

Abstract
This study was conducted to search for the residues of the β3 subunit which affect pentobarbital action on the γ-aminobutyric acid type A (GABAA) receptor. Three chimeras were constructed by joining the GABAA receptor β3 subunit to the ρ1 subunit. For each chimera, the N-terminal sequence was derived from the β3 subunit and the C-terminal sequence from the ρ1 subunit, with junctions located between the membrane-spanning regions M2 and M3, in the middle of M2, or in M1, respectively.
In receptors obtained by the coexpression of α1 with the chimeric subunits, in contrast with those obtained by the coexpression of α1 and β3, pentobarbital exhibited lower potentiation of GABA-evoked responses, and in the direct gating of Cl− currents, an increase in the EC50 together with a marked decrease in the relative maximal efficacy compared with that of GABA.
Estimates of the channel opening probability through variance analysis and single-channel recordings of one chimeric subunit showed that the reduced relative efficacy for gating largely resulted from an increase in gating by GABA, with little change in efficacy of pentobarbital.
A fit of the time course of the response by the predictions of a class of reaction schemes is consistent with the conclusion that the change in the concentration dependence of activation by pentobarbital is due to a change in pentobarbital affinity for the receptor. Therefore, the data suggest that residues of the β3 subunit involved in pentobarbital binding to GABAA receptors are located downstream from the middle of the M2 region.
Pentobarbital affects GABAA receptor-mediated responses in several ways. At low micromolar concentrations it potentiates GABA-evoked responses, at high micromolar concentrations it opens GABAA receptors directly and at millimolar concentrations it reduces the response (Akaike et al. 1987b).
Since the ability to enhance the ion channel activation of GABAA receptors is a common feature of several general anaesthetics (Franks & Lieb, 1994), the action on the GABAA receptor is a probable major molecular mechanism for anaesthetic action in the mammalian central nervous system (Tanelian et al. 1993). However, general anaesthetics do not exert the same effects on all the GABA-gated receptor Cl− ion channels (Thomson et al. 1996). In particular, those composed of ρ1 homomers exhibit little if any response to anaesthetics (Shimada et al. 1992).
A combination of molecular biological, pharmacological and physiological approaches has provided a great deal of information on the portions of the GABAA receptor subunits which affect the response of benzodiazepines (see Smith & Olsen, 1995), of GABA (Amin & Weiss, 1993) and some classes of anaesthetics (Mihic et al. 1997). However, only a few recent studies (Birnir et al. 1997; Krasowski et al. 1998b; Amin, 1999) have provided some initial insights into the regions involved in the physiological effects of barbiturates. In the present study, chimeric subunits were generated between the human β3 subunit of the GABAA receptor and the human ρ1 subunit, with the goal of localizing amino acid residues affecting pentobarbital actions on GABAA receptor channels.
Three constructs were prepared in which the upstream, N-terminal, part of the β3 subunit was joined to the downstream, C-terminal, part of ρ1 (Fig. 1). In each chimera, the junction was located in the region between the N-terminal end of M1 and the N-terminal end of M3. The junction was progressively moved upstream from the middle of the M2-M3 linker (c7), to the middle of M2 (c1), or M1 (c2). If the amino acids of β3 required for pentobarbital responses are replaced by the corresponding residues of ρ1, a decrease in activity should result. Studies on the residues involved in the binding of GABA to GABAA receptors (Amin & Weiss, 1993) and acetylcholine to nicotinic receptors (reviewed in Karlin & Akabas, 1995) indicate that amino acids responsible for the binding of a ligand may be located in several subunits (reviewed in Karlin & Akabas, 1995) and that within each subunit they can be distributed in widely separated regions (Amin & Weiss, 1993). Therefore, pentobarbital effects may not be necessarily eliminated by replacement of the binding residues of one subunit, and in each subunit they may be altered by mutations of residues lying in a long stretch of the subunit primary sequence.
Figure 1. Summary of the chimeric subunits studied.
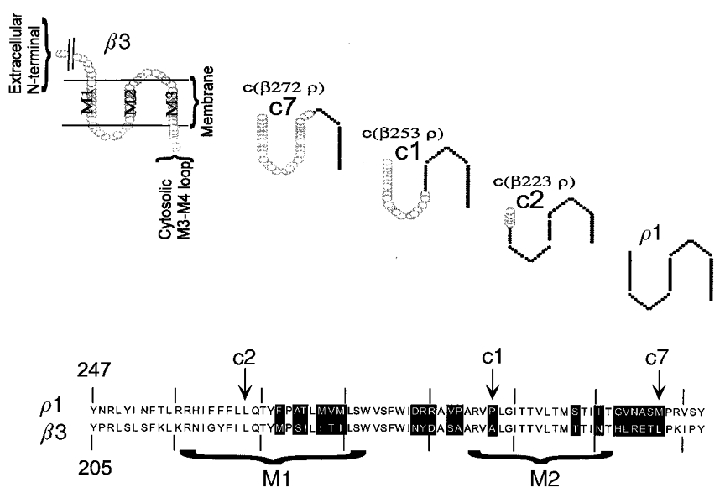
The upper panel shows cartoons of the structures of the chimeric subunits studied. Only the region including the membrane-spanning domains M1, M2 and M3 is shown, as all chimeras were generated by joining subunits within these regions. The residues at the left of M1 belong to the extracellular N-terminal, and the residues at the right of M3 belong to the C-terminal portion (not shown). In the chimeras generated for this work, the β3 subunit contributes the residues at the N-terminal of the joining site, and the ρ1 subunit the residues to the C-terminal. In c7 the joining point is located between residue 272 of β3 and 315 of ρ1, in c1 between β3 253 and ρ1 295, and in c2 between β3 223 and ρ1 265. The lower panel shows an alignment of amino acid residues in this region of the β3 and ρ1 subunits. The membrane-spanning regions are indicated by brackets, and the sites of the joins are indicated by arrows.
Because the hypnotic properties of barbiturates are related to lipid solubility (reviewed in Gallagher & Freer, 1985), binding sites for anaesthetics may be located in a lipophilic pocket of the receptor, and the transmembrane domains may possibly contain residues that bind pentobarbital. However, in the nicotinic acetylcholine receptor, these regions contain residues which can dramatically affect gating mechanisms, as well (reviewed in Karlin & Akabas, 1995). Therefore, to interpret observations we have also attempted to distinguish changes in affinity from changes in pentobarbital efficacy.
Chimeric subunits were expressed in combination with GABAAα1 subunits, and initially concentration-effect curves were obtained for gating by GABA, for pentobarbital potentiation of GABA-evoked responses, for direct gating by pentobarbital and for block by pentobarbital. Then the relative maximal responses elicited by GABA and pentobarbital were compared and the Popen of channel activation by GABA and pentobarbital were estimated. Finally, the time courses of responses to pentobarbital were fitted by the predictions of a class of reaction schemes, to test the adequacy of our analysis. The results indicate that residues affecting pentobarbital potentiation of the GABA-evoked response are localized in a region of the β3 subunit extending from M1 to the M2-M3 linking region. Furthermore, in one chimera (c1, formed in the M2 region), the affinity of pentobarbital for the site involved in direct gating has been reduced. This suggests that residues involved in the binding of pentobarbital are located downstream of the middle of the M2 domain.
The results of the study have been presented in preliminary form (Serafini et al. 1997, 1998).
METHODS
All chemicals were obtained from Sigma Chemical Co. (St Louis MO, USA) unless otherwise specified.
Constructs
The expression construct for the rat α1F pcDNA3 was previously described (Ueno et al. 1996). cDNAs for human ρ1 and human β3 were transferred to the pAlter-1 vector and mutagenized (Altered Sites II, Promega, Madison, WI, USA), to create silent mutations producing restriction endonuclease sites for chimera generation using the following oligonucleotides:
ρ1 X1 site BlnI: CAGAGTCCCCCTAGGTATCAC
ρ1 X2 site Pst I CTTCTTCTTCTTGCTGCAGACTTATTTCCCCG
β3 X2 site Pst I GGATACTTCATTCTGCAGACTTATATGC.
Mutated subunits were transferred to the eucaryotic expression vector pcDNA3 (Invitrogen, San Diego, CA, USA).
The chimeric subunit c1 was made between the rat β3 subunit and the human ρ1 subunit, since the rat subunit has an existing Bln 1 site. There is one amino acid difference between the rat and human β3 subunits in the sequence included; the rat subunit has M at position 231 (in the M1 region), while the human has L (Ymer et al. 1989; Wagstaff et al. 1991). The subunits were digested with Bln 1 and subcloned to form chimera c1 joining the N-terminal residues of β3 to amino acid 253 to the C-terminal residues of ρ1 starting at amino acid 295.
All other chimeras were made between human β3 and human ρ1 subunits. c2 was made by joining human β3 (223) to human ρ1 (265) after Pst 1 digestion. c7 was made by PCR overlap extension (Ho et al. 1989) to produce a fragment containing the chimeric portion. This fragment was subcloned to make c7 joining human β3 (273) to human ρ1 (315)
The chimeras are summarized in Fig. 1. The sequences of the β3 and ρ1 subunits were confirmed, and all chimeric joining regions were resequenced before use.
cDNA constructs for GABAA receptor subunits were provided by A. Tobin, University of California Los Angeles (rat α1), D. Weiss, University of Alabama at Birmingham (rat β3 and human ρ1), and G. White, Neurogen, Brandford, CT, USA (human β3) originally cloned by L. Mahan. Restriction endonucleases were obtained from Boehringer Mannheim Corporation (Indianapolis, IN, USA).
Cell culture and transfection
Quail fibroblasts (QT6 cells; initially provided by Dr J. Merlie, Washington University) were maintained in Medium 199 (Earle's salts) containing 5 % fetal bovine serum (Hyclone), 10 % tryptose phosphate broth (Gibco, Grand Island, NY, USA), 1 % DMSO and penicillin (100 units ml−1) plus streptomycin (100 μg ml−1) in a humidified atmosphere containing 5 % CO2. Calcium phosphate precipitation was used to transfect QT6 cells (Chen & Okayama, 1987; see Ueno et al. 1996), with the additional step of an initial wash to remove tryptose phosphate broth.
Cells which expressed a high level of protein from exogenous cDNA were identified using a bead labelling technique (see Ueno et al. 1997). For all experiments in which both α1 and non-α (β3 or chimeric) subunits were transfected, the α1 subunit was tagged with an inserted FLAG epitope at the N-terminal (Ueno et al. 1996). Previous work has shown that the α1 subunit is not expressed on the surface of the cells as a homomultimer (Ueno et al. 1996), so the presence of bead binding demonstrated that a significant amount of heteromultimeric receptors was present on the surface. We used a mouse monoclonal antibody to the FLAG epitope (M2, Eastman Kodak Scientific Imaging Systems, New Haven, CT, USA), which had been adsorbed to beads with covalently attached goat anti-mouse IgG antibody (Dynal, Lake Success, NY, USA) to identify the FLAG epitope. Control experiments indicated that the epitope had no functional effects on receptors incorporating the tagged α1 subunit (Ueno et al. 1996). For recordings on ρ1 homomers, GABA receptor subunits were co-transfected with cDNA for the CD8 antigen (kindly provided by B. Seed, Massachusetts General Hospital), and cells were identified with beads which had covalently coupled antibody to CD8 (Dynabeads, Dynal, Lake Success, NY, USA).
Electrical recordings
Electrical measurements were obtained through patch-clamp recording. Intracellular (pipette) solution for whole-cell recordings contained (mM): 140 CsCl2, 3 MgCl2, 10 Hepes and 10 BAPTA. The extracellular solution for whole-cell recording contained (mM): 140 NaCl, 5 KCl, 2 mM CaCl2, 10 mM glucose and 10 Hepes. For cell-attached recordings the pipette solution contained (mM): 120 sodium isethionate, 10 TEA-Cl, 5 KCl, 5 4-aminopyridine (4-AP), 0.1 CaCl2, 2 MgCl2, 10 glucose and 10 Hepes. Osmolarity and pH of recording solutions were adjusted to 300–320 mosmol l−1 and 7.2–7.3, respectively.
All whole-cell recordings were obtained at a holding potential of -60 mV. Data were recorded and amplified through an Axopatch 1C and acquired through a Digidata 1200 interface (Axon Instruments, Foster City, CA, USA) to a PC hard drive. Data acquisition and analysis was performed through pCLAMP6 (Axon Instruments).
In the studies of direct gating, for each experiment a normalizing concentration was chosen. Each cell was exposed to the normalizing concentration and one or more test concentrations. The amplitude of the response to the test concentrations was expressed as its ratio to the amplitude of the response to the normalizing concentration. Finally, the maximal evoked response was defined, and data were reported as a fraction of it. In the studies of potentiation, a concentration of 1 μM GABA was used for all constructs, corresponding to 15–20 % of the maximal response to GABA (see Results). In studies of block, the peak of the tail current (see Results) and the current immediately before removal of pentobarbital were measured, and fractional block estimated from the ratio (Itail–Iend)/Itail.
Drugs were applied through a previously described apparatus (Maconochie & Knight, 1989) with some modifications. The head of the solution reservoir was adjusted so that the 10–90 % time for junction potential changes was 2–7 ms with an open pipette tip and ∼50 ms with whole-cell recording. Quicker exchange times dislodged the cell from the recording pipette.
GABA was applied for 10 s in studies of ρ1 homomers, and for 2 s in all other studies. Other drug applications were for 2 s.
Some responses were quite large (for example, to high [GABA] in cells transfected with α1 +β3 or with ρ1 subunit). However, responses were unlikely to have been seriously affected by series resistance. Series resistance was typically 15 MΩ, and was compensated 70–90 % using the amplifier circuitry. The theoretical voltage error for the largest responses ranged between 3 and 24 mV. However, at the highest concentrations no correlation was noticed between the concentration dependence of the response and the expected voltage error. Furthermore, poor series resistance compensation with large amplitude currents would be expected to reduce the relative amplitude of the (large) tail current to the smaller current at the end of the application. However, no correlation between this ratio and the absolute current amplitude was evident (not shown).
Concentration-effect relationships were fitted to the pooled data using the Hill equation:
where R(D) is the response, Rmax is the maximal response, D is the drug concentration, EC50 is the concentration giving half of the maximal response and nHis the Hill coefficient. The fit was performed using Kaleidograph (Synergy Software, Reading, PA, USA). Error estimates on fit parameters are generated during the fitting procedure as the squared roots of the diagonal elements of the covariance matrix.
Non-stationary variance analysis
Whole-cell responses to GABA were analysed to determine the relationship between the variance and the mean of the currents (see Sigworth, 1984). Since at the highest GABA concentration the response fades, the analysis was performed on the portion of the response corresponding to the rise (see Fig. 8). Data were selected from cells which provided data for several concentrations of GABA. The time course of the response was estimated by fitting the response with an exponential function, or the sum of two exponentials. However, only the segment of the response near the peak was analysed because it had the lowest slope and allowed highest accuracy in fitting. This segment included a section of increasing current rising up to the peak. At the higher concentrations a decline from the peak was evident but this part was not analysed. The segments had durations ranging from ∼400 ms to 40–50 ms at the lowest (1 μM) and highest ligand concentrations (100 μM), respectively. At the highest GABA concentrations desensitization had little if any effect on the amplitude of the peak (see Results), and therefore is unlikely to contribute to the variance. For each experimental value of current, i(t), the squared deviation from the fitted mean value, f(t), was calculated:
Figure 8. Non-stationary variance analysis of GABA-gated whole-cell responses.

