

Abstract
Overexpression of cardiac calsequestrin (CSQ) impairs Ca2+ signalling in murine myocytes, leading to marked cardiac hypertrophy. Here we report on contractile, histological and electrophysiological changes accompanying the development of cardiac hypertrophy and failure in CSQ-overexpressing mice.
CSQ mice developed contractile dysfunction after 60 days of age, with only 40% survival at 6 months. Four- to 6-month-old CSQ mice revealed biventricular dilatation, cardiomyocyte hypertrophy, patchy interstitial fibrosis and tissue calcifications.
Cardiac hypertrophy of CSQ mice was accompanied by progressive P-R and Q-T interval prolongation, conduction blocks, 2-fold prolongation of the ventricular action potential and increased cellular membrane capacitance.
Remodelling of ionic currents included marked reduction of both density and absolute magnitude of transient outward (Ito) and inward rectifying (IK1) K+ currents. The density, but not the absolute magnitude, of basal and isoproterenol (isoprenaline)-stimulated Ca2+ current (ICa) was decreased by 42% and the inactivation kinetics of ICa were significantly slowed. Na+ current density was suppressed by 50%, but its steady-state activation and inactivation were shifted to more positive potentials. The density of Na+-Ca2+ exchange current was increased by 35%.
In CSQ but not in control myocytes dialysed with cAMP, isoproterenol continued to enhance ICa. This apparent lower responsiveness of ICa to cAMP could be reversed by the non-hydrolysable cAMP analogue 8-Br-cAMP or the phosphodiesterase inhibitor IBMX, suggesting high phosphodiesterase activity of CSQ myocytes.
In young CSQ mice (< 60 days) with compensated cardiac hypertrophy, only Ito was significantly suppressed. All other currents remained relatively intact.
An increase in cardiac Ca2+-storage capability by overexpression of CSQ results in a dilated cardiomyopathy with tissue fibrosis, calcifications, impaired β-adrenergic signalling and progressive remodelling of ionic currents. The extent of the changes in ionic currents was age dependent.
Human heart failure is characterized by contractile dysfunction, electrophysiological remodelling and sudden death (Tomaselli et al. 1994). At the cellular level, the prolongation of the ventricular action potential, most probably induced by suppression of the transient outward K+ current (Ito) and the inward rectifying K+ current (IK1) and slower inactivation of the calcium current (ICa), as well as the enhancement of the Na+-Ca2+ exchanger are consistently reported for human and animal models of heart failure (Studer et al. 1994; Hatem et al. 1994; Momtaz et al. 1996; Kaab et al. 1996; Thuringer et al. 1996; Hasenfuss et al. 1997; Nabauer & Kaab, 1998; O'Rourke et al. 1999). It has been suggested that alterations in cellular electrophysiology are the most likely cause of the high incidence of ventricular arrhythmias in patients with heart failure.
Recently, the mouse model has become the focus of investigation in the field of cardiac failure. Several transgenic models of dilated cardiomyopathy (D'Angelo et al. 1997; Kubota et al. 1997; Colbert et al. 1997; Fentzke et al. 1998; Mende et al. 1998) have been developed, which also appear to produce ventricular arrhythmias and sudden death (Pan et al. 1998). Comprehensive cellular electrophysiological studies in transgenic mice that develop heart failure, however, are as yet to be reported.
We have recently shown that transgenic mice with cardiac-targeted overexpression of canine calsequestrin (CSQ) develop cardiac hypertrophy, associated with enlargement of ventricular myocytes, impairment of ICa-gated Ca2+ release and downregulation of some of the proteins of the Ca2+-signalling cascade (ryanodine receptor, triadin and junctin) (Jones et al. 1998). In another study of mice overexpressing murine cardiac CSQ, though similar changes in Ca2+ current and impairment of Ca2+ signalling were observed, protein levels of the ryanodine receptor, triadin and junctin remained unchanged (Sato et al. 1998). Both studies, however, concluded that CSQ may serve as both the storage and regulatory protein for cardiac Ca2+ signalling and that its overexpression induces cardiac hypertrophy.
