue and further incubated in the enzyme solution. Isolated myocytes were washed, pelleted in a tabletop centrifuge, and resuspended in 0·5 mM Ca2+ Ringer solution at room temperature at a density of ∼105 cells ml−1. The yield was 1 × 106 to 1·5 × 106 cells per heart, of which typically 85 % were viable rod-shaped cells. Myocytes which displayed clear striations were used for experiments within 8 h of isolation.
Caged diC8 loading in myocytes and photorelease
Myocytes at a density of 3 × 104 to 6 × 104 cells ml−1 were incubated in the dark with 800 μM α-carboxyl caged diC8 dissolved in dimethyl sulfoxide (DMSO; final concentration of DMSO 0·05 %) in a siliconized Eppendorf tube for 45 min at room temperature. The cell pellet was gently suspended twice in order to provide adequate oxygenation and then washed twice with fresh Ringer solution containing 0·5 mM Ca2+ without caged diC8. Ventricular myocytes were then placed in an experimental chamber and perfused at ∼0·5 ml min−1 with a normal Ringer solution with 0·5 mM Ca2+(see above) throughout the experiment.
An inverted Nikon Diaphot 200 microscope (Tokyo, Japan) with Nikon epi-fluorescence attachments was used for the experiments. The UV beam from a HBO 100 W/2 mercury lamp was passed through sequential neutral density filters (ND2 and ND4) and reflected onto the cell via a DM 400 dichroic mirror and Nikon 0·55 LWD × 40 objective lens. After the perforated whole-cell configuration was obtained, illumination was initiated and exposure time (1–30 s as desired) was controlled by hand-switching the light path off or on. Control experiments showed that exposure for up to 3 min to the UV light alone was without effect on ICa (data not shown).
Electrophysiological measurements
The amphotericin B-perforated whole-cell technique was employed to record ICa (Rae et al. 1991). Ventricular myocytes were placed in the experimental chamber mounted on the stage of an inverted microscope (Nikon Diaphot 200). The cells were perfused at 0·5 ml min−1 with 0·5 mM Ca2+ Ringer solution (see above). The pipette solution consisted of (mM): 100 caesium glutamate, 40 CsCl, 10 Hepes, 0·5 CaCl2 (pH adjusted to 7·2 with CsOH). The amphotericin B was prepared as a stock solution (0·1 mg μl−1 in DMSO) and frozen in small aliquots for up to 5 days. The diluted amphotericin B was prepared hourly from the stock solution by diluting in pipette solution to a final concentration of 300 μg ml−1. The pipette solution with amphotericin B was sonicated for 2–5 s. The final solution was used within 1 h after preparation. The tip of the patch pipette was first filled with amphotericin B-free solution by dipping the tip into the solution for 2–5 s. The rest of the pipette was backfilled with amphotericin B-containing solution. Patch electrodes were fabricated from borosilicate glass (TW150F-4, World Precision Instruments, Inc.) with a Flaming/Brown Micropipette Puller Model 87 (Sutter Instruments). The electrode resistance was 1–2 MΩ when filled with the pipette solution. The potential of the electrode was adjusted to zero current between the pipette solution and the bath solution immediately before seal formation. After a giga-seal between the pipette and myocyte had formed, the pipette potential was stepped from -80 mV to -90 mV for 10 ms at 1 Hz. The development of electrical access could be monitored by the appearance of a capacitative current evoked by the -10 mV hyperpolarization test pulse. The uncompensated access resistance of the cell typically dropped to a stable level of 11·8 ± 0·4 MΩ (n = 117) in 10–40 min after seal formation. Series resistance and whole-cell capacitance were analog compensated using the Axopatch 200B circuitry. The series resistance was compensated 70–85 %. Access resistance was periodically monitored during the course of an experiment and the level of compensation was adjusted as needed. Once the perforated whole-cell configuration was formed, the bath solution was switched to a solution containing (mM): 130 NaCl, 10 TEA-Cl, 1 MgCl2, 10 Hepes, 10 glucose, 1·8 CaCl2 (pH adjusted to 7·4 with 20 % TEA-OH), with 1 μM saxitoxin (STX). Currents were recorded at 25 kHz and filtered at 5 kHz using an Axopatch 200B amplifier (Axon Instruments) with pCLAMP 6·04 as acquisition software at room temperature. Under the perforated whole-cell recording conditions, cell contraction was clearly observed during the depolarizing voltage steps. Inclusion of 0·5 mM Ca2+ in the pipette solution ensured that only cells in the perforated whole-cell configuration were studied. The holding potential for these experiments was -80 mV.
Current through L-type Ca2+ channels (ICa) was evoked using 200 ms test pulses over a range of potentials from -30 to +60 mV following a 300 ms prepulse to -40 mV. The prepulse was used to inactivate the Na+ current (INa), the T-type Ca+ current, and the transient outward K+current (Ito, K) (Dukes & Morad, 1991). In addition, 10 mM TEA-Cl and 1 μM STX in the bath solution and 140 mM Cs in the pipette solution were used to inhibit voltage-dependent IK and INa during the measurements of L-type ICa. Only cells which showed no detectable change in ICa during the initial 5 min of observation were used for these experiments, and control experiments revealed that ICa was stable in these cells, with less than 5 % run-down or run-up in 30 min of recording.
The voltage-dependent inactivation relationships were determined using a gapped double-pulse protocol, i.e. a conditioning prepulse for 1000 ms to potentials between -50 and +40 mV, followed by a 10 ms step returning to -50 mV, and then a 200 ms test pulse to +10 mV. These data were fitted to a Boltzmann distribution using the following equation:
where V0·5 is the half-maximum inactivation potential, and k is the slope factor.
Percentage decay of ICa at 40 and 200 ms was compared between control and experimental groups. The depolarization protocol was the same as above, i.e. a prepulse to -40 mV for 300 ms followed by a test pulse to +10 mV for 200 ms. The percentage decay of ICa was established by measuring ICa at 40 and 200 ms as a percentage of the peak current.
Each myocyte response was recorded from a single cell obtained from a fresh aliquot of cells. Myocytes from one heart were used each experimental day, with data collected from one to four myocytes per day. The mean capacitance of cells used in the present study was 105·5 ± 3·3 pF (n = 117).
Chemicals
All reagents were purchased from Sigma Chemical Co. unless otherwise stated. Chelerythrine (chloride), free diC8, phorbol-12-myristate-13-acetate (PMA), and 4α-phorbol-12,13-didecanoate (4αPDD) stock solutions were prepared in DMSO. ET-1 stock solutions were prepared in 5 % acetic acid. PD145065 and STX (Calbiochem, CA) were dissolved in distilled water. All stock solutions were stored at -20°C. The stock solutions were freshly diluted into bath solution immediately prior to experimental recording. The final concentrations of DMSO and acetic acid in the bath were < 0·1 % and < 0·0005 %, respectively, which had no discernible effect on ICa (data not shown). α-Carboxyl caged diC8 was synthesized and purified as described previously (Sreekumar et al. 1997).
Statistics
All values are presented as means ±s.e.m., with n values representing the number of myocytes in the data set. Statistical significance was evaluated using Student's paired or unpaired t test (two-tailed). ANOVA was used for multiple comparisons. Differences with P < 0·05 were considered statistically significant.
RESULTS
ET-1 increases ICa
ET-1 has been shown to have positive inotropic and chronotropic actions in a variety of cardiac preparations. To resolve the role of L-type Ca2+ channels in these responses to ET-1, we used the perforated whole-cell technique to assess the effect of ET-1 on ICa in adult rat ventricular myocytes with or without the specific PKC blocker, chelerythrine. ICa was elicited from a holding potential of -80 mV with a 300 ms prepulse to -40 mV followed by 25 or 200 ms depolarizing pulses to +10 mV every 15 s. The peak ICa was plotted in response to test pulses to +10 mV as a function of time for a representative cell in Fig. 1A. The measured peak ICa slowly increased with a time to half-maximal effect (t½) of 7·9 ± 1·2 min (n = 13) when 10 nM ET-1 was perfused. The inset shows the superimposed control (^) and ET-1-treated (□) current traces. Figure 1B shows the voltage dependence of the effect of 10 nM ET-1 on ICa. The stimulus protocol employed a 300 ms prepulse to -40 mV followed by a family of 200 ms test pulses from -40 mV to +60 mV in 10 mV steps. Representative original current traces in response to these test pulses are shown (^, control; □, during 10 nM ET-1). Peak ICa densities measured in six different myocytes from four rat hearts are plotted as a function of membrane potential. ET-1 (10 nM) increased ICa by 28·2 ± 2·5 % at +10 mV (n = 13, P < 0·001), and a comparable increase was observed across a wide range of membrane potentials from -10 to +60 mV. To confirm the specificity of the ET-1 effect on ICa, a non-selective antagonist of ET receptors, PD145065, was tested (Kelso et al. 1998). Pretreatment with 10 μM PD145065 completely blocked the ET-1 (10 nM)-mediated stimulation of ICa (1·8 ± 2·5 %, n = 4, data not shown). ET-1 also significantly reduced the decay of ICa measured at 40 ms (47·7 ± 4·3 % in control vs. 42·6 ± 3·8 % in ET-1, n = 13, P < 0·01, see Table 1) and at 200 ms (96·1 ± 0·6 % in control vs. 94·2 ± 0·4 % in ET-1, n = 13, P < 0·01, see Table 1).
