

Abstract
The role of myosin light chain kinase (MLCK) in the activation of the noradrenaline-evoked non-selective cation current (Icat) was examined with the whole-cell recording technique in single rabbit portal vein smooth muscle cells.
Intracellular dialysis with 5 μm MLCK(11–19)amide, a substrate-specific peptide inhibitor of MLCK, markedly reduced the amplitude and rate of activation of noradrenaline-evoked Icat. A similar result was obtained when the cells were dialysed with 10 μm AV25, which also inhibits MLCK by an action at the auto-inhibitory domain of MLCK.
Inhibitors of binding of ATP to MLCK, wortmannin and synthetic naphthalenesulphonyl derivatives (ML-7 and ML-9), at micromolar concentrations, also reduced the amplitude of noradrenaline-evoked Icat.
ML-7 and ML-9 (both at 5 μm) reduced the amplitude of Icat induced by both guanosine 5′-O-(3-thiotriphosphate) (GTPγS) and 1-oleoyl-2-acetyl-sn-glycerol (OAG).
MLCK(11–19)amide, AV25 and ML-9 did not inhibit the noradrenaline-evoked Ca2+-activated potassium current at a holding potential of 0 mV. In addition, MLCK(11–19)amide and AV25 did not reduce the non-selective cation current induced by ATP in rabbit ear artery cells.
Intracellular dialysis with 2 μm Ca2+ and 9 μm calmodulin activated Icat, which developed over a period of about 5 min.
Intracellular dialysis with the non-hydrolysable analogue of ATP, 5′-adenylylimidodiphosphate (AMP-PNP), reduced the amplitude and rate of activation of noradrenaline-evoked Icat.
The results indicate that MLCK mediates noradrenaline-activated Icat in rabbit portal vein smooth muscle cells.
In rabbit portal vein smooth muscle cells stimulation of α1-adrenoceptors by noradrenaline evokes calcium-activated chloride and potassium currents (ICl(Ca) and IK(Ca)) and a non-selective cation current (Icat) which does not appear to be mediated by an increase in intracellular calcium concentration (Byrne & Large, 1988; Wang & Large, 1991). It has been proposed that Icat may contribute to the noradrenaline-induced depolarization and may also provide a direct influx of Ca2+ ions in vascular smooth muscle cells (Byrne & Large, 1988; Wang & Large, 1991). There is convincing evidence which suggests that the pathway that links the α1-adrenoceptor to the Ca2+-activated conductances involves G-protein activation with inositol trisphosphate (IP3) production and subsequent release of Ca2+ from an internal store (see Large & Wang, 1996). However, considerably less is known about the transduction mechanism which mediates Icat. Previously we provided evidence to suggest that noradrenaline activates Icat via a G-protein coupled to phospholipase C (PLC) and that the resulting 1,2-diacyl-sn-glycerol (DAG), produced by hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2), plays a pivotal role in the opening of the cation channels (Helliwell & Large, 1997). However, it was found that phorbol esters, which activate protein kinase C (PKC), did not activate Icat. Moreover chelerythrine, an inhibitor of PKC, did not inhibit Icat evoked by noradrenaline or the DAG analogue 1-oleoyl-2-acetyl-sn-glycerol (OAG). Therefore it was concluded that DAG activates Icat by a PKC-independent mechanism (Helliwell & Large, 1997). This finding provokes the question: how does DAG activate Icat? It is possible that DAG may open the channels directly or may involve another mediator.
In the present work we have explored the possibility that myosin light chain kinase (MLCK) may be involved in the activation of Icat. MLCK is a Ca2+-calmodulin-dependent kinase which phosphorylates the regulatory light chain of smooth muscle myosin to produce smooth muscle contraction. Although it is often stated that myosin is the only known substrate for MLCK, it has been shown that agents which affect MLCK activity modulate ionic currents in some cell types. In bullfrog sympathetic neurons it has been shown that inhibitors of MLCK activity reduced the amplitude of the M-current but not the A-type or delayed rectifier potassium currents (Akasu et al. 1993). In guinea-pig gastric myocytes it was shown that inhibitors of MLCK reduced the amplitude of the carbachol-activated non-selective cation current (Kim et al. 1997). In this latter study, the MLCK inhibitors had little effect on the same cationic current induced by guanosine 5′-O-(3-thiotriphosphate) (GTPγS) and it was concluded that the target of MLCK is the muscarinic receptor or a receptor-related protein. Therefore the above data suggest that MLCK may modulate ionic conductances.
In the present experiments we have examined the effects on noradrenaline-evoked Icat of substances that inhibit MLCK activity by three distinct mechanisms and the data suggest a central role for MLCK in the activation of Icat in rabbit portal vein smooth muscle cells.
METHODS
Cell isolation
New Zealand White male rabbits (2-2.5 kg) were killed by intravenous overdose of sodium pentobarbitone. The portal vein was removed and dissected free of connective tissue and fat in normal physiological salt solution (PSS) which contained (mm): NaCl, 126; KCl, 6; CaCl2, 1.5; MgCl2, 1.2; glucose, 10; and Hepes, 11; the pH was adjusted to 7.2 with 10 M NaOH. The tissue was then cut into strips and incubated in low-Ca2+ PSS (PSS with no added CaCl2) for 5 min at 37°C. After preincubation the solution was replaced with low-Ca2+ PSS containing 0.2–0.3 mg ml−1 protease Type XIV (Sigma) for 5 min, after which the tissue was washed with low-Ca2+ PSS. The low-Ca2+ PSS solution was then replaced with 100 μm Ca2+ PSS (PSS containing 100 μm rather than 1.5 mm CaCl2) containing 0.5–1 mg ml−1 collagenase Type IV (Sigma) and the tissue was then incubated for a further 10 min before a final wash in 100 μm Ca2+ PSS. Cells were released by trituration of the tissue through a wide-bore Pasteur pipette in 100 μm Ca2+ PSS. The solution containing dissociated cells was then centrifuged at 100 g for 2 min to form a loose pellet which was resuspended in 0.75 mm Ca2+ PSS. In some experiments single smooth muscle cells were prepared from the rabbit ear artery. The ear artery was dissected free of connective tissue before being cut into strips. The strips were placed in 100 μm Ca2+ PSS containing 5 mg ml−1 bovine serum albumin, 1 mg ml−1 papain (Sigma), 0.5–1.5 mg ml−1 collagenase Type IV (Sigma) and 2.5 mm dl-dithiothreitol, and stored overnight at 4°C. The following day the strips were incubated for 30–50 min at 37°C and then washed in 100 μm Ca2+ PSS. The cells were then released into solution by mechanical agitation of the strips of tissue using a wide-bore Pasteur pipette. The cell suspension was then centrifuged (100 g) to form a loose pellet which was resuspended in 0.75 mm Ca2+ PSS. Cells were stored on coverslips at 4°C and were used within 8 h.
