

Abstract
The active Ca2+ transport properties of malaria-infected, intact red blood cells are unknown. We report here the first direct measurements of Ca2+ pump activity in human red cells infected with Plasmodium falciparum, at the mature, late trophozoite stage.
Ca2+ pump activity was measured by the Co2+-exposure method adapted for use in low-K+ media, optimal for parasitised cells. This required a preliminary study in normal, uninfected red cells of the effects of cell volume, membrane potential and external Na+/K+ concentrations on Ca2+ pump performance.
Pump-mediated Ca2+ extrusion in normal red cells was only slightly lower in low-K+ media relative to high-K+ media despite the large differences in membrane potential predicted by the Lew-Bookchin red cell model. The effect was prevented by clotrimazole, an inhibitor of the Ca2+-sensitive K+ (KCa) channel, suggesting that it was due to minor cell dehydration.
The Ca2+-saturated Ca2+ extrusion rate through the Ca2+ pump (Vmax) of parasitised red cells was marginally inhibited (2-27 %) relative to that of both uninfected red cells from the malaria-infected culture (cohorts), and uninfected red cells from the same donor kept under identical conditions (co-culture). Thus, Ca2+ pump function is largely conserved in parasitised cells up to the mature, late trophozoite stage.
A high proportion of the ionophore-induced Ca2+ load in parasitised red cells is taken up by cytoplasmic Ca2+ buffers within the parasite. Following pump-mediated Ca2+ removal from the host, there remained a large residual Ca2+ pool within the parasite which slowly leaked to the host cell, from which it was pumped out.
Calcium ions are required at two critical stages during the intraerythrocytic asexual life-cycle of P. falciparum: for a few seconds during the invasion of the red cell by the parasite, and continuously for normal parasite development (Wasserman et al. 1982; Tanabe, 1990). The total calcium content of parasitised cells increases with parasite maturation far above the negligible levels observed in uninfected normal red cells (Bookchin et al. 1980). Evidence suggests that this increase is confined to internal parasite compartments (Lee et al. 1988; Tanabe, 1990; Kramer & Ginsburg, 1991; Desai et al. 1996). Calcium homeostasis of malaria-infected red cells has been the subject of intense research in recent years, with substantial advances in our knowledge of stage-related changes in the passive Ca2+ permeability of the membrane (Kramer & Ginsburg, 1991; Desai et al. 1996; Staines et al. 1999). Nothing is known, however, about the performance of the plasma membrane Ca2+ pump (PMCA) in intact, parasitised red cells. The only reported measurements of Ca2+-Mg2+-ATPase activity were done in isolated membranes of murine red cells infected with P. chabaudi (Tanabe, 1990). The results suggested that the pump enzyme was inhibited by about 30 % relative to uninfected controls. There have been no further studies in other Plasmodii infections or on the functional state of the pump in the intact parasitised cell. It is important to bear in mind that the PMCA is a multi-regulated transporter (Schatzmann, 1982) and that ATPase activity in isolated membranes may not reflect the performance of the pump in the intact cell.
We report here the first direct measurements of PMCA-mediated Ca2+ extrusion in intact human red cells infected with P. falciparum at the late trophozoite stage. Assessment of Ca2+ pump function in intact, uninfected red cells is routinely done by the Co2+-exposure method (Dagher & Lew, 1988). However, this method had to be modified to obtain reliable Ca2+ flux measurements in parasitised cells. This required a preliminary feasibility study in normal uninfected red cells.
METHODS
Solutions
Solution A (low-K+, plasma-like medium) contained (mM): KCl, 3; NaCl, 140; MgCl2, 0.2; Hepes-Na (pH 7.4-7.5 at 37°C), 10; and Na-EGTA, 0.1. Solution B was the same as solution A but without Na-EGTA. Solution C (‘clamp’ medium) contained (mM): KCl, 80; NaCl, 70; MgCl2, 0.15; and Hepes-Na (pH 7.4-7.5 at 37°C), 10. Stock solutions of 45CaCl2 were prepared at concentrations of 40–100 mM and with 45Ca2+ specific activity between 107 and 108 c.p.m. (μmol)−1. A23187 was dissolved in ethanol, as a 2 mM stock solution. CoCl2 was prepared as a stock solution (100 mM) in distilled water. Sodium orthovanadate was prepared as a stock solution (100 mM) in solution B. Clotrimazole was prepared as a stock solution (10 mM) in DMSO.
Parasite cultures
The P. falciparum cloned strain A4 (kind gift of B. C. Elford, Institute of Molecular Medicine, Oxford, UK), derived from the P. falciparum ITO4 line (Berendt et al. 1989), was cultured in human red blood cells (type O) by standard methods (Trager & Jensen, 1976), under a low-oxygen atmosphere (1 % O2, 3 % CO2, 96 % N2). The culture medium was RPMI-1640, supplemented with D-glucose (10 mM), glutamine (2 mM), Hepes (40 mM), gentamicin sulphate (25 mg l−1) and 8.5 % (v/v) pooled human serum. Parasites were synchronised by a combination of sorbitol haemolysis (Lambros & Vanderberg, 1979) and gelatin flotation (Jensen, 1978). All experiments with malaria-infected erythrocytes were carried out using red blood cells infected with P. falciparum at the late trophozoite stage (36-40 h post-invasion). Parasitaemia was assessed by microscopic inspection of Giemsa-stain thin blood smears. Cell counts were estimated on suspension samples (5 μl) fixed in formalin (100 μl) using an improved Neubauer counting chamber.
Use of the Co2+-exposure method to measure PMCA-mediated Ca2+ transport in P. falciparum-infected red cells
In the Co2+-exposure method (Dagher & Lew, 1988; Lew & García-Sancho, 1989) a uniform Ca2+ load is rapidly induced in a red cell population by means of the ionophore A23187. After 1–2 min, Co2+ is added to the cell suspension to instantly block ionophore-mediated Ca2+ transport, thereby exposing uphill Ca2+ extrusion by a Ca2+-saturated pump (Vmax). During this procedure the cells become permeabilised to Ca2+ and Mg2+ via the ionophore, and indirectly to K+, by activation of their KCa channels (Gardos, 1958; Lew & Ferreira, 1978). In low-K+, plasma-like media, the cells would hyperpolarise, lose KCl and water, and acidify (Lew & Bookchin, 1986; Freeman et al. 1987). These changes in cell volume, pH, membrane potential and ion content, resulting from the increased ion permeabilities, are prevented by a ‘homeostatic clamp’ obtained by setting the external Mg2+ and K+ concentrations at electrochemical equilibrium with the intracellular ions, to minimise net ion fluxes other than those of Ca2+ (Dagher & Lew, 1988; Lew & García-Sancho, 1989). Application of this procedure to P. falciparum-infected red cells is not feasible. Parasitised cells undergo marked changes in their membrane transport properties due to the early appearance of new permeation pathways (NPPs) (Ginsburg et al. 1986; Lee et al. 1988; Kirk et al. 1991, 1992). Although NPPs are anion selective, they also increase the Na+ and K+ permeabilities resulting in the rapid dissipation of monovalent cation gradients. This predisposes the red cells infected with late trophozoites to rapid haemolysis in high-K+ media, such as those used for clamp conditions in the standard Co2+-exposure method. A suspension of infected cells, however well synchronised, represents an heterogenous mix of cells in different homeostatic states, which is impossible to clamp in a single suspending medium. Since parasitised cells retain optimal volume stability in low-K+ media, such as RPMI media used for in vitro cultures, it would be desirable to measure pump activity in similar low-K+ media. The question would then be whether Ca2+ pump performance would be comparable in and out of clamp conditions, and whether the predicted changes in membrane potential, cell volume, ion content and pH would affect the measurements in ways that precluded proper assessment of pump function. To answer these questions it was necessary to compare Ca2+ pump function in standard clamp conditions and in low-K+ media. This comparison was performed in fresh, uninfected red cells. Besides the value of the results for the general characterisation of Ca2+ pump properties, the study provided the information required for the Co2+-exposure method to be applied to P. falciparum-infected cells. It is worth pointing out that the only alternative to the Co2+-exposure method, i.e. measurement of pump-mediated Ca2+ extrusion after ionophore wash-out (Pereira et al. 1993), cannot be applied to P. falciparum-infected cells because these cells could not withstand the required low-temperature washes.