Figure 1. ET-1 increases ICa.
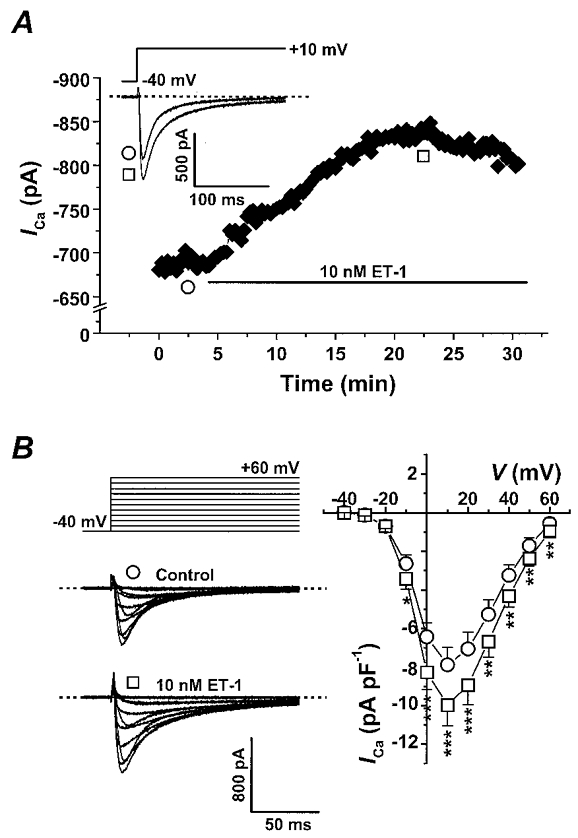
A, the time course of ICa stimulation by 10 nM ET-1 in a representative myocyte. Peak ICa was plotted in response to test pulses to +10 mV every 15 s. The horizontal bar indicates application of 10 nM ET-1. The inset shows the stimulus protocol and the superimposed original current traces in the absence (^) and presence of ET-1 (□). The dashed line indicates the zero current level. The membrane capacitance (Cm) of this cell was 78 pF. B, the stimulus protocol, representative original current traces (Cm= 79 pF), and the mean ICa-V relationship before (^) and during (□) ET-1 exposure. Peak ICa densities (pA pF−1) were measured in 6 different myocytes from 4 rat hearts. Vertical bars in I–V plots indicate the standard errors (*P < 0·05; **P < 0·01; ***P < 0·001).
Table 1.
Decay of ICa at +10 mV after 40 and 200 ms in response to ET-1, photoreleased diC8 and bath-applied diC8
| ICa decayed relative to peak ICa (%) | ||||
|---|---|---|---|---|
| Experimental groups | n | At 40 ms | At 200 ms | |
| ET-1 | Control | 13 | 47·7 ± 4·3 | 96·1 ± 0·6 |
| 10 nm ET-1 | 13 | 42·6 ± 3·8** | 94·2 ± 0·4** | |
| Photoreleased diC8 | Control | 11 | 49·3 ± 3·1 | 96·6 ± 0·9 |
| 10 s UV | 11 | 42·7 ± 3·2** | 89·4 ± 1·3** | |
| Bath-applied diC8 | Control | 7 | 48·5 ± 6·2 | 95·6 ± 1·3 |
| 7·5 μm diC8 | 7 | 59·5 ± 4·0** | 98·2 ± 0·8** | |
P < 0·01, compared to control.
To decide if PKC was involved in the modulation of ICa by ET-1, a specific inhibitor of PKC, chelerythrine, was examined. At the concentrations tested, chelerythrine is a specific inhibitor for PKC compared to other protein kinases (Herbert et al. 1990), and is insensitive to near-UV light (Pi et al. 1997). In control experiments, we did not find any discernible effects of 8 μM chelerythrine on ICa. After 10 min pretreatment with chelerythrine, application of 10 nM ET-1 (□) had no significant effect on ICa, as shown in the time course from a representative cell in Fig. 2A. Figure 2B shows the mean I–V relationship before (^) and during (□) ET-1 exposure. The stimulus protocol was the same as in Fig. 1. Peak ICa densities measured in four different myocytes from four rat hearts were plotted vs. membrane potential. Chelerythrine eliminated the response of myocytes to ET-1 at all potentials examined. These data suggest that the stimulation of ICa by ET-1 is PKC dependent.
Figure 2. Chelerythrine, a specific PKC inhibitor, blocks the effect of ET-1 on ICa.
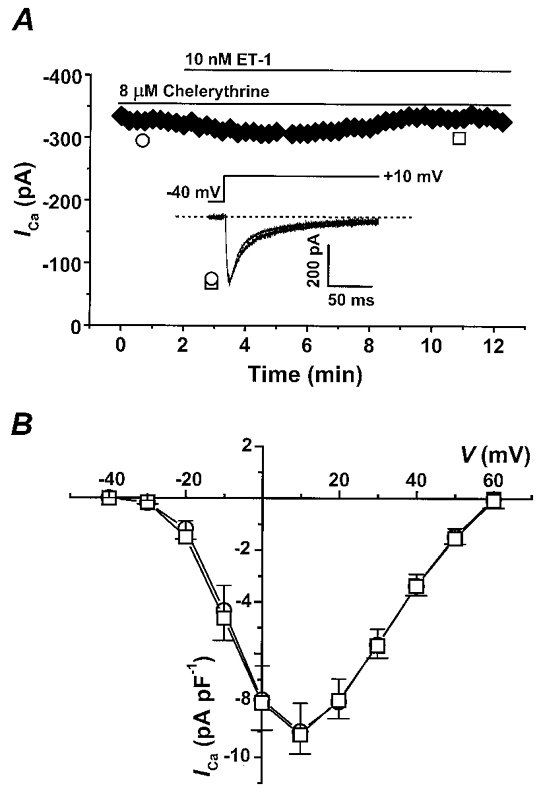
A, the time course of ICa during perfusion of 10 nM ET-1 in a representative myocyte preincubated with 8 μM chelerythrine. Peak ICa is plotted in response to test pulses to +10 mV every 15 s. The inset shows the stimulus protocol and the superimposed original current traces before (^) and during 10 nM ET-1 (□) in a representative myocyte. The dashed line indicates the zero current level. Cm= 39 pF. B, the mean I–V relationship before (^) and during (□) ET-1 exposure in myocytes pre-incubated with 8 μM chelerythrine measured in 4 different myocytes from 4 rat hearts. Upward vertical bars in the I–V plot indicate the standard errors of the control data and the downward bars indicate standard errors of the ET-1 data.
Effect of phorbol esters on ICa
To confirm that the increase in ICa in response to ET-1 was mediated by PKC, two phorbol esters, PMA and 4αPDD, were tested. PMA should be capable of directly activating PKC, bypassing receptor stimulation, while 4αPDD is inactive with regard to PKC stimulation. Perfusion of 100 nM PMA resulted in a small increase in peak ICa, as shown in the time course of ICa from a representative cell in Fig. 3A. The effect was readily reversible with washout of PMA. The mean data from 11 cells treated with 100 nM PMA showed an 11.7 ± 2·2 % increase in ICa at +10 mV (P < 0·001). Figure 3B shows that extracellular perfusion of 100 nM 4αPDD also increased ICa. The mean ICa from four cells treated with 100 nM 4αPDD showed an increase of 10·3 ± 2·1 % (P < 0·05), which was also reversible upon washout. No significant difference was found between the effects of PMA and 4αPDD. Control experiments exposing the cells to 0·1 % DMSO revealed no effect on ICa. Therefore, the small effect of both phorbol esters on ICa suggests PKC-independent modulation of ICa. The rapid reversibility of the effect of the phorbol esters following washout is also consistent with a PKC-independent effect.