Electrophysiology
Whole-cell membrane currents were measured at room temperature (20-25°C) with a List L/M EPC-7 patch clamp amplifier. Patch pipettes were made from borosilicate glass and when filled with normal pipette solution had resistances of about 4 MΩ.
Solutions and drugs
The standard extracellular K+-free PSS used in most experiments contained (mm): NaCl, 126; CaCl2, 1.5; glucose, 10; and Hepes, 11; the pH was adjusted to 7.2 with NaOH. In the present experiments noradrenaline-evoked Icat was recorded in 50 μm external Ca2+ because under these conditions the amplitude of Icat is increased by about eightfold (Helliwell & Large, 1996) thus facilitating analysis of the currents. The increase in amplitude of Icat in reduced [Ca2+]o is thought to be due to relief from channel blockade by Ca2+ ions without change in the current-voltage characteristics (see Helliwell & Large, 1996). Therefore the extracellular PSS [Ca2+] was changed from 1.5 mm to 50 μm about 1 min before noradrenaline was applied. In a few experiments we demonstrated that MLCK inhibitors reduced noradrenaline-evoked Icat when the bathing solution contained 1.5 mm Ca2+. The standard pipette solution contained (mm): CsCl, 18; caesium aspartate, 108; MgCl2, 1.2; glucose, 10; Hepes, 11; BAPTA, 10; and CaCl2, 1 (free [Ca2+]i predicted by EQCAL software (Biosoft, Ferguson, MO, USA) was approximately 14 nm); the pH was adjusted to 7.2 with Trizma base. In some experiments in which noradrenaline-evoked Ca2+-activated potassium currents (IK(Ca)) were measured the external solution contained (mm): NaCl, 131; KCl, 6; MgCl2, 1.2; CaCl2, 0.2; glucose, 11; and Hepes, 10. The pipette solution contained (mm): NaCl, 5; KCl, 126; glucose, 11; Hepes, 10; BAPTA, 0.1; and MgCl2, 1.2. The pH of these solutions was also adjusted to 7.2 with NaOH.
In experiments in which the influence of high intracellular [Ca2+] (i.e. 400 nm and 2 μm ) was studied, 400 nm and 2 μm were achieved by adding, respectively, 8 and 9 mM CaCl2, as estimated with EQCAL software, to the standard pipette solution. GTPγS, OAG (both from Sigma) and calmodulin (from bovine brain; Sigma) were made up as 10 mm stocks in DMSO and diluted to the final concentrations with pipette solution. 5′-Adenylylimidodiphosphate (AMP-PNP) was purchased from Sigma and was made up as a 10 mm stock in distilled water (stored as frozen aliquots) and diluted to final concentrations with the pipette solution on the day of experimentation. 1-(5-Chloronaphthalene-1-sulphonyl) homopiperazine, HCl (ML-9), 1-(5-iodonaphthalene-1-sulphonyl) homopiperazine, HCl (ML-7) (both from Calbiochem) and wortmannin (Sigma) were also made up as 10 mm stocks in DMSO (stored as frozen aliquots) and diluted to final concentrations on the day of experimentation. The concentration of DMSO (0.1 %) had no effect on the amplitude of noradrenaline-evoked Icat.
Synthetic peptide inhibitors of myosin light chain kinase
The myosin light chain kinase inhibitor peptide MLCK(11–19)amide was purchased from Alexis Corporation Ltd (Nottingham, UK). This compound is a substrate-specific inhibitor of MLCK and corresponds to the region around the phosphorylation site (i.e. serine19) of myosin light chain (MLC) from chicken gizzard with the sequence: lysine11-lysine12-arginine13-alanine14-alanine15-arginine16-alanine17-threonine18-serine19. Two substitutions (in bold) were made where proline and glutamine at positions 14 and 15, respectively, were replaced with alanine (Kemp et al. 1983). This was done in order to improve the inhibitor potency of the synthetic peptide (see Kemp et al. 1983; Pearson et al. 1986). Similarly, AV25 (which was a gift from Dr M. P. Walsh, University of Calgary, Canada) is a synthetic peptide which inhibits the removal of the auto-inhibitory particle of MLCK. AV25 is patterned after the auto-inhibitory domain of myosin light chain kinase (Weber et al. 1999). AV25 denotes the first and the last amino acid residues, and the length of the peptide. AV25 corresponds to amino acid residues 783–807 of chicken gizzard (i.e. AKKLAKDRMKKYMARRKLQKAGHAV), with three substitutions (in bold italics). Tryptophan at position 800 was replaced with leucine to remove any influence from interaction of the peptide with calmodulin. Serine at position 787 and threonine at position 803 were replaced by alanine to avoid peptide phosphorylation by other kinases such as Ca2+-calmodulin kinase II (Weber et al. 1999).
