

Abstract
A study was made on the mechanisms by which enkephalins inhibit synaptic transmission at calyx-type presynaptic terminals in the ciliary ganglion of chick embryos at stages 39–40.
Excitatory postsynaptic currents (EPSCs) were recorded by nystatin-perforated patch clamp at low [Ca2+]o and high [Mg2+]o. [Leu5]enkephalin (L-ENK, 1–10 μm) reduced the quantal content (m) without changing the quantal size (q). This effect was antagonized by naloxone (1 μm). Similar results were observed under conventional whole-cell clamp of the postsynaptic neuron.
A specific agonist of the μ-opioid receptor, [D-Ala2, M-Me-Phe4,Gly5]enkephalin-ol (DAMGO) reduced m without changing q. A specific agonist of the δ-opioid receptor, [d-Pen2, d-Pen5]enkephalin (DPDPE) also reduced m without changing q.
Both L-ENK and [Met5]enkephalin (M-ENK) reduced the stimulus-dependent increment of the intraterminal Ca2+ concentration (Δ[Ca2+]t) without affecting the decay time constant of the intraterminal Ca2+ concentration and basal Ca2+ level. This effect was antagonized by naloxone. DAMGO reduced Δ[Ca2+]t more effectively than DPDPE.
When extracellular Ca2+ was replaced by Ba2+, the stimulus-dependent increment of the intraterminal Ba2+ concentration (Δ[Ba2+]t) was also reduced by L-ENK or DAMGO.
L-ENK reduced Δ[Ca2+]t even in the presence of 4-aminopyridine (4-AP), which blocks the transient K+ conductance during the falling phase of the presynaptic action potential. When N-type Ca2+ channels were blocked by ω-conotoxin GVIA (ω-CgTxGVIA), the Δ[Ca2+]t was no longer sensitive to L-ENK and DAMGO.
It is suggested that enkephalins reduce the transmitter release through presynaptic opioid receptors. The μ-opioid receptor may suppress presynaptic Ca2+ influx by selectively inhibiting N-type Ca2+ channels.
Opioid peptides and their receptors are widely distributed throughout the central and peripheral nervous systems (Dun, 1983; Hökfelt et al. 1984; Nicoll et al. 1990). In autonomic ganglia, morphine and opioid peptides suppress the cholinergic transmission by either pre- or postsynaptic mechanisms (Konishi et al. 1979; Cherbini & North, 1985; Margiotta & Berg, 1986; Araujo & Collier, 1987). In the avian ciliary ganglion, opioid peptides have been shown to be present in the cholinergic presynaptic terminal as co-transmitters (Erichsen et al. 1982a). The ganglion consists of two distinct populations of neurons: small choroidal cells which have bouton-type synaptic terminals and large ciliary cells which receive calyx-type presynaptic terminals (Marwitt et al. 1971). [Leu5]enkephalin (L-ENK)-like immunoreactivity has been observed in both populations of presynaptic terminals and in the midbrain preganglionic neurons of the Edinger-Westphal nucleus which send their axons to the ciliary ganglion (Erichsen et al. 1982a, b; Reiner, 1987). Both [Met5]enkephalin (M-ENK) and L-ENK have also been extracted from the ciliary ganglion (White et al. 1985). During embryonic stages 35–40 (Hamburger & Hamilton, 1951) when normal programmed neuronal death occurs in the chick ciliary ganglion (Landmesser & Pilar, 1974), the ganglionic content of M-ENK reaches its maximum (White et al. 1985). The presence of L-ENK immunoreactivity is also highest during the same developmental stages in bouton- and calyx-type presynaptic terminals in the chick ciliary ganglion as well as in the neurons of the Edinger-Westphal nucleus (Meriney et al. 1991).
Neuronal death in the ciliary ganglion of the chick embryo is retarded by exogenous administration of morphine (Meriney et al. 1985) and is enhanced by naltrexone, an opioid receptor antagonist (Meriney et al. 1991). Enkephalins are thus assumed to be endogenous modulators of synaptic transmission in this system (Meriney et al. 1991; Chiappinelli et al. 1993). In this study, we investigated the possibility that enkephalins inhibit the release of transmitter through presynaptic opioid receptors. We show that μ-opioid receptors are present in the calyx-type presynaptic terminal in stage 39–40 chick embryos and that enkephalins reduce the quantal transmitter release. In addition, we show that the μ-opioid receptor preferentially suppresses Ca2+ influx through N-type Ca2+ channels. A preliminary report of these results has appeared elsewhere (Yawo et al. 1994).
METHODS
Preparation
Day 14 chick embryos (stages 39–40; Hamburger & Hamilton, 1951) were decapitated and the ciliary ganglion was removed with the oculomotor nerve. The details of the procedures have been described previously (Yawo & Chuhma, 1994; Yawo, 1999a). Briefly, the collagenous envelope of the ciliary ganglion was enzymatically removed by focal application of a mixture of 2000 U ml−1 collagenase (Type II, Sigma-Aldrich) and 100 U ml−1 thermolysin (Sigma-Aldrich) through a pipette with a 40–50 μm tip diameter for 30 min at room temperature (Yawo, 1999a). The presynaptic oculomotor nerve was attached to a stimulating glass pipette by suction. The postsynaptic ciliary cell was identified by its location and size (Gray et al. 1990). The ganglion was superfused with oxygenated standard saline containing (mM): NaCl, 135; KOH, 5; CaCl2, 5; MgCl2, 1; Hepes, 10; mannitol,; and glucose, 11 (pH 7.4 adjusted with HCl). All experiments were carried out at room temperature (25°C).
Recordings and analyses of the excitatory postsynaptic current (EPSC)
A conventional whole-cell patch-clamp recording (Hamill et al. 1981) was made from a postsynaptic ciliary neuron (Yawo & Chuhma, 1994) using an EPC-7 patch-clamp amplifier (List Electronic, Darmstadt-Eberstadt, Germany). Patch pipettes were made from thin-walled borosilicate glass capillaries (Hilgenberg, Malsfeld, Germany) and coated with silicon resin (Silpot 184W/C, Dow Corning, Midland, MI, USA). The pipettes were fire polished, reducing the tip diameter to 1–2 μm. The pipettes had a resistance of 4–6 MΩ when filled with internal solution containing (mM): CsCl, 130; CsOH, 10; Na2EGTA, 10; Hepes, 20; and MgATP, 5 (pH 7.4 adjusted with HCl). The series resistance was usually less than 10 MΩ throughout the experiment.
To prevent the effects of intracellular dialysis under whole-cell recording conditions, we measured EPSCs by the perforated patch method using a nystatin-fluorescein mixture (Yawo & Chuhma, 1993b). Briefly, a stock solution was made by dissolving 5 mg nystatin (Sigma-Aldrich) and 20 mg fluorescein sodium (Uranine, Nacalai Tesque) in 1 ml methanol. Alternatively, instead of using fluorescein, 0.1 M N-methyl-D-glucamine (Sigma-Aldrich) was first dissolved in methanol, the pH was adjusted by methanesulphonic acid in the presence of 0.01 M phenol red (Sigma-Aldrich), and then the nystatin (5 mg ml−1) was added. Immediately before use, 50 μl of the stock solution was placed in a polyethylene test tube and dried completely with a stream of N2 gas. The tube was then filled with 1 ml of pipette solution containing (mM): KCl, 20; K2SO4, 60; sucrose, 60; MgSO4, 1; and Hepes, 10 (pH 7.4, adjusted with NaOH), and was briefly vortexed. Thus, the pipette solution contained 250 μg ml−1 of nystatin and 2.7 mM fluorescein or 5 mM N-methyl-D-glucamine. It can be expected that bipolar molecules such as fluorescein and N-methyl-D-glucamine will facilitate the dispersion of nystatin in an aqueous solution. In fact, the pipette solution was filtered through a 0.22 μm syringe filter (SLGVL040S, Millipore Japan Co., Osaka, Japan) without additional pressure on the syringe. Nystatin was re-precipitated in 1 h in the fluorescein-containing solution but not in the N-methyl-D-glucamine-containing solution. The tip of the patch electrode was filled with filtered solution first, then backfilled with the same solution. Positive pressure was applied to the pipette as in a conventional patch-clamp technique before and while approaching the cell membrane. A tight seal was immediately formed upon switching to a slight negative pressure. The access resistance decreased rapidly to less than 40 MΩ in 5–10 min.
In both the whole-cell and perforated patch recordings, the capacitative transient was minimized by compensating the input capacitance, then the series conductance was compensated by 50–70 %. The whole-cell currents were low pass-filtered at 3 kHz (-3 dB, eight-pole Bessel filter, P-84P; NF Electronic Instruments, Yokohama, Japan), digitized at 10–20 kHz (ADX-98E; Canopus, Kobe, Japan), and stored in a computer (PC9801Vm21; NEC, Tokyo, Japan).
The quantal content (m) was estimated from the coefficient of variation (c.v.) of EPSCs measured in a low [Ca2+] (0.8–1 mM) and high [Mg2+] (5 mM) solution (Kuno & Weakly, 1972) since the EPSC fluctuation approximately followed Poisson statistics under these conditions (Martin & Pilar, 1964; Yawo & Chuhma, 1994). Because of the infrequent occurrence of miniature EPSCs, the quantal size (q) was estimated as the mean EPSC divided by m.