Figure 3. Effect of phorbol esters on ICa.

A, the effect of 100 nM PMA on peak ICa. The top panel in A plots peak ICa obtained by a depolarization to +10 mV every 15 s during exposure to 100 nM PMA in a representative myocyte. The middle panel in A displays the superimposed original current traces before (^) and during PMA (□) exposure. The dashed line indicates the zero current level. Cm= 86 pF. The bottom panel in A charts the mean ICa densities (pA pF−1) measured in 11 different myocytes from 4 rat hearts before and after 100 nM PMA. B, the same experiment as A, but examining the effects of an inactive form of phorbol ester, 4αPDD. The time course of ICa and current traces during 100 nM 4αPDD in a representative myocyte are shown in the upper and middle panels, respectively (Cm= 141 pF). The mean ICa densities measured in 11 cells from 4 rat hearts are summarized in the bar chart comparing ICa before and after 100 nM 4αPDD. Vertical bars indicate the standard errors (*P < 0·05; ***P < 0·001, compared to control).
Extracellular application of diC8 inhibits ICa
As phorbol esters did not produce a clear PKC-dependent effect on ICa, we investigated another putative direct activator of PKC, diC8. Figure 4A shows the time course of ICa during bath application of 7·5 μM free diC8 in a representative cell. During perfusion of free diC8, ICa was significantly inhibited, with a t½= 6·7 ± 0·7 min (n = 7). Peak ICa densities were measured in five different myocytes from five rat hearts and are plotted as a function of membrane potential (Fig. 4B). With test pulses to +10 mV, 7·5 μM free diC8 decreased mean ICa by 50·4 ± 6·2 % (n = 5, P < 0·01) without changing the voltage dependence of current activation, as shown by the I–V relations. Bath application of diC8 resulted in a significantly greater decay of ICa measured at 40 ms (48·5 ± 6·2 % in control vs. 59·5 ± 4·0 % in diC8, n = 7, P < 0·01, see Table 1) and at 200 ms (95·6 ± 1·3 % in control vs. 98·2 ± 0·8 % in diC8, n = 7, P < 0·01, see Table 1).
Figure 4. Bath application of diC8 inhibits ICa.

A, the time course of ICa inhibited by bath-applied diC8 in a representative myocyte. Peak ICa is plotted in response to test pulses to +10 mV every 15 s before and during application of 7·5 μM diC8. The inset shows the stimulus protocol and the superimposed original current traces of ICa before (^) and after (□) perfusion of free diC8. The dashed line indicates the zero current level. Cm= 117 pF. B, the stimulus protocol, representative original current traces, and mean I–V relationship before (^) and during (□) bath application of diC8. Peak ICa densities (pA pF−1) are measured in 5 different myocytes from 5 rat hearts. Vertical bars in I–V plots indicate the standard errors (**P < 0·01; ***P < 0·001).
To resolve if the effect of free diC8 on ICa is PKC dependent, chelerythrine (8 μM) was pre-incubated with the cells prior to bath application of 7·5 μM diC8. In six cells, ICa was inhibited by diC8 by 65·2 ± 10·5 % at +10 mV (P < 0·01, data not shown), which was not significantly different from the 50·4 ± 6·2 % decrease seen in the absence of chelerythrine. The results show that the inhibitory effect of extracellular perfusion of free diC8 on ICa is likely to be PKC independent.
Intracellular photorelease of caged diC8 increases ICa
As our initial attempts to directly activate PKC failed to produce clear PKC-dependent effects on ICa, we next tested intracellular photorelease of caged diC8, which has recently been demonstrated to stimulate a PKC-dependent positive inotropic response in rat cardiac myocytes (Pi et al. 1997). Isolated adult rat ventricular myocytes were first loaded with caged diC8 (see Methods) and then continuous perfusion was carried out for at least 10 min to remove the extracellular unincorporated caged diC8 prior to studying a given cell. Figure 5A shows the time course of the effect of photoreleased caged diC8 on ICa. The arrow indicates the time point of exposure to 10 s UV light. Photorelease of diC8 produced a gradual increase in ICa with t½= 5·6 ± 1·0 min (n = 13), which plateaued and then gradually declined. The inset displays the stimulus protocol and the superimposed representative current traces illustrated before (^) and after (□) photorelease of diC8. Peak ICa densities measured in 13 different myocytes from 10 rat hearts are plotted as a function of membrane potential in Fig. 5B. Photorelease of diC8 increased ICa 60·8 ± 11·4 % at +10 mV following a 10 s UV light exposure (n = 13, P < 0·01), and a comparable increase was seen throughout the voltage range studied.
Figure 5. Photoreleased diC8 increases ICa.

A, the time course of ICa stimulated by photoreleased diC8 in a representative myocyte. Peak ICa is plotted in response to test pulses to +10 mV every 15 s. The arrow indicates the time point of 10 s UV light exposure. The inset displays the stimulus protocol and the superimposed original current traces of ICa before (^) and after (□) UV light exposure. The dashed line indicates the zero current level. Cm= 72 pF. B shows the stimulus protocol, representative original current traces, and mean I–V relationship before (^) and after (□) photorelease of diC8. Peak ICa densities (pA pF−1) were measured in 13 different myocytes from 10 rat hearts. Vertical bars in I–V plots indicate the standard errors (*P < 0·05; **P < 0·01; ***P < 0·001).
To confirm that the measured currents before and after UV photorelease of diC8 were through L-type Ca2+ channels, we investigated the sensitivity of these currents to Ca2+ channel blockers. First, we examined the effect of the inorganic Ca2+ channel blocker, Cd2+, which potently blocks L-type Ca2+ channels. Figure 6A displays that, in cells loaded with caged diC8, 0·2 mM Cd2+ can completely inhibit the inward current at +10 mV before and after UV photorelease of diC8. The inhibitory effect of the Cd2+ is also largely reversible. We next examined the effect of the dihydropyridine nifedipine, which is a selective and voltage-dependent blocker of L-type Ca2+ channels. Figure 6B shows the effect of nifedipine on peak inward currents at +10 mV, which have been enhanced by photorelease of diC8. Application of 1·5 μM nifedipine at the holding potential of -80 mV resulted in about a 40 % decrease in the inward current. Changing the holding potential to -40 mV from -80 mV produced no significant change in ICa measured under control conditions (data not shown); however, in the presence of 1·5 μM nifedipine, there was a large enhancement of block. Application of 10 μM nifedipine further blocked the currents, again in a voltage-dependent fashion. From a holding potential of -40 mV, 10 μM nifedipine produced near complete block of the measured inward current. These data confirm that under the present ionic and experimental conditions, the measured currents are through L-type Ca2+ channels with minimal contamination by other currents.
Figure 6. Cadmium and nifedipine block ICa stimulated by photoreleased diC8.
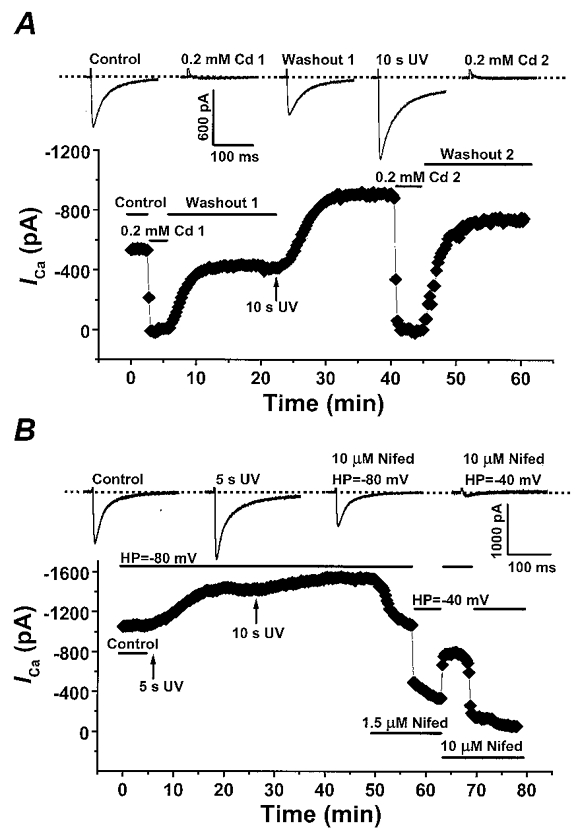
A, the inhibition of ICa by 0·2 mM cadmium (Cd2+) before and after photorelease of diC8. Peak ICa is plotted in response to test pulses to +10 mV every 15 s. The vertical arrow indicates the time point of 10 s UV light exposure and the horizontal bars describe the experimental conditions. The inset displays the original current traces in response to the each treatment during experiment. The dashed line indicates the zero current level. Cm= 90 pF. B, the voltage-dependent inhibition of ICa by nifedipine (Nifed). The vertical arrows indicate the time of 5 s and 10 s UV light exposure. The horizontal bars indicate the experimental conditions. The inset displays the original current traces in response to each treatment. The dashed line indicates the zero current level. HP, holding potential. Cm= 140 pF.