We performed non-stationary variance analysis of GABA-evoked whole-cell responses for α1β3 and α1c1 receptors to estimate the probability that a channel is open (Popen). The upper panel shows the experimental approach (see Methods). A response to 10 μM GABA from a cell expressing α1β3 receptors is shown on the left. The variance near the peak was analysed, as shown at the top right. This portion of the response was fitted with the sum of two exponentials (smooth line through the data) to approximate the mean response. The variance was estimated from the squared deviations of the data from the smooth fit. This analysis was performed at the peak of the responses over a range of GABA concentrations (1, 3, 10, 30, 100, 300 and 1000 μM). Difference traces (not squared deviations) are shown in the middle and lower left panels. Finally, data were accumulated for several cells with similar maximal responses to GABA (4.3–4.7 nA) with each subunit combination. For a given subunit combination, the estimated variance and fitted mean currents were then averaged in bins by pooling all of the responses. The variance versus mean current plots constructed are shown in the middle and lower right panels. Data from α1β3 receptors show a linear relationship, suggesting that Popen remains below 0.5 even at high concentrations of GABA. In contrast, data from α1c1 receptors show a parabolic form consistent with the idea that the maximal Popen approaches 0.85 at a high GABA concentration. Activation by GABA has an EC50 value of less than 10 μM for both of these constructs (see Fig. 4), so the data indicate that the maximal Popen value has been increased in the c1 construct relative to β3. The inferred unitary conductance values are 5 pS in α1β3 and 1 pS in α1c1.
The values were binned and averaged over intervals of mean current (f(t)) to provide average values for the residual and mean current. The average value of the residuals of each binned interval represent an estimate of the variance for that interval. Finally, the binned data were pooled for all responses for cells expressing a given type of receptor at different GABA concentrations.
A simple quantitative estimate of the channel open probablility (Popen) from the current-variance plot is established only for recordings of channels with one single-channel conductance but a qualitative estimate can be made even for channels exhibiting multiple-conductance states. The data we obtained with single-channel recording indicate that channel opening can be reliably approximated as a channel with one single-channel conductance (see Appendix II).
Estimate of the probability of being open (Popen) from single-channel recordings
Single-channel recordings were performed in the cell-attached configuration. Previous experience with cell-attached recordings indicated that recordings with a good signal-to-noise ratio can be obtained by using pipette solutions with potassium channel blockers (Serafini et al. 1995) at hyperpolarized patch potentials and that linear single-channel current-voltage plots can be obtained with Cl− concentrations in the pipette solution approximating the intracellular concentration. All single-channel recordings were obtained with the low Cl− pipette solution (see solutions for electrical recordings). In these ionic conditions, the reversal of the GABA-evoked current is expected to occur at a patch potential close to 0 mV, that is, at some negative pipette potential. Conversely, at positive pipette potentials the amplitude is expected to increase. In fact, this type of activity was seen in 17 out of 35 patches when GABA or pentobarbital was in the recording pipette. Furthermore, those channels, whose amplitude increased at positive pipette potential, exhibited a time-dependent decrease in activity, if recording pipettes contained high desensitizing GABA concentrations. Of 12 control recordings, none exhibited any similar activity.
The current record shows the channel activity in the patch. On average, this is the product of the number of channels in the patch multiplied by the Popen of the individual channel. To analyse the records, the single-channel current amplitude was first estimated from the all-points histograms for short segments of data, by fitting with the sum of multiple Gaussian distributions separated by a constant step amplitude (see Fig. 9).
Figure 9. Analysis of cell-attached recordings of patches showing a high activity of low amplitude channels.
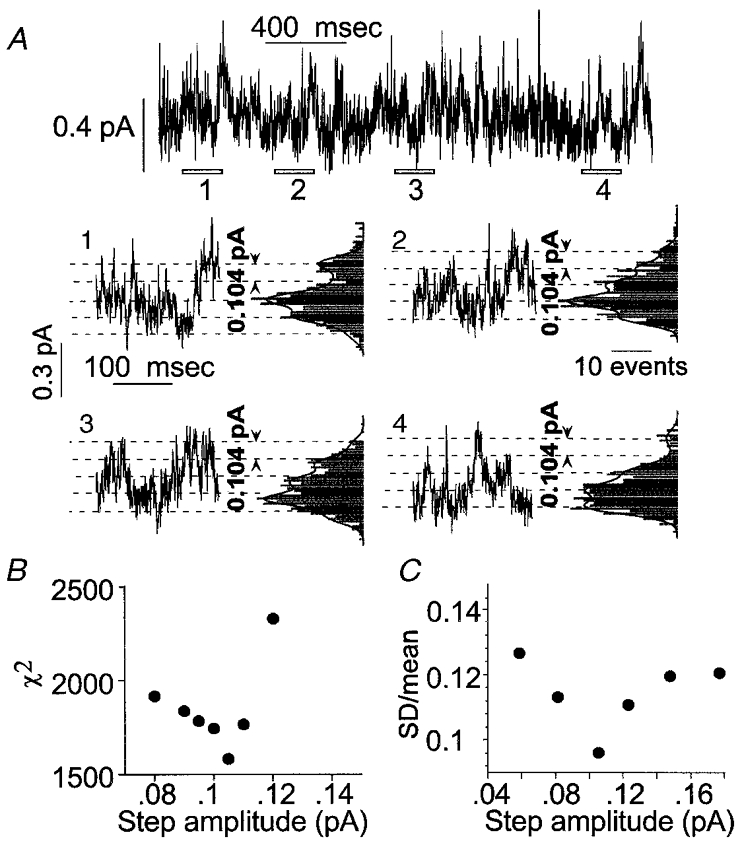
The record shown in the top line of A was obtained from a cell-attached patch on a cell transfected with α1 and c1 subunits. It was difficult to identify current steps corresponding to the opening or closing transitions of individual channels in these records, as they reflect the aggregate activity of a large number of channels of low unitary conductance. In order to define the amplitude of channel currents, each recording was divided into short consecutive stretches. The open bars marked 1 to 4 indicate a selection of such stretches at different times of the recording. The data segments are shown in A1 to A4, with corresponding all point current histograms. The histograms show regularly spaced peaks or shoulders. The lines show the sums of multiple Gaussian distributions with peaks separated by a constant value of 0.104 pA for all histograms. The unitary current will correspond to the peak spacing for the fitted Gaussian functions. Two tests were made of the idea that the all-points histograms could be described by the sum of multiple, equally spaced Gaussian components. The first was to fit all four distributions simultaneously with the sum of multiple Gaussian components. The total number of components was set to 6, and the step amplitude was varied in the fitting but kept constant for all histograms. For each histogram, there were additional parameters including the weight for each of the 6 components fitted, the s.d. for the components in each histogram (the same for all Gaussians fitted to a given histogram) and the baseline offset for each histogram. The fit was performed by minimization of χ2, using a Simplex algorithm. The best fitting value for the step amplitude was 0.104 pA, as illustrated. B shows values for χ2 for step amplitudes near this value, showing the minimum. The second approach was to analyse the original data segments using different assumed values for the step amplitude. For each assumed value, the data were divided into windows centred on integral multiples of the assumed step amplitude. The means and s.d.s were calculated in each window. If the appropriate step size were selected, each window would be centred on a single current level and the variance would be generated by open channel noise and transient values captured during opening or closing transitions. If, on the other hand, the step size were so large as to include more than one integral multiple of the single-channel current, the variance would be increased. Alternatively, at small step sizes the variance relative to the mean would also be increased because a larger proportion of the values would probably reflect transient values during transitions. The data shown in C indicate that coefficient of variation (s.d./mean) is minimized by step amplitudes of ≈0·1 pA (cell-attached recordings obtained with low Cl− extracellular saline at a pipette potential of 60 mV).
To infer the Popen of an individual channel, the number of channels in each patch were estimated through the GC Bayesian algorithm described by Horn (1991). The GC Bayesian algorithm is not model dependent and utilizes a beta function for the estimate of Popen and a gamma function to estimate the number of channels. Both the beta and the gamma function contain two parameters (a and b) which are related to the initial range of estimates for N (number of channels) and Popen (Horn, 1991). Although a and b can be given values in the absence of any previous information on N and Popen, a more efficient estimate may be achieved by utilizing insight from independent observations. In our experiments, an independent estimate of the channel Popen was given by the ratio between the steady-state response and the maximal evoked response, in the whole-cell recording. For example, in the whole-cell recordings the steady-state response to 1 μM GABA in cells transfected with α1c1 was 0·12-fold the maximal evoked response, and therefore the Popen of single-channel recordings was unlikely to be higher than 0·12. Simulations of single-channel recordings with known Popen and number of channels were run, with Popen values between 0·04 and 0·2; channel amplitudes, background noise and number of channels were adjusted to obtain recordings resembling the experimental ones. These single-channel simulations were performed through Strathclyde Electrophysiology Software (provided by J. Dempster of the University of Strathclyde, Glasgow, UK) using a simple model with two states. The GC estimator was tested in these simulations and the parameters a and b of the GC estimator were optimized to reduce the error of estimates for the simulations. After optimization of a and b, the estimated N value would have been 80 % of the real value for Popen= 0·04, ∼95–105 % for Popen between 0·08 and 0·15, and ∼120 % for Popen= 0·2. Finally, calculations were applied to the experimental recording. Similar principles and criteria were used with recordings from cells transfected with other constructs. However, in the responses evoked by GABA in cells transfected with α1β3, data from non-stationary noise analysis indicated a maximal Popen lower than 0.5. The GABA concentrations used for the cell-attached recording determined in the whole-cell recordings a steady-state response which was 0·1-fold the maximal peak evoked response. Therefore a and b were optimized to expect Popen lower than 0.05. Finally to infer the maximal Popen from these data, we assumed that different levels of activation of the receptor channel would affect the probability of the open state without modifying the unitary single-channel amplitude (see Appendix 2), and we calculated a rough correction using the whole-cell concentration-response curves for GABA- and pentobarbital-evoked responses. For example, in α1c1 the GABA-evoked response (1 μM GABA) at steady state is 8-fold less than the maximal evoked response. At steady state, with 1 μM GABA the Popen is 0·11. Therefore the maximal Popen is expected to approximate 0·9.
How accurate are rise times of the response in the whole-cell recording in reflecting channel kinetics?
In the whole-cell recordings the speed of the solution exchange on the cell surface might alter the time course of the response. Before attempting an estimate of the binding Kd (dissociation constant; see Appendix 1) by fitting the time course of the response to different reaction schemes, we evaluated how and to what extent the speed of the solution exchange could result in unreliable estimates of kinetic parameters, and especially of those related to binding.
The time course of the response is affected by both the speed of the application and the time course of the cellular response. We modelled the system by assuming that the cell was approximated by a line segment parallel to the flow with uniformly distributed receptors, and that the solution swept over the line segment at a uniform rate. The proportion of receptors activated, therefore, increased linearly from 0 to 1 during the assumed exchange time (tex) for the solution and can be divided into two parts; the interval necessary for the solution to cover the entire segment (0 < t < tex) and the following development of the response (t > tex), respectively. The observed response can be calculated in each of these two parts by integrating the theoretical response through the corresponding time intervals. In Fig. 2, predicted responses are shown for the effect of the perfusion time course on an intrinsic response consisting of a single exponential time course (τR). The response shows a sigmoidal start, as a result of the increasing proportion of receptors exposed to agonist, then an inflection point followed by an exponential approach to the final steady level. Clearly, if the intrinsic response is very rapid (τR≪tex) the response will follow the solution exchange, while if the intrinsic response is very slow (τR≫tex) the response will follow the intrinsic response time course. In cases in which τR and tex are approximately equal, the initial part of the response up to the inflection point is determined by both the perfusion and intrinsic response, while the final approach to the peak provides a good estimate of the time constant for the intrinsic response. The results also indicate that a correct estimate of the time constant of the response can be obtained even with τR 2–4 times smaller than tex. For τR smaller than one fifth tex the final exponential approach is too small to be accurately fitted.
Figure 2. Effect of the speed of the perfusion system on the time course of the response.
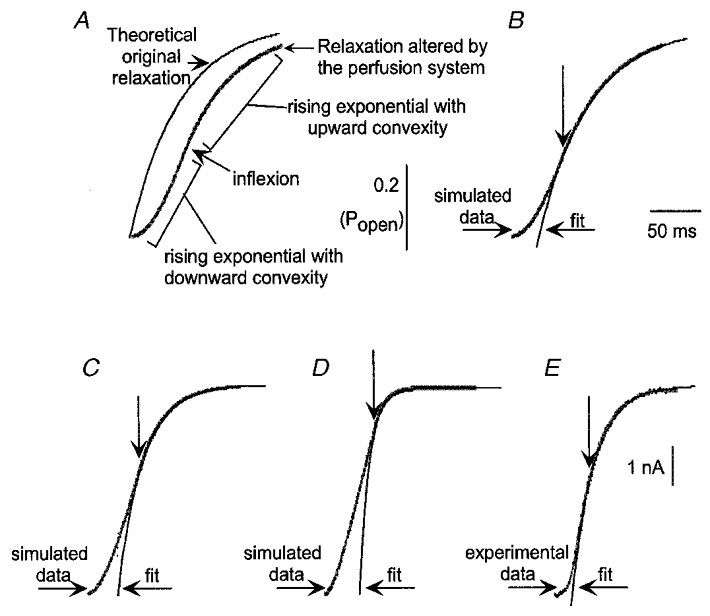
This figure shows calculated responses (A, B, C and D) and the rising phase of a recorded tail current on removal of pentobarbital (E). For the calculations, it was assumed that the perfusion resulted in a linear increase in the fraction of receptors exposed to ligand over a time period of 50 ms (tex= 50 ms; see Methods). The response of receptors was modelled as a simple exponential response with a time constant τR as (1 − exp(−t/τR). In A and B, τR= 50 ms (τR/tex= 1); in C, τR= 25 ms (τR/tex= 0·5); and for D, τR= 10 ms (τR/tex= 0·2). A shows the response predicted for instantaneous perfusion (thin line), and that predicted for tex= 50 ms (thick line). The initial sigmoidal start of the response is clear, followed by an inflection into a final approach to the peak. The responses were fitted with a single exponential, beginning at the times indicated by the downward arrows. For B, C and D the time constant fit (τfit) values were 50, 25 and 10 ms, respectively (identical to τR), although in D the amplitude of the relaxation was so small that in actual data the fit might have been much less accurate. E shows the initial portion of the tail current recorded at the end of an application of 1 mM pentobarbital to a cell transfected with α1β3 subunits (the current has been inverted to make it the same sign as in A-D. The basic shape is the same as in the simulated responses. The interval between the start of the tail current and the inflection was ≈35 ms. The fit was started after the inflexion point and yielded a τfit value of 25 ms.
The rate of development of the pentobarbital-evoked response was slower than the exchange time of the solutions and therefore is expected to reflect the intrinsic channel kinetics (at low concentrations the initial development had a time constant greater than 100 ms, while even at the highest concentrations it decreased to about 30–50 ms). The portion of the response in our data which exhibited the greatest speed was the rise of the tail at the end of an application of a high concentration of pentobarbital. The tail current had an initial foot lasting up to 50 ms, likely to reflect the solution exchange, followed by an inflexion into an exponential relaxation with a time constant of ∼20 ms. The peak of the tail also is unlikely to be significantly distorted, since the fast component of decay of the current has a time constant of ∼100 ms. Accordingly, the time course and amplitude of the tail current are likely to have been determined with sufficient accuracy for fitting kinetic models to the time course. The analysis was directed to estimating values for the affinity of pentobarbital to the activation sites on the receptor. The association and dissociation rates for binding to these sites should be reflected most strongly in the development of the initial response and the decay of the tail current, which were relatively unaffected by perfusion speed.