Experiments with fresh, uninfected red cells
Venous blood from healthy volunteers was drawn into heparinised syringes after written consent. The cells were washed 3 times by centrifugation (2500 g, 5 min) and resuspension in 6–8 volumes of solution A to remove Ca2+ loosely bound to the outer cell surface (Harrison & Long, 1968). The cell pellet was divided into two aliquots. One aliquot was washed 3 more times in solution B to remove EGTA from the medium, and the other aliquot 3 more times in solution C. After each spin, the supernatant and the top cell layer containing white cells and platelets were removed. After the washes the cells were suspended at ∼10 % haematocrit in solution B or C, supplemented with inosine (10 mM) and the additives indicated in each case. The haematocrit of cell suspensions was estimated from spectrophotometric haemoglobin (Hb) measurements at 540 nm by the cyanmethaemoglobin method. In the experiments where clotrimazole was used as a specific inhibitor of the red cell KCa channels (Alvarez et al. 1992), clotrimazole was added to a final concentration of 10 μM in the cell suspension, sufficient to inhibit over 99 % of KCa-mediated K+ fluxes (Brugnara et al. 1994).
Experiments with P. falciparum-infected red cells, cohort and co-cultured cells
Cells from cultures were separated by centrifugation (400 g, 10 min) on Percoll diluted with ×10 phosphate-buffered saline and water to a density of 1.090 (∼66 % v/v) and osmolality of 320 mosmol (kg H2O)−1 (Kirk et al. 1996). Suspensions with 50–96 % parasitaemia were obtained by this method. Uninfected red blood cells from the same culture (cohorts) were harvested from the cell pellet under Percoll. The cohort pellet had < 5 % parasitised red cells, mainly young, immature parasites. Uninfected red cells from the same donors were incubated in parallel (co-cultured cells) under identical conditions, for at least 48 h prior to the experiment. These cells and cohorts were used as controls.
Since the Co2+-exposure protocol had to be carried out consecutively for each of the different conditions, it was important to ensure that the various suspensions of Percoll-harvested parasitised cells reached the flux assay in optimal state. Percoll-harvested parasitised cells were washed twice by centrifugation (600 g, 5 min) and resuspension in culture medium without serum to remove residual Percoll; after the washes the cells were resuspended in culture medium (< 0.5 % haematocrit) and kept at room temperature, under low PO2, until use. This procedure gave comparable results for the first and last processed control groups within each experiment, and preserved the normal morphology of the parasitised cells without extra lysis. Haematocrits were estimated from cell counts per unit volume (see below). All suspensions with Percoll-harvested parasitised cells were inspected to assess the morphological condition of the infected cells. Five to 10 μl of fresh, unfixed cell suspensions in RPMI were placed between slide and coverslip, observed by phase contrast under oil at ×1000, and video-recorded. If there was no evidence of Brownian motion of haemozoin crystals within the parasites’ food vacuoles the sample was discarded.
Measurement of PMCA-mediated Ca2+ extrusion in parasitised red cells, cohorts, co-cultured cells, and fresh, uninfected red cells
Immediately before use the cells were washed twice in solution B and resuspended (3-10 % haematocrit) in solution B with 10 mM inosine and 5 mM glucose. 45CaCl2 was added to 1.5-2.0 ml aliquots of cell suspensions to a final Ca2+ concentration in the suspensions ([CaT]s) of 124–170 μM (specific details in the corresponding figure legends). The low-haematocrit conditions chosen in these experiments ensured minimal dilution of the original 45Ca2+ specific activity by the parasites’ endogenous cold Ca2+ pools. At least 108 parasitised cells were used for each condition. The suspensions were pre-incubated at 37°C for 5–10 min under magnetic stirring before the Co2+-exposure protocol was carried out. At time (t)= 0, the ionophore A23187 was added from a concentrated stock solution to final concentrations intended to ensure rapid 45Ca2+ redistribution to equilibrium in both infected and uninfected red cells. Normal, uninfected human red cells are virtually Ca2+ free (Harrison & Long, 1968; Bookchin & Lew, 1980; Engelmann & Duhm, 1987) but P. falciparum parasites contain a substantial endogenous Ca2+ pool which increases the mean total intracellular calcium concentration ([CaT]i) of the infected red cells by more than tenfold. Bookchin et al. (1980) reported a [CaT]i of 52 μmol (l cells)−1 in a 60 % parasitised cell suspension. Ionophore-induced Ca2+ permeabilisation mixes all membrane-bound compartments containing Ca2+ pools (Bookchin et al. 1980). At t = 1-2 min, CoCl2 was added to the suspension to a concentration of 400 μM and uphill Ca2+ extrusion was followed for 6–8 min, thus minimising both the ATP-depleting effect caused by the pump ATPase activity (Dagher & Lew, 1988; García-Sancho & Lew, 1988) and the toxic effects of long-term parasite exposure to ionophore (Krungkrai & Yuthavong, 1983). Samples (50 μl) were taken throughout the procedure at the times indicated in the figures for measurements of [CaT]i, and processed as reported before (Dagher & Lew, 1988; Tiffert & Lew, 1997). In the experiments with orthovanadate, sampling for [CaT]i was continued for an extended period of 20 min. Ionophore and pump-mediated Ca2+ fluxes were estimated from the slope of the curves reporting [CaT]i as a function of time. Initial Ca2+ extrusion after Co2+ addition is usually linear. It represents the maximal Ca2+ extrusion rate through a Ca2+-saturated pump; the initial slope reports the pump Vmax and was estimated by linear regression.
For consistency with data in the literature and for internal comparability, Ca2+ fluxes in fresh, uninfected cells, from suspensions in which haematocrits were estimated from spectrophotometric Hb measurements, were expressed in units of millimoles per 340 grams of haemoglobin per hour (mmol (340 g Hb)−1 h−1). In the experiments with cultured cells, from suspensions in which haematocrits were estimated from cell counts, Ca2+ fluxes were expressed in units of millimoles per 1013 cells per hour (mmol (1013 cells)−1 h−1). Within a 10–15 % variation, 340 g Hb may be assumed to represent the 1013 cells present in 1 l of normal volume, packed red blood cells. When estimated from cell counts per unit volume, haematocrits were calculated as fractions of 1013 cells per litre, assumed to represent a haematocrit of 100 %.