As the photorelease of diC8 is proportional to UV exposure time (Huang et al. 1996), we next examined whether there was a concentration-dependent effect of diC8 on ICa. Concentration-dependent effects of photoreleased diC8 were previously shown for the positive inotropic effect of this compound on isolated rat ventricular myocytes (Pi et al. 1997). The composite time courses for the response of ICa to 3 s and 10 s UV exposure are shown in Fig. 7A. The peak ICa gradually increased in response to diC8 and reached a plateau. The kinetics of current increase were comparable for both 3 s and 10 s UV light exposure as the t½ values were not significantly different, 4·2 ± 0·7 min (n = 8) and 5·6 ± 1·0 min (n = 13), respectively. However, 10 s UV exposure did cause a significantly greater increase in ICa compared to 3 s UV exposure, 60·8 ± 11·4 %vs. 20·7 ± 8·5 %, respectively (P < 0·01). These data suggest that there is a concentration-dependent effect of intracellular diC8 on ICa, and that the rate-limiting steps in this effect are downstream from diC8 production.
Figure 7. Average time course of ICa in response to UV photorelease of diC8.

A, the mean percentage increase of ICa induced by 3 s (▪) and 10 s (•) of UV light. Peak ICa is plotted in response to test pulses to +10 mV every 15 s. The vertical arrow indicates the time point of UV light exposure. B, the mean maximum responses to 3 s (□) and 10 s (▪) of UV light exposure. Vertical bars in both A and B indicate the standard errors (*P < 0·05; **P < 0·01, compared to control).
We also investigated whether photorelease of diC8 altered the kinetics of ICa decay. The percentage of ICa decay at 40 and 200 ms was measured at a test potential of +10 mV. The decay of ICa has previously been demonstrated to be due to both Ca2+- and voltage-dependent inactivation. We anticipated an acceleration of current decay due to the larger currents after photorelease of diC8 and greater Ca2+-dependent inactivation. However, we found that the percentage of ICa decay at 40 ms was reduced from 49·3 ± 3·1 % in control to 42·7 ± 3·2 % after 10 s UV photorelease of diC8 (n = 11, P < 0·01, see Table 1). Likewise, the percentage of ICa decay measured at 200 ms was reduced from 96·6 ± 0·9 % in control to 89·4 ± 1·3 % after photorelease (n = 11, P < 0·01, see Table 1). The slowing of the current decay suggests alterations in Ca2+- or voltage-dependent gating of the channels following modulation by diC8.
The initial experiments revealed that photorelease of diC8 caused a concentration-dependent increase in ICa, similar in characteristics to that stimulated by ET-1. The following experiment examined whether this increase in ICa was sensitive to the PKC inhibitor chelerythrine, like the response to ET-1. Figure 8 plots the time course of the effect of photoreleased diC8 on ICa without (left) or with a pre-incubation with 4 μM (middle) and 8 μM (right) chelerythrine. Photorelease of diC8 for 10 s only resulted in a 22·2 ± 12·9 % (n = 6) increase in ICa of myocytes pretreated with 4 μM chelerythrine compared to a 60·8 ± 11·4 % (n = 13) increase in control. However, following pre-incubation with 8 μM chelerythrine, 10 s of UV exposure led to a 14·0 ± 16 % decrease in ICa relative to the baseline (n = 8, P < 0·001). Control experiments with chelerythrine alone or chelerythrine plus 10 s UV light exposure did not show any significant effect on ICa over the period of time studied. We conclude that the concentration-dependent inhibition of the effect of photoreleased diC8 on ICa produced by chelerythrine is consistent with this effect being mediated by a PKC-dependent pathway. The inhibition of ICa in the presence of 8 μM chelerythrine following photorelease of diC8 may reflect an unmasking of the PKC-independent blocking effect demonstrated by bath application of diC8 (Fig. 4).
Figure 8. Chelerythrine blocks the effect of photoreleased diC8 on ICa.

A, the mean percentage increase in ICa induced by 10 s UV light exposure in the absence (left) and presence of 4 μM (middle) or 8 μM chelerythrine (right). Peak ICa is plotted in response to test pulses to +10 mV every 15 s. The insets display the normalized superimposed original current traces of control ICa (^) and photoreleased diC8 (□). The vertical arrows indicate the time point of UV light exposure. The dashed line indicates the zero current level. B, the sum of the maximal effect of photoreleased diC8 on ICa in the absence (□) and presence of 4 μM (▪) or 8 μM ( ) chelerythrine. Vertical bars in A and B indicate the standard errors (**P < 0·01; ***P < 0·001, compared to 10 s UV exposure alone).
) chelerythrine. Vertical bars in A and B indicate the standard errors (**P < 0·01; ***P < 0·001, compared to 10 s UV exposure alone).
If ET-1 and photorelease of diC8 act by the same PKC-dependent pathway to modulate ICa, then we predicted that following a saturating response to photoreleased diC8, application of ET-1 would not further upregulate ICa. Figure 9 displays the response of ICa measured in a myocyte following five successive exposures to UV light. ICa was increased by 38, 66, 83 and 100 % in response to successive exposures of 1, 5, 10 and 20 s of UV light, respectively. An additional exposure of 30 s UV light failed to further increase ICa, suggesting that the response was saturated. Addition of 10 nM ET-1 to the cell after saturating the photoreleased diC8 effect not only failed to further increase ICa, but actually resulted in a decrease in ICa. This result suggests that the regulation of ICa by diC8 and ET-1 is complex, but is consistent with a PKC-dependent upregulation of ICa sharing a final common pathway for photoreleased diC8 and ET-1. The observed inhibition by ET-1 may represent the unmasking of an inhibitory influence of ET-1 on ICa, perhaps acting through a distinct ET-1 receptor subtype. The effect of ET-1 was reversible upon washout and actually resulted in an overshoot of ICa compared to the pre-ET-1 levels.
Figure 9. Effect of successive UV photorelease of diC8 and ET-1 on ICa.

Peak ICa is plotted in response to test pulses to +10 mV every 15 s. The vertical arrows indicate the times of each UV light exposure. Exposure of 1, 5, 10 and 20 s UV light, in the presence of caged diC8, gradually increased ICa by 38, 66, 83 and 100 %, respectively. A final 30 s UV light exposure failed to increase ICa further, and ET-1 (10 nM) was then applied where indicated. The original traces corresponding to each UV light exposure are shown above. The dashed lines indicates the zero current level. Cm= 188 pF.
To investigate the mechanism for the proposed PKC-dependent modulation of ICa by photoreleased diC8, we examined possible changes in the inactivation of L-type Ca2+ channels. For example, a shift in the voltage dependence of Ca2+ channel inactivation to more positive potentials by photoreleased diC8 could underlie the observed increase in ICa. A gapped double-pulse protocol (see Fig. 10A) was employed to examine the voltage dependence of current inactivation following a 1000 ms prepulse over a range of potentials from -50 to +40 mV. Figure 10B displays the mean data from four cells studied before and after photorelease of diC8. The peak current from the test pulse normalized to the maximal test pulse current was plotted as a function of membrane potential and then fitted to a Boltzmann distribution. The mean V0·5 and k were unchanged (-22 ± 0·77 mV and 4·1 ± 0·15 mV in control and -22·6 ± 0·57 mV and 4·4 ± 0·12 mV after photorelease of diC8). These data argue that photoreleased diC8 does not result in a significant voltage shift in the inactivation of ICa.
Figure 10. Photorelease of diC8 does not change steady-state inactivation of ICa.