Measurement of the intraterminal Ca2+ and Ba2+ concentrations
The method of measuring the intraterminal Ca2+ concentration ([Ca2+]t) was almost the same as that described previously (Yawo & Chuhma, 1994). The oculomotor nerve was cut at its point of exit from the orbital bone in Ca2+-free saline containing 1 mM EGTA. Crystals of fura-2-conjugated dextran (fura-dextran, MW 10 000, Molecular Probes Inc.) were applied to the cut end of the distal stump as described previously (Yawo, 1999a). After 30 min of incubation at 10°C, the ganglion was superfused with the oxygenated standard saline and further incubated at 36°C for 1.5 h. Fura-dextran was anterogradely transported (Glover et al. 1986) and was confined to the presynaptic axons and their terminals. A whole ganglion was mounted in a chamber, the oculomotor nerve was attached by suction to the stimulating glass pipette, and the collagenous envelope was enzymatically removed by focally applying a collagenase-thermolysin mixture (see above; Yawo, 1999a). A conventional epifluorescence system equipped with a water-immersion objective (× 40, NA 0.7, Olympus, Tokyo, Japan) and xenon lamp (150 W) was focused on calyx-type presynaptic terminals at the surface of the ciliary ganglion. Fluorescence was excited alternately at wavelengths of 340 and 380 nm through the minimum iris orifice (diameter, 100 μm). Because there is a window (diameter, 50 μm) in front of the photomultiplier tube (OSP-3, Olympus), the fluorescence from one to three terminals was measured simultaneously. The intracellular Ca2+ concentration was calculated from the ratio of fluorescence intensities at wavelengths of 340 and 380 nm (Grynkiewicz et al. 1985) using the dissociation constant (KD) determined by the manufacturer (350 nM) and the system-dependent parameters determined by the conventional in situ calibration method. The signal was integrated for 50 ms and sampled at 20 Hz by a computer (PC-9801RS, NEC) with software for measuring intracellular Ca2+ concentration (MiCa, provided by Drs K. Furuya & K. Enomoto, National Institute of Physiological Science, Japan). Twelve records were averaged, using the computer-generated stimulating pulse as a trigger for summation.
The intraterminal Ba2+ concentration ([Ba2+]t) was also measured as the 340 nm/380 nm ratio of fura-dextran fluorescence when extracellular Ca2+ was replaced with Ba2+ in the presence of 1 mM EGTA. The method of calibration was similar to that for Sr2+ (Yawo, 1999b), although the intracellular [Ca2+] was ignored. We did not directly measure the Kd of fura-dextran for Ba2+; instead, the Kd of fura-2 for Ba2+, 1360 nM (Kwan & Putney, 1990), was adopted. To calibrate the Ba2+ concentration, the minimum fluorescence ratio and the maximum fluorescence at 380 nm were measured in a divalent cation-free solution containing 5 mM BAPTA and 0.1 mM ionomycin. Thereafter the solution was changed to that containing 10 mM BaCl2, and the maximum fluorescence ratio and the minimum fluorescence at 380 nm were measured.
Reagents
Pharmacological agents were usually applied in a superfusing solution over the ganglion, and were bath applied through a perfusing line. The solution in the chamber (∼1 ml) was completely replaced in less than 2 min. Agents used in this study and their sources are as follows: [Met5]enkephalin (M-ENK, Sigma-Aldrich); [Leu5]enkephalin (L-ENK, Peptide Institute Inc., Minoh, Japan); [d-Pen2, D-Pen5]enkephalin (DPDPE, Peninsula Laboratory Inc., Belmont, CA, USA); [D-Ala2, M-Me-Phe4,Gly5]enkephalin-ol (DAMGO, Penisula Laboratory Inc.); and naloxone (Sigma-Aldrich). ω-Conotoxin GVIA (ω-CgTxGVIA, Peptide Institute Inc.) was prepared at a concentration of 1 mM in a solution of 100 mg ml−1 bovine serum albumin (Sigma-Aldrich). The stock solution of ω-CgTxGVIA was added directly to the bath while perfusion was halted. In these experiments, a flow of O2 was directly applied over the bathing fluid.
All the above experiments were carried out in accordance with the guiding principles of the Physiological Society of Japan. The values in the text and figures are means ±s.e.m. (n = number of experiments). Statistically significant differences between various parameters were determined using Student's two-tailed t test for paired data. When the variability of raw data was large, normalized data were compared with unity using Student's two-tailed t test for one variable. Otherwise, the Mann-Whitney U test was used. P < 0.05 was considered significant.
RESULTS
Synaptic depression by enkephalins
When the nystatin-containing patch pipettes were sealed onto the ciliary cell membrane, the access resistances gradually decreased and reached a minimum (19–26 MΩ, n = 4) in 5–10 min. When the patch pipettes contained K+ intracellular solution, the resting potential was in the range -57 to -61 mV (n = 4). EPSCs were recorded from the ciliary cell by clamping at -60 mV. Figure 1A shows the mean EPSCs in response to stimulation pulses applied to the presynaptic oculomotor nerve. Although the amplitude of the EPSCs fluctuated between 8 and 116 pA at 1 mM [Ca2+]o and 5 mM [Mg2+]o, the stimulation artifact was faithfully followed by the capacitative coupling response and the EPSC before exposure to L-ENK (Fig. 1A and B). In the presence of 10 μm L-ENK, which inhibits the cholinergic synaptic transmission in autonomic ganglia (Konishi et al. 1979; Margiotta & Berg, 1986), the EPSC amplitude of the chick ciliary cell synapse was, on average, reduced (Fig. 1A), and synaptic failure was occasionally observed (Fig. 1B). Figure 1C and D shows the amplitude distribution of the first EPSCs before and during the application of 10 μm L-ENK, respectively. Based on Poisson statistics (Martin & Pilar, 1964; Yawo & Chuhma, 1994), m and q were calculated from the c.v. In the control experiment of Fig. 1C, m and q were 4.2 and 12.5 pA, respectively. In the presence of L-ENK (Fig. 1D), m and q were 2.5 and 10.1 pA, respectively. The expected occurrence of failure was 4 in 50 trials, which was very close to the observed occurrence (Fig. 1D). In all four similar experiments, L-ENK consistently decreased m to a mean of 45 ± 12 % of control without changing q (111 ± 15 % of control), and the difference was significant (P < 0.05, paired t test).
Figure 1. Inhibition of quantal transmitter release by enkephalins.
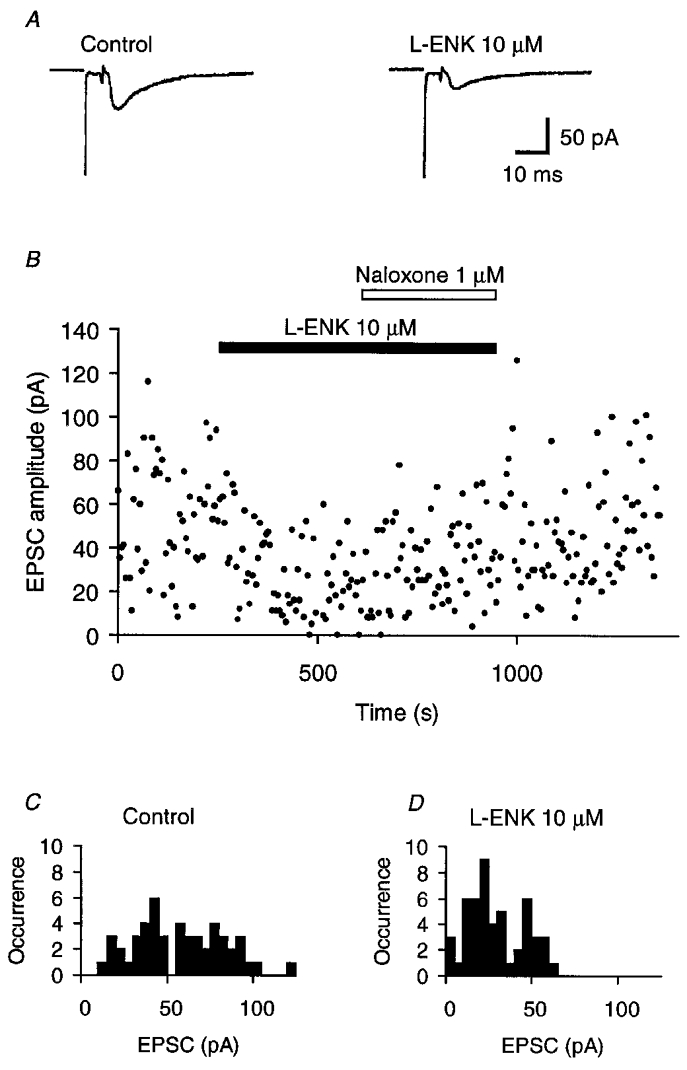
A, the effect of 10 μm [Leu5]enkephalin (L-ENK) on the mean EPSC; representative experimental data measured by the perforated-patch method using a nystatin-fluorescein mixture. The presynaptic oculomotor nerve was electrically stimulated at 0.2 Hz. The biphasic current between the stimulus artifact and the EPSC is the capacitative coupling response indicating the invasion of the action potential into the presynaptic terminal. Left, mean EPSC of 50 consecutive records in control conditions. Right, mean EPSC of 50 consecutive records during bath application of L-ENK. B, time-dependent plots of EPSC amplitude for the experiment shown in A. L-ENK was bath applied during the indicated period (filled bar). Naloxone was added to L-ENK as indicated (open bar). C, EPSC amplitude histogram for 50 consecutive records before the application of L-ENK, the mean of which is shown in A (left). The quantal content (m) and the quantal size (q) were estimated from the c.v., and were 4.2 and 12.5 pA, respectively. D, EPSC amplitude histogram of 50 consecutive records during L-ENK-induced depression, the mean of which is shown in A (right). The values of m and q were 2.5 and 10.1 pA, respectively.