Use of the red cell model to estimate the value of red cell homeostatic variables
Estimates of the approximate value of relevant homeostatic variables (membrane potential, cell volume and pH), and analysis of the changes induced by the different conditions set up in the experiments with fresh uninfected cells, were done using the Lew-Bookchin red cell model (Lew & Bookchin, 1986). For the range of conditions studied here, this model was shown to predict the value of those variables with a precision level of 5–10 % (Freeman et al. 1987; Tiffert et al. 1993; Raftos & Lew, 1995; Etzion et al. 1996). The simulations followed the experimental protocols applied here and the relevant values obtained in each case are reported in Results.
The Lew-Bookchin red cell model is available as a free-standing program for PC platforms from http://www.physiol.cam.ac.uk/staff/lew/index.htm
Materials
All chemicals were analytical reagent quality. Plasmagel (Bellon, Neuilly Sur Seine, France) was a kind gift from Barry C. Elford, Institute of Molecular Medicine, Oxford (UK). EGTA, Hepes, glucose, inosine, clotrimazole, sodium orthovanadate, CoCl2, DMSO, RPMI-1640 medium (R0883), gentamicin sulphate, glutamine and formalin solution were from Sigma-Aldrich Co. (UK). A23187 was from Calbiochem-Novabiochem (UK) Ltd. CaCl2, MgCl2, NaCl and KCl were from FSA Laboratory Supplies (Loughborough, UK). 45Ca2+ was from Amersham International plc (Little Chalfont, UK). PBS was from Life Technologies Ltd (UK). Percoll was from Amersham Pharmacia Biotech. (Sweden). Human blood and serum used in cultures were from the Eastern Anglia Blood Centre, Cambridge (UK).
RESULTS
Comparison of PMCA performance in uninfected red cells in and out of homeostatic clamp conditions
In the experiment illustrated in Fig. 1, typical of four others with similar results, fresh normal red cells were suspended either in low-K+ solution B, or in high-K+, clamp-solution C, with or without clotrimazole. Addition of the ionophore A23187 caused rapid loading of the cells with [45CaT]i to a level of ∼600 μmol (340 g Hb)−1, which was similar for both media. This Ca2+ load fully activates the KCa channels of the cells, effectively increasing their K+ permeability by over three orders of magnitude (Lew & Ferreira, 1978). In the experiment of Fig. 1, the Ca2+ pump Vmax values were 15.8 and 16.3 mmol (340 g Hb)−1 h−1 for control clamp conditions, without and with clotrimazole, respectively, indicating that clotrimazole per se has no effect on pump-mediated fluxes. In low-K+ media, the Ca2+ pump Vmax values were 14.9 and 16.4 mmol (340 g Hb)−1 h−1 in the absence and presence of clotrimazole, respectively. In the four experiments of this series, the Vmax values of the Ca2+ pump in low K+ were 3.5-7.0 % lower than those in clamp conditions, and 6.5-9.0 % lower than those in low K+ with clotrimazole. Though hardly significant within each experiment, the pump inhibitory effects of low-K+ media were present in all experiments. The effect was prevented by clotrimazole, a KCa channel inhibitor, suggesting that it was due to minor cell dehydration caused by the activation of the KCa channels in the low-K+ medium.
Figure 1. Measurement of Ca2+ pump-mediated Ca2+ extrusion from intact fresh red cells in both high- and low-K+ media: effects of clotrimazole.
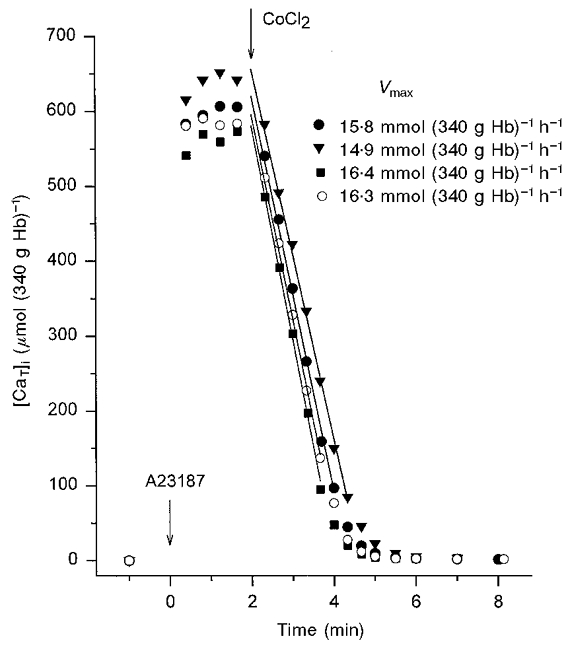
Red cells from freshly drawn blood were suspended at 10 % haematocrit in either high-K+, clamp-solution C (circles), or in low-K+ solution B (triangles and squares), both supplemented with 10 mM inosine, and with (open circles, squares) or without (filled circles, triangles) clotrimazole (10 μM). All cell suspensions contained 45Ca2+. The 45Ca2+ concentration added to the cell suspensions ([CaT]s) was ≈150 μM. Arrows indicate additions of ionophore A23187 (t = 0, 10 μM) and CoCl2 (t = 2 min, 400 μM). The Vmax values correspond to the slope of the linear regression lines through the indicated points, chosen by eye for each curve.
For the low-K+ conditions of these experiments, with the Cl− permeability rate-limiting to net KCl loss and dehydration, the red cell model predicted membrane potential hyperpolarisation to about -90 mV, less than 10 % dehydration within the 8 min duration of the experiments and insignificant cytoplasmic acidification. The results in Fig. 1 show that Ca2+ pump performance was not significantly affected by any of the following: large changes in membrane potential and external Na+/K+ concentrations, isotonic cell dehydration (< 10 %), or by the associated minor changes in cell and medium pH (Freeman et al. 1987). Moreover, the different conditions had no effect on ionophore-induced Ca2+ influx or on the ionophore-induced equilibrium distribution of Ca2+ between cells and medium (Fig. 1). Thus, the Co2+-exposure protocol used here may be safely applied to investigate pump-mediated Ca2+ extrusion from uninfected and infected cells in non-clamp, low-K+ media, as long as the KCa channels are blocked or the measurements completed before significant cell dehydration.
PMCA performance in P. falciparum-infected red cells
PMCA function in infected cells was explored in low-K+, non-clamp media only. Cell counts before and after the Co2+-exposure protocol showed less than 5 % haemolysis for both infected and uninfected cells under these conditions, comparable to that observed under clamp conditions. In the experiment of Fig. 2, the ionophore concentration was the same for parasitised and cohort cell suspensions (10 μM). The pattern of changes in [CaT]i with time was markedly different. The main difference was in the ionophore-induced Ca2+ influx which was relatively slow in the parasitised cells. It was clear that an ionophore concentration sufficient to induce Ca2+ equilibration by 1 min in uninfected cells failed to induce a significant Ca2+ load in infected cells. This reduced ionophoric effect has been observed before (Krungkrai & Yuthavong, 1983). Its possible origin is considered in Discussion.
Figure 2. Comparative response of parasitised and cohort cells to the Co2+-exposure protocol using identical ionophore concentrations.