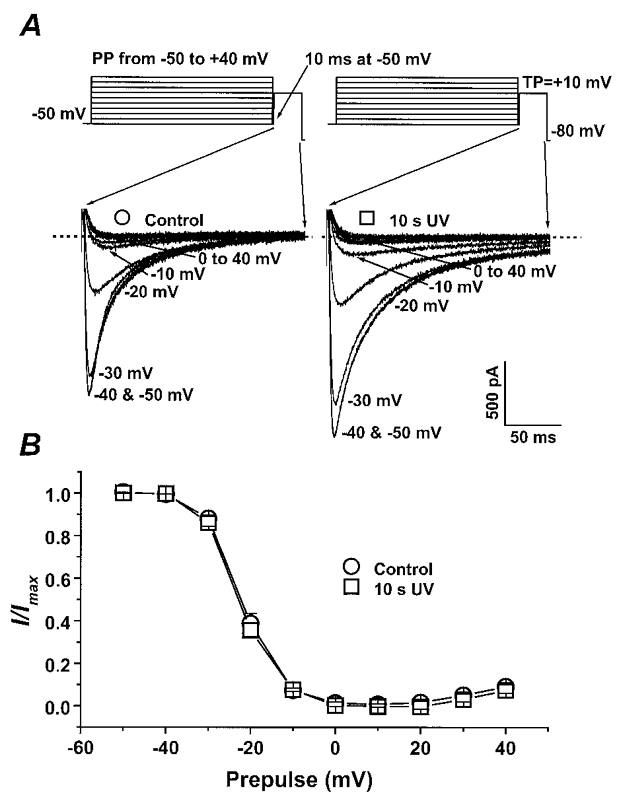
A, the gapped double-pulse voltage protocol with 1000 ms prepulses (PP) over a range of potentials followed by a 200 ms test pulse (TP) to +10 mV. Representative current traces during the test pulses before (^) and after (□) photorelease of diC8 are displayed, and the dashed lines indicate the zero current level. Cm= 142 pF. B, the mean normalized current (I/Imax) during the test pulse to +10 mV as a function of the 1000 ms prepulse potential before (^) and after photoreleased diC8 (□). This inactivation relationship was examined in 4 different cells from 3 rat hearts. The data are fitted to a Boltzmann distribution using a non-linear least-squares regression. Vertical bars indicate standard errors of the data.
DISCUSSION
ET-1 regulation of ICa
ET-1 was initially described as a potent vasoconstrictor, but further investigations showed that ET-1 also produces a positive inotropic effect when applied to many isolated cardiac muscle preparations (Ishikawa et al. 1988; Takanashi & Endoh, 1991; Pi et al. 1997). Multiple studies have also demonstrated that an increase in the intracellular Ca2+ transient contributes to this positive inotropic effect (Qiu et al. 1992; Damron et al. 1993; Ebihara et al. 1996). It was originally postulated that the basis for the increased Ca2+ transient was a stimulation of ICa (Ishikawa et al. 1988), and the results of the present study are consistent with this hypothesis, as 10 nM ET-1 caused a reproducible increase in ICa in adult rat ventricular myocytes. However, previous investigations examining the effect of ET-1 on L-type Ca2+ channels have produced conflicting results with decreases (Ono et al. 1994; Xie et al. 1996), increases (Lauer et al. 1992; Bkaily et al. 1995), and no effect (Thomas et al. 1997) on basal ICa being reported. Comparison of these different studies reveals differences in species, differences in concentrations of ET-1 tested, and differences in experimental techniques.
Recent pharmacological studies have suggested that multiple subtypes of ET receptors, ETA and ETB, exist, which may have opposing effects on ICa (Kelso et al. 1998). For example, ETB receptors have been implicated in the stimulation of ICa in rabbit ventricular myocytes based on the greater potency of ET-3 as an agonist and on the sensitivity to specific ET receptor antagonists (Kelso et al. 1998). Based on our results, we suggest that such an upregulation of ICa may be mediated by ETB receptor coupling to the Gq-PLC-DAG-PKC pathway. In contrast, the majority of studies characterizing the role of ET-1 in regulation of ICa have exhibited either no effect on basal currents or an inhibition of ICa (Ono et al. 1994; Xie et al. 1996; Thomas et al. 1997). In addition, several studies have demonstrated a clear inhibition of β-adrenergic (isoproterenol (isoprenaline))-stimulated ICa (Cheng et al. 1995; Xie et al. 1996; Thomas et al. 1997). Based on pharmacological studies (Ono et al. 1994; Thomas et al. 1997), the inhibition of basal currents and isoproterenol-stimulated currents by endothelin has been suggested to be mediated by ETA receptors. For example, BQ123, a specific ETA receptor antagonist, blocks the observed inhibition in several different preparations. Our results also reveal that ET-1 can inhibit ICa when it is maximally upregulated by photoreleased diC8 (Fig. 9). Therefore, it is possible that multiple ET receptor subtypes can be present in ventricular muscle, which may exert opposing actions on L-type Ca2+ channels, and, furthermore, the receptor subtypes present may be dependent on the species studied. The system is probably even more complex, as subtypes of ETA and ETB receptors have also been proposed.
Differences in experimental techniques may also contribute to the diversity of results observed. Perhaps the most important variable is related to the type of patch clamp technique used. When the ruptured patch clamp technique is used, dialysis of large molecules from the intracellular compartment occurs, while use of the perforated patch clamp technique, as in the present study, allows electrical access to the cell and dialysis of monovalent cations and anions with no exchange of larger molecules, which may be involved in regulating the channels. A previous study using isolated rabbit ventricular myocytes showed that 1 nM ET-1 caused a 25 % increase in ICa when studied using the nystatin-perforated patch clamp technique, but had no effect when the ruptured patch clamp technique was used (Kelso et al. 1996). One potential regulatory molecule which may be affected by cellular dialysis is GTP. Using the ruptured patch technique, Lauer and colleagues reported an increase in ICa in response to 10 nM ET-1 only if GTP was included in the pipette; otherwise ET-1 caused a decrease in ICa (Lauer et al. 1992). Others have not confirmed that GTP is the critical intracellular constituent required for regulation (Cheng et al. 1995), but use of the amphotericin-perforated patch technique in the present study should preserve intracellular GTP as well as other potential regulatory molecules.
Many other neurohormone receptors in the heart are coupled via Gq to PLC and the resulting liberation of IP3 and DAG. The regulation of ICa by these agents has also been investigated by others, with apparently conflicting results present in the literature. The best studied is the effect of α1-adrenergic receptor stimulation on ICa. An initial study using bovine trabeculae and the sucrose-gap voltage clamp demonstrated an increase in ICa in response to α1-adrenergic stimulation (Bruckner & Scholz, 1984); however, many subsequent studies using the ruptured whole-cell patch clamp technique failed to show any clear modulation of cardiac ICa in several species including the rat (Boutjdir et al. 1992; Fedida & Bouchard, 1992). More recent evidence has revealed, in rat ventricular myocytes, that if the perforated patch technique is employed, α1-adrenergic stimulation causes an increase in ICa (Liu & Kennedy, 1998). Cell-attached single channel studies have confirmed that phenylephrine can increase the open probability of single L-type Ca2+ channels and enhance the ensemble Ca2+ channel currents, and these effects are blocked by the PKC inhibitor chelerythrine (Zhang et al. 1998). In aggregate, these findings suggest that regulation of ICa in rat ventricular myocytes by the Gq-DAG-PKC pathway is altered by the intracellular dialysis that occurs using the ruptured patch clamp technique, whereas the upregulation of ICa in response to PKC activation is clearly evident when the intracellular environment is preserved using the cell-attached or perforated patch clamp technique.