As shown in Fig. 1B, the inhibitory effect of L-ENK was reversed by the opioid receptor antagonist naloxone. When naloxone (1–10 μm) was added to 10 μm L-ENK, m was increased to 120 ± 9 % of the control before the application of L-ENK (n = 4), and was significantly larger than in the presence of L-ENK alone (P < 0.05, Mann-Whitney U test). In another series of experiments, naloxone (1–10 μm) was applied first, and the effect of subsequent application of L-ENK (10 μm) was investigated; the mean EPSC amplitude in the presence of both naloxone and L-ENK was 91–110 % that in the presence of naloxone alone (n = 3). These results suggest that L-ENK reduced quantal transmitter release through opioid receptors in the presynaptic terminal. Although opioid receptors are also distributed in the postsynaptic membrane and reduced both an inward Ca2+ current and transient and sustained K+ currents (Polo-Parada & Pilar, 1999), the postsynaptic opioid receptors appear not to affect the synaptic transmission (see also Margiotta & Berg, 1986).
Similarly, the mean EPSC was again reversibly reduced by 10 μm L-ENK when the EPSC was measured under conventional whole-cell clamp of ciliary cells. At low [Ca2+]o and high [Mg2+]o, the reduction was again accompanied by the increased occurrence of failure responses. For nine experiments, L-ENK (10 μm) reduced m to a mean of 42 ± 6 % of control (Fig. 2A), i.e. to the same extent as in the nystatin-perforated patch recordings (P > 0.4, Mann-Whitney U test). On the other hand, q was unaffected by L-ENK (125 ± 9 % of control, Fig. 2B), being significantly different from m (P < 0.001, paired t test).
Figure 2. Summary of the effects of opioid peptides on the quantal transmitter release.
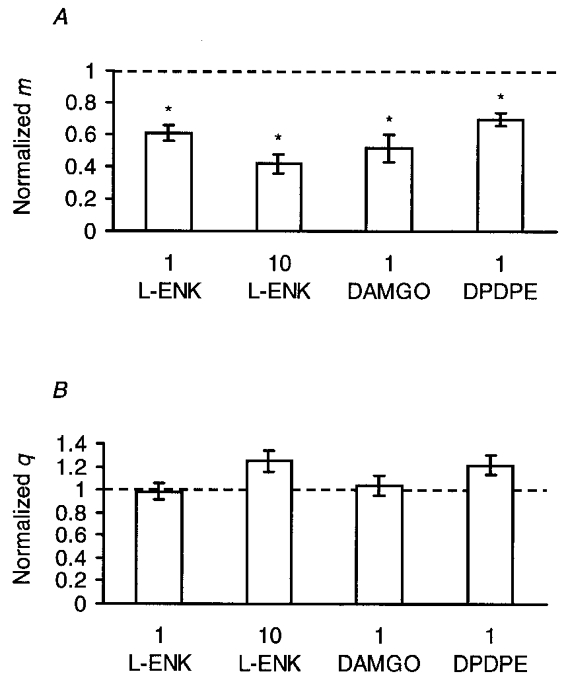
A, effects of opioid peptides on the quantal content (m). In each experiment, the EPSC amplitudes were recorded in an extracellular solution containing low [Ca2+] and high [Mg2+], and m was calculated from the c.v. of the EPSC amplitude fluctuation based on Poisson statistics. Each column and bar are the mean ±s.e.m. of m normalized to the control value before the application of enkephalins. From left to right: 1 μm L-ENK (n = 6); 10 μm L-ENK (n = 9); 1 μm DAMGO, a μ-opioid receptor-selective agonist (n = 8); and 1 μm DPDPE, a δ-opioid receptor-selective agonist (n = 8). Asterisks indicate that the effect was statistically significant (P < 0.05, two-tailed t test between raw data). B, effects of opioid peptides on the quantal size (q). q was calculated as the mean EPSC amplitude divided by m. Each column and bar are the mean ±s.e.m. of q normalized to the control value before the application of enkephalins. From left to right: 1 μm L-ENK (n = 6), 10 μm L-ENK (n = 9), 1 μm DAMGO (n = 8), and 1 μm DPDPE (n = 8). In each case the effect was not statistically significant (P > 0.05, two-tailed t test between raw data).
In another series of experiments the effects of 1 μm L-ENK were investigated. In a summary of data from six whole-cell experiments, 1 μm L-ENK reduced m to 61 ± 5 % of control (Fig. 2A) with a negligible change in q (99 ± 0.07 % of control, Fig. 2B); the difference was significant (P < 0.05, paired t test). The effect of 1 μm L-ENK was significantly less than that of 10 μm L-ENK (Fig. 2A, P < 0.05, Mann-Whitney U test), indicating that the effect was dose dependent. We did not test whether L-ENK was effective at lower concentrations; instead, 1–100 μm was used in the following study for convenience.
Opioid receptor subtypes
Among opioid receptor subtypes, the μ- and δ-opioid receptors are activated by L-ENK (Reisine, 1995). To determine which receptor subtype was involved in the L-ENK-dependent reduction of transmitter release, the effects of subtype-specific agonists were investigated. DAMGO is expected to be selective for μ-opioid receptors at 1 μm, and DPDPE is expected to be selective for δ-opioid receptors at 1 μm (Reisine, 1995). Figure 2 summarizes the effects of 1 μM DAMGO and those of 1 μM DPDPE on m (Fig. 2A) and q (Fig. 2B). DAMGO reduced m to 51 ± 9 % of control without changing q (104 ± 9 % of control), the difference being significant (n = 8, P < 0.05, paired t test). DPDPE reduced m to 70 ± 4 % of control without changing q (122 ± 8 % of control); the difference was significant (n = 9, P < 0.01, paired t test). Therefore, the L-ENK-dependent reduction of m involves both μ- and δ-opioid receptors.
Effects of opioid peptides on [Ca2+]t
The [Ca2+]t in a calyx-type nerve terminal was monitored by the 340 nm/380 nm ratio of fura-dextran fluorescence. The basal [Ca2+]t was 40 ± 11 nM (n = 12) in 5 mM [Ca2+]o solution, and a single nerve stimulus increased [Ca2+]t by 15 ± 4 nM (n = 12). As shown in Fig. 3A, L-ENK (10 μM) reduced the stimulus-dependent peak increment of [Ca2+]t (Δ[Ca2+]t) by 33 %. The effects of enkephalins on Δ[Ca2+]t are summarized in Fig. 3B. M-ENK reduced Δ[Ca2+]t to the same extent as L-ENK. The magnitude of the reduction was dependent on the dose of L-ENK although a significant Δ[Ca2+]t remained in the presence of L-ENK at concentrations as high as 100 μm.
Figure 3. Effects of enkephalins on the intraterminal Ca2+ transient.