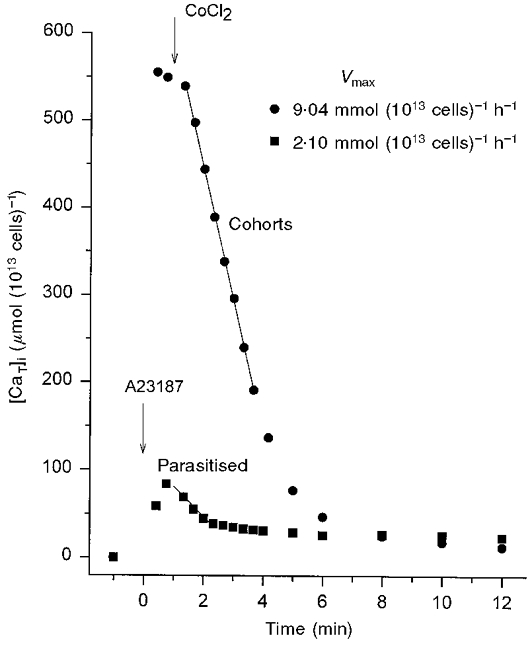
Cells were suspended in low-K+ solution B with 10 mM inosine and 5 mM glucose. Ionophore A23187 and Co2+ were added as described in Fig. 1, except for the timing of Co2+ addition which was 1 min after addition of the ionophore. Parasitaemia was 87 % in the parasitised cell suspension and ≈4.5 % (mostly immature, ring-stage parasites) in the cohort cell suspension. The 45Ca2+ concentration added to the cell suspensions ([CaT]s) was 124 μM. The haematocrits of parasitised and cohort cell suspensions were 3.9 and 6.2 %, respectively, relative to 100 %= 1013 cells l−1. Vmax values were calculated from the slope of the linear regression lines through the indicated points, chosen by eye for each curve. Note that the parasitised cells show a slow ionophore-induced Ca2+ influx, a limited Ca2+ load, a large apparent Vmax inhibition and a slightly higher residual calcium content than the cohorts.
Further experiments indicated that doubling of the ionophore concentration was required to elicit comparable loading patterns between infected and uninfected cells without causing additional haemolysis. Figure 3 shows the results obtained using a higher ionophore concentration (20 μM) in the parasitised cell suspensions. Parasitaemia in the parasitised cell suspensions was 50 % in Fig. 3a and 96 % in Fig. 3b. The results are representative of two other similar experiments, with 56 and 91 % infected cells, respectively. Ionophore-induced Ca2+ loads reached steady [CaT]i levels before Co2+ addition in all conditions. Estimates of the minimal net Ca2+ influx induced by the ionophore may be obtained from the Ca2+ gained by the cells in the first 20 s following ionophore addition. These varied between 100 and 200 mmol (1013 cells)−1 h−1, far higher than the maximal pump Vmax in any condition (see below). Thus, the near-plateau [CaT]i levels attained must have been close to the Ca2+ equilibrium distributions induced by the ionophore. The effect of vanadate is illustrated in Fig. 3b. As with normal human red cells (Tiffert & Lew, 1997), 1 mM vanadate fully inhibited uphill Ca2+ extrusion from parasitised cells. The similarity between the Ca2+ load induced by the ionophore and the level of [CaT]i sustained in the presence of vanadate confirms that the 2 min [CaT]i load is close to the ionophore-induced equilibrium. In each experiment, the Ca2+ load at equilibrium was higher in the parasitised cells than in the uninfected controls. The higher the parasitaemia the higher the [CaT]i load at equilibrium (compare Fig. 3a with B). Fig. 3b also shows that both parasitised and uninfected cells responded with increased loads to increased [CaT]s.
Figure 3. Comparative response of parasitised, co-cultured and cohort cells to the Co2+-exposure protocol, using a higher ionophore concentration in parasitised cells: Ca2+ pump Vmax and effect of vanadate.
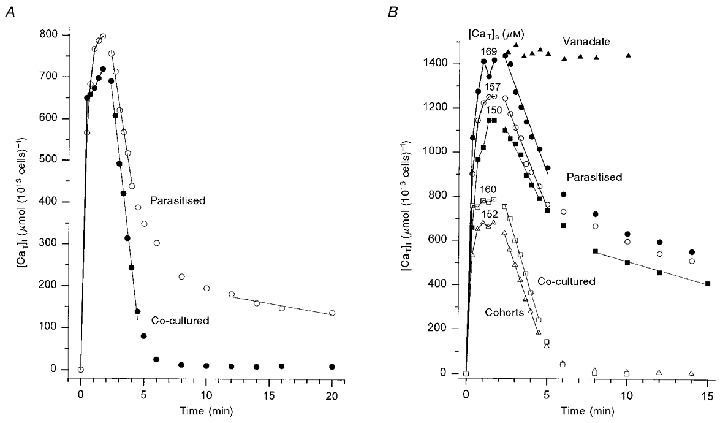
Cells were suspended in low-K+ solution B with 10 mM inosine and 5 mM glucose. Ionophore A23187 and Co2+ were added as described in Fig. 1. The ionophore concentration was 20 μM in the parasitised cell suspensions and 10 μM in co-cultured and cohort cell suspensions. A, parasitaemia in the parasitised cell suspension was 50 %; [CaT]s= 150 μM in parasitised and co-cultured cell suspensions; haematocrits of parasitised and co-cultured cells were 3.72 and 4.54 %, respectively. Pump Vmax values (slope ±s.e.m.) were 11.4 ± 0.5 mmol (1013 cells)−1 h−1 for parasitised cells, and 15.5 ± 0.4 mmol (1013 cells)−1 h−1 for co-cultured cells. The residual Ca2+ extrusion rate from parasitised cells, estimated by linear regression through the points between 12–20 min, was about 0.3 mmol (1013 cells)−1 h−1, equivalent to 0.6 mmol (1013 cells)−1 h−1, when corrected to 100 % parasitaemia. B, parasitaemia in the parasitised cell suspension was 96 %; parasitaemia in the cohort cell suspension was < 5 % (immature, ring-stage parasites). [CaT]s for each condition is indicated above the corresponding ionophore-induced equilibrium [CaT]i level. The Ca2+-loading stage for the condition with vanadate was omitted from the figure; it showed a similar equilibrium load as the condition with [CaT]s= 169 μM. The vanadate concentration in the medium was 1 mM. Haematocrits for each of the six curves were (from top to bottom): 4.3 (vanadate), 4.8, 4.4, 7.1, 6.5 and 7.7%. The Vmax values obtained for the five curves showing uphill Ca2+ extrusion were (from top to bottom, slope ±s.e.m.): 11.70 ± 0.73; 10.91 ± 0.54; 9.08 ± 0.64; 14.38 ± 0.32; and 12.50 ± 0.26 mmol (1013 cells)−1 h−1. Note the lower coefficient of variation of the last two curves corresponding to uninfected cells. The residual Ca2+ extrusion rate from parasitised cells is reported here for the condition with [CaT]s= 150 μM; the linear regression through points between 8–15 min gave a rate of about 1.2 mmol (1013 cells)−1 h−1.
The Vmax of the Ca2+ pump was estimated from the linear Ca2+ extrusion rate immediately after addition of Co2+. The pump Vmax of infected and uninfected cells varied between 8.1 and 15.5 mmol (1013 cells)−1 h−1 in the four experiments of this series. These values fell well within the range of normal variation observed before in fresh human red cells (Dagher & Lew, 1988; Tiffert et al. 1993). The Vmax value in cohort cells was lower than that in co-cultured controls (Fig. 3b). The Vmax values in the infected cell samples were 2–27 % lower than those in cohort and co-cultured samples in the different experiments.