MLCK(11–19)amide and AV25 were made up as 1 mm stock solutions in distilled water (stored as frozen aliquots) and diluted to the final concentrations with the pipette solution on the day of experimentation. After the whole-cell configuration was achieved, 5–10 min was allowed for sufficient dialysis of the peptides and AMP-PNP before any recordings were made. Control currents were also obtained using a pipette solution which contained the equivalent amount of distilled water.
The values in the text are means ±s.e.m. (n, number of cells examined) and the test for statistical significance was Student's t test. P < 0·05 was considered significant.
RESULTS
Effect of specific peptide inhibitors of MLCK on noradrenaline-evoked Icat
Figure 1A shows the non-selective cation current produced by bath-applied noradrenaline (100 μm) in an external solution containing 50 μm Ca2+. In this cell the peak amplitude of Icat was about 175 pA; the current then decayed spontaneously in the continued presence of noradrenaline due to an unknown inactivation/desensitization process. Figure 1B illustrates the response to 100 μm noradrenaline in a cell where the patch pipette contained the MLCK substrate-specific inhibitor MLCK(11–19)amide (5 μm). It can be clearly seen that the current was markedly reduced compared with the control response shown in Fig. 1A. The mean data from three populations of cells are shown in Fig. 1C, where it can be seen that intracellular dialysis with 5 μm MLCK(11–19)amide reduced the amplitude of Icat by about 80 %.
Figure 1. The effect of MLCK(11–19)amide on noradrenaline-activated Icat.
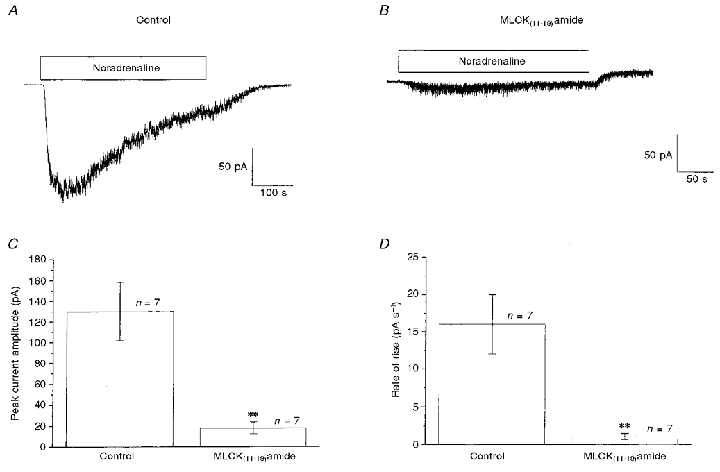
A, an example of a control Icat activated by bath-applied 100 μm noradrenaline, applied during the period denoted by the horizontal bar. B, Icat activated by bath-applied noradrenaline 5 min after achieving whole-cell configuration with a pipette solution containing 5 μm MLCK(11–19)amide. C and D show quantitatively the effects of MLCK(11–19)amide on the peak amplitude (pA; C) and the activation rate (pA s−1; D) of Icat. In this and subsequent figures error bars represent the s.e.m. and the numbers (n) above the columns indicate the number of cells examined. Records are from different cells. **P < 0·01.
Another feature of the inhibitory effect of MLCK(11–19)amide was that the rate of activation of Icat, as estimated from 10–90 % of the rise time, was also markedly decreased from a control value of 16 pA s−1 to 1 pA s−1 (Fig. 1D). It should be noted that in these experiments control currents were obtained using a pipette solution which contained the equivalent amount of distilled water required to dissolve the peptides MLCK(11–19)amide and AV25.
In another series of experiments we investigated the effects of intracellular dialysis with AV25, which also inhibits MLCK activity but by binding to a different site on the MLCK molecule (see Methods). Figure 2 shows noradrenaline-induced Icat under control conditions (Fig. 2A) and in another cell after 5 min dialysis with 10 μm AV25 in the patch pipette solution, where the amplitude of Icat was greatly decreased (Fig. 2B). In three populations of cells intracellular dialysis with 10 μm AV25 reduced the amplitude of Icat by about 72 % (Fig. 2C). Also, as with MLCK(11–19)amide, the rate of activation of Icat was markedly depressed by AV25 (Fig. 2D).
Figure 2. The effect of AV25 on noradrenaline-activated Icat.
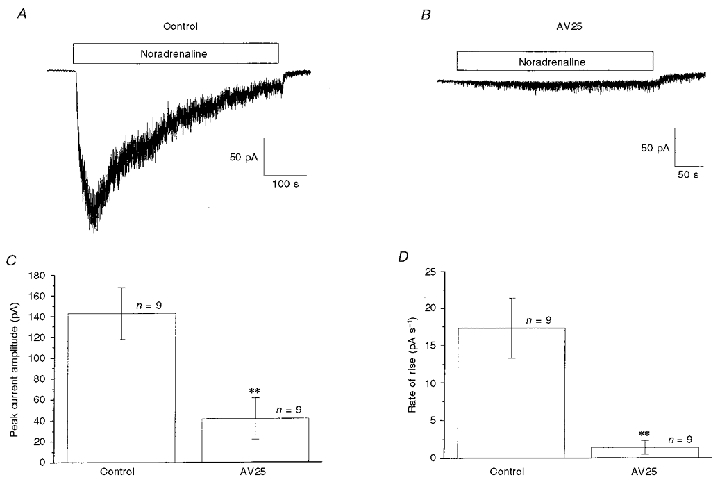
A, control Icat evoked by bath-applied noradrenaline (100 μm, horizontal bar). B illustrates Icat recorded 5 min after achieving whole-cell configuration with a pipette solution containing 10 μm AV25. C and D show the effect of 10 μm AV25 on peak amplitude (C) and the activation rate (D) of Icat. Records are from different cells. **P < 0·01.