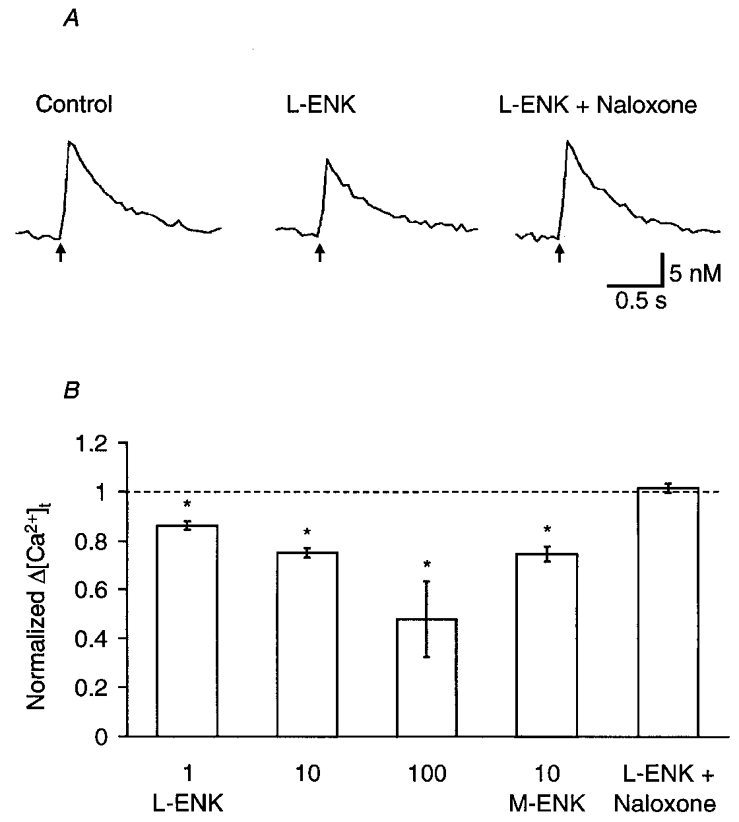
A, sample records of the intraterminal Ca2+ ([Ca2+]t) transient in response to electrical stimulation of the oculomotor nerve at 0.5 Hz (indicated by arrows). From left to right: mean control response before the application of L-ENK, response in the presence of 10 μM L-ENK, and response in the presence of both 10 μm L-ENK and 0.1 μm naloxone. In each case, 12 records were averaged using the computer-generated stimulating pulse as a trigger for summation. B, summary of the effects of enkephalins on the stimulus-dependent increment of [Ca2+]t (Δ[Ca2+]t). Each column and bar are the mean ±s.e.m. of Δ[Ca2+]t normalized to the control value before the application of enkephalins. From left to right: 1 μm L-ENK (n = 5), 10 μm L-ENK (n = 6), 100 μm L-ENK (n = 3), 10 μ [Met5]ENK (M-ENK, n = 5), and 10 μm L-ENK plus 0.1 μm naloxone (n = 5). Asterisks indicate that the effect was statistically significant (P < 0.05, two-tailed t test between raw data). The difference between the 10 μm L-ENK and 10 μM L-ENK + 0.1 μM aloxone columns is also significant (P < 0.01, Mann-Whitney U test).
The opioid receptor antagonist naloxone (0.1 μM) completely reversed the depressing effect of L-ENK on Δ[Ca2+]t (Fig. 3A). When 10 μM L-ENK was applied in the presence of 0.1 μM naloxone, the reduction of Δ[Ca2+]t was not observed (Fig. 3B). Therefore, naloxone-sensitive opioid receptor subtypes should be involved in the reduction of Δ[Ca2+]t. To determine which opioid receptor subtype was involved, the effects of a δ-opioid receptor-selective agonist, DPDPE (0.1–1 μM), and a μ-opioid receptor-selective agonist, DAMGO (0.1–1 μM), were compared. As shown in Fig. 4A, the Δ[Ca2+]t was little affected by 0.1 μM DPDPE whereas it was reduced by the subsequent application of the same concentration of DAMGO. In summary, DAMGO attenuated the Δ[Ca2+]t at both 0.1 and 1 μM (Fig. 4B). Although the effect of 0.1 μM DPDPE was negligible, DPDPE significantly reduced the Δ[Ca2+]t at 1 μM (Fig. 4B, P < 0.01, two-tailed t test between raw data). Therefore, both μ- and δ-opioid receptors appear to be involved in the enkephalin-dependent suppression of Δ[Ca2+]t.
Figure 4. Reduction of Δ[Ca2+]t by μ-opioid receptor activation.
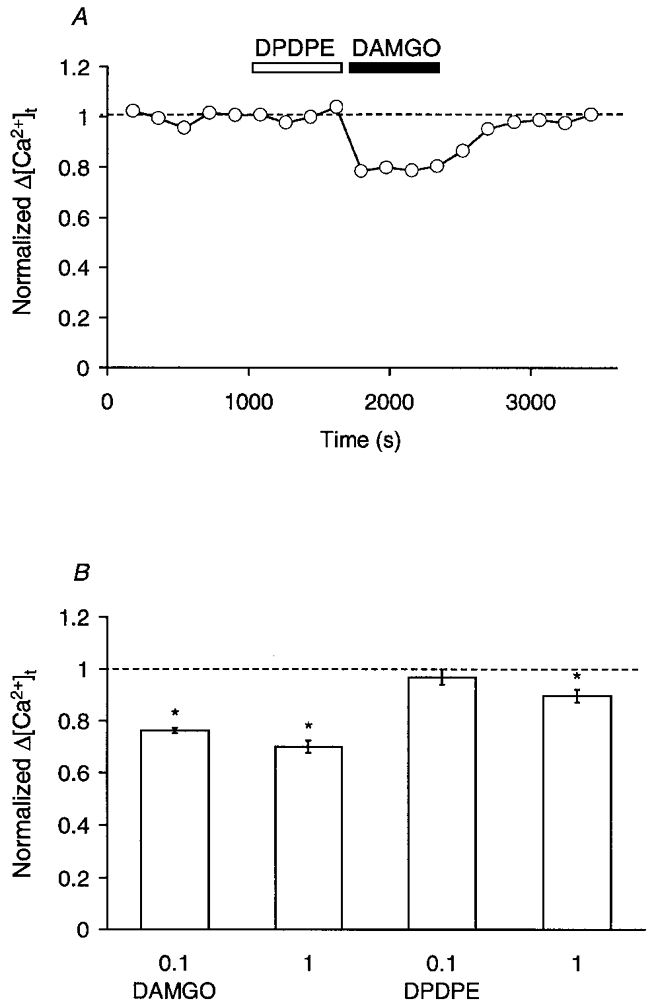
A, time-dependent plots for a representative experiment comparing the effects of DPDPE and DAMGO. The Δ[Ca2+]t was normalized to the mean control value before the application of DPDPE, and was plotted against time. DPDPE (0.1 μM, open bar) and DAMGO (0.1 μM, filled bar) were bath applied during the indicated periods. B, summary of the effects of DPDPE and DAMGO on the Δ[Ca2+]t. Each column and bar are the mean ±s.e.m. of Δ[Ca2+]t normalized to the control value before the application of the agonists. From left to right: 0.1 μM DAMGO (n = 4), 1 μM DAMGO (n = 4), 0.1 μM DPDPE (n = 6) and 1 μM DPDPE (n = 5). Asterisks indicate that the effect was statistically significant (P < 0.01, two-tailed t test between raw data).
Reduction of Ca2+ influx by enkephalins
Upon the invasion of the action potential, a focal increase of intracellular Ca2+ was observed in the presynaptic terminal (Zucker, 1996; Neher, 1998). This is explained by the clustering of Ca2+ channels around active zones (Robitaille et al. 1990; Cohen et al. 1991; Yawo & Chuhma, 1994) and the slow diffusion of intracellular Ca2+ (Zucker, 1996; Neher, 1998). Although [Ca2+]t transients should be much larger and faster in the vicinity of Ca2+ channel clusters (Zucker, 1996; Neher, 1998), the activity-dependent increment of the volume-averaged fura-2 signal would be proportional to the changes in the local concentration (Sinha et al. 1997; Yawo, 1999a).
Following a single nerve stimulus, the [Ca2+]t increased with a short delay, peaked within 50–150 ms and decayed slowly (Yawo & Chuhma, 1993a). The falling phase of Δ[Ca2+]t did not follow a simple exponential function. For convenience, the falling phase between 90 and 10 % of the peak [Ca2+]t was fitted to the sum of a single exponential function and constant (Fig. 5A). That is,
| (1) |
where α is the asymptote of the peak Δ[Ca2+]t and β is the basal [Ca2+]t. At 5 mM [Ca2+]o, the falling phase time constant, τ, was 417 ± 20 ms (n = 9). If enkephalins accelerate intracellular Ca2+ sequestration, a reduction of τ would be expected. However, as shown in Fig. 5B, the change of τ was negligible with either 10 μM L-ENK (106 ± 7 % of control, n = 5) or 10 μM M-ENK (99 ± 4 % of control, n = 4), the effect being insignificant (P > 0.5, paired t test between raw data). The basal [Ca2+]t, calculated as β of eqn (1), was also unaffected by the enkephalins (Fig. 5C).
Figure 5. Effects of enkephalins on the falling phase of Δ[Ca2+]t.