Another systematic and more subtle difference in Vmax between infected and uninfected cells concerned the scatter of the experimental points. The coefficients of variation of the linear regression slopes obtained from uninfected cells were usually less than 2.5 %, whereas those from infected cells varied between 4.5 and 7.5 % (5.0-7.1 % in the experiments of Fig. 3b). Also, the choice of the number of points to use for Vmax estimates by linear regression was less clear in infected cells. This was due to the marked increase in Ca2+ retention within the parasitised cells relative to uninfected controls (Fig. 3). In the experiment with 96 % parasitaemia (Fig. 3b), the level of [CaT]i at which Ca2+ efflux slowed down after the initial rapid Ca2+ extrusion period was between 700 and 900 μmol (1013 cells)−1 in infected cells, and about 200 μmol (1013 cells)−1 in uninfected controls. In the two experiments with lower parasitaemia, the difference was smaller but still substantial. The residual Ca2+ extrusion rate from the 96 % parasitaemia suspensions was about 1.2 mmol (1013 cells)−1 h−1 (Fig. 3b) whereas that from the suspension with 50 % parasitaemia was 0.6 mmol (1013 cells)−1 h−1 (corrected to 100 % parasitaemia, Fig. 3a).
DISCUSSION
The present results report the first direct measurements of Ca2+ pump performance in P. falciparum-infected, intact red cells. The preliminary investigation (Fig. 1) showed that the activity of the Ca2+ pump in uninfected, normal red cells was minimally affected by large variations in membrane potential and external Na+ and K+ concentrations, or by minor cell dehydration. This comparability of pump Vmax rates established the feasibility of measuring Ca2+ pump Vmax by the Co2+-exposure method outside the usual clamp constraining conditions, provided the measurements were performed in the presence of KCa channel blockers, or completed before significant cell dehydration, as in the present study. Comparison of results between normal, infected, cohort and co-cultured cells revealed substantial differences in the Ca2+-permeabilising efficiency of the ionophore, the [CaT]i load at equilibrium and the Ca2+-desaturation pattern after Co2+ addition.
To interpret these results it is necessary to analyse the events which take place in parasitised cells during the Co2+-exposure protocol. When ionophore is added to isolated cells in suspension, it partitions extremely rapidly and reversibly into all plasma and organelle membranes, with partition ratios membrane/medium of > 60 (Lew & Simonsen, 1981). In the membranes, the ionophore performs electroneutral M2+:2H+ exchange, driving the distribution of divalent cations (M2+; mainly Ca2+ and Mg2+) across each membrane towards an equilibrium given by [M2+]i/[M2+]o= ([H+]i/[H+]o)2 (Pressman, 1976). The magnitude of the ionophoric effect is an exponential function of the ionophore concentration in the membrane (Lew & Simonsen, 1980; Simonsen & Lew, 1980). Thus, the more membrane area available for ionophore distribution the more diluted its ionophoric effect would be at each total ionophore concentration. This provides a clear and simple explanation for the results observed in Fig. 2, where an ionophore concentration sufficient to induce rapid Ca2+ equilibration across the membrane of uninfected cells, failed to do so in a suspension of infected cells with comparable haematocrit and 87 % parasitaemia (Fig. 2). The tentative explanation from the analysis above is that the extra membrane area provided by parasites effectively diluted the ionophore concentration in parasite and host cell membranes relative to its concentration in the plasma membrane of the uninfected cells, causing an exponential reduction in the ionophoric effect. Thus, increasing the ionophore concentration in parasitised cells, to levels which would normally destabilise uninfected red cell membranes and enhance their lysis, simply increased the ionophoric effect in the parasitised cell without further lysis (Fig. 3). Krungkrai & Yuthavong (1983) reported that the capacity of the ionophore A23187 to induce Ca2+ uptake was much reduced in mice red cells infected with P. berghei relative to uninfected red cells, despite a substantial increase in ionophore uptake by the infected cells. The reduced ionophoric capacity was attributed to abnormal interactions between the ionophore and the membrane of the infected cells. However, their observation of increased ionophore uptake in infected cells is precisely what would be expected from the increased membrane area. The consequent dilution effect provides a simple explanation of all the experimental results in terms of known ionophore properties.
At adequate ionophore concentrations, both parasitised and uninfected red cells attained Ca2+ equilibration in less than 2 min (Fig. 3). The total Ca2+ content at equilibrium was substantially higher in parasitised cells than in uninfected cells. To interpret the significance of this difference it is necessary to analyse the distribution of Ca2+ induced by the ionophore in uninfected controls and in infected cells. The equilibrium distribution of free ionised Ca2+ between cells and medium in uninfected cells is given by:
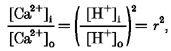 |
(1) |
where r represents the proton ratio set by the operation of the Jacobs-Stewart mechanism (Jacobs & Stewart, 1947), usually about 1.4 (r2∼2) in clamp conditions (Lew & García-Sancho, 1989). The Lew-Bookchin model predicts that outside clamp conditions the cells would dehydrate by less than 6 % by the time the ionophore-induced equilibrium is attained (< 2 min; Fig. 1), and that r would increase also by less than 6 %. Thus, the clamp value of r may be considered largely conserved in non-clamp conditions as long as cell dehydration does not exceed 6 %.
The Ca2+ concentration added to the cell suspension ([CaT]s) becomes distributed between cells ([CaT]i) and medium ([Ca2+]o) according to:
| (2) |
where Hct is expressed here as the cell volume fraction (haematocrit/100). At high-Ca2+ loads, cytoplasmic Ca2+ buffering in human red cells is well approximated by the equation (Ferreira & Lew, 1977; Tiffert & Lew, 1997):
| (3) |
where α (fraction of total cell calcium which is in ionised form in cell water) is approximately 0.3 (Tiffert & Lew, 1997). It is now possible to estimate the intracellular free Ca2+ concentration ([Ca2+]i) and [Ca2+]o, at equilibrium, in the uninfected cells using: (i) eqns (2) and (3); (ii) measured [CaT]i; (iii) known [CaT]s and Hct; and (iv) the approximate value of α. For instance, the values of [CaT]s, [CaT]i and Hct for the co-cultured cells in Fig. 3b were: 160 μM, 780 μmol (1013 cells)−1 and 0.065, respectively. With these values, [Ca2+]o, from eqn (2), renders 116 μM; and [Ca2+]i, from eqn (3), gives 234 μM. The ratio [Ca2+]i/[Ca2+]o is thus approximately 2.0. From eqn (1), we calculate r2= 2, which corresponds to the clamp value of r2. This coincidence, repeated in all the experiments with uninfected cells, confirms that the normal proton concentration ratio persists largely unchanged in non-clamp conditions, at least for 2 min after ionophore addition, as expected from the predicted negligible extent of dehydration (< 6 %).
To analyse the equilibrium distribution of Ca2+ in the infected cells it is necessary to consider that the total measured cell Ca2+ content ([CaT]i) is now divided between host ([CaT]H) and parasite ([CaT]P). Thus:
| (4) |
where fV is the mean volume fraction of red cell cytosol remaining in the parasitised cell sample under study. As defined, this comprises the mean residual volume fraction of host cell cytosol in the infected cells as well as the full volume fraction of uninfected cells in the population. Assuming that cytoplasmic Ca2+ buffering in the host cells is not altered, eqn (3) becomes:
| (5) |
where [Ca2+]H represents the free ionised Ca2+ concentration in the host cytosol. Equation (1) becomes:
| (6) |
For precise solutions of the four unknowns [Ca2+]H, [Ca2+]o, [CaT]H and [CaT]P, using eqns (2), (4), (5) and (6), it would have been necessary to measure r and fV. This was outside the scope of the present investigation. For the analysis below it is sufficient to rely on approximate estimates of r and test alternative values of fV. Recent non-invasive fluorimetric pH measurements by Wünsch et al. (1998) have indicated that host cell pH is about 7.12 ± 0.02, very near normal. In the absence of more detailed stage-related data it will be assumed that the normal value of r, of about 1.4, is conserved in the parasitised cells throughout the parasite's asexual cycle.