Phorbol ester modulation of ICa
Phorbol esters can directly activate PKC, bypassing surface membrane receptors, and therefore these agents can be useful tools for determining the role of PKC in regulatory pathways. In the present experiments, we found that both the active form of phorbol ester, PMA, and the inactive form, 4αPDD, produced comparable, small increases in ICa in rat ventricular myocytes. The similar effects of PMA and 4αPDD argue against a PKC-specific effect of these compounds on ICa. In a previous study using guinea-pig ventricular myocytes a small inhibition by both PMA and 4αPDD was found (Asai et al. 1996), which also suggested PKC-independent effects on ICa. However, our results are in direct contrast to the results of Zhang et al. (1997) which demonstrated that 100 nM PMA produced a 40 % inhibition of basal ICa in adult rat ventricular myocytes while 4 αPDD was without effect. In addition, the specificity of the inhibition of PKC in their study was further confirmed by the use of peptide inhibitors of PKC. The most obvious difference between the studies is the use of the ruptured patch clamp technique by Zhang et al., but the use of the perforated patch clamp in the present study. In addition, the complexity of the response of ICa to phorbol esters has been demonstrated in studies of neonatal rat ventricular myocytes and adult canine ventricular myocytes which show a biphasic effect on ICa, with an initial stimulation followed by an inhibition (Lacerda et al. 1988; Tseng & Boyden, 1991). Studies in neurons and endocrine cells have also revealed both stimulatory and inhibitory effects of phorbol esters on L-type Ca2+ channels, depending on the preparation (Di Virgilio et al. 1986; Yang & Tsien, 1993). It is possible that PMA is capable of activating different isoforms of PKC which may have opposing effects on L-type Ca2+ channels. The effect of phorbol esters on the chronotropic state of neonatal rat ventricular myocytes has previously been attributed to opposing effects of different PKC isoforms (Johnson & Mochly-Rosen, 1995). The net result of stimulation by phorbol esters probably depends on the PKC isoforms which are present in a given preparation and their ability to regulate ICa, which may be in part altered by intracellular dialysis.
diC8 modulation of ICa
Several synthetic DAG analogs can directly activate PKC, and these agents have been used as tools to examine the role of PKC in the regulation of myocardial contraction and excitability. Previous studies by Walker and colleagues have shown that bath application of diC8 to isolated adult rat ventricular myocytes engendered a marked negative inotropic effect, as measured by cell shortening which was not blocked by chelerythrine (Pi et al. 1997). In contrast, the intracellular release of caged diC8 led to a large increase in cell shortening which was mainly due to an increase in the intracellular Ca2+ transient, and this response was abolished by chelerythrine suggesting a PKC-dependent positive inotropic effect (Pi et al. 1997; Pi & Walker, 1998). In the present study, a similar modulation of ICa by diC8 has been revealed, as bath application of 7.5 μM diC8 resulted in a large decrease in ICa while photorelease of intracellular diC8 greatly upregulated ICa. Furthermore, chelerythrine did not affect the inhibition produced by bath-applied diC8, but did block the stimulation of ICa produced by photorelease of diC8. These parallel results suggest that the inotropic effects mediated by diC8 are in large part due to modulation of L-type Ca2+ channels. A PKC-independent inhibition of ICa by bath-applied diC8 is consistent with the results of others obtained using the same rat ventricular myocyte preparation (Schreur & Liu, 1996), embryonic chick cardiomyocytes (Conforti et al. 1995), rat myometrial cells (Kusaka & Sperelakis, 1995), and chick dorsal root ganglion neurons (Hockberger et al. 1989). The potential mechanism for this modulation of ICa remains unknown. However, it has become increasingly evident that multiple proteins besides PKC contain a DAG binding motif (Newton, 1997). Does DAG interact directly with a Ca2+ channel subunit or associated protein? Interestingly, previous studies have provided evidence that another synthetic DAG analogue, OAG, does not inhibit ICa (Conforti et al. 1995; Schreur & Liu, 1996). Future studies will be needed to clarify this mechanism of modulation and its physiological relevance.
Photorelease of diC8, in direct contrast to the effect of bath-applied diC8, stimulates an increase in ICa which is inhibited by the PKC inhibitor chelerythrine. This effect is specific for photolysis of caged diC8, as control experiments exposing cells loaded with vehicle (DMSO) to prolonged UV light failed to show any effect on ICa. Previous work by Walker and colleagues has demonstrated that the effect of photoreleased diC8 on cell shortening is stereospecific, as only the enantiomer S-diC8 stimulated cell shortening, arguing that the observed effects are due to the liberated diC8 and not other by-products of photolysis (Pi et al. 1997). The present study used racemic caged diC8, as did the majority of the previous experiments (Huang et al. 1996; Pi et al. 1997; Pi & Walker, 1998), and we assume the effect that we observed is primarily due to release of S-diC8. The effect of photoreleased diC8 was also shown to be concentration dependent, since increased photolysis time (3 s vs. 10 s) produced a greater stimulation of ICa (∼20 %vs.∼60 %). Furthermore, the nearly identical kinetics for the increase in ICa by photorelease of diC8 for either 3 s or 10 s suggest that the rate-limiting step in this regulation is downstream from diC8 binding to its effectors. We propose that a major effector which binds photoreleased diC8 is PKC and this activated kinase is then responsible for upregulation of ICa. A role for PKC in this process is supported by the ability of chelerythrine to concentration dependently inhibit the upregulation of ICa.
The finding that the same molecule, diC8, can produce opposite effects on ICa when applied extracellularly or photoreleased intracellularly is a central paradox of these studies. The specific membrane pools of DAG liberated may be critically important in determining the action of this second messenger. Does intracellularly liberated DAG have preferential access to the critical PKC binding sites required for enhancement of ICa? Does diC8 applied extracellularly directly interact with the Ca2+ channel or associated proteins, by virtue of entering via the surface membrane of the cell, to inhibit the channel prior to any potential effects on PKC? Can diC8 produce both types of modulation on the channel which are additive, or are the effects exclusive in nature? In fact, we observed that 8 μM chelerythrine not only blocked the stimulation of ICa by 10 s UV photorelease of diC8, but also unmasked a small but significant inhibition of ICa. This suggests that if the PKC-mediated enhancement of ICa is blocked, photoreleased diC8 is capable of inhibiting the channel just as bath-applied diC8 does. Our working hypothesis is that the two methods of applying diC8 result in binding of diC8 to different sites involved in two distinct mechanisms of modulation of ICa, causing opposite effects on ICa. How these two forms of modulation by diC8 interact at the level of the Ca2+ channel will require future evaluation.
ET-1 and photoreleased diC8 stimulate ICa through a common pathway
Both ET-1 and photorelease of diC8 caused an increase in ICa in the present study, and we conclude that both of these agents lead to modulation of ICa by a common final pathway requiring activation of PKC. There are many similarities between the modulation of ICa by ET-1 and photoreleased diC8 which lead to this conclusion. First, the increases in ICa in response to both ET-1 and photorelease of diC8 are blocked by the specific PKC inhibitor chelerythrine. Second, the currents stimulated by ET-1 or photoreleased diC8 show similar changes in the kinetics of current decay, with both treatments resulting in a statistically significant slowing of current decay. Third, if the effect of photoreleased diC8 is maximal, ET-1 causes no further stimulation of ICa, suggesting a common final pathway which mediates upregulation of ICa (Fig. 9). Fourth, the slower time course for upregulation of ICa mediated by ET-1 relative to that mediated by photoreleased diC8 is consistent with ET-1 action requiring additional steps (i.e. receptor binding, Gq stimulation, PLC activation) prior to PKC stimulation. The major difference between the effects of ET-1 and photoreleased diC8 that we observed was the relative magnitude of the effects. Photorelease of diC8 with 10 s UV light exposure produced about a two-fold greater increase in ICa than did 10 nM ET-1. It is possible that ET-1 at an optimal concentration may exert comparable effects, but 10 nM was tested as our previous study had demonstrated that this concentration produced the maximal positive inotropic effect in this preparation (Pi et al. 1997). The present observations imply that 10 nM ET-1 does not fully activate the DAG-PKC pathway or that competing inhibitory influences are also activated on by this agonist.
Molecular mechanisms of PKC regulation of L-type Ca2+ channels
The present study and previous studies suggest that PKC-activating pathways can upregulate the L-type Ca2+ channel in cardiac muscle; however, the substrate(s) for PKC and the underlying molecular mechanisms of this regulation remain unknown. The subunits of the L-type Ca2+ channel represent potential targets for PKC phosphorylation, and in vitro biochemical studies have demonstrated that both the α1C and β2a subunits of the L-type Ca2+ channel can be substrates for PKC (Puri et al. 1997). Heterologous expression studies of the cardiac α1C subunit in Xenopus oocytes have revealed that the amino terminus of α1C may be critical for upregulation of α1C by PKC (Bouron et al. 1995; Shistik et al. 1998). Bouron et al. (1995) proposed that the cloned human α1C subunit is not upregulated by PKC because it lacks the initial 41 amino acids present in the cloned rabbit α1C subunit which is significantly stimulated by PKC in heterologous systems (Bouron et al. 1995). However, no studies have produced direct evidence for PKC-dependent phosphorylation of the α1C subunit or other subunits in intact cells. It remains possible that the substrate for PKC modulation may be associated regulatory proteins rather than channel subunits.