Fit of data to a kinetic scheme
To calculate the time course of the response we used previously established algorithms (Colquhoun & Hawkes, 1977). For a k × k Q matrix (k is the number of states) the time course of the response I(t) is the following:
where N is the number of channels, V is the driving force on ion movement through the channel, p (0) is the vector of initial probabilities for the k states, Ai are the matrices calculated for spectral expansion of Q, λi are the eigenvalues of the Q matrix; Γ is the vector of conductances of the different states; and u is a post-multiplying k× 1 unit vector allowing the sum of all the terms. Parameter optimization was performed by χ2 minimization through an ‘amoeba’ downhill simplex algorithm in multiple dimensions (Press et al. 1996a) coupled to Q-matrix calculations written in Mathematica (Wolfram, Champaign, IL, USA) by R. Serafini. Parameters were not constrained for the reaction mechanisms to obey the principle of microscopic reversibility. Complicated models, such as the ones used to interpret pentobarbital effects, often can result in trapping of the routine in local minima. For this reason, several dozen sets of parameters had to be tested as initial values before a set of values was obtained which gave a good fit of the data by eye. Preliminary attempts to find parameters providing a good fit of experimental data indicated that the decay of the tail current was affected both by the dissociation rate constant for pentobarbital at the activation site (koff) and by the closing rate of the channel (α), but that it was not possible to define in an unambiguous way both these parameters. We found also that the data could be fitted by a wide range of values for the association rate constant for pentobarbital at the activation site (kon) and koff and of α and β (channel opening rate) provided that the ratios koff/kon and α/(α+β) remained constant. Since the goal of the kinetic analysis was to provide insight on whether the mutation affected the binding affinity, koff/kon and α/(α+β) were the effective parameters used by the iterations of the simplex routine.
In order to define the errors in the parameter estimates we performed a simulation with the reaction scheme shown in Fig. 12 and the numeric microscopic rate constants estimated for α1β3 and we re-estimated parameters by fitting the simulated data to the theoretical predictions of the response time course.
Figure 12. The ability of one kinetic model to describe the time courses of pentobarbital action.
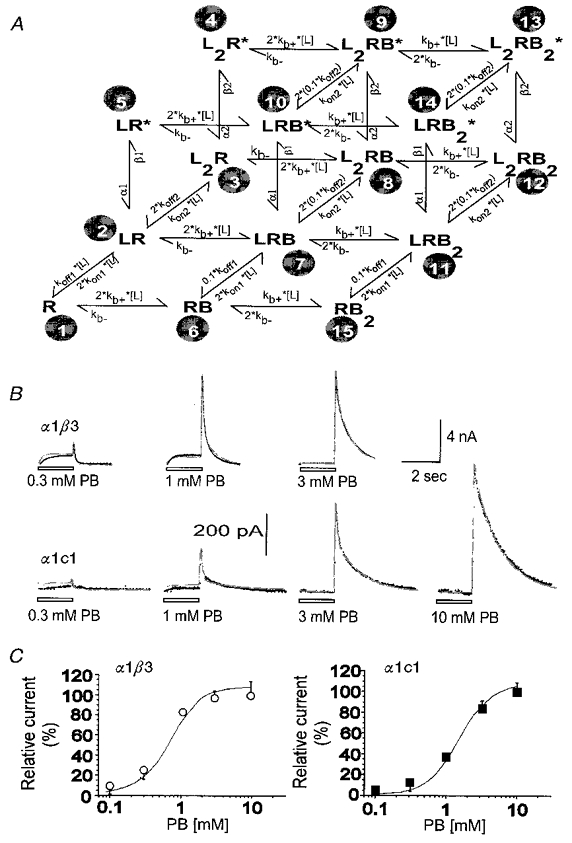
This figure shows the ability of a variant of model P8 (termed the ‘independent model’ in the text) to predict experimental data. A shows the independent kinetic scheme used to fit the time courses of responses. B shows responses obtained from a cell transfected with α1 and β3 subunits (upper row) and a cell transfected with α1 and c1 subunits (lower row). In each case responses to several concentrations of pentobarbital were obtained from the same cell. Superimposed on the data are predicted responses generated by fitting the predictions of the scheme shown in A simultaneously to all of the responses recorded from that cell. The quality of the fits is reasonable over the concentration range examined. The fit obtained with the independent model is very similar to that obtained with the coupled model both in α1β3 (see Fig. 11) and in α1c1 (not shown). The best-fitting parameter values for all of the cells analysed are summarized in Table 2. C shows the dose-response data of the peak of tail current (shown also in Fig. 5) fitted with the predictions of the scheme of A and the Kd values shown in Table 3.
To generate simulated data we proceeded as following. The method of Colquhoun & Hawkes (1977) was used to calculate the sum of exponential functions describing the theoretical time course of the response. The effect of perfusion delay was considered only in the response off-rate, and the exponential functions corresponding to the off-rate were manipulated, in order to model the effect of a perfusion exchange time of 50 ms (see the paragraph ‘How accurate are the rise times of the response…’ above). Finally, the probability values of being open, Popen (t1y2), Popen (t2), … Popen (tj), … Popen (tn), were calculated at successive time intervals (t1, t2, …tj, …tn) matching the experimental sampling rate. The simulated time course was supposed to originate from channels with a unitary current amplitude of 0.6 pA. Such a value should be close to that expected for recordings with α1β3, that is to the amplitude of channels with a unitary conductance of 10 pS under a 60 mV driving force. The number of functional ion channels expressed was arbitrarily set to 20 000. For each Popen (tj) the expected macroscopic current (in pA) is m(tj) = 0·6 × 20 000 Popen (tj). In order to add noise fluctuations we calculated a set of n pseudorandom numbers R(t1), R(t2), …R(tj), …R(tn) each of them belonging to a normal distribution with mean value μ(tj) =m(tj) and standard deviation δ(tj) =[0·6 m(tj) – (m(tj)2/20 000)]0·5. This procedure allows each R(tj) to be randomly scattered around an average value m(tj) with errors normally distributed around the expected value. The variance of the fluctuations is related to the macroscopic current by a parabolic, bell-shaped relationship similar to that expected for real channels. Baseline noise was not added because the noise corresponding to baseline fluctuations of recordings in α1β3 was far smaller than that of ion-channel openings. The simulated time course corresponds to the plot of number pairs {[t1, R(t1)], [t2, R(t2)], …[tj, R(tj)], …[tn, R(tn) ]} and exhibits a close resemblance by eye with the experimental recordings. Finally, the simulated data were analysed through time course fitting in a manner identical to that applied to experimental recordings. Confidence limits of parameters were estimated by defining contours of constant Δχ2 (see Press et al. 1996b). This analysis was performed also with other reaction schemes (such as Scheme III of Appendix 1) to test the possibility of generalizing the reliability of the measurements obtained with this procedure (not shown).
RESULTS
General overview
We tested GABA and pentobarbital sensitivity in homomultimeric receptors expressed in cells transfected with only β3, c7, c1, c2 or ρ1 subunits. Our attention, however, was focussed on experiments performed on heteromultimeric receptor channels expressed in cells transfected with wild-type α1 subunit plus β3 or a chimeric subunit. We will present the results in two parts: a descriptive characterization of whole-cell responses followed by an interpretation of the differences in pentobarbital evoked-responses observed between α1β3 and α1c1 receptors.
Concentration-effect curves were obtained for gating by GABA, for the potentiation by pentobarbital of the GABA-evoked response, for the direct gating by pentobarbital curves, and for the block by pentobarbital. The channel Popen for GABA- and pentobarbital-activated currents was estimated by variance analysis and single-channel recording. Finally the binding affinity for pentobarbital was estimated by fitting several kinetic models to observations.
Descriptive characterization of whole-cell responses
Spontaneous currents and differential responses to GABA and pentobarbital distinguish homomultimeric from heteromultimeric receptors
We measured the holding currents in cells expressing the different constructs shortly after breaking the patch into the whole-cell configuration (see Table 1). The holding currents were largest in cells expressing homomultimeric receptors containing β3 or chimeric subunits. In those constructs for which larger spontaneous current was evident, such as β3, c2 and α1c2, this was markedly reduced by application of 1 mM picrotoxinin, and the reduction in current was paralleled by a decrease in the membrane conductance (data not shown). The sensitivity of these spontaneous currents to an inhibitor of GABA-gated currents suggested that they are due to unliganded gating of transfected channel subunits, as previously shown for β3 (Woolworton et al. 1997)
Table 1.
Summary of the functional properties of the constructs studied as defined by whole-cell recordings
| GABA gating | Pentobarbital gating | Pentobarbital block | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Ihold (pA) | EC50 (μm) | nH | EC50 (mm) | nH | IC50 (mm) | nH | Potentiation | Relative gating | |
| α1β3 | 101 ± 19 (16) | 6·2 ± 0·9 | 0·9 ± 0·05 | 0·6 ± 0·04 | 2·3 ± 0·3 | 0·58 ± 0·06 | 1·7 ± 0·1 | 9·5 ± 2·7 (3) | 2·0 ± 0·9 (3) |
| α1c7 | 44 ± 10 (33) | 9·2 ± 3·0 | 0·6 ± 0·1 | 1·1 ± 0·1 | 2·3 ± 0·6 | 1·23 ± 0·13 | 1·9 ± 0·3 | 4·9 ± 1·6 (3) | 2·8 ± 0·6 (4) |
| α1c1 | 142 ± 28 (43) | 5·3 ± 0·7 | 1·0 ± 0·1 | 1·1 ± 0·1 | 1·9 ± 0·4 | 0·47 ± 0·01 | 1·7 ± 0·05 | 1·8 ± 0·4 (3) | 0·3 ± 0·06 (4) |
| α1c2 | 617 ± 69 (26) | 3·1 ± 0·8 | 0·9 ± 0·05 | NR | * | (> 1) | * | 1·01 ± 0·0 (3) | (0) |
| p1 | 82 ± 9 (140) | 7·3 ± 0·4 | 1·6 ± 0·3 | NR | * | (> 1) | * | 1·0 | (0) |
| β3 | 241 ± 71 (23) | NR | * | (<0·05, > 1) | * | * | * | * | * |
| c7 | 110 ± 38 (6) | NR | * | (> 1) | * | * | * | * | * |
| c1 | 214 ± 71 (7) | NR | * | (∼0·7) | * | * | * | * | * |
| c2 | 720 ± 91 (8) | NR | * | NR | * | * | * | * | * |
Data are shown for the heteromeric receptors (upper 4 rows) and homomeric receptors (lower 5 rows) examined. The first column gives the composition of the subunits transfected. The second column gives the initial holding current at -60 mV in the absence of applied drugs (means ±s.e.m., with numbers of cells in parentheses). The third and fourth columns give values obtained from fitting the Hill equation to the concentration-response data for activation by GABA (best fit parameter value ± 95% confidence limit on the fit). The fifth and sixth columns give similar fit parameters for activation by pentobarbital. The values in parentheses for the EC50(β3, c1, c7) are estimates from the concentration-response curves (Fig. 3), since in the presence of spontaneous activity and channel block by pentobarbital no effort was made to fit the data. The concentration-response curve for β3 showed two apparent components. The response in c7 and in the low affinity component of β3 is not saturated at 10 mm pentobarbital and therefore it is not possible to define the EC50. The seventh and eighth columns give parameters for block by pentobarbital. No estimate was made for the IC50 for β3, c1 or c7 receptors, due to spontaneous activity, and block of α1c2 and p1 receptors was not fully characterized. The ninth column gives the potentiation by 100 μm pentobarbital, expressed as the ratio of the response produced by co-application of 1 μm GABA plus 100 μm pentobarbital to that produced by application of 1 μm GABA alone. The tenth column gives the relative maximal response to pentobarbital, expressed as the ratio of the response to 10 mm pentobarbital to the response in the same cell to 100 μm GABA. NR indicates no activation, (0) indicates that since gating was not observed the relative gating was set to 0, and* indicates that the parameter was not estimated.
No homomultimeric receptor (except ρ1) responded to 100 μM GABA, while the heteromultimeric receptors formed with the α1 subunit were activated by GABA. We conclude that in heteromultimeric receptors GABA-evoked currents reflect the responses of heteromultimers and responses are not contaminated by the coexpression of homomultimers.
Cells expressing α1c2 subunits had a large holding current (see Table 1), but also responded to application of GABA with an increased inward current (Fig. 4). Interestingly, at the end of an application to a high concentration of GABA, the current actually overshot the initial holding current, then gradually returned to the initial level. These observations suggest that the α1c2 receptor has a measurable probability of having a channel open in the absence of GABA, but has a higher probability of being open after binding of GABA. Further, the spontaneously open channels can be desensitized by the application of a high concentration of GABA.
Figure 4. Activation by GABA is similar in receptors containing α1β3, α1c7, α1c1 and α1c2 subunits.
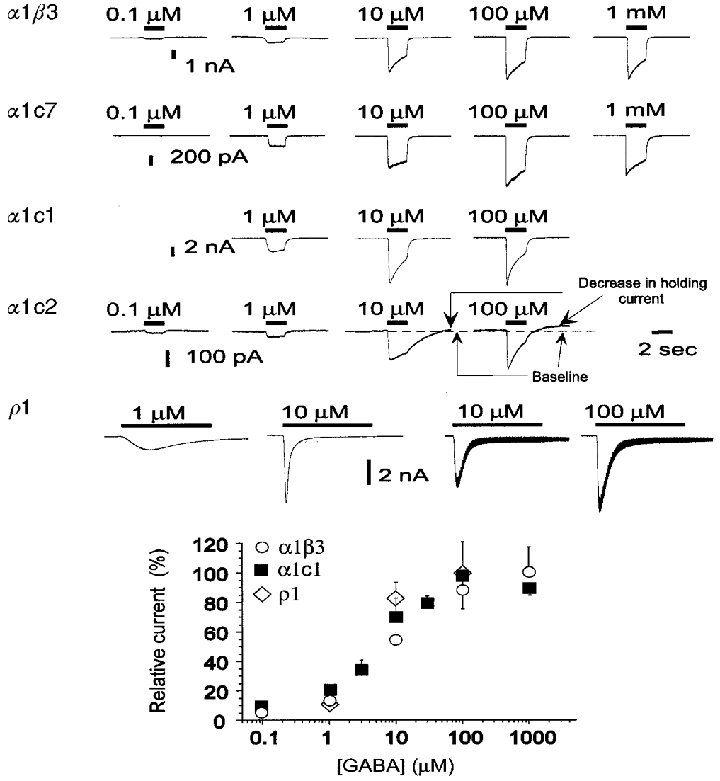
The upper panel shows traces of GABA-gated responses. In α1β3, α1c7, α1c1 and α1c2 responses were evoked by the application of GABA for 2 s. Preliminary observations indicated that in ρ1 homomultimers maximal responses are obtained only after 5–7 s at low GABA concentrations. Therefore in ρ1 homomultimers GABA was applied for 10 s. Responses shown for α1β3, α1c7, α1c1 and α1c2 were evoked in the same cell for each construct. In contrast, responses shown for ρ1 were evoked in two distinct cells: 1 and 10 μM traces on the left from one cell, and 10 and 100 μM traces on the right from another cell. In all constructs the maximal response is evident at concentrations of 100 μM GABA. The baseline conductance of cells transfected with α1c2 is higher than that of cells transfected with other constructs, and the holding current exhibits spontaneous fluctuations which probably correspond to spontaneous activity of unliganded channels. Also, the application of desensitizing concentrations of GABA causes a reduction of the holding current after removing GABA (see responses to 10 and 100 μM GABA), indicating an interaction between desensitizing GABA concentrations and the spontaneous activity. In ρ1 homomultimers application of 10–100 μM GABA evokes a large current that fades quickly. Most of this decline is the result of a redistribution of Cl− ions as a result of the large evoked currents. Redistribution is indicated by the observation that the conductance (measured by the change in current produced by 10 mV steps superimposed on the holding potential) reaches a peak at the peak of the current response, but remains high during the subsequent rapid fade of the current (see right-hand traces in bottom row, upper panel). The lower panel shows the concentration-response plots for the current evoked by GABA. In each experiment the measured values have been normalized to the response evoked at a determined concentration (10 μM in all constructs except α1c1 where 1 μM GABA was used). Finally, results have been renormalized to the maximal response. Plotted data are the means ±s.e.m. of values from 5 cells in α1β3 and α1c1 and 6 cells in ρ1. Data were fitted by the Hill equation, resulting in parameters shown in Table 1.