In the suspension of parasitised cells with [CaT]s= 150 μM (Fig. 3b), for instance, the mean [CaT]i at equilibrium was 1150 μmol (1013 cells)−1, with a Hct of 0.071. From eqns (2), (6) and (5), we successively calculate [Ca2+]o= 73.6 μM, [Ca2+]H= 147 μM (using r2= 2) and [CaT]H= 490 μmol (1013 cells)−1 (using α= 0.3). Assuming fV= 0.4, eqn (4) renders [CaT]P= 1590 μmol (1013 cells)−1. If fV= 0.6, then [CaT]P= 2140 μmol (1013 cells)−1. These rough estimates indicate that in suspensions of red cells infected with P. falciparum at the mature trophozoite stage, in conditions of ionophore-induced Ca2+ equilibration, most of the ionophore-induced [CaT]i load is within the parasite ((1 –fV)[CaT]P >> fV[CaT]H), the total Ca2+ concentration of the parasite is much higher than that of the host ([CaT]P >> [CaT]H), and the real differences in total Ca2+ concentrations between host and parasite are much larger than apparent from the observed differences between infected and uninfected cells (Fig. 3b). Since the ionophore permeabilises to Ca2+ all membrane-bound compartments of the host-parasite system, the elevated total Ca2+ concentration within the parasite indicates either that the parasite has a large Ca2+-binding capacity associated with cytoplasmic Ca2+-buffering systems, or that large pH gradients persist across internal parasite membranes, causing an elevated Ca2+ partition within acidic compartments (according to eqn (1)), or both. The pH of the parasite cytoplasm is similar to that of the host cell (Bosia et al. 1993; Wünsch et al. 1998). Thus, the concentration of free Ca2+ would not be expected to differ much between host and parasite cytosols. This analysis, together with the results obtained so far, establish the feasibility of investigating the cytoplasmic Ca2+ buffering of P. falciparum parasites in situ, within their host red cells.
Pump-mediated Ca2+ extrusion from parasitised cells is considered next. Addition of Co2+ instantly exposed uphill Ca2+ extrusion by the PMCA. Co2+ blocks ionophore-mediated Ca2+ transport and is itself rapidly transported to equilibrium (Tiffert et al. 1984; Brown & Simonsen, 1985). Within red cells Co2+ acts as a low-affinity Mg2+ substitute for the Na+ and Ca2+ pumps (Richards, 1988; Raftos & Lew, 1995), with no detectable effect on the PMCA at physiological [Mg2+]i levels. In parasitised cells the rapid build up of the internal Co2+ concentration would block ionophore-mediated Ca2+ transport between parasite and host. Thus, almost immediately after Co2+ addition, Ca2+ traffic to and from the red cell Ca2+ pool would be entirely dependent on endogenous active and passive Ca2+ transport across parasite and red cell plasma membranes. The parasitophorous vacuolar membrane which surrounds the parasite membrane is assumed to be devoid of active Ca2+ transport mechanisms and to represent no permeability barrier (Desai & Rosenberg, 1997). Active Ca2+ extrusion by a PMCA with normal Vmax would be expected to rapidly empty the host cell Ca2+ pool (Vmax measurement stage in Fig. 3), exposing residual Ca2+ trapped within parasite compartments. To a first approximation, this is the pattern observed in all the experiments of this series (Figs 2 and 3).
In suspensions of parasitised cells, pump Vmax was marginally but systematically lower than in cohort or co-cultured cell suspensions (Fig. 3). It is doubtful whether this minor difference reflects a genuine pump inhibition in parasitised cells. Factors which could account for a minor apparent Vmax inhibition are: Ca2+ redistribution between the rather limited Ca2+ pool of the host cell and that of the parasite, after Co2+ addition; the increased coefficient of variation resulting from the large residual Ca2+ pools; and the increased variability observed in repeated Vmax estimates on the same suspension (9.08-11.7 mmol (1013 cells)−1 h−1 in the experiment of Fig. 3b). The conclusion from this analysis is that the performance of the Ca2+ pump in parasitised red cells is not significantly affected by P. falciparum parasites up to the late trophozoite stage. The documented conservation of PMCA function also removes any concerns about the estimates of host cell Vmax arising from variable proportions of uninfected cells or from minor differences in parasite developmental stage as a result of imperfect synchronisation.
A final consideration on pump Vmax concerns the comparison between results obtained with cell suspensions of different parasitaemia levels. In the experiment of Fig. 3a, the parasitaemia was 50 %. Pump Vmax appeared similar in co-cultured and infected cells, and a superficial interpretation of this result may attribute the normal Vmax to the 50 % cohort cells in the parasitised cell suspension, with minor or null contribution from the parasitised cells. Expression of mean cell Ca2+ content and flux rates per 1013 cells precludes that interpretation. If only 50 % of cells had contributed a normal Vmax rate, the measured Vmax would have been half-normal if expressed as millimoles per 1013 cells per hour. Thus, in the experiment of Fig. 3a, the similarity of Vmax between co-cultured and parasitised cell suspensions suggests that pump performance was similar in the cohort and parasitised cells. That this is so, however, could only be confirmed at very high parasitaemia, as in the experiment of Fig. 3b, because the result in Fig. 3a may also reflect compensated differences between cohort and parasitised cells.
After the initial Vmax, the residual Ca2+ retained within parasite compartments continued to be extruded from the cells, but at a much reduced rate. This extrusion was also uphill and therefore PMCA mediated. The emptying of the red cell Ca2+ pool during the Vmax period changed the environment provided to the parasite, from a high-Ca2+ condition immediately after Co2+ addition, to its physiological low-Ca2+ condition. Residual Ca2+ traffic from parasite to host was therefore very probably down-gradient. After the initial Vmax period, extrusion of residual Ca2+ settled to rates of between 0.6 and 1.2 mmol (1013 cells)−1 h−1 (Fig. 3). This presumed flow of Ca2+ from parasite to host may reflect properties of Ca2+ gradients and endogenous Ca2+ transport pathways at the parasite plasma membrane, amenable to further investigation.