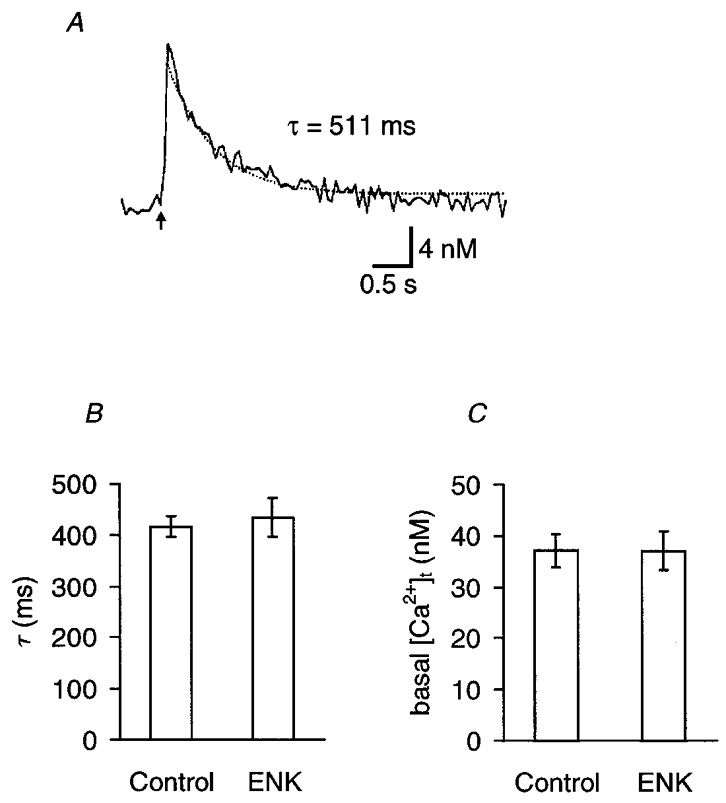
A, a sample record of [Ca2+]t in response to an electrical stimulus to the oculomotor nerve (indicated by the arrow). The falling phase of Δ[Ca2+]t between 90 and 10 % of its peak was fitted to eqn (1) in the text: [Ca2+]t=α exp(-t/τ) +β, where α is the asymptote of the peak Δ[Ca2+]t and β is the basal [Ca2+]t. In this example, τ= 511 ms, α= 13.0 nM and β= 42.2 nM. B, the decay time constant (τ) of [Ca2+]t. Summary of nine experiments in control and in the presence of 10 μM enkephalin (5 experiments with L-ENK and 4 experiments with M-ENK). The difference was not significant (P > 0.5, two-tailed t test). C, basal [Ca2+]t (β). Summary of nine similar experiments in control and in the presence of 10 μM enkephalin (5 experiments with L-ENK and 4 experiments with M-ENK). The difference was not significant (P > 0.8, two-tailed t test).
To investigate whether enkephalins reduce Ca2+ influx through Ca2+ channels, we measured the change in [Ba2+]t instead of [Ca2+]t. When extracellular Ca2+ was replaced with Ba2+ in the presence of 1 mM EGTA, nerve stimulation increased the 340 nm/380 nm ratio of fura-dextran fluorescence, indicating an increase of [Ba2+]t. The rate of rise of [Ba2+]t was as fast as that of [Ca2+]t, whereas the decline of the action potential-dependent increase of [Ba2+]t to the basal level occurred much more slowly than that of Δ[Ca2+]t (Fig. 6) with a time constant of 20–75 s (n = 4; Yawo & Chuhma, 1993a). With a stimulation interval of 120 s, the peak increment of [Ba2+]t (Δ[Ba2+]t) was 0.7–1.9 μM (n = 5), but this interval was not enough to allow full recovery to the baseline fluorescence ratio. The large Δ[Ba2+]t and the slow decline of Δ[Ba2+]t indicate that intracellular Ba2+ is buffered more weakly and more slowly than Ca2+ (Schilling et al. 1989; Kwan & Putney, 1990; Yawo & Chuhma, 1993a). Therefore, we considered that the change in Δ[Ba2+]t should be more dependent on the change of Ba2+ influx through Ca2+ channels than on that of divalent cation buffering. As shown in Fig. 6, L-ENK (10 μM) reduced the Δ[Ba2+]t to 81 ± 4 % of control (P < 0.05, single value t test). The effect of L-ENK was reversible within 10 min and the subsequent application of DAMGO (1 μM) again reduced the Δ[Ba2+]t (Fig. 6). When the declining phase of [Ba2+]t was fitted to eqn (1), τ was 37 ± 7 s in control and 33 ± 8 s in the presence of L-ENK, the difference being insignificant (n = 4, P > 0.4, paired t test). These results indicate that the μ-opioid receptor reduces Ba2+ influx through Ca2+ channels.
Figure 6. Effects of enkephalins on the intraterminal Ba2+ transient.
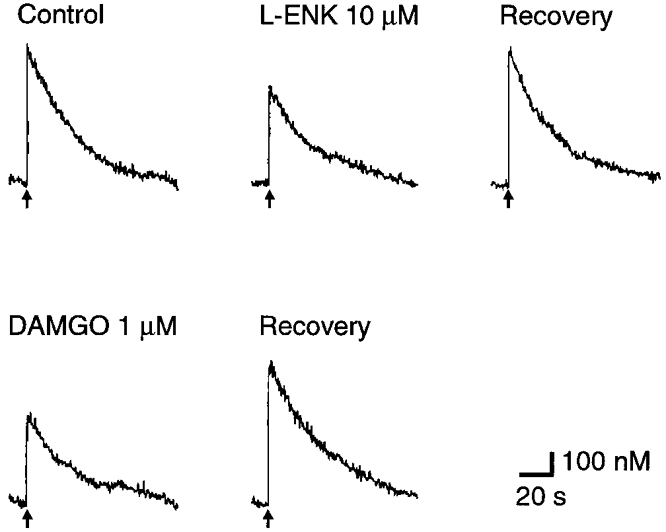
Records of the intraterminal Ba2+ ([Ba2+]t) transient in response to electrical stimulation of the oculomotor nerve (indicated by arrows). Top, a representative experiment. From left to right: control record before the application of L-ENK, after bath application of 10 μM L-ENK, and after 9 min recovery. Bottom, the same series of experiments as above, after bath application of 1 μM DAMGO (left) and after 9 min recovery (right).
Preferential inhibition of N-type Ca2+ channels by enkephalins
Since the presynaptic action potential has been shown to be prolonged by 4-aminopyridine (4-AP; 0.4 mM), the transient K+ current would be involved in the falling phase of the action potential in the calyx-type presynaptic terminal of chick ciliary ganglion (Yawo & Chuhma, 1994). The Δ[Ca2+]t was enhanced by 4-AP (Yawo & Chuhma, 1993a) because of the slow activation of the presynaptic Ca2+ conductance (Yawo & Momiyama, 1993). As shown in Fig. 7A, the Δ[Ca2+]t was reversibly reduced by L-ENK in the presence of 4-AP. In five experiments in the presence of 0.4 mM 4-AP, L-ENK (10 μM) reduced Δ[Ca2+]t to 79 ± 5 % of control (Fig. 7B, P < 0.05, paired t test between raw data), a magnitude comparable to that without 4-AP (P > 0.3, Mann-Whitney U test). Therefore, the 4-AP-sensitive transient K+ channels appear not to be involved in the enkephalin-dependent reduction of Ca2+ influx in the calyx-type presynaptic terminal. It is possible that enkephalins directly inhibit Ca2+ channel activity (see Discussion).
Figure 7. Inhibition of ω-CgTxGVIA-sensitive Ca2+ influx by enkephalins.
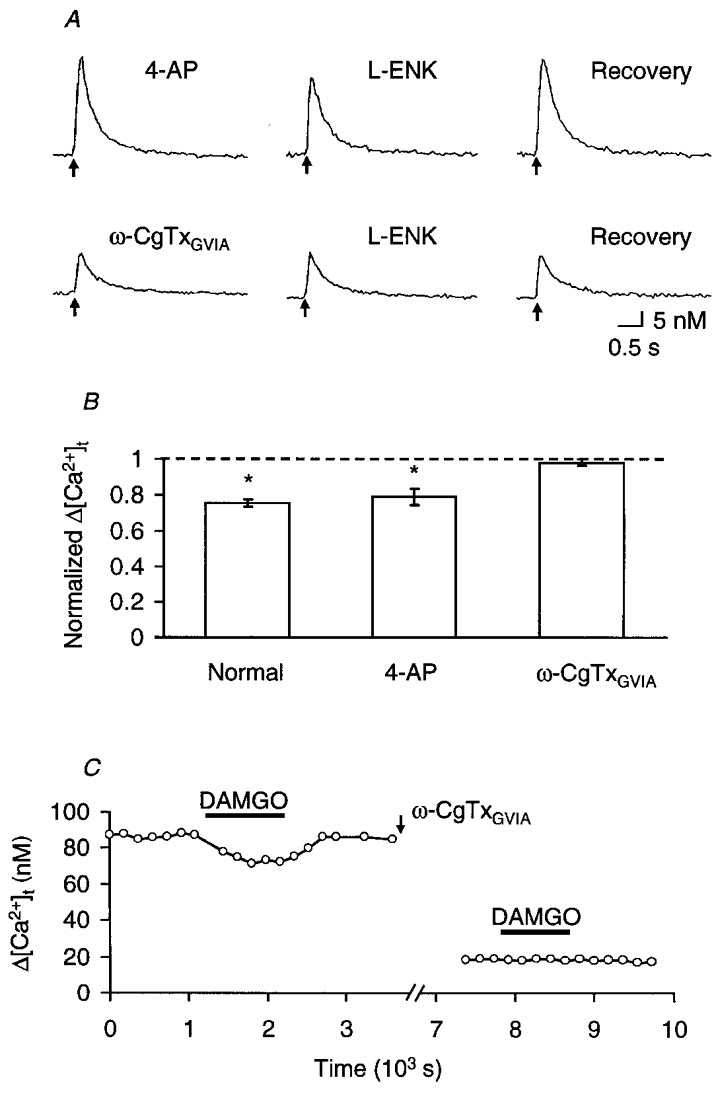
A, a series of sample records of [Ca2+]t in response to a single presynaptic stimulus (indicated by arrows) in a solution containing 0.4 mM 4-AP. To remove the voltage-dependent inactivation of ω-CgTxGVIA-resistant Ca2+ conductance, the external [K+] was reduced to 1 mM. Top, control experiment before the application of ω-CgTxGVIA. From left to right: control record before the application of L-ENK, the effect of 10 μM L-ENK and recovery. Bottom, the effect of L-ENK (10 μM) on the same presynaptic terminal after 30 min treatment with 10 μM ω-CgTxGVIA. B, summary of the effects of 10 μM L-ENK on Δ[Ca2+]t, normalized to the control value before the application of L-ENK. Each column and bar are the mean ±s.e.m. From left to right: normal extracellular solution (n = 6, the same as that shown in Fig. 3B, 10 μM L-ENK column), in the presence of 0.4 mM 4-AP (n = 5), and in the presence of 4-AP after treatment with 10 μm ω-CgTxGVIA (n = 6). Asterisks indicate that the effect was statistically significant (P < 0.05, two-tailed t test between raw data). The difference between the 4-AP and ω-CgTxGVIA columns is also significant (P < 0.01, Mann-Whitney U test). C, effects of DAMGO on Δ[Ca2+]t before and after pre-treatment with ω-CgTxGVIA (10 μm, arrow). DAMGO (0.1 μm) was bath applied during the indicated period (filled bars).