Pentobarbital could activate and block some homomultimeric receptors (Fig. 3). Receptor block will be discussed in more detail below, but was manifest for these receptors by the appearance of an apparent outward current during the application of high concentrations of pentobarbital (Fig. 3). The outward current was very pronounced in those constructs with a high spontaneous holding current such as β3 or c1. In contrast, it was small with the constructs with low spontaneous holding current such as c7 and at 10 mM pentobarbital we did not observe any outward current in heteromultimers (see later). Therefore, the pentobarbital-induced outward current is not due to a non-specific effect on the membrane, but rather, it correlates with the expression of a holding current. The outward current might be preceded by an inward current, indicating activation by pentobarbital, but was always followed by the appearance of an inward tail current when pentobarbital was removed (Fig. 3). This pattern was seen for receptors composed of β3, c1 or c7 subunits (Fig. 3). In contrast, receptors composed of c2 subunits showed no response to 1 mM pentobarbital (data not shown). We did not study responses of homomultimeric receptors extensively, since the presence of spontaneous activity complicated analyses of activation and block by pentobarbital.
Figure 3. Activation of currents by pentobarbital in homomultimeric receptors.
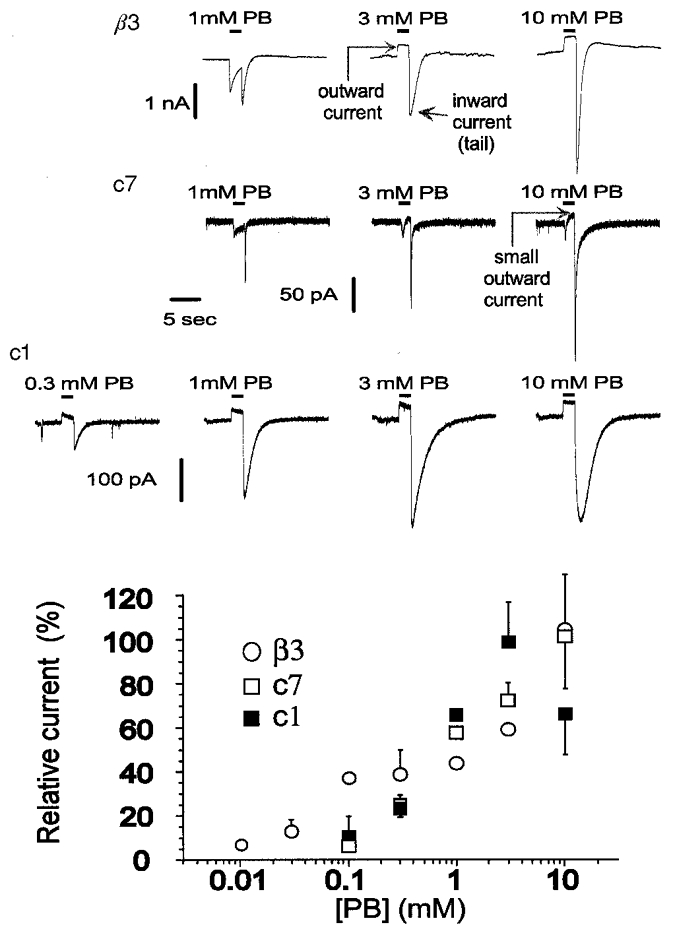
The upper panel shows current traces of gated responses evoked by application of pentobarbital for 2 s. During the application of high concentrations of pentobarbital there is an apparent outward current, resulting from block of spontaneous activity. Upon removal of pentobarbital there is an inward ‘tail’ current, demonstrating activation of receptors. The lower panel shows the concentration-response relationships for activation by pentobarbital for these homomultimeric receptors. The activation was measured by subtracting the initial holding current (before the application of pentobarbital) from the peak tail current, and normalizing it to the peak of the tail after application of 1 mM pentobarbital. The response is estimated from the tail peak values in c1 and from the larger of the peak current or the tail current in c7 and in β3. For the plot, the relative responses have been rescaled to the maximal current seen. Data are means ±s.e.m. of values obtained from 9, 7 and 3 cells from α1β3, α1c7 and α1c1, respectively. Data for this and all subsequent figures were acquired at a holding potential of -60 mV and filtered for the figure at 1 kHz.
The ability of pentobarbital to activate homomultimeric receptors raises the possibility that responses from cells transfected with both α1 and a second subunit might include some contribution from homomultimeric receptors. We believe that this possibility, while it exists, is unlikely to have significantly affected our observations. One reason for this is the finding that cells transfected with single subunits did not respond to GABA, while cells transfected with α1 plus a second subunit did. This observation demonstrates that a significant number of heteromultimeric receptors was present on the cell surface. Similarly, cells transfected with α1 plus a second subunit had low holding currents, indicating that few receptors with spontaneously open channels (that is, homomultimeric receptors) were present on the surface. The final reason is that cells transfected with α1 plus another subunit were identified for study by use of beads which bound to an epitope expressed on the α1 subunit (see Methods). Previous work has shown that the α1 subunit is not expressed on the surface of these cells when it is transfected by itself (Ueno et al. 1996). Accordingly, the individual cells studied expressed a high level of α1 subunit on the surface, in association with the non-α subunit.
In summary, the responses of heteromultimeric receptors largely reflect the properties of the heteromultimers, rather than a heterogeneous response reflecting both homomultimeric and heteromultimeric receptors.
The concentration dependence for activation by GABA
GABA evoked responses in all heteromultimeric receptors (Fig. 4). At higher GABA concentrations, the current decreased after the peak response. This effect is relatively slow to reverse, in that at the end of the application, no rebound was observed. However, a second GABA application after 1 min may evoke again a full response, indicating that the reduction of the response amplitude is temporary and that the channel may recover. This fading of current can be attributed to the development of a non-conducting ‘desensitized’ state, with recovery slower than channel deactivation. In addition, however, Cl− redistribution has been reported to occur during large evoked currents and produce a reduction in current with no corresponding reduction in conductance (Akaike et al. 1987a). Indeed, during the largest responses we found that the current could fade with little if any reduction in conductance (for example, the responses of a cell expressing ρ1 receptors to 10 and 100 μM GABA, Fig. 4). Thus, the fade of response to GABA may reflect both desensitization and Cl− redistribution. In our experiments Cl− redistribution is unlikely to significantly affect the peak response. Even when redistribution is most apparent (e.g. cell transfected with ρ1, Fig. 4), when a 10 mV step was applied to measure the conductance at the peak of the response, the extrapolated reversal potential was close to 0 mV, indicating that significant chloride ion redistribution takes place only after the observed peak. Furthermore, the conductance reached its maximal value at the observed peak current, rather than continuing to increase while the current faded. To determine whether the fade of current (from either cause) affected our estimate of the peak response, the responses were fitted with the sum of two exponentials, one to describe the rise and the other the fade. The measured peak was always at least 90 % of the amplitude of the exponential describing activation, and we did not correct our estimates of peak response. In cells transfected with α1c7 we did not observe fading in all the cells recorded. These differences were not characterized in more detail; in fact the low amplitude of responses with α1c7 made it difficult to obtain concentration-response plots at the lowest ligand concentration, and therefore the attention was rather focussed on α1β3 and α1c1.
In all chimeric constructs Hill coefficients for activation by GABA were close to one (Table 1). There is no clear correlation between the amount of ρ1 replacement into β3 and either the EC50 or the Hill coefficient for activation by GABA.
Pentobarbital potentiation of responses to a low concentration of GABA
The ability of pentobarbital to potentiate responses evoked by 1 μM GABA was studied. This GABA concentration produced a relatively small response, ranging from 10 to 15 % of the maximal peak response in the different constructs. Pentobarbital was co-applied with GABA. In α1β3 receptors, an increase was observed in the maximal evoked response with pentobarbital concentrations as low as 10 μM (Fig. 5). At pentobarbital concentrations of 100 μM or more, at the end of the application a transient inward current (the tail current) was noticed. At the highest pentobarbital concentrations the tail current was several times larger than the peak current during the co-application. Tail currents have been previously observed with pentobarbital, and probably reflect the recovery of active receptors from a non-conducting state with a faster rate than the deactivation of the channel (Akaike et al. 1987b). The non-conducting conformation is rapidly reversible and is called ‘blocked’ to distinguish it from the desensitized state where no rebound current is observed and the recovery from the non-conducting state is far slower. Since the tail current reveals the activation of the receptor channel hidden in non-conducting states, in order to study the concentration dependence for activation of the receptor channel, tail currents were measured rather than maximal responses during the drug application (Fig. 5). The peak amplitude of the tail current is unlikely to have been affected by chloride redistribution during the preceding application, because at the highest concentrations the response during the application was mostly blocked and had very low amplitude. In ρ1 homomers, the co-application of 1–10 mM pentobarbital induced no consistent effects on the current amplitude during the application. However, at the end of the co-application of 10 mM pentobarbital with 1 μM GABA a large tail current was evident. Smaller tail currents were seen also with 1 mM pentobarbital, but not at lower concentrations (Fig. 5). In summary, the data indicate that ρ1 homomers possess receptors for barbiturates, occupation of which is capable of potentiating GABA-evoked responses, although the apparent affinity is much less than that of α1β3, α1c7 or α1c1.
Figure 5. The insertion of longer stretches of ρ subunit results in a corresponding loss in the ability of pentobarbital to potentiate GABA-evoked responses.
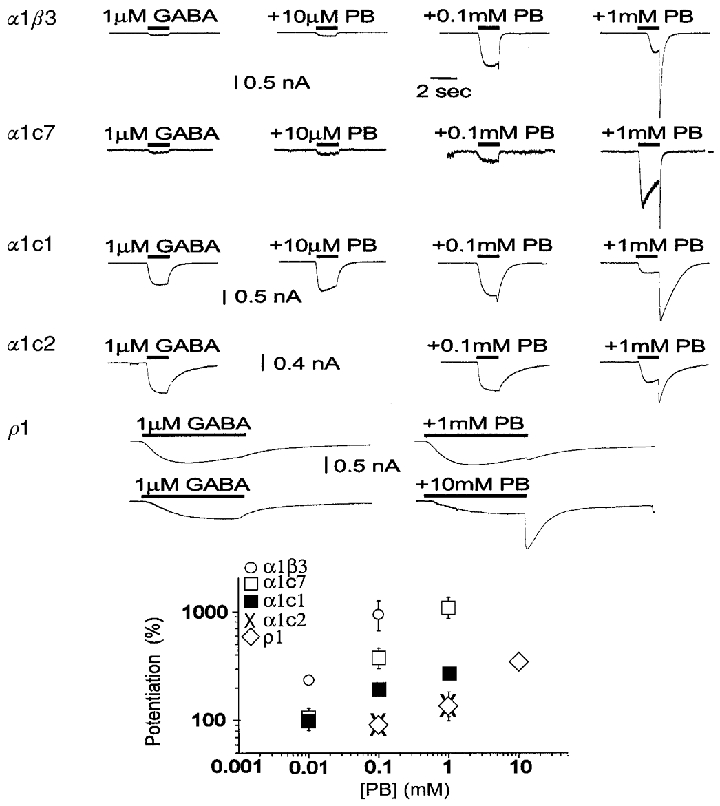
The upper panel shows the ability of pentobarbital (PB) to potentiate responses to 1 μM GABA. Pentobarbital has two effects. At lower pentobarbital concentrations the peak response to GABA is increased. However, at higher pentobarbital concentrations the peak may be reduced, and a large response is seen when GABA plus pentobarbital are rapidly removed (the ‘tail’). The tail results from rapid recovery from block of response by pentobarbital, so that the active and unblocked receptors cause a large conductance increase. Accordingly, the response is estimated from the larger of the peak current or the tail current. Receptors composed of α1β3 subunits are potentiated at low concentrations of pentobarbital (10 μM), while other constructs require higher concentrations. All constructs, however, are both potentiated and blocked by higher pentobarbital concentrations, as shown by the tail currents. Even the responses from homomultimeric ρ1 receptors are potentiated and blocked, although only at millimolar concentrations of pentobarbital. The lower panel shows concentration-effect plots for the relative currents elicited by co-applications of pentobarbital with 1 μM GABA, compared with the response to GABA alone. Data are means ±s.e.m. of observations obtained by recording from 5, 11, 3, 4 and 9 cells transfected with α1β3, α1c7, α1c1, α1c2 and ρ1, respectively. Note that in α1β3, α1c7 and α1c1, pentobarbital can gate GABAA channels directly (see Fig. 6). Therefore the current increase observed with co-application of GABA and pentobarbital is the result of both gating and potentiation by pentobarbital. However, gating by pentobarbital is minimal at a pentobarbital concentration of 0.1 mM. At this concentration of pentobarbital, receptors containing α1and β3 subunits show a higher potentiation than the others tested. α1c2 receptors are very similar to homomultimeric ρ1 receptors, showing both a reduced amount of potentiation under these conditions, and an apparent increase in the EC50 for potentiation.
In α1β3 and α1c7 pentobarbital activates a rather large current at concentrations higher than 1 mM (see Fig. 6), and direct gating may constitute a large fraction of the response to co-applications. In α1β3, the current evoked by 1 μM GABA was increased by 35 ± 3-fold (n = 5) by 10 mM pentobarbital, but the response evoked by 10 mM pentobarbital without GABA was a large fraction (66 ± 22 %; n = 3) of the response evoked by pentobarbital and GABA; the observed potentiated response at 10 mM pentobarbital must largely reflect the direct gating effect. In contrast, in α1β3, 0.1 mM pentobarbital is only the EC5 of the dose- response curve for direct gating and at this concentration the fraction of the response to GABA + pentobarbital due to the contribution of direct pentobarbital gating is practically negligible (< 10 % of the potentiating effect). Because of the overlap of the direct gating and potentiation, for the purpose of studying the ability of potentiating GABA-evoked currents, our investigation did not consider the effects of pentobarbital concentrations above 0.1 mM.
Figure 6. The insertion of progressively longer stretches of ρ subunit changes the ability of pentobarbital to directly gate GABAA receptors.