A main additional factor which may influence the rate of residual Ca2+ efflux is the availability of ATP to the pump. The sudden ionophore-induced Ca2+ load fully activated a normal functioning PMCA which would have hydrolysed ATP with a Ca2+:ATP stoichiometry of 1:1 (Rega & Garrahan, 1986). Normal uninfected red cells lack the metabolic capacity to restore ATP at that rate, whatever the glycolytic substrate (McManus, 1967; Dagher & Lew, 1988). The combined operation of the PMCA ATPase, adenylate kinase and AMP deaminase enzymes leads to irreversible ATP depletion by conversion of AMP to IMP (inosine monophosphate) (Lew, 1971; Almaraz et al. 1988), processes which may, or may not, be conserved in cells with mature stage parasites. In normal red cells, ATP depletion becomes limiting to Ca2+ pump function only after about 20–30 min of pump activation, but in parasitised cells, with reduced host cell volume and ATP pool, ATP may become rate-limiting earlier. On the other hand, parasitised cells are known to have hugely increased glycolytic rates, with transport of ATP from parasite to host via a nucleotide translocase in the parasite plasma membrane (Kanaani & Ginsburg, 1989). The extra ATP consumption by the Ca2+-saturated PMCA represents a small fraction of the overall glycolytic rate of cells with mature parasites, but the relevant open question here is whether the rate of ATP translocation can match the rate of ATP consumption by the Ca2+-saturated pump of the host cell. It is worth noting that the residual Ca2+ pools generated by the Co2+-exposure protocol enable direct access to parasite Ca2+ pools with 45Ca2+ of known specific activity, thus opening new ways of investigating the nature and dynamic behaviour of parasite Ca2+ pools.
This analysis suggests that the different experimental stages of the Co2+-exposure protocol can be refined to open new areas of investigation on the Ca2+ homeostasis of P. falciparum-infected cells, such as stage-related growth in parasite membrane area, cytoplasmic Ca2+ buffering within the parasite, the dynamics of parasite Ca2+ pools, effects of pharmacological agents on these functions, and study of the participation of Ca2+ in the mechanism of action of antimalarial drugs.
Acknowledgments
We are very grateful to B. C. Elford for much help and advice, to H. Ginsburg for helpful discussions and to the anonymous reviewers for their very helpful comments. This work was supported by funds from the Wellcome Trust (grant no. 053657) and the Department of Physiology, University of Cambridge.
References
- Almaraz L, García-Sancho J, Lew VL. Calcium-induced conversion of adenine nucleotides to inosine monophosphate in human red cells. The Journal of Physiology. 1988;407:557–567. doi: 10.1113/jphysiol.1988.sp017431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez J, Montero M, García-Sancho J. High affinity inhibition of Ca2+-dependent K+ channels by cytochrome P-450 inhibitors. Journal of Biological Chemistry. 1992;267:11789–11793. [PubMed] [Google Scholar]
- Berendt AR, Simmons DL, Tansey J, Newbold CI, Marsh K. Intercellular adhesion molecule-1 is an endothelial cell adhesion receptor for Plasmodium falciparum. Nature. 1989;341:57–59. doi: 10.1038/341057a0. [DOI] [PubMed] [Google Scholar]
- Bookchin RM, Lew VL. Progressive inhibition of the Ca pump and Ca:Ca exchange in sickle red cells. Nature. 1980;284:561–563. doi: 10.1038/284561a0. [DOI] [PubMed] [Google Scholar]
- Bookchin RM, Lew VL, Nagel RL, Raventos C. Increase in potassium and calcium transport in human red cells infected with Plasmodium falciparum in vitro. The Journal of Physiology. 1980;312:65P. [Google Scholar]
- Bosia A, Ghigo D, Turrini F, Nissani E, Pescarmona GP, Ginsburg H. Kinetic characterization of Na+/H+ antiport of Plasmodium falciparum membrane. Journal of Cellular Physiology. 1993;154:527–534. doi: 10.1002/jcp.1041540311. [DOI] [PubMed] [Google Scholar]
- Brown AM, Simonsen LO. Intracellular cobalt buffering by intact human red cells – evidence for time-dependent and metabolism-dependent changes. The Journal of Physiology. 1985;361:26P. [Google Scholar]
- Brugnara C, Armsby CC, Sakamoto M, Rifai N, Alper SL, Platt O. In vivo blockade of Ca2+-activated K+ transport in normal human erythrocytes by oral administration of clotrimazole. Journal of General Physiology. 1994;104:15a. [Google Scholar]
- Dagher G, Lew VL. Maximal calcium extrusion capacity and stoichiometry of the human red cell calcium pump. The Journal of Physiology. 1988;407:569–586. doi: 10.1113/jphysiol.1988.sp017432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai SA, McCleskey EW, Schlesinger PH, Krogstad DJ. A novel pathway for Ca2+ entry into Plasmodium falciparum-infected blood cells. American Journal of Tropical Medicine and Hygiene. 1996;54:464–470. doi: 10.4269/ajtmh.1996.54.464. [DOI] [PubMed] [Google Scholar]
- Desai SA, Rosenberg RL. Pore size of the malaria parasite's nutrient channel. Proceedings of the National Academy of Sciences of the USA. 1997;94:2045–2049. doi: 10.1073/pnas.94.5.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelmann B, Duhm J. Intracellular calcium content of human erythrocytes: relation to sodium transport systems. Journal of Membrane Biology. 1987;98:79–87. doi: 10.1007/BF01871047. [DOI] [PubMed] [Google Scholar]
- Etzion Z, Lew VL, Bookchin RM. K(86Rb) transport heterogeneity in the low-density fraction of sickle cell anemia red blood cells. American Journal of Physiology. 1996;271:C1111–1121. doi: 10.1152/ajpcell.1996.271.4.C1111. [DOI] [PubMed] [Google Scholar]
- Ferreira HG, Lew VL. Passive Ca transport and cytoplasmic Ca buffering in intact red cells. In: Ellory JC, Lew VL, editors. Membrane Transport in Red Cells. London: Academic Press; 1977. pp. 53–91. [Google Scholar]
- Freeman CJ, Bookchin RM, Ortiz OE, Lew VL. K-permeabilized human red cells lose an alkaline, hypertonic fluid containing excess K over diffusible anions. Journal of Membrane Biology. 1987;96:235–241. doi: 10.1007/BF01869305. [DOI] [PubMed] [Google Scholar]
- García-Sancho J, Lew VL. Heterogeneous calcium and adenosine triphosphate distribution in calcium-permeabilized human red cells. The Journal of Physiology. 1988;407:523–539. doi: 10.1113/jphysiol.1988.sp017429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardos G. The function of calcium in the potassium permeability of human erythrocytes. Biochimica et Biophysica Acta. 1958;30:653–654. doi: 10.1016/0006-3002(58)90124-0. [DOI] [PubMed] [Google Scholar]
- Ginsburg H, Handeli S, Friedman S, Gorodetsky R, Krugliak M. Effects of red blood cell potassium and hypertonicity on the growth of Plasmodium falciparum in culture. Zeitschrift Fur Parasitenkunde. 1986;72:185–199. doi: 10.1007/BF00931146. [DOI] [PubMed] [Google Scholar]
- Harrison DG, Long C. The calcium content of human erythrocytes. The Journal of Physiology. 1968;199:367–381. doi: 10.1113/jphysiol.1968.sp008658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs MH, Stewart DR. Osmotic properties of the erythrocyte. XII. Ionic and osmotic equilibria with a complex external solution. Journal of Cellular and Comparative Physiology. 1947;30:79–103. doi: 10.1002/jcp.1030300106. [DOI] [PubMed] [Google Scholar]