At least two Ca2+ channel subtypes have been identified in the calyx-type presynaptic terminal; one is sensitive to ω-CgTxGVIA but insensitive to dihydropyridines (N-type); the other is resistant to both ω-CgTxGVIA and dihydropyridines (Yawo & Momiyama, 1993). To determine which subclass of Ca2+ channels is involved in the modulation through opioid receptors, the Δ[Ca2+]t was compared before and after treatment with 10 μm ω-CgTxGVIA for 30 min, which fully blocks the N-type Ca2+ channel subpopulation (Yawo & Momiyama, 1993; Yawo & Chuhma, 1994). As a result, the ω-CgTxGVIA treatment reduced Δ[Ca2+]t (Fig. 7A) on average to 22 ± 4 % of control (n = 6, P < 0.005, paired t test between raw data). After treatment with ω-CgTxGVIA, the addition of L-ENK (10 μm) was no longer effective (Fig. 7A and B). Figure 7C shows another example in which DAMGO (0.1 μm) was used instead of L-ENK. Before treatment with ω-CgTxGVIA, the Δ[Ca2+]t was reversibly reduced by DAMGO whereas the ω-CgTxGVIA-resistant component was completely insensitive to DAMGO. It is therefore suggested that the μ-opioid receptor preferentially inhibits the ω-CgTxGVIA-sensitive Ca2+ influx through N-type channels.
DISCUSSION
Inhibition of transmitter release by enkephalins
The present study was focused on the effects of enkephalins on the synaptic transmission between the calyx-type presynaptic terminal and the ciliary cell in the ciliary ganglion of the chick embryo. Since the calyx-type terminal contains L-ENK as a co-transmitter (Erichsen et al. 1982a; Meriney et al. 1991), the endogenous enkephalins would modulate the cholinergic synaptic transmission at the presynaptic terminal, the postsynaptic membrane, or both. In the present work, we studied the effects of exogenous L-ENK on the quantal transmitter release under a nystatin-perforated patch clamp. Since L-ENK reduced m with negligible changes in q and the inhibitory effect of L-ENK was antagonized by naloxone (Fig. 1), it is suggested that opioid receptors are present in the presynaptic terminal and inhibit transmitter release. On the other hand, the postsynaptic junctional acetylcholine receptors appear not to be sensitive to L-ENK (Margiotta & Berg, 1986).
In sympathetic ganglia conditioning tetanic stimulation applied to the presynaptic nerve releases the endogenous enkephalins and inhibits acetylcholine release from the presynaptic terminal (Konishi et al. 1981). Were the endogenous enkephalins released following stimulation of the presynaptic nerve in our preparation? Did they inhibit transmitter release? Further experiments are necessary to address these issues.
Opoid receptor subtypes
In the present study, both DAMGO and DPDPE reduced transmitter release from calyx-type presynaptic terminals (Fig. 2). Therefore, it is suggested that L-ENK would activate both μ- and δ-opioid receptors in the presynaptic terminal. Although the endogenous agonist of the κ-opioid receptor has not been identified in the chick ciliary ganglion, the excitability of the calyx-type presynaptic terminal is modulated by a κ-opioid receptor-selective agonist, U-50,488 (Fletcher & Chiappinelli, 1993). U-50,488 inactivates at least three membrane conductances: an inward rectifier permeable to Na+ and K+, a K+ conductance which is active at the resting potential and is blocked by Ba2+, and a Ca2+-dependent K+ conductance (Fletcher & Chiappinelli, 1993). How these modulations of membrane conductances influence the transmitter release should be examined experimentally.
The reduction of Ca2+ influx
The enkephalin-dependent reduction of Δ[Ca2+]t (Figs 3 and 4) can be attributed to the inhibition of Ca2+ influx, the enhancement of Ca2+ sequestration or both. However, the modulation of Ca2+ sequestration by opioid peptides is unlikely for the following three reasons. First, the rate of the falling phase of Δ[Ca2+]t was not accelerated by the enkephalins (Fig. 5) although the early transient of local [Ca2+]t in the vicinity of Ca2+ channel clusters was not detectable in our system. Second, the Δ[Ba2+]t, which may not be influenced by the Ca2+-buffering mechanisms, was also reduced by the enkephalins to the same extent as Δ[Ca2+]t (Fig. 6). Finally, the Δ[Ca2+]t was not reduced by the enkephalins after treatment with ω-CgTxGVIA (Fig. 7). Because DAMGO and DPDPE reduced Δ[Ca2+]t (Fig. 4), it is suggested that the enkephalins inhibit the Ca2+ influx during the presynaptic action potential through both μ- and δ-opioid receptors.
At 5 mM [Ca2+]o, 10 μm L-ENK reduced Δ[Ca2+]t to 75 ± 2 % of control. Therefore, the Ca2+ influx should be reduced to 75% of control or less by L-ENK because of the non-linear relationship between [Ca2+]o and Δ[Ca2+]t (Yawo, 1999a). If L-ENK (10 μm) could reduce the Ca2+ influx to a maximum of 75 % at 1 mM [Ca2+]o, the EPSC would be expected to be reduced to a maximum of 55 % of control based on the non-linear relationship between [Ca2+]o and the EPSC (Yawo, 1996). Since this value is comparable to the experimental observation at 1 mM [Ca2+]o (51 ± 10 % of control, n = 7, data not shown), most of the effects of L-ENK could be explained by the reduction of Ca2+ influx. We cannot, however, exclude the possibility that the opioid receptors downregulate an exocytotic mechanism other than Ca2+ influx (Neher, 1998; Yawo, 1999a).
Preferential inhibition of N-type Ca2+ channels
The reduction of Ca2+ influx could be attributed to the inactivation of Ca2+ channels, the activation of K+ channels, or both. One candidate among such K+ channels is the 4-AP-sensitive transient K+ channel activated during the falling phase of the presynaptic action potential (Yawo & Chuhma, 1994), as suggested for the GABAergic synaptic transmission in the midbrain (Vaughan et al. 1997). However, L-ENK reduced Δ[Ca2+]t to the same extent in the presence of 4-AP as in its absence (Fig. 7). Alternatively, enkephalins might inhibit the Ca2+-dependent K+ channels (Twitchell & Rane, 1993) co-distributing with the N-type Ca2+ channels (Robitaille et al. 1993). However, this notion conflicts with the fact that enkephalins reduced the Δ[Ba2+]t to the same extent as the Δ[Ca2+]t when extracellular Ca2+ was replaced by Ba2+ (Fig. 6), which does not, in general, activate Ca2+-dependent K+ conductances (Hille, 1992).
It has been reported that the activation of a certain subclass of K+ channels is the major function of μ-opioid receptors in some neuronal somata (North, 1986; Williams et al. 1988) as well as in the presynaptic terminal of myenteric plexus. (Cherubini & North, 1985). This K+ conductance is active around the resting potential and is blocked by Ba2+. As a result, opioid peptides would hyperpolarize the membrane potential and reduce the excitability of the membrane, thereby effectively suppressing neuronal activity. On the other hand, enkephalins depolarize the resting membrane potential of the ciliary presynaptic terminal of the hatched chick, although they hyperpolarize the Edinger-Westphal neuronal soma which sends its axon to the ciliary ganglion (Chiappinelli et al. 1993). When the resting membrane potential of the calyx-type presynaptic terminal was monitored by the nystatin-perforated patch method, it was actually unaffected by DAMGO (1 μM, n = 2, data not shown). We also tested the effects of membrane hyperpolarization by reducing [K+]o from 5 to 1 mM. However, the hyperpolarization per se did not reduce the EPSC (Yawo & Chuhma, 1994) as has been suggested in some other synapses (Cohen et al. 1992; Capogna et al. 1993). Therefore, the hyperpolarization of the presynaptic membrane potential appears not to be the principal mechanism of the μ-opioid receptor-dependent inhibition of transmitter release in the chick ciliary presynaptic terminal. Because of the prominent reduction of Δ[Ba2+]t by L-ENK and DAMGO (Fig. 6), a modulation of the Ba2+-sensitive K+ conductance would be unlikely (Hori et al. 1992; Vaughan & Christie, 1997). All the above observations strongly suggest that the μ-opioid receptor reduces the Ca2+ influx directly through the inhibition of Ca2+ channels rather than through the activation of K+ channels.