Effects of agents that inhibit binding of ATP to MLCK on noradrenaline-evoked Icat
Experiments were then carried out to ascertain whether agents that inhibit the binding of ATP to MLCK, and hence prevent its action, affect Icat. First, we investigated the effects of wortmannin, which is also known to inhibit phosphoinositide 3-kinase (PI 3-kinase) at nanomolar concentrations (Shepherd et al. 1996). Figure 3A illustrates a control current to noradrenaline and Fig. 3B shows that the current activated by noradrenaline was not reduced in the presence of 50 nm wortmannin. In three populations of cells the control amplitude of noradrenaline-evoked Icat was 309 ± 70 pA (n = 7) compared with 310 ± 56 pA (n = 7) in the presence of 50 nm wortmannin. These results indicate that PI 3-kinase is not involved in the activation of Icat and therefore we investigated whether micromolar concentrations of wortmannin, at which it inhibits MLCK, affect Icat.
Figure 3. Effects of wortmannin on Icat.
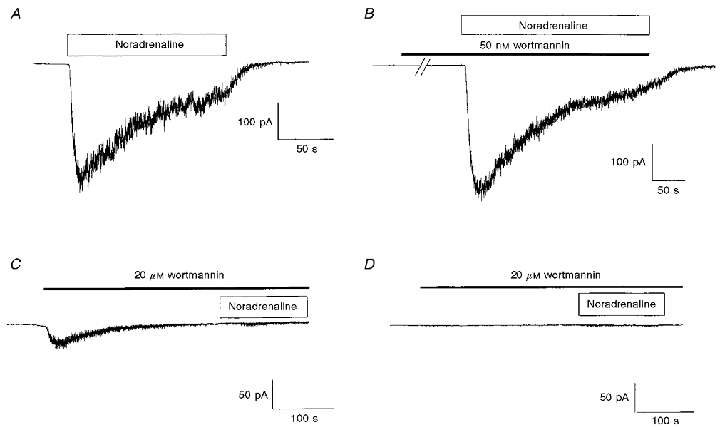
A, example of Icat activated by bath application of 100 μm noradrenaline in the absence of wortmannin. B, Icat activated by bath-applied noradrenaline 5 min after preincubation with 50 nm wortmannin. The break in the record represents about 4 min. C, in another cell bath application of 20 μm wortmannin evoked a ‘noisy’ inward current similar to the noradrenaline-activated Icat. However, subsequent application of noradrenaline in the continued presence of wortmannin failed to activate a current. D illustrates a cell where 20 μm wortmannin did not evoke a current and subsequent application of noradrenaline (in the continued presence of wortmannin) failed to activate a current. The duration of application of noradrenaline and wortmannin is indicated by the horizontal open and filled bars, respectively.
A higher concentration of wortmannin (20 μm), when applied on its own, activated in some cells a ‘noisy’ inward current similar to noradrenaline-evoked Icat (Fig. 3C). In five cells the mean amplitude of the wortmannin-induced current was 60 ± 17 pA, compared with a value of 158 ± 25 pA (n = 5) produced by noradrenaline (100 μm) in the same population of cells. The rate of activation of the wortmannin-evoked current (5 ± 1 pA s−1) was slower than that of the current produced by noradrenaline (18 ± 1 pA s−1). Addition of noradrenaline in the presence of 20 μm wortmannin did not activate a current, although sometimes there was a small increase in ‘noise’ (e.g. Fig. 3C).
In five other cells, 20 μm wortmannin did not evoke a current but almost totally blocked the response to noradrenaline applied subsequently (Fig. 3D). Thus, the control response to noradrenaline was 148 ± 27 pA (n = 5) and in the presence of wortmannin (20 μm) the amplitude of noradrenaline-evoked Icat was 6 ± 2 pA (n = 5). Overall these data show that wortmannin at micromolar concentrations inhibits noradrenaline-evoked Icat but that in some cells Icat is activated by wortmannin.
Subsequently, the effects of the synthetic napthalenesulphonyl compounds ML-7 and ML-9, which also inhibit the binding of ATP to MLCK, were investigated. Figure 4A–C shows that application of ML-9 and ML-7 (both at 5 μm) during activation of Icat by noradrenaline almost completely abolished the current. The relationship between the inhibitory effect on Icat and the concentration of ML-7 and ML-9 is illustrated in Fig. 4D. The IC50 values (concentration required to produce 50 % inhibition of Icat) were 0.8 and 2 μm for ML-7 and ML-9, respectively. It is apparent from Fig. 4D that the slope of the curves for ML-7 (0.9) and ML-9 (2.5) were different and also that in some cells these compounds produced greater than 100 % inhibition, i.e. they also reduced the holding current (see Discussion). Interestingly, in seven out of 25 cells, after the initial inhibition produced by ML-7 and ML-9 there was partial recovery of the noradrenaline-evoked Icat, although there was still marked inhibition of the current. There is no obvious explanation for this observation.
Figure 4. Effects of ML-9 and ML-7 on Icat.
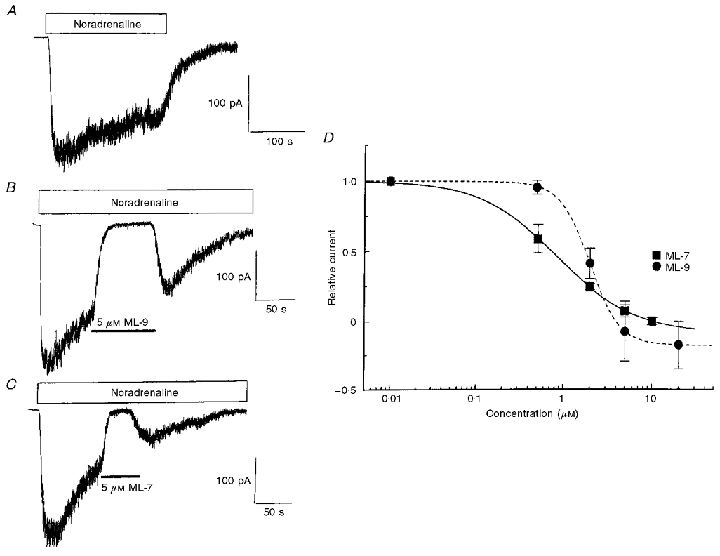
A, Icat recorded by bath application of 100 μm noradrenaline in the absence of ML-9 and ML-7. B and C show the inhibition of Icat by 5 μm ML-9 (B) and 5 μm ML-7 (C) applied in the continued presence of noradrenaline. D shows the concentration-dependent inhibitory effects of ML-9 and ML-7, where the current amplitude (pA) in the presence of ML-9 or ML-7 was normalized to the current amplitude immediately prior to application (Relative current). The relative current is plotted against the concentrations of ML-9 and ML-7 on a logarithmic scale. The data could be fitted with a logistic equation of the following form: y =ymax[xnH/(IC50nH+xnH)], where x denotes the concentration of ML-9 or ML-7 and the slope (nH) was 2.5 and 0.9 for ML-9 and ML-7, respectively. This gave estimated IC50 values (the concentrations of ML-9 and ML-7 required to inhibit the current amplitude by 50 %) of 2 and 0.8 μm for ML-9 and ML-7, respectively. Each data point represents the mean of 5–6 cells.