The goal of the current study was to quantify the cardiac phenotype and the electrophysiological changes that accompany the development of hypertrophy and failure in CSQ-overexpressing mice. Our data show that the progressive cardiac hypertrophy of transgenic mice leads to dilated cardiomyopathy accompanied by extensive electrophysiological remodelling. Pronounced prolongation of the action potential and downregulation of Ito already occur in young transgenic mice, when the cardiac hypertrophy is still well compensated. Further, the progression of the disease is accompanied by atrioventricular conduction abnormalities, suppression of all ionic currents, upregulation of the Na+-Ca2+ exchanger and phosphodiesterase activity. Our findings demonstrate for the first time that extensive electrical remodelling of ionic currents develops with age in transgenic mice overexpressing cardiac CSQ, where cardiac Ca2+ signalling is the primary molecular site of the defect. Preliminary reports of these results have appeared (Knollmann et al. 1998a,b).
METHODS
Transgenic mice
The generation of calsequestrin-overexpressing transgenic mice (CSQ mice) has been described (Jones et al. 1998). Briefly, the canine cardiac CSQ cDNA clone IC3A (Scott et al. 1988) was used to generate a transgene containing the mouse cardiac α-myosin heavy chain promoter, the entire protein coding region for CSQ and the SV40 polyadenylation signal sequence. The transgene was microinjected in embryos from C3H/DBA bred mice and transgene-positive mice were identified by PCR assay of toe digests.
General experimental procedures
All studies were carried out according to NIH guidelines and approved by the institutional animal care and use committee. Transgenic animals and their non-transgenic littermates were matched for both age and gender. All animals were housed under the same conditions and on the same diet. The mouse to be used was taken from the housing facility and assessed for its mobility and the presence of cyanosis or oedema. The animal was anaesthetized with methoxyflurane vapour until a surgical level of anaesthesia was confirmed by loss of withdrawal reflex to toe pinch. A thoracotomy was then performed and the heart removed, resulting in exsanguination. The presence of atrial thrombi, pleural fluid or ascites was recorded. The heart wet weight was measured after trimming of the major heart vessels. Atrial dimensions were measured.
ECG and echocardiography were carried out in a subgroup of animals. For these experiments, animals were sedated with 15 mg kg−1 diazepam by intraperitoneal injection. At this dose, animals are sedated enough to tolerate these non-invasive procedures and have relatively normal heart and respiratory rates. A heating lamp was used to prevent hypothermia, and the animals were allowed to recover for 2–3 days before the heart removal and cell isolation procedures.
ECG measurements
Diazepam-sedated mice were positioned prone in a shielded box, with all four extremities immersed in 3 M KCl-filled wells to reduce skin resistance. A custom-built ECG amplifier (Vibraspec, Bear Island, ME, USA) with a high-frequency filter of 100 Hz and 10000-fold gain was used to record six bipolar limb leads in standard fashion. Signals were digitized at 2 kHz using pCLAMP software (Axon Instruments, Inc.). For each animal, P-R and R-R intervals and the QRS complex were measured in a blinded fashion from three consecutive beats in leads I and II and averaged. The Q-T interval was measured similarly in the lead with the longest and most prominent T-wave.
Echocardiography measurements
In diazepam-sedated mice, echocardiography was performed with a 1 cm gel standoff. An Acuson Sequoia C256 system with a linear transducer in 13 MHz mode was used to obtain standard two-dimensional (2-D) short axis views. A 2-D guided M-mode was performed of the left ventricle (LV) at the tip of the mitral leaflets and through the centre of the LV cavity. LV end-systolic and end-diastolic internal diameters (LVIDS and LVIDD, respectively) and LV wall thickness were measured for each animal from the M-mode image in a blinded fashion. LV fractional shortening was calculated as (LVIDD – LVIDS)/LVIDD.