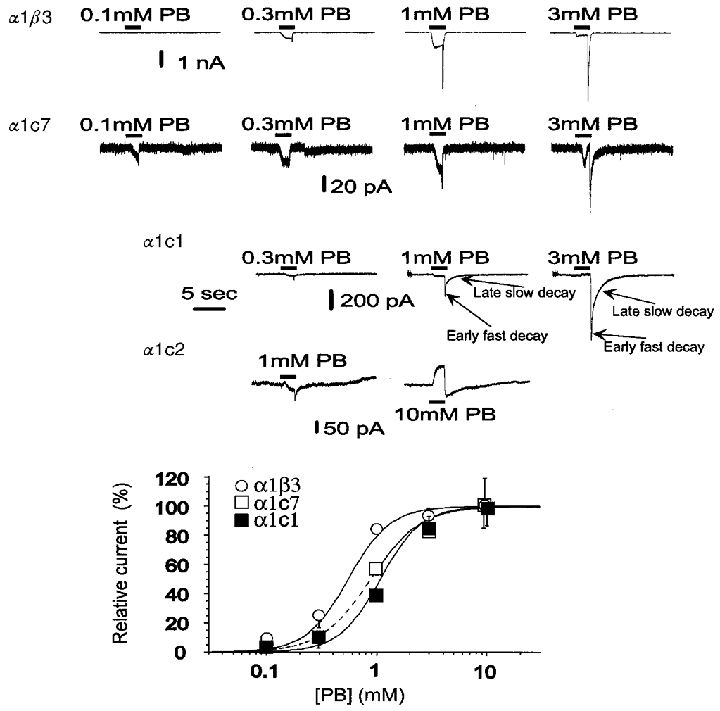
The upper panel shows responses elicited by pentobarbital. Pentobarbital was applied for 2 s. Note that at higher concentrations of pentobarbital block is evident, seen as a reduction in peak current, the appearance of a tail on removal of pentobarbital, or both. Accordingly, activation was measured from the larger of the peak response or the tail. In receptors containing α1β3, α1c7 or α1c1 subunits, the tail currents are much larger than the peak responses, indicating that block by pentobarbital is quite effective in these constructs. There was no observable activation of ρ1 receptors by pentobarbital (data not shown). In α1c2 at 1 mM PB little, if any, effect was observed during the application but a small tail (downward deflection) after removal of pentobarbital could be seen (see figure); at 10 mM PB the current exhibited a decrease (outward current evident as an upward delection) during the application followed by a rebound increase after the drug application had stopped. The outward current seen with 10 mM PB reflects a reduced membrane conductance (not shown) and can be mimicked by 1 mM picrotoxinin (not shown). This observation suggests that spontaneously active channels are blocked by pentobarbital. The concentration-response relationships are shown in the lower panel. Data are means ±s.e.m. of values obtained from 15, 5 and 18 cells in α1β3, α1c7 and α1c1, respectively. The amplitude of the response to each concentration was expressed as a ratio to the amplitude of the tail peak evoked by 1 mM pentobarbital and for the figure data were reported as a percentage of the maximal evoked response. The values for the Hill coefficient (Table 1) were larger than 1 for all relationships, suggesting that most channels opened by pentobarbital are in receptors with more than one pentobarbital molecule bound. In α1c1 and α1c7 the activation curves are shifted to higher pentobarbital concentrations, suggesting a reduced affinity of pentobarbital.
We also noticed that the current evoked by 1 μM GABA from α1β3 receptors, was ∼1/7 of the maximal GABA-evoked response, and the 35-fold enhancement of the GABA-evoked response by 10 mM pentobarbital suggested that the maximal response after GABA application must be much lower than the maximal response obtainable by the receptor channel. If the unitary conductance of the GABA-activated Cl− channels does not change in the potentiation effect, the maximal Popen of the GABA-evoked response should be lower than 0.2 (7-fold/35-fold). This conclusion is consistent with measurements obtained by variance analysis and single-channel recording (see later).
In summary, at a low pentobarbital concentration (100 μM), the data demonstrate a difference in the ability of pentobarbital to potentiate responses to GABA, with a rank order inversely related to the amount of ρ1 substitution into β3. The gradual, progressive loss of potentiation, correlating with the amount of ρ1 substitution suggests that residues affecting potentiation are probably distributed over a relatively long stretch of the primary sequence of the subunit.
The concentration dependence of activation by pentobarbital
Pentobarbital activated α1β3, α1c7 and α1c1 receptors (Fig. 6). Tail currents were apparent at the end of the application of high pentobarbital concentrations. The concentration-dependent amplitude of the tail peak reached a plateau at 1 mM in α1β3 while in α1c7 and α1c1 it increased nearly 3-fold between 1 and 10 mM (Fig. 6). No direct activation of ρ1 receptors was observed even with 10 mM pentobarbital. We also saw that the tail decay exhibited a biphasic time course with an early fast component followed by a late slow one. This was more evident in the recordings from α1c1 (Fig. 6). Indeed, the tail decay could be fitted by the sum of two exponential functions (see Table 2). In α1β3, comparing the tail induced by 1 and 3 mM pentobarbital, at 3 mM we observed a pronounced increase in the area of the slow component of the tail decay (Table 2). This change suggested a positive co-operativity in the mechanisms generating the tail. In a later stage of our research (see later) we fitted experimental data with the predictions of several theoretical models and found that such a steeply concentration-dependent change of the tail helped to discriminate among models.
Table 2.
Concentration-dependent effects on the kinetics of pentobarbital-induced tail current
| α1β3 | α1c1 | |||||
|---|---|---|---|---|---|---|
| 0·3 mm | 1 mm | 3 mm | 1 mm | 3 mm | 10 mm | |
| τfast (ms) | 66 ± 12 | 135 ± 28 | 166 ± 60 | 74 ± 8 | 109 ± 9 | 156 ± 50 |
| Areafast | 0·58 ± 0·05 | 0·44 ± 0·08 | 0·06 ± 0·03 | 0·27 ± 0·07 | 0·13 ± 0·07 | 0·07 ± 0·03 |
| Δslow (ms) | 390 ± 93 | 491 ± 97 | 664 ± 310 | 1450 ± 93 | 1092 ± 214 | 1149 ± 266 |
| Areaslow | 0·4 ± 0·05 | 0·56 ± 0·08 | 0·94 ± 0·03 | 0·73 ± 0·07 | 0·86 ± 0·07 | 0·91 ± 0·04 |
This table shows the parameters estimated by fitting the tail current observed at different pentobarbital concentrations (0·3–10 mm) with a function consisting of the sum of two exponentials of the type Afastexp[-t/τfast]+Aslowexp[-t/τslow]. Areas were calculated by integrating the area under each component and normalizing it to the total area. Data are means ±s.e.m., calculated from 4 cells in α1β3 and 8 cells in α1c1. Each determination is the average of 3–8 values. Data show a concentration-dependent increase in the time constant of the fast component in both α1β3 and α1c1. However, the largest and most evident concentration-dependent effect is on the area of each component.
In cells expressing α1c2 receptors 1 and 10 mM pentobarbital were applied to 3 and 5 cells, respectively. With 1 mM pentobarbital variable effects were observed during the application with a small decrease (n = 2) or increase (n = 1) (Fig. 6) in the holding current, but at the end of the application a small rebound current increase was observed in all cells (Fig. 6). With 10 mM pentobarbital, the current decreased during the drug application and showed a rebound increase at the end of the application (Fig. 6).
Although chloride redistribution may possibly have affected responses induced by pentobarbital, the evidence indicates that any possible effect is unlikely to be significant. No current fading during the application of pentobarbital was observed even with current amplitudes as high as 1–2 nA and at the highest pentobarbital concentrations the response was mostly blocked. In principle, chloride redistribution could result in a quicker current fading after the peak of the tail. However, at 1 mM pentobarbital in α1β3 the tail amplitude of pentobarbital-evoked responses was markedly larger than in α1c1 and yet the decay of the early tail current was slower in α1β3 (∼140 ms) than in α1c1 (∼70 ms) (Table 2). Finally, in α1β3, at the highest pentobarbital concentrations, in spite of a larger amplitude of the tail peak, we saw a dose-dependent increase in the duration of the tail exponential decay (Table 2). Taken together, these data are not consistent with the Cl− redistribution affecting significantly the amplitude or the time course of the tail. Rather, the difference in the decay of the tail current between α1β3 and α1c1 should reflect the kinetic properties of the receptors.
Fits of the Hill equation to the concentration-response curves for activation by pentobarbital produced the values shown in Table 1. The EC50 values for the receptors containing α1c7 or α1c1 are larger than that for receptors containing α1β3. This observation provides an initial indication of qualitative differences in the properties of the pentobarbital-gated response in the different constructs. In addition, the Hill coefficient is close to 2. All of the receptors studied were blocked by high concentrations of pentobarbital (Fig 5 and Fig 6). The amount of block was estimated by calculating the ratio between amplitudes of the current at the end of the application and the tail current. This was done for tails seen either after co-applications of pentobarbital and GABA, or after applications of pentobarbital alone. (The results were indistinguishable for receptors for which both measurements could be made, and results were pooled for analysis.) When the data were fitted with the Hill equation, the Hill coefficient was greater than 1. The Hill coefficient value higher than 1 for both activation and block suggested a complex mechanism with more than one molecule of pentobarbital binding to each receptor channel to produce its effects.
The insertion of longer stretches of ρ1 into β3 results in a decrease of the ratio of the maximal response for pentobarbital to that for GABA
The relative maximal efficacies of GABA and pentobarbital were estimated by comparing the responses elicited in a single cell to concentrations of pentobarbital or GABA which produce a maximal response. The relative maximal efficacy for pentobarbital is about 2 in α1β3 and 3 in α1c7 (Fig. 7). In contrast, the relative maximal efficacy is markedly reduced to only about 0.3 in α1c1 (Fig. 7). Gating by pentobarbital was also greatly reduced in α1c2 (see above), to a very low level. These observations might suggest that gating by pentobarbital is greatly reduced in α1c1 receptors. However, the interpretation of these data is difficult because the change in relative maximal efficacy can be due to a decrease in the efficacy of pentobarbital, to an increase in the maximal efficacy of GABA, or both. We also observed that the relative maximal efficacy correlates with the ability of pentobarbital to potentiate the GABA-evoked current. In fact α1β3 and α1c7 exhibit the highest values of the maximal relative efficacy and the largest potentiation by 0.1 mM pentobarbital, whilst α1c1 and α1c2 had the lowest values of maximal relative efficacy and the lowest potentiation by 0.1 mM pentobarbital.
Figure 7. The insertion of progressively longer stretches of the ρ subunit changes the relative maximal gating by pentobarbital and GABA.
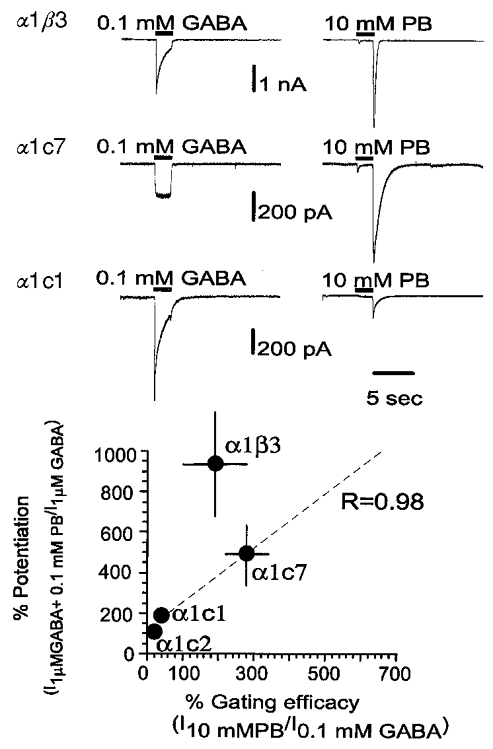
Traces in the upper panel show the response of a given cell to a saturating concentration of GABA (100 μM) and pentobarbital (10 mM). As can be seen, the maximal response to pentobarbital is larger than that to GABA for α1β3 receptors (2.0 ± 0.9; mean ±s.e.m., n = 3 cells) and α1c7 receptors (2.8 ± 0.6, n = 4). However, with α1c1 receptors the response to pentobarbital is smaller than that to GABA (0.27 ± 0.06, n = 4). These values suggest that for α1c1 receptors the gating efficacy of pentobarbital is reduced and/or the gating efficacy for GABA is increased. As shown in Fig. 6, gating by pentobarbital is negligible for α1c2 receptors. The lower panel shows a plot of the ability of 0.1 mM pentobarbital to potentiate responses to 1 μM GABA for a type of receptor against the relative maximal current gated by pentobarbital and GABA. The regression coefficient is close to 1 (R = 0.98) for data from receptors containing the 3 chimeric subunits (α1c7, α1c1 and α1c2). This relationship suggests that the regions of the subunit involved in gating and potentiation by pentobarbital are probably close together.
Differences in the primary structure of c2, c1 and c7 are present only between M1 and the M2-M3 loop. A regression analysis of the potentiation ability on the relative gating efficacy in the constructs composed by α1 + each of the three chimeras exhibits a correlation coefficient close to 1 (Fig. 7). This suggests that, within the region between the M1 domain and the M2-M3 loop, the residues involved in potentiation might be located close to those affecting the relative gating efficacy. The point corresponding to α1β3 does not lie close to the regression line of c2, c1 and c7. However, the primary structures of c2, c1 and c7 differ from that of β3 by the presence in the chimeras of a long ρ stretch in the region downstream from the junction points. Therefore, either potentiation or the relative gating may be affected by residues expressed in the M3 domain, the M3-M4 loop and the M4 domain too.
Interpreting the differences in pentobarbital-evoked responses between α1β3 and α1 c1
How to infer estimates for the binding Kd from dose- response curves
While clear effects were observed on the ability of pentobarbital to activate receptors containing chimeric subunits, the data do not distinguish whether the alterations reflect changes in affinity (potency) or in conformational alterations (efficacy) following binding. On face value, the observation that the EC50 for gating by pentobarbital is shifted to higher concentrations in α1c1 might suggest a change in affinity. However, at the same time the maximal response relative to that for GABA is reduced, suggesting a change in efficacy as well.
For responses interpretable in terms of a linear reaction scheme with three states (see Appendix 1, Scheme 1), the binding Kd can be estimated from the activation EC50 if the maximal channel Popen (Ψ=β/(β+α)) is known. The binding Kd can be calculated by dividing the EC50 by (1 –Ψ). A somewhat similar relationship can be derived for reaction schemes with more than one binding step. In general, a rightward shift in the concentration-response curve may reflect a decrease in Ψ, or a decrease in the binding affinity if no significant effect on Ψ is found. For more complex schemes it is no longer possible to simply calculate the Kd, but an estimate of the binding may still be possible through determinations of Ψ and a global fit of the time course of the corresponding responses (Appendix 1).
Accordingly, we estimated Ψ for GABA- and pentobarbital-evoked responses, defined a kinetic scheme able to describe pentobarbital effects, and finally estimated the binding Kd by fitting the scheme to the time course of experimental responses.
The maximal Popen for GABA-evoked responses is larger for α1c1 receptors than for α1β3 receptors
For GABA-evoked responses, two approaches were used to estimate the maximal probability that a channel will be open: non-stationary variance analysis of whole-cell responses and single-channel recordings. Either approach has several experimental limitations, but the use of two complementary approaches provides some additional confidence in the results.
To perform non-stationary noise analysis, we analysed the data near the peak of the whole-cell response. Because we obtained only a few records from each cell, we estimated the mean response by fitting the time course of the response and analysed the variance of the data points from the fit (see Methods). In recordings from cells expressing α1β3 receptors, the deviations from the fit showed slow fluctuations whose amplitudes increased with the GABA concentration (Fig. 8). In contrast, in α1c1 the deviations exhibited faster oscillations. Further, at the highest GABA concentration the amplitude of the oscillations did not increase. Accordingly the plot of variance against the mean is linear for α1β3 and bell-shaped in α1c1 (Fig. 8). The largest currents were recorded during applications of saturating concentrations of GABA (greater than 100 μM, while the EC50 for activation by GABA is less than 10 μM). In summary, through analysis of fluctuations the maximal Popen for α1β3 is inferred to be less than 0.5, whilst for α1c1 it is inferred to be higher than 0.5 and possibly to approach 1. The inferred unitary conductance is ∼5 pS in α1β3 and ∼1 pS in α1c1 in our ionic conditions (Fig. 8). A quantitative estimate of the channel properties by variance analysis may be distorted by bandwidth limitations. Nevertheless, these data suggest a qualitative difference in the properties of GABAA receptors composed by α1β3 and α1c1. These results predict that the GABAA channels of α1β3 should have a low Popen and a larger conductance, while those of α1c1 should have a smaller amplitude and a larger Popen.