Effect of ML-9 on Icat evoked by GTPγS and OAG
Previously we have demonstrated that intracellular dialysis with GTPγS and bath-applied OAG evoked Icat (Helliwell & Large, 1997), and we carried out experiments to assess whether ML-9 inhibits these currents. Figure 5A and C shows control Icat activated by, respectively, intracellular dialysis with 500 μm GTPγS and bath application of 10 μm OAG, as described previously (Helliwell & Large, 1997). Application of 5 μm ML-9 produced a marked decrease in the amplitude of Icat evoked by GTPγS (Fig. 5B) and OAG (Fig. 5D). ML-9, at a concentration of 5 μm, inhibited the GTPγS- and the OAG-induced Icat by, respectively, 100 ± 4 % (n = 5) and 92 ± 5 % (n = 6). Also, 5 μm ML-7 reduced the GTPγS-evoked current by 93 ± 7 % (n = 5; not shown).
Figure 5. Effect of ML-9 on GTPγS- and OAG-activated Icat.
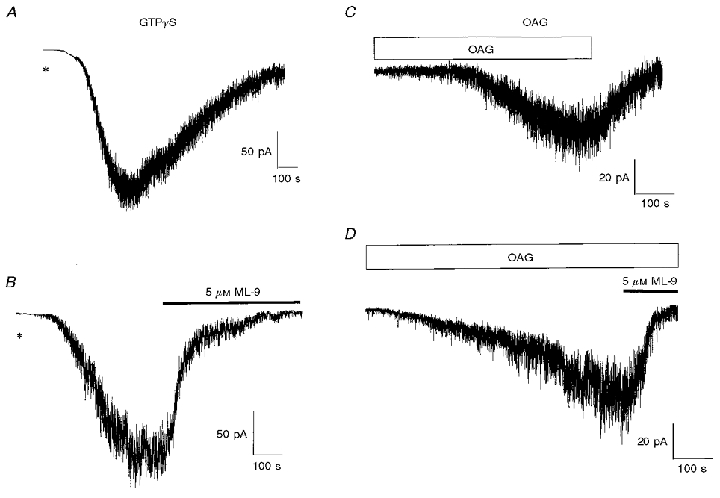
A, representative trace of Icat evoked by inclusion of 500 μm GTPγS in the pipette solution 2–4 min after achieving whole-cell configuration (denoted by *). B illustrates the effect of ML-9 applied during the sustained phase of the GTPγS-evoked Icat. C, bath application of OAG (10 μm, horizontal bar) activated a slowly developing ‘noisy’ inward current. D illustrates the effect of ML-9 applied during the sustained phase of the OAG-activated Icat.
Effect of MLCK inhibitors on noradrenaline-evoked Ca2+-activated K+ conductance
We carried out experiments to test whether the inhibitors of MLCK have general non-specific effects on cell function. For these studies we investigated whether these agents inhibited the Ca2+-activated K+ current evoked by 100 μm noradrenaline with K+-containing pipette and bathing solutions at a holding potential of 0 mV. It was observed that MLCK(11–19)amide (5 μm, in the pipette), AV25 (10 μm, in the pipette) and 5 μm ML-9 had no effect on the noradrenaline-induced IK(Ca). Thus the control and test amplitudes of noradrenaline-induced IK(Ca), respectively, were 0.85 ± 0.2 nA (n = 4) and 0.86 ± 0.1 nA (n = 5) for MLCK(11–19)amide, 2.0 ± 0.6 nA (n = 4) and 2.0 ± 0.3 nA (n = 4) for AV25, and 2.0 ± 0.5 nA (n = 4) and 2.0 ± 0.2 nA (n = 5) for ML-9.
In order to test whether the peptide inhibitors of MLCK had an inhibitory effect on non-selective cation currents we investigated their action on the inward current activated by ATP in rabbit ear artery cells, using the same bathing and pipette solutions as those used to record Icat in portal vein cells. The response to ATP is mediated by a ligand-gated receptor but is similar to noradrenaline-evoked Icat in portal vein cells in respect of a significant permeability to divalent cations (cf. Benham et al. 1987; Benham & Tsien, 1987; Wang & Large, 1991). With 5 μm MLCK(11–19)amide in the pipette, the amplitude of the ATP-induced current (IATP) in ear artery cells was 340 ± 70 pA (n = 3), compared with a control current of 322 ± 74 pA (n = 3). With 10 μm AV25 in the pipette solution, IATP was 168 ± 20 pA (n = 4), compared with a control IATP of 161 ± 20 pA (n = 4). It can be concluded that the inhibitory peptides of MLCK do not have a non-specific effect on non-selective cation channels.