We have previously shown that two distinct subtypes of Ca2+ channel coexist in the calyx-type presynaptic terminal of embryonic chick ciliary ganglion (Yawo & Momiyama, 1993). Because the major one was sensitive to ω-CgTxGVIA but insensitive to dihydropyridines, it was classified as the N-type Ca2+ channel (Stanley, 1991). The other subtype gave rise to a high-voltage activated current resistant to ω-CgTxGVIA, dihydropyridines and ω-agatoxin IVA (Yawo & Momiyama, 1993; Yawo, 1994), and was classified as the R-type Ca2+ channel. The N-type Ca2+ channels are closely coupled with exocytosis (Stanley, 1993, 1997) and release transmitters more efficiently than the R-type (Yawo & Chuhma, 1994). In the present study, the L-ENK- and DAMGO-dependent reduction of Δ[Ca2+]t completely disappeared after ω-CgTxGVIA treatment (Fig. 7). This conflicts with the notion that enkephalin-sensitive Ca2+ influx could occur through R-type channels. It is suggested that the μ-opioid receptor would preferentially inhibit N-type Ca2+ channels.
Pivotal role of N-type Ca2+ channels in the regulation of transmitter release
Accumulating evidence indicates that the N-type Ca2+ channel is one of the common sites of presynaptic modulation (Wu & Saggau, 1997). At least four inhibitory receptors, A1-adenosine (Yawo & Chuhma, 1993a), α2-adrenergic (Yawo, 1996), and μ- and δ-opioid receptors, appear to converge at the N-type Ca2+ channel in the calyx-type presynaptic terminal of the chick ciliary ganglion. In fact, the presynaptic N-type Ca2+ channels are the targets of G-protein-mediated modulation in this system (Stanley & Mirotznik, 1997). N-type Ca2+ channels are suggested to cluster near the release sites (Miller, 1987; Robitaille et al. 1990; Cohen et al. 1991; Yawo & Chuhma, 1994; Stanley, 1997) and are the principal pathways of Ca2+ influx during the presynaptic depolarization (Miller, 1987; Yawo & Momiyama, 1993). The transmitter release appears to be tightly coupled with Ca2+ influx through the N-type Ca2+ conductance (Miller, 1987; Yawo & Chuhma, 1994; Stanley, 1997), although in some mammalian peripheral and central synapses a significant part of transmitter release is resistant to ω-CgTxGVIA (Dunlap et al. 1995; Takahashi et al. 1998). Since the N-type Ca2+ channels are rapidly and reversibly modulated by G-proteins (Kasai, 1992; Taussig et al. 1992; Wilding et al. 1995; Dolphin, 1998), the manipulation of these channels would be the most reliable mechanism for the rapid and reversible modulation of transmitter release.
Acknowledgments
We thank M. Fukao for technical support, Dr H. Ohmori for comments on the manuscript and Mr B. Bell for reading the revised manuscript. This work was supported by Grants-in-Aid from the Ministry of Education, Science and Culture of Japan.
References
- Araujo DM, Collier B. Effect of endogenous opioid peptides on acetylcholine release from the cat superior cervical ganglion: selective effect of a heptapeptide. Journal of Neuroscience. 1987;7:1698–1704. doi: 10.1523/JNEUROSCI.07-06-01698.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogna M, Gähwiler BH, Thompson SM. Mechanism of μ-opioid receptor-mediated presynaptic inhibition in the rat hippocampus in vitro. The Journal of Physiology. 1993;470:539–558. doi: 10.1113/jphysiol.1993.sp019874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherubini E, North RA. μ and κ opioids inhibit transmitter release by different mechanisms. Proceedings of the National Academy of Sciences of the USA. 1985;82:1860–1863. doi: 10.1073/pnas.82.6.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappinelli VA, Wolf KM, Feng C, Yum L, McMahon LL. Different responses to opioids measured in terminals and somas of Edinger-Westphal neurons. Neuroscience. 1993;57:425–432. doi: 10.1016/0306-4522(93)90074-p. [DOI] [PubMed] [Google Scholar]
- Cohen GA, Doze VA, Madison DV. Opioid inhibition of GABA release from presynaptic terminals of rat hippocampal interneurons. Neuron. 1992;9:325–335. doi: 10.1016/0896-6273(92)90171-9. [DOI] [PubMed] [Google Scholar]
- Cohen MW, Jones OT, Angelides KJ. Distribution of Ca2+ channels on frog motor nerve terminals revealed by fluorescent ω-conotoxin. Journal of Neuroscience. 1991;11:1032–1039. doi: 10.1523/JNEUROSCI.11-04-01032.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. Mechanisms of modulation of voltage-dependent calcium channels by G proteins. The Journal of Physiology. 1998;506:3–11. doi: 10.1111/j.1469-7793.1998.003bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dun NJ. Peptide hormones and transmission in sympathetic ganglia. In: Elfvin L-G, editor. Autonomic Ganglia. Chichester: John Wiley & Sons Ltd; 1983. pp. 345–366. [Google Scholar]
- Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends in Neurosciences. 1995;18:89–98. [PubMed] [Google Scholar]
- Erichsen JT, Karten HJ, Eldrad WD, Brecha NC. Localization of substance P-like and enkephalin-like immunoreactivity within preganglionic terminals of the avian ciliary ganglion: light and electron microscopy. Journal of Neuroscience. 1982a;2:994–1003. doi: 10.1523/JNEUROSCI.02-07-00994.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erichsen JT, Reiner A, Karten HJ. Co-occurrence of substance P-like and Leu-enkephalin-like immunoreactivities in neurones and fibres of avian nervous system. Nature. 1982b;295:407–410. doi: 10.1038/295407a0. [DOI] [PubMed] [Google Scholar]
- Fletcher GH, Chiappinelli VA. The action of κ1 opioid agonist U-50,488 on presynaptic nerve terminals of the chick ciliary ganglion. Neuroscience. 1993;53:239–250. doi: 10.1016/0306-4522(93)90302-v. [DOI] [PubMed] [Google Scholar]
- Glover JC, Petursdottir G, Jansen KS. Fluorescent dextran-amines used as axonal tracers in the nervous system of the chicken embryo. Journal of Neuroscience Methods. 1986;18:243–254. doi: 10.1016/0165-0270(86)90011-7. [DOI] [PubMed] [Google Scholar]
- Gray DB, Zalazny D, Manthay N, Pilar G. Endogenous modulation of ACh release by somatostatin and the differential roles of Ca2+ channels. Journal of Neuroscience. 1990;10:2687–2698. doi: 10.1523/JNEUROSCI.10-08-02687.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. Journal of Morphology. 1951;88:49–92. [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. 2. Sunderland MA, USA: Sinauer Associates Inc; 1992. [Google Scholar]
- Hökfelt T, Johansson O, Goldstein M. Chemical anatomy of the brain. Science. 1984;225:1326–1334. doi: 10.1126/science.6147896. [DOI] [PubMed] [Google Scholar]
- Hori Y, Endo K, Takahashi T. Presynaptic inhibitory action of enkephalin on excitatory transmission in superficial dorsal horn of rat spinal cord. The Journal of Physiology. 1992;450:673–685. doi: 10.1113/jphysiol.1992.sp019149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H. Voltage and time-dependent inhibition of neuronal calcium channels by a GTP-binding protein in a mammalian cell line. The Journal of Physiology. 1992;448:189–209. doi: 10.1113/jphysiol.1992.sp019036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi S, Tsunoo A, Otsuka M. Enkephalins presynaptically inhibit cholinergic transmission in sympathetic ganglia. Nature. 1979;282:515–516. doi: 10.1038/282515a0. [DOI] [PubMed] [Google Scholar]
- Konishi S, Tsunoo A, Otsuka M. Enkephalin as a transmitter for presynaptic inhibition in sympathetic ganglia. Nature. 1981;294:80–82. doi: 10.1038/294080a0. [DOI] [PubMed] [Google Scholar]
- Kuno M, Weakley JN. Quantal components of the inhibitory synaptic potential in spinal motoneurones of the cat. The Journal of Physiology. 1972;224:287–303. doi: 10.1113/jphysiol.1972.sp009895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan C-Y, Putney W., Jr Uptake and intracellular sequestration of divalent cations in resting and methacholine-stimulated mouse lacrimal acinar cells. Journal of Biological Chemistry. 1990;265:678–684. [PubMed] [Google Scholar]
- Landmesser L, Pilar G. Synaptic transmission and cell death during normal ganglionic development. The Journal of Physiology. 1974;241:737–749. doi: 10.1113/jphysiol.1974.sp010681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margiotta JF, Berg DK. Enkephalin and substance P modulate synaptic properties of chick ciliary ganglion neurons in cell culture. Neuroscience. 1986;18:175–182. doi: 10.1016/0306-4522(86)90186-7. [DOI] [PubMed] [Google Scholar]