We also estimated the maximal Popen for GABA from single-channel recordings. The approach was to record steady-state activity from cell-attached patches. First, the amplitude of the single-channel current was estimated from histograms of current levels (Fig. 9). Then, the number of channels in the patch and the Popen were estimated from the recordings, using a Bayesian estimator procedure (Horn, 1991). Because the accuracy of the estimate of the number of channels is more precise when the Popen is high, the agonist concentrations used were selected in order to give the highest Popen consistent with high steady-state activity. A qualitative evaluation of the whole-cell responses indicated that in α1β3, 3 μM GABA was the concentration giving the highest maintained response at steady state. The corresponding concentration used for α1c1 was 1 μM GABA. In whole-cell recordings of the GABA-evoked response from α1c7 receptors, even at 100 μM GABA we typically observed responses with no fading (see Fig. 7) or lower fading than with the other constructs (see Fig. 4). Therefore, 100 μM GABA was used for cell-attached recordings from these receptors.
The estimated Popen values were: α1β3, 0·023 ± 0·005 (3 μM GABA, mean ±s.e.m., n = 3 patches); α1c1, 0·12 ± 0·02 (1 μM GABA, n = 4); and α1c7, 0·29 ± 0·05 (100 μM GABA, n = 3). Analysis of simulated ion channel recordings indicated that the GC Bayesian estimator allows estimates of Popen in this range of low values if the parameters a and b of the beta and gamma functions are appropriately optimized (see Methods). The maximal Popen was then estimated by scaling these Popen values by a factor derived from the whole-cell concentration-response curve for activation to provide estimates for the Popen at a concentration of GABA producing the maximal response: α1β3, 0·19 ± 0·1; α1c1, 0·94 ± 0·13; and α1c7, 0·29 ± 0·05. This procedure assumes that differences in the current evoked by different ligand concentrations are due only, or at least mostly, to differences in the channel opening probability rather than in the single-channel conductance (see Appendix 2).
The unitary conductance elicited by GABA was estimated from plots of single-channel currents obtained at different pipette potentials (Fig. 10). For α1β3 receptors the conductance was 7·0 ± 1·2 pS (n = 3) while for α1c1, it was 3·6 ± 0·9 pS (n = 3). In α1c1, the definition of the size of the unitary current steps was more difficult, because of the small amplitude and the high activity. The conductances differ between α1β3 and α1c1. The single-channel conductance values for α1β3 are smaller than those reported for heterodimers composed of α1β2 (Verdoorn et al. 1990), or α1β1 (Angelotti & Macdonald, 1993; Amato et al. 1999) which range between 11 and 16 pS. This difference could possibly be explained considering differences in experimental approaches used (symmetrical 140 mM Cl− in outside out vs. our cell-attached recordings with symmetrical 30 mM Cl−, or in the β subunit expressed (β1 and β2 vs.β3). The single-channel conductance of α1c1 is even lower than that of α1β3 and this could possibly be due to the insertion of a stretch of ρ subunit in c1. In fact, ρ subunits assemble in channels with very low unitary conductance (see Chang & Weiss, 1999) and in c1 much of the M2 region, known to be involved in ion permeability (Xu & Akabas, 1996), is composed of the residues of the ρ subunit.
Figure 10. Single-channel currents activated by GABA or pentobarbital.
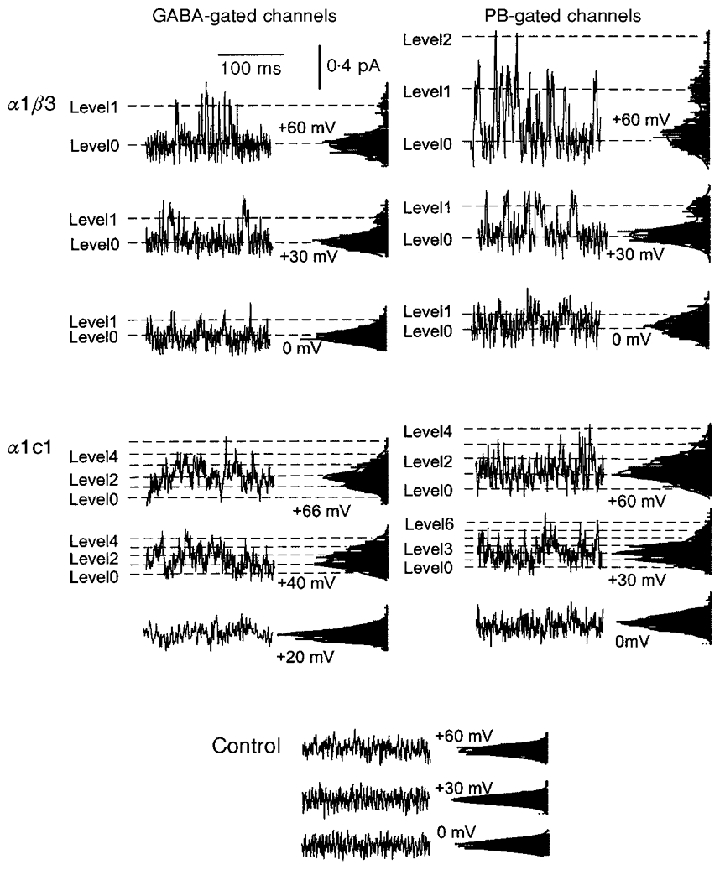
The figure shows data recorded from cell-attached patches on two cells transfected with α1 and β3 subunits (top row) and two cells transfected with α1 and c1 subunits (middle row). The pipette solution contained 3 μM GABA (α1β3, top left), 1 μM GABA (α1c1, middle left), 300 μM pentobarbital (α1β3, top right) or 1 mM pentobarbital (α1c1, middle right), respectively. The control traces (bottom row) were obtained from a patch with neither GABA nor pentobarbital in the pipette solution. Each section shows short current traces at different pipette potentials, with a corresponding all-points histogram to the right of the trace. The vertical axis of each histogram corresponds to the current amplitude, while the horizontal axis corresponds to the number of events. Each histogram is fitted by the sum of multiple Gaussian functions with mean values spaced by a constant value, corresponding to the channel step amplitude. In each histogram, the lowest amplitude component corresponds to the baseline noise. In recordings with α1β3 relatively large amplitude channels are evident, while in α1c1 receptors the unitary current is very small and it is difficult to visualize transitions of individual channels. Furthermore, in α1β3 the activity is low, and the recording exhibits stretches of silent baseline. In contrast, in α1c1 a baseline is not clearly evident, indicating a high activity. Note that the all-points histograms are relatively narrow and symmetrical in the control records, while they show a skew to larger currents and exhibit multiple peaks in records obtained in the presence of GABA or pentobarbital.
The two independent approaches to estimating the maximal Popen provided qualitatively similar results for activation by GABA. Popen is larger for receptors composed of α1c1 than for α1β3, while conductance is smaller. The agreement between the results increases our confidence that the differences are real. The estimate of channel maximal Popen in α1β3 of ∼0·2 is also in excellent agreement with the estimates obtained by comparing GABA maximal response with the maximal response that can be evoked in this construct after potentiation with 10 mM pentobarbital (see above).
Previous estimates of the maximal Popen for GABA-evoked Cl− current response obtained through analysis of single-channel recordings indicated values higher than 0·2. For example, Newland et al. (1991) estimated the maximal Popen as 0·83 in recordings from neurones of the superior cervical ganglion. The disagreement between the previous estimate and the one of our study might possibly reflect differences in the construct studied.
The maximal Popen for pentobarbital-evoked responses does not differ for α1c1 receptors and α1β3 receptors
We estimated the maximal Popen for pentobarbital from cell-attached recordings of single-channel activity. The pentobarbital concentrations used were 0·3 mM in α1β3 and 1 mM in α1c1. The estimated Popen values for pentobarbital-gated channels were 0·05 ± 0·02 (n = 5) in α1β3 and 0·06 ± 0·006 (n = 7) in α1c1. The Popen values were then scaled by a factor derived from the whole-cell concentration curve for activation to provide estimates for the Popen at a pentobarbital concentration producing the maximal response: 0·52 ± 0·12 in α1β3 and 0·79 ± 0·08 for α1c1.
For α1β3 receptors the unitary conductance was 9·3 ± 1·3 pS (n = 3) while for α1c1, it was 2·0 ± 0·5 pS (n = 3). In agreement with what was observed for GABA-gated channels, the single-channel conductance is reduced in α1c1 receptors. The conductances of channels activated by GABA and pentobarbital from receptors of a given type do not show statistically significant differences.
The results of the single-channel current analysis indicate that for pentobarbital-evoked responses the insertion of a ρ1 sequence into β3 does not result in a decrease of the maximal Popen. Indeed, the difference in relative maximal currents elicited from α1c1 and α1β3 receptors appears to result from a change in efficacy for GABA, rather than for pentobarbital.
This observation, in turn, indicates that the increase in the EC50 for activation by pentobarbital between α1β3 receptors and α1c1 receptors might possibly be as the result of an increase in the binding Kd.
The estimated Kd for activation by pentobarbital is larger for α1c1 receptors than for α1β3 receptors
The final steps in the analysis were the definition of a class of kinetic schemes able to explain pentobarbital effects, and the estimates of the binding Kd values by optimization of the parameters of each scheme, to yield theoretical responses fitting experimental observations. To obtain a first, qualitative estimate of the difference in dissociation constants for the α1β3 and α1c1 receptors, we made the following assumptions. We assumed that at the peak of the tail current all receptors had unblocked and no receptors had deactivated, so the concentration-response relationship for the tail current reflected the pseudosteady-state activation curve. We then scaled the concentration-response curves (Fig. 6) by the respective estimated maximal Popen values. The resulting steady-state activation curves, in the absence of block, were fitted with the simplest model which would describe the data. This is a linear activation scheme in which pentobarbital binds to two identical and independent sites on the receptor, and the diliganded receptor opens (Scheme II of Appendix 1). The only free parameters are the dissociation constant and the maximal Popen. The dissociation constant of pentobarbital binding for the fit to the data from α1β3 was 0·5 ± 0·1 mM (standard error of the fit) while the value for the fit to the data from α1c1 was 2·5 ± 0·6 mM. However, this simple analysis makes a number of assumptions which are very difficult to justify, and uses a kinetic model which clearly does not describe the data.
We next adopted a more appropriate approach in which the entire time courses of responses to pentobarbital were fitted by the predictions of models including blocked states. The goal was to determine the simplest scheme which could describe the responses over the entire concentration range studied, and to use that scheme to estimate the dissociation constants for pentobarbital at the activation sites. We tested a number of simple schemes (Fig. 11), including schemes with only one blocking site and schemes which had only open channel block. Whilst it was possible to obtain adequate dose-response curves for the peak of the tail current corresponding to experimental data with simple schemes (Schemes P1–7), the simplest mechanism that adequately described the whole time course of the experimental response, required two sites of activation, with two block sites connected in sequence to each of the activated states (Scheme P8). In Scheme P8 each receptor has two binding sites for pentobarbital, occupation of which can lead to activation (indicated by L in LxR). There are also two additional sites, occupation of which leads to block (indicated by B in LxRBx). The two classes of sites are independent of each other, so that the blocking site can be occupied (and the blocked state can develop) even when the channel is closed (coupled model).
Figure 11. Global fit of the predictions of several theoretical models to the experimental data obtained at multiple concentrations.
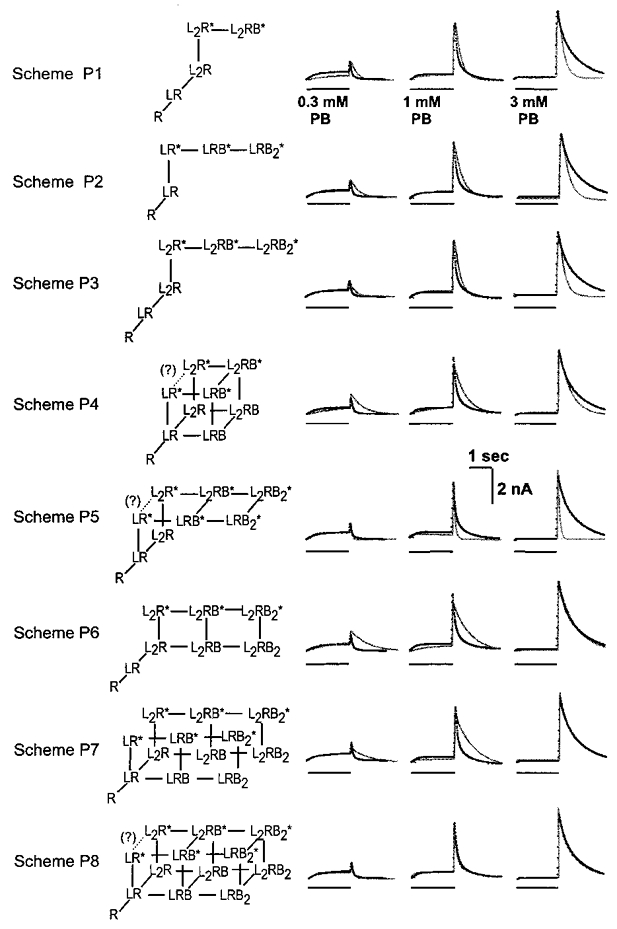
This figure shows 8 hypothetical reaction schemes to describe the activation and block of GABAA receptors by pentobarbital (left column). The traces on the right show the responses of a cell expressing α1β3 receptors to applications of 0·3, 1 and 3 mM pentobarbital (dotted lines), with the predictions of each scheme superimposed (thin lines). The predictions of each scheme were fitted to response and parameter values producing the best obtainable fit, using the approach described in the Methods. The predicted Popen values were constrained, for each scheme, to the experimentally determined values (see Results). Note that the responses at all concentrations were fitted simultaneously. For each scheme, R represents a receptor with a closed channel. Pentobarbital bound to an activation site is represented by L (e.g. L2R is a receptor with pentobarbital bound to 2 activation sites). Pentobarbital bound to a blocking site is represented as B (e.g. LRB is a receptor with pentobarbital bound to one activation site and one blocking site). An asterisk indicates a receptor in the open channel conformation (e.g. LR*). However, it is assumed that when one or more blocking sites are occupied the channel does not conduct ions. That is, LRB* is non-conducting while LR* is conducting. In general, the two activation sites (if present) were assumed to be identical but possibly interacting, while the two blocking sites (if present) were assumed to be identical and independent. Furthermore, it was assumed that the interaction with the blocking site did not change when the channel opened or closed. However, it was necessary to assume that dissociation of pentobarbital from the activation sites was slowed when the blocking site was occupied (see below). The models are presented in general order of complexity. It is of some interest that each model is able to produce a reasonable description of the concentration dependence of the peak tail amplitude, indicating the inadequacies of the steady-state analysis for model discrimination. Models P1-P7 fail to produce bi-exponential decays of the tail current with concentration-dependent prevalence of the components. The simplest model which can produce this behaviour, of the ones we tested, is model P8 (or the variant shown in Fig. 12). Some qualitative features of the results are that two activation and two blocking sites are necessary to produce the bi-exponential decay with the strong concentration dependence observed. A strict open channel block (P1, P2 or P6) produced a tail current, but was not able to describe the bi-exponential decays appropriately. A single open state (L2R*), even with two blocking sites, also did not describe the decays. Binding of pentobarbital to the activation sites had to be allowed when the blocking site is occupied (compare P7 to P8). However, in those models which allowed both monoliganded and di-liganded open states (P4, P5, P7 and P8), it was necessary to assume that the association/dissociation steps between LR* and L2R* were so slow as to be negligible in comparison with the other rates (dotted connections in Fig. 11). Finally, it was necessary to assume that dissociation of pentobarbital from the activation site when the channel was both open and blocked was slowed by an arbitrary factor of 10-fold (for example, the step L2RB* to LRB*). Of these kinetic models, only P8 was able to describe the time courses of pentobarbital action across the concentration range tested. In the text, P8 is termed the ‘coupled model,’ as the blocking site is not available until after at least one activation site is occupied. The best fitting parameter values for this scheme are shown in Table 3.