Effect of increasing intracellular Ca2+ and including calmodulin in the pipette solution on noradrenaline-evoked Icat
The above data suggest that MLCK may be involved in the activation of noradrenaline-evoked Icat. With regard to smooth muscle contraction MLCK is activated by an increase in the intracellular calcium concentration, with subsequent binding of Ca2+ to the Ca2+-binding protein calmodulin. This sequence leads to activation of the kinase and therefore we investigated whether increasing the pipette Ca2+ concentration ([Ca2+]i) in the presence of calmodulin could activate Icat. Figure 6A illustrates noradrenaline-evoked Icat with [Ca2+]i at 14 nm. When 400 nm Ca2+ and 9 μm calmodulin were included in the pipette solution there was an increase in the background ‘noise’ but no net inward current over a period of 5–10 min. Subsequent application of noradrenaline evoked Icat (Fig. 6B). With 400 nm and 9 μm calmodulin the amplitude of noradrenaline-evoked Icat was 109 ± 15 pA (n = 6), compared with 148 ± 25 pA (n = 8) with 14 nm Ca2+ in the pipette solution.
Figure 6. Effect of high intracellular calcium concentration and calmodulin on noradrenaline-evoked Icat.
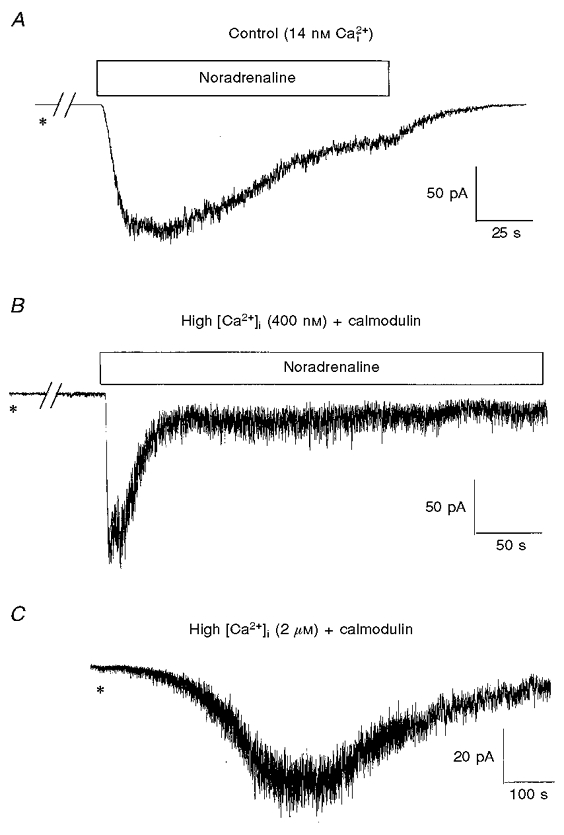
A, example of Icat activated by bath application of noradrenaline 5 min after achieving whole-cell configuration (*) using a pipette solution containing a free calcium concentration of 14 nm. B, Icat evoked with 400 nm Ca2+ plus 9 μm calmodulin in the pipette solution. C illustrates the activation of Icat with a pipette solution containing 2 μm Ca2+ and 9 μm calmodulin. In A and B, noradrenaline (100 μm) was applied for the period denoted by the horizontal bars. The breaks in the records in A and B represent about 4 min. Note the differences in time scale.
Intracellular dialysis with 2 μm Ca2+ and 9 μm calmodulin induced a large ‘noisy’ inward current with a mean amplitude of 60 ± 14 pA (n = 7). However, this current developed relatively slowly and had a mean time to peak of 334 ± 40 s (n = 7). Thus it can be concluded that Icat can be activated by increasing the intracellular calcium concentration.
Role of phosphorylation in activation of noradrenaline-evoked Icat
In the final series of experiments we carried out a simple test to ascertain whether phosphorylation was important in the activation of noradrenaline-evoked Icat, which would be predicted if a kinase was involved in the transduction mechanism. In this study cells were dialysed with a pipette solution containing 5 mm AMP-PNP, a non-hydrolysable analogue of ATP. It can be seen that after 5 min dialysis with AMP-PNP the amplitude of the noradrenaline-induced Icat (Fig. 7B) was much smaller than the control response (Fig. 7A, no AMP-PNP). The mean data from three cell populations are shown in Fig. 7C and as with the peptide inhibitors AMP-PNP also significantly decreased the rate of activation of Icat (Fig. 7D). Therefore it can be concluded that protein phosphorylation plays a role in the activation of Icat.
Figure 7. Effect of AMP-PNP on Icat.
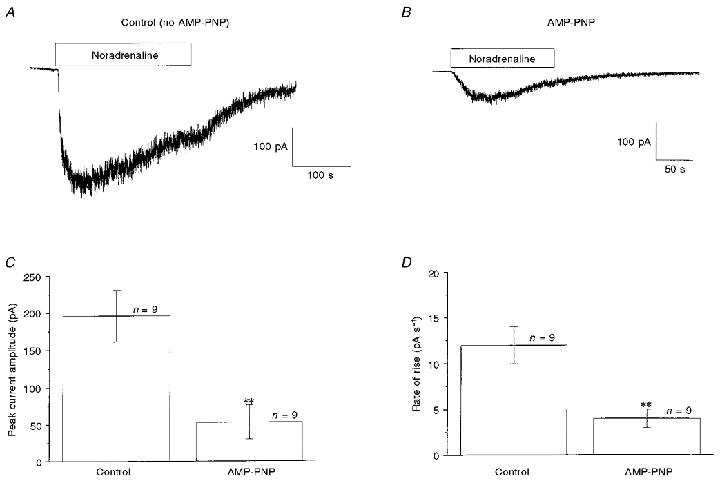
A, Icat evoked by bath-applied noradrenaline (100 μm, horizontal bar) in the absence of AMP-PNP. B, Icat evoked 5 min after achieving whole-cell configuration with a pipette solution containing 5 mm AMP-PNP. C and D show the quantitative effect of AMP-PNP on the peak amplitude (C) and the rate of activation (D) of Icat. **P < 0·01.