A second important issue regarding the molecular mechanisms of the upregulation of cardiac L-type Ca2+ channels is what isoform(s) of PKC are responsible for the regulation. Our experiments using chelerythrine supported a role for PKC, but did not provide any evidence for the isoform of PKC responsible. Adult rat ventricular myocytes are known to express several PKC isoforms, which probably have distinct cellular targets (Puceat & Vassort, 1996). ET-1 has been suggested to cause the translocation of PKCε (Jiang et al. 1996), and therefore it is a potential candidate for regulation of ICa. Another possibility is that different PKC isoforms may exert opposing effects on L-type Ca2+ channels. This may explain some of the conflicting data in the literature. For example, Zhang et al. (1997) reported that inhibitory peptides for conventional PKC isoforms blunted the downregulation of ICa by PKC. Future studies using isoform-specific antagonists and agonists will be necessary to establish which isoform(s) of PKC are responsible for the upregulation of ICa.
In summary, the present work suggests that activation of PKC by either receptor stimulation using ET-1 or intracellular photorelease of diC8 significantly upregulates ICa studied using the perforated patch technique in rat ventricular myocytes. In contrast, a strong inhibition of ICa by bath-applied diC8 occurs independently of PKC activation and represents a distinct mechanism by which DAG can modulate L-type Ca2+ channels. Therefore, DAG and PKC can exert strong regulatory influences on ICa, but the nature of the modulation is critically dependent on the pools of DAG liberated and the techniques used to study ICa.
Acknowledgments
This study was funded by NIH P01HL47053 for Dr J. W. Walker. and Dr T. J. Kamp. Dr Y. Q. Pi was supported by a Postdoctoral Fellowship from the American Heart Association, Wisconsin Affiliate. We thank Dr Johannes Hell for helpful discussions.
References
- Asai T, Shuba LM, Pelzer DJ, Mcdonald TF. PKC-independent inhibition of cardiac L-type Ca2+ channel current by phorbol esters. American Journal of Physiology. 1996;270:H620–627. doi: 10.1152/ajpheart.1996.270.2.H620. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. The 1996 Massry Prize. Inositol trisphosphate and calcium: two interacting second messengers. American Journal of Nephrology. 1997;17:1–11. doi: 10.1159/000169064. [DOI] [PubMed] [Google Scholar]
- Bkaily G, Wang S, Bui M, Menard D. ET-1 stimulates Ca2+ currents in cardiac cells. Journal of Cardiovascular Pharmacology. 1995;26(suppl. 3):S293–S296. [PubMed] [Google Scholar]
- Bouron A, Soldatov NM, Reuter H. The beta 1-subunit is essential for modulation by protein kinase C of a human and a non-human L-type Ca2+ channel. FEBS Letters. 1995;377:159–162. doi: 10.1016/0014-5793(95)01327-x. [DOI] [PubMed] [Google Scholar]
- Boutjdir M, Restivo M, El-Sherif N. Alpha 1- and beta-adrenergic interactions on L-type calcium current in cardiac myocytes. Pflügers Archiv. 1992;421:397–399. doi: 10.1007/BF00374231. [DOI] [PubMed] [Google Scholar]
- Bruckner R, Scholz H. Effects of alpha-adrenoreceptor stimulation with phenylephrine in the presence of propranolol on force of contraction, slow inward current and cyclic AMP content in the bovine heart. British Journal of Pharmacology. 1984;82:223–232. doi: 10.1111/j.1476-5381.1984.tb16462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng TH, Chang CY, Wei J, Lin CI. Effects of endothelin 1 on calcium and sodium currents in isolated human cardiac myocytes. Canadian The Journal of Physiology and Pharmacology. 1995;73:1774–1783. doi: 10.1139/y95-242. [DOI] [PubMed] [Google Scholar]
- Conforti L, Sumii K, Sperelakis N. Dioctanoyl-glycerol inhibits L-type calcium current in embryonic chick cardiomyocytes independent of protein kinase C activation. Journal of Molecular and Cellular Cardiology. 1995;27:1219–1224. doi: 10.1016/0022-2828(95)90058-6. [DOI] [PubMed] [Google Scholar]
- Damron DS, Van Wagoner DR, Moravec CS, Bond M. Arachidonic acid and endothelin potentiate Ca2+ transients in rat cardiac myocytes via inhibition of distinct K+ channels. Journal of Biological Chemistry. 1993;268:27335–27344. [PubMed] [Google Scholar]
- Di Virgilio F, Pozzan T, Wollheim CB, Vicentini LM, Meldolesi J. Tumor promoter phorbol myristate acetate inhibits Ca2+ influx through voltage-gated Ca2+ channels in two secretory cell lines, PC12 and RINm5F. Journal of Biological Chemistry. 1986;261:32–35. [PubMed] [Google Scholar]
- Dorn GW, Brown JH. Gq signaling in cardiac adaptation and maladaptation. Trends in Cardiovascular Medicine. 1999;9:26–34. doi: 10.1016/s1050-1738(99)00004-3. [DOI] [PubMed] [Google Scholar]
- Dosemeci A, Dhallan RS, Cohen NM, Lederer WJ, Rogers TB. Phorbol ester increases calcium current and simulates the effects of angiotensin II on cultured neonatal rat heart myocytes. Circulation Research. 1988;62:347–357. doi: 10.1161/01.res.62.2.347. [DOI] [PubMed] [Google Scholar]
- Dukes ID, Morad M. The transient K+ current in rat ventricular myocytes: evaluation of its Ca2+ and Na+ dependence. The Journal of Physiology. 1991;435:395–420. doi: 10.1113/jphysiol.1991.sp018516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara Y, Haist JV, Karmazyn M. Modulation of endothelin-1 effects on rat hearts and cardiomyocytes by nitric oxide and 8-bromo cyclic GMP. Journal of Molecular and Cellular Cardiology. 1996;28:265–277. doi: 10.1006/jmcc.1996.0025. [DOI] [PubMed] [Google Scholar]
- Endoh M, Fujita S, Yang HT, Talukder MA, Maruya J, Norota I. Endothelin: receptor subtypes, signal transduction, regulation of Ca2+ transients and contractility in rabbit ventricular myocardium. Life Sciences. 1998;62:1485–1498. doi: 10.1016/s0024-3205(98)00094-0. [DOI] [PubMed] [Google Scholar]
- Fedida D, Bouchard RA. Mechanisms for the positive inotropic effect of α1-adrenoreceptor stimulation in rat cardiac myocytes. Circulation Research. 1992;1992:673–688. doi: 10.1161/01.res.71.3.673. [DOI] [PubMed] [Google Scholar]
- Habuchi Y, Tanaka H, Furukawa T, Tsujimura Y, Takahashi H, Yoshimura M. Endothelin enhances delayed potassium current via phospholipase C in guinea pig ventricular myocytes. American Journal of Physiology. 1992;262:H345–354. doi: 10.1152/ajpheart.1992.262.2.H345. [DOI] [PubMed] [Google Scholar]
- He JQ, Pi YQ, Walker JW, Kamp TJ. Photoreleased diacylglycerol increases L-type Ca2+ currents in rat ventricular myocytes. Biophysical Journal. 1999;76:A339. doi: 10.1111/j.1469-7793.2000.00807.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochemical and Biophysical Research Communications. 1990;172:993–999. doi: 10.1016/0006-291x(90)91544-3. [DOI] [PubMed] [Google Scholar]
- Hockberger P, Toselli M, Swandulla D, Lux HD. A diacylglycerol analogue reduces neuronal calcium currents independently of protein kinase C activation. Nature. 1989;338:340–342. doi: 10.1038/338340a0. [DOI] [PubMed] [Google Scholar]
- Huang XP, Sreekumar R, Patel JR, Walker JW. Response of cardiac myocytes to a ramp increase of diacylglycerol generated by photolysis of a novel caged diacylglycerol. Biophysical Journal. 1996;70:2448–2457. doi: 10.1016/S0006-3495(96)79816-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Yanagisawa M, Kimura S, Goto K, Masaki T. Positive inotropic action of novel vasoconstrictor peptide endothelin on guinea pig atria. American Journal of Physiology. 1988;255:H970–973. doi: 10.1152/ajpheart.1988.255.4.H970. [DOI] [PubMed] [Google Scholar]