This model is the simplest one whose predictions adequately fitted the experimental data. We will briefly review the main difficulties in obtaining an adequate fit through models involving a lower number of states. With maximal Popen values lower than 1 it was difficult to obtain an adequate fit with the predictions of an open channel block mechanism. The bi-exponential shape of the tail decay and the dose dependence of their relative contribution, was very difficult to fit unless we postulated two activation sites (R ⇌ LR ⇌ L2R) with the blocked states connected to both the monoliganded (LR and LR*) and bi-liganded states (L2R and L2R*). Furthermore, the contribution of the slow component of the tail decay was modest at 1 mM, whilst it became prevalent at 3 mM; the large enhancement by only a modest pentobarbital increase suggested a high co-operativity. In fact, we found that this effect could be fitted only with the predictions of a mechanism involving the sequential binding to two block sites (for example L2R ⇌ L2RB ⇌ L2RB2).
Previous observations based on single-channel recording reported that pentobarbital-induced block of GABAA receptor channels is associated with a dose-dependent decrease in the mean open time (Rho et al. 1996). This effect, and the observation of a tail current, so far have been explained by hypothesizing a mechanism in which pentobarbital binds to its block site when the channel is open. The two reaction schemes whose predictions allowed a fit of experimental data differ from those previously proposed for pentobarbital action because they allow pentobarbital to bind to its blocking site, not only when the channel is open but also when the channel is closed. However, even in the two models proposed by our study, an increase in pentobarbital concentration shifts the equilibrium from the open to the blocked states and reduces the open-time duration.
Our model did not consider desensitized states for pentobarbital activation, because the evidence was against them playing a significant role in the data we were analysing. First, tail current peaks at high pentobarbital concentrations correspond to Popen values of ∼0·6 in α1β3 and ∼0·8 in α1c1. The tail reveals the activation of the receptor channel hidden in rapidly recovering non-conducting states (which we define as blocked states). Therefore, the Popen level obtained in the tail should reflect the probability of the blocked states: at the end of pentobarbital application the probability of a blocked state must be approximately 0·6 and 0·8 in α1β3 and α1c1, respectively. The probability of being in any other states, including desensitized states, must be lower than ∼0·4 in α1β3 and ∼0·2 in α1c1. Furthermore, in potentiation experiments on α1β3 with 10 mM pentobarbital, the Popen of the tail current peak should probably approach a value close to 1 (see above) and the probability of blocked states should be close to 1. The interpretation of these potentiation data may be difficult because of a possible interaction in the mechanisms of GABA and pentobarbital. However, the results suggested that even after a further activity enhancement, those activated channels which are not conducting ions in the presence of pentobarbital, would be largely in a blocked state. Furthermore, in both α1β3 and α1c1, the time course of the pentobarbital-evoked current did not show any evidence of desensitization, that is of a current peak followed by a fading. Finally, desensitized states were not necessary to obtain an adequate fit of the time course of the current and therefore were not considered because of the principle of minimal complexity.
An alternative kinetic scheme (Fig. 12) allows the binding sites involved in producing block to be exposed even when the activation site is empty (independent model). For the independent blocking model (Fig. 12), fit parameters for the dissociation constant of pentobarbital at the two activation sites were ∼22 and ∼8 mM for α1β3 and ∼230 and ∼40 mM for α1c1 (Table 3). The fit values were very similar to those of the coupled blocking scheme (Table 3). In order to define the errors in the parameter estimates we also simulated data using parameters for the fit of the independent model to data from α1β3 receptors and performed time course fitting of the simulated data (see Methods). Fit parameters were very close to the input values: Kd1= 23 ± 3 mM and Kd2= 9 ± 2·5 mM (95 % confidence limits).
Table 3.
Estimates of pentobarbital affinity at activation and block sites
| Kd1 (mm) | Kd2 (mm) | Kblock (mm) | Popen tail | |
|---|---|---|---|---|
| Independent model | ||||
| α1β3 | 22 ± 7 | 8 ± 3 | 0·7 ± 0·1 | 0·67 ± 0·12 |
| α1c1 | 231 ± 34 | 38 ± 5 | 0·7 ± 0·1 | 0·86 ± 0·02 |
| Coupled model | ||||
| α1β3 | 19 ± 5 | 8 ± 3 | 0·8 ± 0·1 | 0·66 ± 0·02 |
| α1c1 | 324 ± 39 | 40 ± 6 | 0·7 ± 0·1 | 0·85 ± 0·03 |
This table presents parameter estimates for the affinity of pentobarbital at the activation (Kd1=koff,1/kon,1 and Kd2=koff,2/kon,2) and block sites (Kblock=kb-/kb+) in α1β3 and αc1 receptors (see Fig. 12). Data are means ±s.e.m., calculated from the best fitting parameter values for data from 8 cells (α1c1) or 4 cells (α1β3). Data from each each cell comprised responses to 2–4 different concentrations of pentobarbital. The final column (Popen tail) gives the calculated maximal probability that channels are open at the peak of the largest tail currents for comparison to the values obtained from single-channel analyses.
In summary, the data obtained are consistent with the hypothesis that the affinity of pentobarbital is reduced in the α1c1 (and probably the α1c7) receptor.
DISCUSSION
Recapitulation of salient findings
The data indicate that: (1) when the joining point used in producing the chimeric subunits was moved towards the N-terminus of the β3 subunit, there was a marked decrease in the maximal response gated by pentobarbital relative to that gated by GABA. This effect correlated with a reduction in the ability of pentobarbital to potentiate the GABA-evoked current; (2) in the α1β3 receptor, the pentobarbital-gated response was nearly saturated at 1 mM pentobarbital, whereas in α1c1 and in α1c7 the response continued to increase at 3 or 10 mM; (3) more detailed comparisons of the properties of α1β3 and α1c1 receptors indicated that in the chimera there had been an increase in the maximal Popen for GABA and a decrease in the unitary conductance of GABA-gated Cl− ion channels. The maximal Popen for pentobarbital was less affected. The conductances for GABA and pentobarbital did not differ significantly for either type of receptor.
These data indicate that the reduction in apparent affinity for pentobarbital seen in whole-cell recordings from cells expressing α1c1 receptors corresponds to a decrease in the binding affinity to the sites involved in receptor activation. In addition, fits of the time course of the response by a minimal kinetic scheme allowed an estimate of the dissociation constants for pentobarbital at the activation sites of the α1β3 and α1c1 receptors, indicating that the affinity was about 10-fold lower in the chimeric subunit.
Where are the binding sites for GABA and for anaesthetics?
Previous work performed in oocytes indicates that residues binding GABA are located in two regions of the extracellular N-terminal part of the β3 subunit, around residues 153 and 200–210 (Amin & Weiss, 1993). Because mutations of these residues do not affect pentobarbital responses (Amin & Weiss, 1993; Ueno et al. 1996), the binding sites for GABA and pentobarbital are unlikely to coincide.
The anaesthetic properties of most anaesthetics are related to their lipophilicity (reviewed in Kennedy & Longnecker, 1995; Franks & Lieb, 1994). It has long been debated whether this relationship is due to the action of anaesthetics on membrane lipids, or whether the lipophilic properties actually reflect the nature of the binding site on a protein target. More recent data, including observations of enantioselectivity in anaesthetics, anaesthetic actions on soluble proteins (Franks & Lieb, 1994), and studies of mutated receptors (Mihic et al. 1997), support the second alternative.
Several regions of the GABAA receptor subunits are markedly more lipophilic than the rest, and have been termed the putative membrane-spanning regions, M1 to M4. The M2 region contributes residues which line the wall of the ion-conducting channel of the GABAA receptor (Xu & Akabas, 1996), and also is involved in the channel gating process (Amin & Weiss, 1993). Our work focussed on the M2 and M3 regions of the β3 subunit.
Previous studies have already identified some amino acid residues in this part of GABAA receptor subunits which can affect the actions of anaesthetics and anticonvulsants, although this work has not yet determined whether affinity or efficacy is altered.
In the M2 region, a critical residue appears to be located at position 270 (in α1 or α2) and a homologous residue at 265 of the β1 subunit. In the α1 subunit, mutation of this residue from S (serine) to I (isoleucine) removes the ability of isoflurane or enflurane to potentiate GABA responses or to directly gate responses, when the mutated α subunit is expressed with β1 (Mihic et al. 1997; Krasowski et al. 1998b). This mutation in α was considered to be specific for halogenated ether anaesthetics, since the actions of the halogenated alkane halothane, or other drugs such as propofol, etomidate, methohexital, alfaxalone or trichloroethanol, were not affected (Mihic et al. 1997; Krasowski et al. 1998a, b). Mutation of the homologous residue in the β1 subunit from S265 to I had little effect on potentiation by enflurane when the mutated β1 subunit was expressed with α1 (Mihic et al. 1997), although it completely removed potentiation by isoflurane when the mutated β1 subunit was expressed with α2 (Krasowski et al. 1998b). Earlier studies of the anticonvulsant drug loreclezole had demonstrated that the ability of loreclezole to both potentiate GABA responses and to directly activate GABAA receptors was strongly dependent on the specific β subunit expressed. Analysis of mutated subunits demonstrated that the critical residue is located at the homologous position to that studied by Mihic et al. (1997); the less sensitive β1 subunit has S while the more sensitive β2 and β3 subunits have N, and sensitivity can be switched by reciprocal mutations in the subunits (Wingrove et al. 1994). Further studies have found that the ability of etomidate to potentiate and gate responses is also strongly influenced by this residue in the β subunits in a similar fashion to loreclezole (Belelli et al. 1997), in contrast to the results of Krasowski et al. (1998b). It should be noted, however, that these studies by Belelli et al. used different specific mutations in the β subunits, and also expressed the mutated subunit with both wild-type α6 and γ2 subunits, rather than α2 alone. None of these studies have found any effect on the actions of barbiturates. However, mutation of threonine 262 in the M2 region of the β1 subunit into a glutamine greatly reduced the ability of pentobarbital to potentiate responses to GABA when the mutated subunit was expressed with wild-type α1 (Birnir et al. 1997). Since this mutation also altered desensitization and gating by GABA, the change was likely to have altered channel gating properties, rather than specifically affected pentobarbital binding or a unique barbiturate-induced conformational change.
In the M3 region, interest has focussed on a residue at location 291 in the α2 subunit, and homologous residues in β and ρ1 subunits. The mutation α1 A291 to W removes potentiation and gating by enflurane and isoflurane (but not halothane) when the mutated subunit is expressed with β1 (Mihic et al. 1997; Krasowski et al. 1998b). Interestingly, the mutation of the homologous residue in β1, M286 to W, also has these effects when the mutated subunit is expressed with α2 (Mihic et al. 1997; Krasowski et al. 1998). These residues also affect the actions of other anaesthetics – the mutation in α2 reduces potentiation by etomidate and methohexital but not propofol or alfaxalone while the mutation in β1 completely removes potentiation by propofol and trichloroethanol, reduces potentiation by etomidate and methohexital, but does not affect potentiation by alfaxalone (again, mutated subunits were expressed with a complementary wild-type α2 subunit; Krasowski et al. 1998a, b). Of the greatest interest to our studies, a recent study has found that mutations of the homologous residue in the ρ1 subunit has major effects on the ability of pentobarbital to act on this receptor. When ρ1 W328 is changed to the corresponding M residue of the β2 subunit, pentobarbital could both activate and potentiate currents gated by GABA from the homomultimeric receptors composed of mutated subunits (Amin, 1999). The reciprocal mutation in the β2 subunit also removes the ability of pentobarbital to act on homomultimeric receptors expressed in Xenopus oocytes, but does not affect the actions of pentobarbital when the mutated β2 subunit is co-expressed with the wild-type α1 subunit (Amin, 1999). In contrast, we note that in our studies homomultimeric receptors composed of c7 or c1 subunits could be gated by pentobarbital, although they also contain the residues from the ρ1 subunit in this region of the subunit.
In our studies there was a strong correlation between potentiation and direct gating by pentobarbital in the chimeric subunits. This observation suggests that residues involved in potentiation are located close to those required for direct gating. In general, previous studies have also seen this correlation (see above). One clear exception, however, has been reported for a mutation in the M3 region of the β1 subunit: when β1 M286W is expressed with wild-type α1 potentiation by propofol is removed, while gating remains essentially normal (Krasowski et al. 1998b).
Overall, these results have demonstrated that specific residues in the M2 and M3 regions of several subunits can affect the ability of a variety of drugs to act on the GABAA receptor. It appears likely that there are more than one residue in this region which interact to influence drug actions, and there may be interactions between the residues contributed by different subunits in heteromultimeric receptors. At present, it is not known whether the studied mutations alter drug binding or conformational changes, although it has been argued that the evidence for some specificity indicates a change in a recognition (binding) site on the receptor (Mihic et al. 1997). Our studies also implicate these regions in the actions of barbiturates, and provide direct evidence that affinity of pentobarbital can be altered in our chimeric subunits.
Acknowledgments
J.H.S is the Russel and Mary Shelden Professor of Anesthesiology. We thank G. Akk, D. Covey, H. Li, C. Lingle and C. Zorumski for comments and advice. This work was supported by grant NIH P01-47969 to J.H.S.
APPENDIX 1
We will briefly explain, in several reaction schemes, the relationship between the EC50 of the observed response and the corresponding binding Kd.
In a simple model, such as the kinetic scheme consisting of three states:
scheme I.
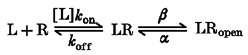
The channel open probability (Popen) as a function of the ligand concentration [L] is:
where Ψ=β/(α+β), and in Scheme I is the maximal Popen, and Kd is the binding dissociation constant (koff/kon). The EC50 of the macroscopic response is determined by the binding Kd and by the maximal Popen:
For ligands with low efficacy (that is, low β/α) little of the activity is shifted from the closed liganded state to the open liganded state, and the EC50 approximates the binding Kd. For ligands with high efficacy the EC50 corresponds to the Kd multiplied by fraction of channels closed in saturation conditions.
For a scheme involving two binding steps, such as the following one:
scheme II.
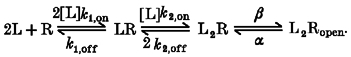
The Popen as a function of the ligand concentration for Scheme II is:
The analytical solution for the EC50 is:
where C = 0·5+ 0·5√{1 +Kd1/((Kd2)(1 –Ψ))}. In summary, even in this reaction scheme the EC50 is a function of the Kd values and of the maximal Popen. The two reaction schemes mentioned above do not involve non-conducting states along the pathway of activation other than LR and L2R. However, for the GABAA receptor it has been suggested that there are additional non-conducting states connected with the pathway of activation of the receptor channels (Jones & Westbrook, 1996). These non-conducting states may prolong the activated state, while decreasing the Popen. For such schemes more complicated relationships are to be expected and analytical solutions can seldom, if ever found.
The following reaction scheme is based on Scheme I, but incorporates a non-conducting state of the receptor connected to the liganded-closed state:
scheme III.
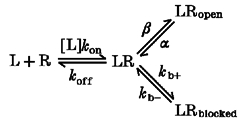
The Popen as a function of the ligand concentration for Scheme III is:
where the unblocked equilibrium constant KB=kb-/kb+.