DISCUSSION
The present work provides evidence for a role for MLCK in the activation of noradrenaline-evoked Icat in rabbit portal vein smooth muscle cells. This conclusion is based on the observed marked reduction in amplitude and rate of activation of Icat by several chemically different inhibitors which act at different sites on the MLCK molecule. In our experiments we used concentrations of the MLCK inhibitors that are close to the estimated Ki values obtained in biochemical studies where the ability of MLCK to phosphorylate myosin was assessed. Thus when myosin kinase activity was assayed the Ki values of MLCK(11–19)amide and AV25 were about 10 μm (Pearson et al. 1986) and 0.2 μm (Weber et al. 1999), respectively. In our studies we used 5 μm MLCK(11–19)amide and 10 μm AV25 in the pipette solution. Also the Ki values for smooth muscle MLCK of ML-7 and ML-9 were 0.3 and 3.8 μm, respectively (Saitoh et al. 1987). In the present study the IC50 values of ML-7 and ML-9 for inhibition of noradrenaline-evoked Icat were 0.8 and 2.0 μm, respectively, i.e. similar to the Ki values obtained in the biochemical experiments. Consequently, the concentrations of inhibitors used in the present study are in the range expected to produce inhibition of MLCK. Wortmannin at micromolar concentrations also inhibits MLCK and the concentration used (20 μm) has little effect on cAMP-dependent protein kinase, cGMP-dependent protein kinase and calmodulin-dependent protein kinase II (Nakanishi et al. 1992).
MLCK(11–19)amide, AV25 and ML-9 did not reduce the noradrenaline-evoked IK(Ca). Since this conductance involves the well-known α1-adrenoceptor-G-protein-phosphoinositide-IP3-Ca2+-store pathway (see Large & Wang, 1996), it can be deduced that these compounds do not compromise (a) binding of noradrenaline to α1-adrenoceptors, (b) G-protein activation, (c) hydrolysis of PIP2 by PLC to produce IP3 and DAG, (d) release of Ca2+ ions from the sarcoplasmic reticulum and (e) the opening of Ca2+-activated K+ channels. Therefore these inhibitors of MLCK do not have general non-specific effects on cell function. Moreover the inhibitory peptides did not inhibit the non-selective cation currents induced by ATP in ear artery cells. Therefore it is apparent that the compounds used inhibited noradrenaline-evoked Icat in portal vein cells without compromising cellular function or blocking non-selective cation channels. It is not possible to state unequivocally that the compounds used did not affect other kinases, but since MLCK(11–19)amide, AV25 and the ML-compounds (and wortmannin) act at three distinct sites on the MLCK molecule, namely the substrate site (MLCK(11–19)amide), the auto-inhibitory site (AV25) and the ATP-binding site (ML-compounds and wortmannin), the most likely explanation for our data is that activation of Icat by noradrenaline involves MLCK. It has also been suggested that MLCK is involved in the bradykinin-stimulated Ca2+ influx in endothelial cells (Watanabe et al. 1998).
If MLCK is involved in the activation of Icat it might be expected that the introduction of high concentrations of Ca2+ and calmodulin into the cell via the pipette would activate Icat. It was observed that 2 μm Ca2+ in the pipette solution (with calmodulin) did induce Icat, indicating that an increase in [Ca2+]i does activate Icat. However, the relative slowness of this response (a time to peak of about 5 min) shows that this is not a classical Ca2+-activated conductance. For example, with 1 μm Ca2+ in the pipette solution a Ca2+-activated chloride current is evoked immediately (within 1 s) after rupture of the cell membrane in rabbit portal vein cells (I. A. Greenwood, personal communication). Therefore, our data indicate that Icat may be activated by a Ca2+-activated process (e.g. a calcium-sensitive kinase) although we feel that noradrenaline can evoke Icat without a rise in [Ca2+]i. Interestingly, a high molecular weight isoform of MLCK has been identified in bovine endothelial and smooth muscle cells, which can be activated in the absence of a rise in cytosolic [Ca2+] (Verin et al. 1998; Gilbert-McClain et al. 1998).
It was observed that the ML-compounds also inhibited Icat evoked by GTPγS and OAG. This is in contrast to the result of Kim et al. (1997), who demonstrated that whereas the carbachol-induced non-selective cation current in guinea-pig gastric myocytes was inhibited by micromolar concentrations of ML-7, the same current evoked by GTPγS was not inhibited by ML-7. These authors concluded that the inhibitory site of ML-7 was upstream of the G-protein and was either the muscarinic receptor or a closely related molecule. Our data indicate that the target for the ML-compounds is downstream of GTPγS and OAG. The lack of effect of the inhibitors on noradrenaline-evoked IK(Ca) also suggests that the inhibitory effect on Icat occurs at a stage between the production of DAG, which is produced simultaneously with IP3, and the opening of the non-selective cation channel. Taking together the previous results of Helliwell & Large (1997; see Introduction) and the present work a plausible transduction mechanism is: α1-adrenoceptor → G-protein → PLC activation → DAG → MLCK → cation channel opening.
At present we are uncertain of the links between DAG and MLCK, and between MLCK and the ion channel, but AMP-PNP also produced a substantial reduction of the amplitude of Icat, which indicates that protein phosphorylation is involved in activation of the conductance. MLCK may phosphorylate myosin light chain leading to channel opening by some unknown mechanism or alternatively MLCK may phosphorylate directly the channel protein, or perhaps an intermediate substrate, to produce channel opening.
There are some effects of the inhibitors of the binding of ATP to MLCK that require further comment. Firstly, in some cells wortmannin itself activated a current as well as inhibiting the response to noradrenaline applied subsequently and it is possible that wortmannin may act as a partial agonist at MLCK, i.e. possess the ability to stimulate and block the kinase. Secondly, in some cells the ML-compounds appeared to reduce the holding current. This observation suggests that these agents may also affect conductances other than Icat but it should be noted that in some unstimulated cells there seemed to be a low resting activity of Icat (as manifested by the background ‘noise’) before noradrenaline was applied. Thus Icat may be active before application of noradrenaline in some cells, which may, in part at least, account for the reduction of the holding current by ML-compounds, although other actions cannot be ruled out.