- Martin AR, Pilar G. Quantal components of the synaptic potential in the ciliary ganglion of the chick. The Journal of Physiology. 1964;175:1–16. doi: 10.1113/jphysiol.1964.sp007499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marwitt R, Pilar G, Weakly JN. Characterization of two ganglion cell populations in avian ciliary ganglia. Brain Research. 1971;25:317–334. doi: 10.1016/0006-8993(71)90441-0. [DOI] [PubMed] [Google Scholar]
- Meriney SD, Ford MJ, Oliva D, Pilar G. Endogenous opioids modulate neuronal survival in the developing avian ciliary ganglion. Journal of Neuroscience. 1991;11:3705–3717. doi: 10.1523/JNEUROSCI.11-12-03705.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meriney SD, Gray DB, Pilar G. Morphine-induced delay of normal cell death in the avian ciliary ganglion. Science. 1985;228:1451–1453. doi: 10.1126/science.2990029. [DOI] [PubMed] [Google Scholar]
- Miller RJ. Multiple calcium channels and neuronal function. Science. 1987;235:46–52. doi: 10.1126/science.2432656. [DOI] [PubMed] [Google Scholar]
- Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC, Kauer JA. Functional comparison of neurotransmitter receptor subtypes in mammalian central nervous system. Physiological Reviews. 1990;70:513–565. doi: 10.1152/physrev.1990.70.2.513. [DOI] [PubMed] [Google Scholar]
- North RA. Opioid receptor types and membrane ion channels. Trends in Neurosciences. 1986;9:114–117. [Google Scholar]
- Polo-Parada L, Pilar G. κ- and μ-Opioids reverse the somatostatin inhibition of Ca2+ currents in ciliary and dorsal root ganglion neurons. Journal of Neuroscience. 1999;19:5213–5227. doi: 10.1523/JNEUROSCI.19-13-05213.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A. A VIP-like peptide co-occurs with substance P and enkephalin in cholinergic preganglionic terminals of the avian ciliary ganglion. Neuroscience Letters. 1987;78:22–28. doi: 10.1016/0304-3940(87)90555-6. [DOI] [PubMed] [Google Scholar]
- Reisine T. Opiate receptors. Neuropharmacology. 1995;34:463–472. doi: 10.1016/0028-3908(95)00025-2. [DOI] [PubMed] [Google Scholar]
- Robitaille R, Adler EM, Charlton MP. Strategic location of calcium channels at transmitter release sites of frog neuromuscular synapses. Neuron. 1990;5:773–779. doi: 10.1016/0896-6273(90)90336-e. [DOI] [PubMed] [Google Scholar]
- Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron. 1993;11:645–655. doi: 10.1016/0896-6273(93)90076-4. [DOI] [PubMed] [Google Scholar]
- Schilling WP, Rajan L, Strobl-Jager E. Characterization of the bradykinin-stimulated calcium influx pathway of cultured vascular endothelial cells. Journal of Biological Chemistry. 1989;264:12838–12848. [PubMed] [Google Scholar]
- Sinha SR, Wu L-G, Saggau P. Presynaptic calcium dynamics and transmitter release. Proceedings of the National Academy of Sciences of the USA. 1997;94:5888–5893. [Google Scholar]
- Stanley EF. Single calcium channels on a cholinergic presynaptic nerve terminal. Neuron. 1991;7:585–591. doi: 10.1016/0896-6273(91)90371-6. [DOI] [PubMed] [Google Scholar]
- Stanley EF. Single calcium channels and acetylcholine release at a presynaptic nerve terminal. Neuron. 1993;11:1007–1011. doi: 10.1016/0896-6273(93)90214-c. [DOI] [PubMed] [Google Scholar]
- Stanley EF. The calcium channel and the organization of the presynaptic transmitter release face. Trends in Neurosciences. 1997;20:404–409. doi: 10.1016/s0166-2236(97)01091-6. [DOI] [PubMed] [Google Scholar]
- Stanley EF, Mirotznik RR. Cleavage of syntaxin prevents G-protein regulation of presynaptic calcium channels. Nature. 1997;385:340–343. doi: 10.1038/385340a0. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kajikawa Y, Tsujimoto T. G-protein-coupled modulation of presynaptic calcium currents and transmitter release by a GABAB receptor. Journal of Neuroscience. 1998;19:3138–3146. doi: 10.1523/JNEUROSCI.18-09-03138.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taussig R, Sanchez S, Rifo M, Gilman AG, Belardetti F. Inhibition of the ω-conotoxin-sensitive calcium current by distinct G proteins. Neuron. 1992;8:799–809. doi: 10.1016/0896-6273(92)90100-r. [DOI] [PubMed] [Google Scholar]
- Twitchell WA, Rane SG. Opioid peptide modulation of Ca2+-dependent K+ and voltage-activated Ca2+ currents in bovine adrenal chromaffin cells. Neuron. 1993;10:701–709. doi: 10.1016/0896-6273(93)90171-m. [DOI] [PubMed] [Google Scholar]
- Vaughan CW, Christie MJ. Presynaptic inhibitory action of opioids on synaptic transmission in the rat periaqueductal grey in vitro. The Journal of Physiology. 1997;498:463–472. doi: 10.1113/jphysiol.1997.sp021872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Connor MA, Christie MJ. How opioids inhibit GABA-mediated neurotransmission. Nature. 1997;390:611–614. doi: 10.1038/37610. [DOI] [PubMed] [Google Scholar]
- White JD, Krause JE, Karten HJ, McKelvy JF. Presence and ontogeny of enkephalin and substance P in the chick ciliary ganglion. Journal of Neurochemistry. 1985;45:1319–1322. doi: 10.1111/j.1471-4159.1985.tb05562.x. [DOI] [PubMed] [Google Scholar]
- Wilding TJ, Womack MD, McCleskey EW. Fast, local signal transduction between the μ opioid receptor and Ca2+ channels. Journal of Neuroscience. 1995;15:4124–4132. doi: 10.1523/JNEUROSCI.15-05-04124.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, North RA, Tokimasa T. Inward rectification of resting and opiate-activated potassium currents in rat locus coeruleus neurons. Journal of Neuroscience. 1988;8:4299–4306. doi: 10.1523/JNEUROSCI.08-11-04299.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L-G, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends in Neurosciences. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- Yawo H. Diversity of calcium channel subtypes in presynaptic terminals of the chick ciliary ganglion. Annals of the New York Academy of Sciences. 1994;707:379–381. doi: 10.1111/j.1749-6632.1993.tb38076.x. [DOI] [PubMed] [Google Scholar]
- Yawo H. Noradrenaline modulates transmitter release by enhancing the Ca2+ sensitivity of exocytosis in the chick ciliary presynaptic terminal. The Journal of Physiology. 1996;493:385–391. doi: 10.1113/jphysiol.1996.sp021390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yawo H. Protein kinase C potentiates the transmitter release from the chick ciliary presynaptic terminal by increasing exocytotic fusion probability. The Journal of Physiology. 1999a;515:169–180. doi: 10.1111/j.1469-7793.1999.169ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yawo H. Two components of transmitter release from the chick ciliary presynaptic terminal and their regulation by protein kinase C. The Journal of Physiology. 1999b;516:461–470. doi: 10.1111/j.1469-7793.1999.0461v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yawo H, Chuhma N. Preferential inhibition of ω-conotoxin-sensitive presynaptic Ca2+ channels by adenosine autoreceptors. Nature. 1993a;365:256–258. doi: 10.1038/365256a0. [DOI] [PubMed] [Google Scholar]
- Yawo H, Chuhma N. An improved method for perforated patch recordings using nystatin-fluorescein mixture. Japanese Journal of Physiology. 1993b;43:267–273. doi: 10.2170/jjphysiol.43.267. [DOI] [PubMed] [Google Scholar]
- Yawo H, Chuhma N. ω-Conotoxin-sensitive and -resistant transmitter release from the chick ciliary presynaptic terminal. The Journal of Physiology. 1994;477:437–448. doi: 10.1113/jphysiol.1994.sp020205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yawo H, Chuhma N, Endo K. Modulation of presynaptic calcium channels by neurotransmitters. Biomedical Research. 1994;15(suppl. 1):9–16. [Google Scholar]
- Yawo H, Momiyama A. Re-evaluation of calcium currents in pre- and postsynaptic neurons of the chick ciliary ganglion. The Journal of Physiology. 1993;460:153–172. doi: 10.1113/jphysiol.1993.sp019464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Exocytosis: a molecular and physiological perspective. Neuron. 1996;17:1049–1055. doi: 10.1016/s0896-6273(00)80238-x. [DOI] [PubMed] [Google Scholar]