This equation differs from the corresponding one for the three state scheme without block (Scheme I above) by the presence of a factor describing block. The EC50 for the observed response is:
The EC50 of the response is determined by the binding Kd, by Ψ and by the equilibrium constant for the unblocked state. That is, to estimate the binding Kd it is not sufficient to determine only the maximal Popen and the EC50 for the response, because the estimate of KB is required. This conclusion can be generalized in the statement that, in principle, the presence of blocked states may not always allow a reliable inference of the binding Kd from only the EC50 and the maximal Popen. In the present study, pentobarbital effects suggested the existence of blocked states and therefore estimates of binding Kd values by the EC50 and the maximal Popen were uncertain; rather, Kd values were estimated through the time course fitting of the experimental data to the predictions of a reaction mechanism, with the fit parameters adjusted to obtain maximal Popen values close to those inferred through single-channel recordings.
APPENDIX 2
We will briefly show that the analysis and interpretation of the variance-mean plots should not be affected by multiple conductance states because in α1β3 the activity can be approximated to that of one single-channel conductance and in α1c1 the bell-shaped current-variance plot must be indicative of a high Popen.
A simple quantitative inference of the channel Popen from the current-variance plot is established only for recordings of channels with one conductance state. However, interpretations for channels with openings to multiple conductance states can be made if the relative occurrence of the different conductance states is constant at different levels of activation of the receptor channel. In theory, for a channel with multiple conductance states, a linear current-variance plot may not exclude a Popen higher than 0·5 for a subconductance state with an extremely low amplitude which makes a small contribution to the overall charge transfer. A bell-shaped variance-mean plot indicates that the sum of the Popen of the individual conductance states must be at least higher than 0·5, though a precise and accurate quantitative estimate on the maximal Popen may be difficult.
In practice, a simple quantitative interpretation may still be made when channel openings have multiple conductances: when the unitary conductance amplitudes exhibit only small differences, or when one conductance state is responsible for the large majority of the charge transfer, the activity can be considered to approximate that of channels with one single-channel conductance.
Several studies on GABAA receptors indicate the presence of multiple conductance states (see, for example, Newland et al. 1991). However, especially with data obtained from neural cells, the evidence may not always be conclusive in excluding the possibility that the distinct conductance states actually represent distinct channel types and some studies on native membranes show only one single conductance (Zhang & Jackson, 1995). In fact, in data from recombinant receptors composed of α+β subunits (Verdoorn et al. 1990; Angelotti & Macdonald, 1993), one main conductance state is responsible for the large majority of charge transfer.
In our recordings with α1β3 with GABA, the low single-channel activity and the signal-to-noise ratio may have reduced the possibility of resolving subconductance states. However, we did not see indications of multiple conductance states in single-channel recordings (see Fig. 10). This observation suggests that supra- or sub-conductance states are likely to make only a negligible contribution to the overall charge transfer.
Statements on subconductance states of the α1c1 receptor channels are very difficult because their average amplitude is extremely small. If channel openings approximated those of a channel with only one conductance the superimposition of the openings of various conductance states should result in histograms of amplitudes of all points composed of multiple Gaussian functions spaced by constant step amplitudes. If this approximation were not valid, the superimposition of the openings of many channels to different conductances should result in histograms of amplitudes of all points deviating from such shape, and this deviation should be more evident the larger the number of channels opening simultaneously. However, even with the data exhibiting the largest excursion of amplitude values, which probably reflected the simultaneous openings of many channels, histograms of all points could indeed be fitted by multiple Gaussian functions spaced by constant step amplitudes. This observation is consistent with the hypothesis that the channel activity should approximate that of one single-channel conductance. Finally, the bell-shaped current-variance plot of α1c1 indicates that, if multiple conductance states are present, the arithmetic sum of the Popen of the individual conductance states should be higher than 0·5.
Both variance analysis and the single-channel interpretation assume that the current increase at different levels of activation is due to changes in the opening probability without changing the unitary amplitude. For channels with openings distributed in multiple conductance states, the ratios between the probabilities of all the conductance states must remain constant at different levels of activation of the receptor channel.
Observations on GABAA receptors of other preparations are consistent with this assumption. For example, in recordings of the GABAA receptors of cultured mouse spinal cord neurons, two conductance states were seen. However, no change in the relative occurrence of the conductance states was seen at different GABA concentrations (Macdonald et al. 1989a) or after addition of pentobarbital (Macdonald et al. 1989b).
References
- Akaike N, Inomata N, Tokutomi N. Contribution of chloride shifts to the fade of γ-aminobutyric acid-gated currents in frog dorsal root ganglion cells. The Journal of Physiology. 1987a;391:219–234. doi: 10.1113/jphysiol.1987.sp016735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike N, Maruyama T, Tokutomi N. Kinetic properties of the pentobarbitone-gated chloride currents in frog sensory neurones. The Journal of Physiology. 1987b;394:85–98. doi: 10.1113/jphysiol.1987.sp016861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato A, Connolly CN, Moss SJ, Smart TG. Modulation of neuronal and recombinant GABAA receptors by redox agents. The Journal of Physiology. 1999;517:35–50. doi: 10.1111/j.1469-7793.1999.0035z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin J. A single hydrophobic residue confers barbiturate sensitivity to γ-aminobutyric acid type C receptor. Molecular Pharmacology. 1999;55:411–423. [PubMed] [Google Scholar]
- Amin J, Weiss DS. GABAA receptor needs two homologous domains of the β subunit for activation by GABA but not by pentobarbital. Nature. 1993;366:565–569. doi: 10.1038/366565a0. [DOI] [PubMed] [Google Scholar]
- Angelotti TP, Macdonald RL. Assembly of GABAA receptor subunits. α1β1 and α1β1γ2 subunits produce unique ion channels with dissimilar single-channel properties. Journal of Neuroscience. 1993;13:1429–1440. doi: 10.1523/JNEUROSCI.13-04-01429.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Lambert JJ, Peters JA, Wafford K, Whiting PJ. The interaction of the general anaesthetic etomidate with the gamma-aminobutyric acid type A receptor is influenced by a single-amino acid. Proceedings of the National Academy of Sciences of the USA. 1997;94:11031–11036. doi: 10.1073/pnas.94.20.11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnir B, Tierney ML, Dalziel JE, Cox GB, Gage PW. A structural determinant of desensitization and allosteric regulation by pentobarbitone of the GABAA receptor. Journal of Membrane Biology. 1997;155:157–166. doi: 10.1007/s002329900167. [DOI] [PubMed] [Google Scholar]
- Chang Y, Weiss DS. Channel opening locks agonist onto the GABAC receptor. Nature Neuroscience. 1999;2:219–225. doi: 10.1038/6313. [DOI] [PubMed] [Google Scholar]
- Chen C, Okayama H. High efficiency transformation of cells by plasmid DNA. Molecular and Cellular Biology. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Hawkes AG. Relaxation and fluctuations of membrane currents that flow through drug-operated channels. Proceedings of the Royal Society B. 1977;199:231–262. doi: 10.1098/rspb.1977.0137. [DOI] [PubMed] [Google Scholar]
- Franks NP, Lieb WR. Molecular and cellular mechanisms of general anaesthesia. Nature. 1994;367:607–614. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- Gallagher BB, Freer LS. Barbituric acid derivatives. In: Frey HH, Yanz D, editors. Antiepileptic Drugs. Berlin: Springer Verlag; 1985. pp. 421–447. [Google Scholar]
- Ho S, Hunt HD, Horton RM, Pullen JK, Pease LR. Site directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Horn R. Estimating the number of channels in patch recordings. Biophysical Journal. 1991;60:433–439. doi: 10.1016/S0006-3495(91)82069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M, Westbrook G. The impact of receptor desensitization on fast synaptic transmission. Trends in Neurosciences. 1996;19:96–101. doi: 10.1016/s0166-2236(96)80037-3. [DOI] [PubMed] [Google Scholar]
- Karlin A, Akabas M. Towards a structural basis for the function of nicotinic acetylcholine receptors and their cousins. Neuron. 1995;15:1231–1244. doi: 10.1016/0896-6273(95)90004-7. [DOI] [PubMed] [Google Scholar]
- Kennedy SK, Longnecker DE. Goodman and Gilman's The Pharmacological Basis of Therapeutics. Toronto: McGraw-Hill; 1995. History and principles of anesthesiology; pp. 295–305. [Google Scholar]
- Krasowski MD, Finn SE, Ye Q, Harrison NL. Trichloroethanol modulation of recombinant GABAA, glycine and GABA ρ1 receptors. Journal of Pharmacology and Experimental Therapeutics. 1998a;284:934–942. [PubMed] [Google Scholar]
- Krasowski MD, Koltchine VV, Rick CE, Ye Q, Finn SE, Harrison NL. Propofol and other intravenous anesthetics have sites of action on the γ aminobutyric acid type A receptor distinct from that for isoflurane. Molecular Pharmacology. 1998b;53:530–538. doi: 10.1124/mol.53.3.530. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Rogers CJ, Twyman RE. Kinetic properties of the GABAA receptor main conductance state of mouse spinal cord neurones in culture. The Journal of Physiology. 1989a;410:479–499. doi: 10.1113/jphysiol.1989.sp017545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Rogers CJ, Twyman RE. Barbiturate regulation of kinetic properties of the GABAA receptor channel of mouse spinal neurones in culture. The Journal of Physiology. 1989b;417:483–500. doi: 10.1113/jphysiol.1989.sp017814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maconochie DJ, Knight DE. A method for making solution changes in the submillisecond range at the tip of a patch pipette. Pflügers Archiv. 1989;414:589–596. doi: 10.1007/BF00580996. [DOI] [PubMed] [Google Scholar]
- Mihic SJ, Ye W, Wick MJ, Koltchine VV, Krasowski MA, Finn SE, Mascia MP, Valuenzuela CF, Hanson KK, Greenblatt EP, Harris RA, Harrison NL. Sites of alcohol and volatile anaesthetics action on GABAA and glycine receptors. Nature. 1997;389:385–389. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- Newland CF, Colquhoun D, Cull-Candy SG. Single-channels activated by high concentrations of GABA in superior cervical ganglion neurones of the rat. The Journal of Physiology. 1991;432:203–233. doi: 10.1113/jphysiol.1991.sp018382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press WH, Teukolsky SA, Vetterling WT, Flannery BP. Numerical Recipes in C. Cambridge: Cambridge University Press; 1996a. Downhill Simplex method in multidimensions; pp. 408–412. [Google Scholar]
- Press WH, Teukolsky SA, Vetterling WT, Flannery BP. Numerical Recipes in C. Cambridge: Cambridge University Press; 1996b. Confidence limits on estimated model parameters; pp. 689–699. [Google Scholar]
- Rho JM, Donevan SD, Rogawski MA. Direct activation of GABAA receptors by barbiturates in cultured rat hippocampal neurons. The Journal of Physiology. 1996;497:509–522. doi: 10.1113/jphysiol.1996.sp021784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini R, Bracamontes J, Steinbach JH. Structural domains involved in the effects of general anesthetics on recombinant GABAA receptors. Society for Neuroscience Abstracts. 1997;23:51.8. [Google Scholar]
- Serafini R, Bracamontes J, Steinbach JH. Characterization of pentobarbital-induced block of GABA-gated Cl− ion channels in recombinant receptors. Society for Neuroscience Abstracts. 1998;24:233.16. [Google Scholar]
- Serafini R, Valeyev A, Barker JL, Poulter M. Depolarizing GABA-activated Cl− channels in embryonic rat spinal and olfactory bulb cells. The Journal of Physiology. 1995;488:371–386. doi: 10.1113/jphysiol.1995.sp020973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada S, Cutting G, Uhl GR. γ-Aminobutyric acid A or C receptor?γ-Aminobutyric acid ρ1 receptor RNA induces bicuculline-, barbiturate-, and benzodiazepine-insensitive γ-aminobutyric acid responses in Xenopus oocytes. Molecular Pharmacology. 1992;41:683–687. [PubMed] [Google Scholar]
- Sigworth FJ. Non stationary noise analysis of membrane current. In: Eisemberg RS, Frank M, Stevens CF, editors. Membranes, Channels and Noise. New York: Plenum Press; 1984. pp. 21–48. [Google Scholar]
- Smith GB, Olsen RW. Functional domains of GABAA receptors. Trends in Pharmacological Sciences. 1995;16:162–168. doi: 10.1016/s0165-6147(00)89009-4. [DOI] [PubMed] [Google Scholar]
- Tanelian DL, Kosek P, Mody I, Maciver B. The role of the GABAA receptor/chloride channel complex in anesthesia. Anesthesiology. 1993;78:757–776. doi: 10.1097/00000542-199304000-00020. [DOI] [PubMed] [Google Scholar]
- Thomson SA, Whiting PJ, Wafford KA. Barbiturate interactions at the human GABAA receptor: dependence on receptor subunit combination. British Journal of Pharmacology. 1996;117:521–527. doi: 10.1111/j.1476-5381.1996.tb15221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Bracamontes J, Zorumski C, Weiss DS, Steinbach JH. Bicuculline and gabazine are allosteric inhibitors of channel opening of the GABAA receptor. Journal of Neuroscience. 1997;17:625–634. doi: 10.1523/JNEUROSCI.17-02-00625.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Zorumski C, Bracamontes J, Steinbach JH. Endogenous subunits can cause ambiguities in the pharmacology of exogenous γ-aminobutyric acid A receptors expressed in human embryonic kidney 293 cells. Molecular Pharmacology. 1996;50:931–938. [PubMed] [Google Scholar]
- Verdoorn T, Draghun A, Ymer S, Seeburg PH, Sackmann B. Functional properties of recombinant rat GABAA receptors depend upon subunit composition. Neuron. 1990;4:919–928. doi: 10.1016/0896-6273(90)90145-6. [DOI] [PubMed] [Google Scholar]
- Wagstaff J, Chaillet JR, Lalande M. The GABAA receptor β3 subunit gene; characterization of a human cDNA from chromosome 15q11q13 and mapping to a region of conserved synteny on mouse chromosome 7. Genomics. 1991;11:1071–1078. doi: 10.1016/0888-7543(91)90034-c. [DOI] [PubMed] [Google Scholar]
- Wingrove PB, Wafford KA, Bain C, Whiting PJ. The modulatory action of loreclezole at the gamma-aminobutyric acid type A receptor is determined by a single aminoacid in the β2 and β3 subunit. Proceedings of the National Academy of Sciences of the USA. 1994;91:4569–4573. doi: 10.1073/pnas.91.10.4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolworton JR, Moss J, Smart TG. Pharmacological and physiological characterization of murine homomultimeric β3 GABAA receptors. European Journal of Neuroscience. 1997;9:2225–2235. doi: 10.1111/j.1460-9568.1997.tb01641.x. [DOI] [PubMed] [Google Scholar]
- Xu M, Akabas MH. Identification of channel-lining residues in the M2 membrane-spanning segment of the GABAA receptor α1 subunit. Journal of General Physiology. 1996;107:195–205. doi: 10.1085/jgp.107.2.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ymer S, Schofield PR, Draguhn A, Werner P, Kohler M, Seeburg PH. GABAA receptor β subunit heterogeneity; functional expression of cloned cDNAs. EMBO Journal. 1989;8:1665–1670. doi: 10.1002/j.1460-2075.1989.tb03557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SJ, Jackson MB. Properties of the GABAA receptor of rat posterior pituitary nerve terminals. Journal of Neurophysiology. 1995;73:1135–1144. doi: 10.1152/jn.1995.73.3.1135. [DOI] [PubMed] [Google Scholar]