In conclusion, these experiments suggest that MLCK is involved in the activation of noradrenaline-evoked non-selective cation channels in rabbit portal vein smooth muscle cells.
Acknowledgments
We are grateful to Dr M. P. Walsh for the gift of AV25 and helpful discussions. The work was supported by The British Pharmacological Society and The British Heart Foundation.
References
- Akasu T, Ito M, Nakano T, Schneider CR, Simmons MA, Tanaka T, Tokimasa T, Yoshida M. Myosin light chain kinase occurs in bullfrog sympathetic neurons and may modulate voltage-dependent potassium currents. Neuron. 1993;11:1133–1145. doi: 10.1016/0896-6273(93)90226-h. [DOI] [PubMed] [Google Scholar]
- Benham CD, Bolton TB, Byrne NG, Large WA. Action of externally applied adenosine triphosphate on single smooth muscle cells dispersed from rabbit ear artery. The Journal of Physiology. 1987;387:473–488. doi: 10.1113/jphysiol.1987.sp016585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham CD, Tsien RW. A novel receptor-operated Ca2+-permeable channel activated by ATP in smooth muscle. Nature. 1987;328:275–278. doi: 10.1038/328275a0. [DOI] [PubMed] [Google Scholar]
- Byrne NG, Large WA. Membrane ionic mechanisms activated by noradrenaline in cells isolated from the rabbit portal vein. The Journal of Physiology. 1988;404:557–573. doi: 10.1113/jphysiol.1988.sp017306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert-McClain LI, Verin AD, Shi S, Irwin RP, Garcia JGN. Regulation of endothelial cell myosin light chain phosphorylation and permeability by vanadate. Journal of Cellular Biochemistry. 1998;70:141–155. doi: 10.1002/(sici)1097-4644(19980701)70:1<141::aid-jcb14>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Helliwell RM, Large WA. Dual effect of external Ca2+ on noradrenaline-activated cation current in rabbit portal vein smooth muscle cells. The Journal of Physiology. 1996;492:75–88. doi: 10.1113/jphysiol.1996.sp021290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helliwell RM, Large WA. α1-Adrenoceptor activation of a non-selective cation current in rabbit portal vein by 1,2-diacyl-sn-glycerol. The Journal of Physiology. 1997;499:417–428. doi: 10.1113/jphysiol.1997.sp021938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp BE, Pearson RB, House C. Role of basic residues in the phosphorylation of synthetic peptides by myosin light chain kinase. Proceedings of the National Academy of Sciences of the USA. 1983;80:7471–7475. doi: 10.1073/pnas.80.24.7471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CY, Kim JS, Kang MT, Suh HS, So I, Kim WK. Effects of myosin light chain kinase inhibitors on carbachol-activated nonselective cationic current in guinea-pig gastric myocytes. Pflügers Archiv. 1997;434:346–353. doi: 10.1007/s004240050407. [DOI] [PubMed] [Google Scholar]
- Large WA, Wang Q. Characteristics and physiological role of the calcium-activated chloride conductance in smooth muscle. American Journal of Physiology. 1996;271:C435–454. doi: 10.1152/ajpcell.1996.271.2.C435. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, Kakita S, Takahashi I, Kawahara K, Tsukuda E, Sano T, Yamada K, Yoshida M, Kase H, Matsuda Y, Hashimoto Y, Nonomura Y. Wortmannin, a microbial product inhibitor of myosin light chain kinase. Journal of Biological Chemistry. 1992;267:2157–2163. [PubMed] [Google Scholar]
- Pearson RB, Misconi LY, Kemp BE. Smooth muscle myosin kinase requires residues on the COOH-terminal side of the phosphorylation site. Journal of Biological Chemistry. 1986;261:25–27. [PubMed] [Google Scholar]
- Saitoh M, Ishikawa T, Matsushima S, Naka M, Hidaka H. Selective inhibition of catalytic activity of smooth muscle myosin light chain kinase. Journal of Biological Chemistry. 1987;262:7796–7801. [PubMed] [Google Scholar]
- Shepherd PR, Reaves BJ, Davidson HW. Phosphoinositide 3-kinases and membrane traffic. Trends in Cell Biology. 1996;6:92–97. doi: 10.1016/0962-8924(96)80998-6. [DOI] [PubMed] [Google Scholar]
- Verin AD, Lazar V, Torry RJ, Labarrere CA, Patterson CE, Garcia JGN. Expression of a novel high molecular-weight myosin light chain kinase in endothelium. American Journal of Respiratory Cell and Molecular Biology. 1998;19:758–766. doi: 10.1165/ajrcmb.19.5.3125. [DOI] [PubMed] [Google Scholar]
- Wang Q, Large WA. Noradrenaline-evoked cation conductance recorded with the nystatin whole-cell method in rabbit portal vein cells. The Journal of Physiology. 1991;435:21–39. doi: 10.1113/jphysiol.1991.sp018496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Takahashi R, Zhang XX, Goto Y, Hayashi H, Ando J, Isshiki M, Seto M, Hidaka H, Niki I, Ohno R. An essential role of myosin light-chain kinase in the regulation of agonist- and fluid flow-stimulated Ca2+ influx in endothelial cells. FASEB Journal. 1998;12:341–348. doi: 10.1096/fasebj.12.3.341. [DOI] [PubMed] [Google Scholar]
- Weber LP, Van Lierop JE, Walsh MP. Ca2+-independent phosphorylation of myosin in rat caudal artery and chicken gizzard myofilaments. The Journal of Physiology. 1999;516:805–824. doi: 10.1111/j.1469-7793.1999.0805u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]