- Jiang T, Pak E, Zhang HL, Kline RP, Steinberg SF. Endothelin-dependent actions in cultured AT-1 cardiac myocytes. The role of the epsilon isoform of protein kinase C. Circulation Research. 1996;78:724–736. doi: 10.1161/01.res.78.4.724. [DOI] [PubMed] [Google Scholar]
- Johnson JA, Mochly-Rosen D. Inhibition of the spontaneous rate of contraction of neonatal cardiac myocytes by protein kinase C isozymes. A putative role for the epsilon isozyme. Circulation Research. 1995;76:654–663. doi: 10.1161/01.res.76.4.654. [DOI] [PubMed] [Google Scholar]
- Kelso E, Spiers P, McDermott B, Scholfield N, Silke B. Dual effects of endothelin-1 on the L-type Ca2+ current in ventricular cardiomyocytes. European Journal of Pharmacology. 1996;308:351–355. doi: 10.1016/0014-2999(96)00366-4. [DOI] [PubMed] [Google Scholar]
- Kelso EJ, Spiers JP, McDermott BJ, Scholfield CN, Silke B. Receptor-mediated effects of endothelin on the L-type Ca2+ current in ventricular cardiomyocytes. Journal of Pharmacology and Experimental Therapeutics. 1998;286:662–669. [PubMed] [Google Scholar]
- Kurata S, Ishikawa K, Iijima T. Enhancement by arginine vasopressin of the L-type Ca2+ current in guinea pig ventricular myocytes. Pharmacology. 1999;59:21–33. doi: 10.1159/000028302. [DOI] [PubMed] [Google Scholar]
- Kusaka M, Sperelakis N. Direct block of calcium channels by dioctanoylglycerol in pregnant rat myometrial cells. Molecular Pharmacology. 1995;47:842–847. [PubMed] [Google Scholar]
- Lacerda AE, Rampe D, Brown AM. Effects of protein kinase C activators on cardiac Ca2+ channels. Nature. 1988;335:249–251. doi: 10.1038/335249a0. [DOI] [PubMed] [Google Scholar]
- Lauer MR, Gunn MD, Clusin WT. Endothelin activates voltage-dependent Ca2+ current by a G protein-dependent mechanism in rabbit cardiac myocytes. The Journal of Physiology. 1992;448:729–747. doi: 10.1113/jphysiol.1992.sp019067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Kennedy AR. Alpha1-adrenergic activation of L-type Ca current in rat ventricular myocytes: perforated patch-clamp recordings. American Journal of Physiology. 1998;274:H2203–2207. doi: 10.1152/ajpheart.1998.274.6.H2203. [DOI] [PubMed] [Google Scholar]
- Newton AC. Regulation of protein kinase C. Current Opinion In Cell Biology. 1997;9:161–167. doi: 10.1016/s0955-0674(97)80058-0. [DOI] [PubMed] [Google Scholar]
- Ono K, Tsujimoto G, Sakamoto A, Eto K, Masaki T, Ozaki Y, Satake M. Endothelin-A receptor mediates cardiac inhibition by regulating calcium and potassium currents. Nature. 1994;370:301–304. doi: 10.1038/370301a0. [DOI] [PubMed] [Google Scholar]
- Pi YQ, Sreekumar R, Huang XP, Walker JW. Positive inotropy mediated by diacylglycerol in rat ventricular myocytes. Circulation Research. 1997;81:92–100. doi: 10.1161/01.res.81.1.92. [DOI] [PubMed] [Google Scholar]
- Pi YQ, Walker JW. Role of intracellular Ca2+ and pH in positive inotropic response of cardiomyocytes to diacylglycerol. American Journal of Physiology. 1998;44:H1473–1481. doi: 10.1152/ajpheart.1998.275.4.H1473. [DOI] [PubMed] [Google Scholar]
- Puceat M, Vassort G. Signalling by protein kinase C isoforms in the heart. Molecular and Cellular Biochemistry. 1996;157:65–72. doi: 10.1007/BF00227882. [DOI] [PubMed] [Google Scholar]
- Puri TS, Gerhardstein BL, Zhao XL, Ladner MB, Hosey MM. Differential effects of subunit interactions on protein kinase A- and C-mediated phosphorylation of L-type calcium channels. Biochemistry. 1997;36:9605–9615. doi: 10.1021/bi970500d. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Wang J, Perreault CL, Meuse AJ, Grossman W, Morgan JP. Effects of endothelin on intracellular Ca2+ and contractility in single ventricular myocytes from the ferret and human. European Journal of Pharmacology. 1992;214:293–296. doi: 10.1016/0014-2999(92)90134-p. [DOI] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. Journal of Neuroscience Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Schreur KD, Liu S. 1,2-Dioctanoyl-sn-glycerol depresses cardiac L-type Ca2+ current: independent of protein kinase C activation. American Journal of Physiology. 1996;270:C655–662. doi: 10.1152/ajpcell.1996.270.2.C655. [DOI] [PubMed] [Google Scholar]
- Shistik E, Ivanina T, Blumenstein Y, Dascal N. Crucial role of N terminus in function of cardiac L-type Ca2+ channel and its modulation by protein kinase C. Journal of Biological Chemistry. 1998;273:17901–17909. doi: 10.1074/jbc.273.28.17901. [DOI] [PubMed] [Google Scholar]
- Sreekumar R, Pi YQ, Huang XP, Walker JW. Stereospecific protein kinase C activation by photolabile diglycerides. Bioorganic & Medicinal Chemistry Letters. 1997;7:341–346. [Google Scholar]
- Stiles GL. Multifunctional G proteins. Searching for functions in the heart. Circulation. 1996;94:602–603. doi: 10.1161/01.cir.94.4.602. [DOI] [PubMed] [Google Scholar]
- Takanashi M, Endoh M. Characterization of positive inotropic effect of endothelin on mammalian ventricular myocardium. American Journal of Physiology. 1991;261:H611–619. doi: 10.1152/ajpheart.1991.261.3.H611. [DOI] [PubMed] [Google Scholar]
- Thomas GP, Sims SM, Karmazyn M. Differential effects of endothelin-1 on basal and isoprenaline-enhanced Ca2+ current in guinea-pig ventricular myocytes. The Journal of Physiology. 1997;503:55–65. doi: 10.1111/j.1469-7793.1997.055bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng GN, Boyden PA. Different effects of intracellular Ca and protein kinase C on cardiac T and L Ca currents. American Journal of Physiology. 1991;261:H364–379. doi: 10.1152/ajpheart.1991.261.2.H364. [DOI] [PubMed] [Google Scholar]
- Van Heugten HA, Eskildsen-Helmond YE, De Jonge HW, Bezstarosti K, Lamers JM. Phosphoinositide-generated messengers in cardiac signal transduction. Molecular and Cellular Biochemistry. 1996;157:5–14. doi: 10.1007/BF00227875. [DOI] [PubMed] [Google Scholar]
- Walsh KB, Kass RS. Regulation of a heart potassium channel by protein kinase A and C. Science. 1988;242:67–69. doi: 10.1126/science.2845575. [DOI] [PubMed] [Google Scholar]
- Xie LH, Horie M, James AF, Watanuki M, Sasayama S. Endothelin-1 inhibits L-type Ca currents enhanced by isoproterenol in guinea-pig ventricular myocytes. Pflügers Archiv. 1996;431:533–539. doi: 10.1007/BF02191900. [DOI] [PubMed] [Google Scholar]
- Yang J, Tsien RW. Enhancement of N- and L-type calcium channel currents by protein kinase C in frog sympathetic neurons. Neuron. 1993;10:127–136. doi: 10.1016/0896-6273(93)90305-b. [DOI] [PubMed] [Google Scholar]
- Zhang S, Hirano Y, Hiraoka M. Effects of alpha1-adrenergic stimulation on L-type Ca2+ current in rat ventricular myocytes. Journal of Molecular & Cellular Cardiology. 1998;30:1955–1965. doi: 10.1006/jmcc.1998.0758. [DOI] [PubMed] [Google Scholar]
- Zhang ZH, Johnson JA, Chen L, El-Sherif N, Mochly-Rosen D, Boutjdir M. C2 region-derived peptides of beta-protein kinase C regulate cardiac Ca2+ channels. Circulation Research. 1997;80:720–729. doi: 10.1161/01.res.80.5.720. [DOI] [PubMed] [Google Scholar]