- Jensen JB. Concentration from continuous culture of erythrocytes infected with trophozoites and schizonts of Plasmodium falciparum. American Journal of Tropical Medicine and Hygiene. 1978;27:1274–1276. doi: 10.4269/ajtmh.1978.27.1274. [DOI] [PubMed] [Google Scholar]
- Kanaani J, Ginsburg H. Metabolic interconnection between the human malarial parasite Plasmodium falciparum and its host erythrocyte. Regulation of ATP levels by means of an adenylate translocator and adenylate kinase. Journal of Biological Chemistry. 1989;264:3194–3199. [PubMed] [Google Scholar]
- Kirk K, Elford BC, Ellory JC, Newbold CI. A transport pathway responsible for the increased permeability of malaria-infected erythrocytes shows characteristics of a Cl− channel. The Journal of Physiology. 1992;452:342P. [Google Scholar]
- Kirk K, Horner HA, Kirk J. Glucose uptake in Plasmodium falciparum-infected erythrocytes is an equilibrative not an active process. Molecular and Biochemical Parasitology. 1996;82:195–205. doi: 10.1016/0166-6851(96)02734-x. [DOI] [PubMed] [Google Scholar]
- Kirk K, Wong HY, Elford BC, Newbold CI, Ellory JC. Enhanced choline and Rb+ transport in human erythrocytes infected with the malaria parasite Plasmodium falciparum. Biochemical Journal. 1991;278:521–525. doi: 10.1042/bj2780521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer R, Ginsburg H. Calcium transport and compartment analysis of free and exchangeable calcium in Plasmodium falciparum-infected red blood cells. Journal of Protozoology. 1991;38:594–601. [PubMed] [Google Scholar]
- Krungkrai J, Yuthavong Y. Reduction of Ca2+ uptake induced by ionophore A23187 of red cells from malaria (Plasmodium berghei)-infected mice. Cell Biology International Reports. 1983;7:237–244. doi: 10.1016/0309-1651(83)90231-x. [DOI] [PubMed] [Google Scholar]
- Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. Journal of Parasitology. 1979;65:418–420. [PubMed] [Google Scholar]
- Lee P, Ye Z, Van Dyke K, Kirk RG. X-ray microanalysis of Plasmodium falciparum and infected red blood cells: effects of qinghaosu and chloroquine on potassium, sodium, and phosphorus composition. American Journal of Tropical Medicine and Hygiene. 1988;39:157–165. doi: 10.4269/ajtmh.1988.39.157. [DOI] [PubMed] [Google Scholar]
- Lew VL. On the ATP dependence of the Ca2+-induced increase in K+ permeability observed in human red cells. Biochimica et Biophysica Acta. 1971;233:827–830. doi: 10.1016/0005-2736(71)90185-4. [DOI] [PubMed] [Google Scholar]
- Lew VL, Bookchin RM. Volume, pH, and ion-content regulation in human red cells: analysis of transient behavior with an integrated model. Journal of Membrane Biology. 1986;92:57–74. doi: 10.1007/BF01869016. [DOI] [PubMed] [Google Scholar]
- Lew VL, Ferreira HG. Calcium transport and the properties of a calcium-activated potassium channel in red cell membranes. In: Kleinzeller A, Bronner F, editors. Current Topics in Membranes and Transport. Vol. 10. NY: Academic Press; 1978. pp. 217–277. [Google Scholar]
- Lew VL, García-Sancho J. Measurement and control of intracellular calcium in intact red cells. In: Fleischer S, Fleischer B, editors. Methods in Enzymology, Biomembranes, Cellular and Subcellular Transport: Eukaryotic (Nonepithelial) Cells. Vol. 173. San Diego, CA: Academic Press, Inc.; 1989. pp. 100–112. part T. [DOI] [PubMed] [Google Scholar]
- Lew VL, Simonsen LO. Ionophore A23187-induced calcium permeability of intact human red blood cells. The Journal of Physiology. 1980;308:60P. doi: 10.1113/jphysiol.1980.sp013346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew VL, Simonsen LO. A23187-induced 45Ca-flux kinetics reveal uniform ionophore distribution and cytoplasmic calcium buffering in ATP-depleted human red cells. The Journal of Physiology. 1981;316:6–7P. [Google Scholar]
- McManus TJ. Comparative biology of red cells. Federation Proceedings. 1967;26:1821–1826. [Google Scholar]
- Pereira AC, Samellas D, Tiffert T, Lew VL. Inhibition of the calcium pump by high cytosolic Ca2+ in intact human red cells. The Journal of Physiology. 1993;461:63–73. doi: 10.1113/jphysiol.1993.sp019501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pressman BC. Biological applications of ionophores. Annual Review of Biochemistry. 1976;45:501–530. doi: 10.1146/annurev.bi.45.070176.002441. [DOI] [PubMed] [Google Scholar]
- Raftos JE, Lew VL. Effect of intracellular magnesium on calcium extrusion by the plasma membrane calcium pump of intact human red cells. The Journal of Physiology. 1995;489:63–72. doi: 10.1113/jphysiol.1995.sp021030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rega AF, Garrahan PJ. The Ca2+ Pump of Plasma Membranes. Boca Raton: CRC Press; 1986. [Google Scholar]
- Richards DE. Occlusion of cobalt ions within the phosphorylated forms of the Na+-K+ pump isolated from dog kidney. The Journal of Physiology. 1988;404:497–514. doi: 10.1113/jphysiol.1988.sp017302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatzmann HJ. The plasma membrane calcium pump of erythrocytes and other animal cells. In: Carafoli E, editor. Membrane Transport of Calcium. London: Academic Press; 1982. pp. 41–108. [Google Scholar]
- Simonsen LO, Lew VL. The correlation between ionophore A23187 content and calcium permeability of ATP-depleted human red blood cells. In: Lassen UV, Ussing HH, Wieth JO, editors. Membrane Transport in Erythrocytes. Copenhagen: Munksgaard; 1980. pp. 208–212. [Google Scholar]
- Staines HM, Chang W, Ellory JC, Tiffert T, Kirk K, Lew VL. Passive Ca2+ transport and Ca2+-dependent K+ transport in Plasmodium falciparum-infected red cells. Journal of Membrane Biology. 1999;172:13–24. doi: 10.1007/s002329900579. [DOI] [PubMed] [Google Scholar]
- Tanabe K. Ion metabolism in malaria-infected erythrocytes. Blood Cells. 1990;16:437–449. [PubMed] [Google Scholar]
- Tiffert T, Etzion Z, Bookchin RM, Lew VL. Effects of deoxygenation on active and passive Ca2+ transport and cytoplasmic Ca2+ buffering in normal human red cells. The Journal of Physiology. 1993;464:529–544. doi: 10.1113/jphysiol.1993.sp019649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffert T, García-Sancho J, Lew VL. Irreversible ATP depletion caused by low concentrations of formaldehyde and of calcium-chelator esters in intact human red cells. Biochimica et Biophysica Acta. 1984;773:143–156. doi: 10.1016/0005-2736(84)90559-5. [DOI] [PubMed] [Google Scholar]
- Tiffert T, Lew VL. Cytoplasmic calcium buffers in intact human red cells. The Journal of Physiology. 1997;500:139–154. doi: 10.1113/jphysiol.1997.sp022005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Wasserman M, Alarcón C, Mendoza PM. Effects of Ca2+ depletion on the asexual cell cycle of Plasmodium falciparum. American Journal of Tropical Medicine and Hygiene. 1982;31:711–717. doi: 10.4269/ajtmh.1982.31.711. [DOI] [PubMed] [Google Scholar]
- Wünsch S, Sanchez CP, Gekle M, Große-Wortmann L, Wiesner J, Lanzer M. Differential stimulation of the Na+/H+ exchanger determines chloroquine uptake in Plasmodium falciparum. Journal of Cell Biology. 1998;140:335–345. doi: 10.1083/jcb.140.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]