Morphology and histology
A subset of animals was specifically killed to obtain histological sections from heart, lung, liver and kidneys. Necropsy was also performed on each mouse that died unexpectedly. Age, sex, body weight, heart weight and the presence of ascites, pleural effusion or atrial/ventricular thrombi were recorded. The organs were fixed with formalin, sectioned and slides were stained with haematoxylin-eosin or Masson trichrome for microscopic analysis.
Isolation of ventricular myocytes
Ventricular myocytes were isolated by a modification of the collagenase/protease method (Mitra & Morad, 1985). The excised hearts were placed in ice-cold incubation solution of the following composition (mmol l−1): taurine, 30; NaCl, 90; KCl, 5.4; Hepes, 10; glucose, 10; MgCl2, 1; pH 7.2 with NaOH. The aorta was cannulated and perfused for 5 min with an oxygenated incubation solution at 36°C containing 0.5 mmol l−1 EGTA. The solution was then switched to an isolation solution, containing 1 mg ml−1 albumin (Sigma Chemical Co.), 0.12 mg ml−1 protease (Type XIV, Sigma Chemical Co.) and 0.33 mg ml−1 collagenase (Type IV, Worthington Biochemical Corp., 204 U mg−1) for 8–10 min. The heart was removed and the ventricles were minced and digested in the same isolation solution for an additional two to three digestion periods (5-10 min each) in a shaking water bath (37°C). The resulting cell suspension was collected after each digestion and stored at room temperature in incubation solution containing an additional 0.2 mmol l−1 CaCl2. This procedure yielded 30 −50 % rod-shaped myocytes that were used for up to 8 h.
Electrophysiological measurements
Whole-cell electrophysiological measurements and rapid solution exchanges were performed as previously described (Cleemann & Morad, 1991). All measurements were carried out at 36°C. Ventricular action potentials were measured using pipette solutions containing (mmol l−1): potassium glutamate, 110; NaCl, 10; KCl, 10; EGTA, 2; Hepes, 10; MgATP, 5; pH adjusted to 7.2 with KOH. Whole-cell patches were established in control Tyrode solution containing (mmol l−1): NaCl, 137; KCl, 5.4; MgCl2, 1; CaCl2, 2; Hepes, 10; glucose, 10; pH 7.4. Holding currents were adjusted to maintain the membrane potential at approximately −80 mV. Only cells with an input resistance of > 1 GΩ were used. Cells were paced at 1 Hz for 3 min prior to measurements. Action potentials were elicited by application of a 4 ms current injection of about 20 % above threshold. Resting potential, overshoot potential and action potential duration measured at 50% and 90% repolarization (APD50 and APD90, respectively) were averaged from three beats at stimulation frequencies of 1 Hz.
To measure K+ currents, the pipette solution contained (mmol l−1): KCl, 155; NaCl, 10; EGTA, 14; CaCl2, 1; Hepes, 10; MgATP, 5; pH 7.2. The external solution was Tyrode solution containing 0.2 mmol l−1 CdCl2 to block L-type Ca2+ current. Depolarization-activated outward K+ currents were measured from a holding potential of −80 mV in response to 400 ms voltage steps ranging from −40 to +40 mV. Inward rectifier K+ currents (IK1) were measured as the maintained currents at the end of 500 ms voltage steps ranging between −150 and −40 mV, from a holding potential of −80 mV. To facilitate the analysis of the small outward component of IK1, a linear leak component defined by a line connecting the origin and the current at −80 mV was subtracted from the net current values at each potential (Kaab et al. 1996).
To effectively measure Na+ current (INa), a number of precautions were employed. The magnitude of INa was drastically reduced by making the measurements in symmetrical (5 mmol l−1) Na+ concentrations. Large 1 MΩ patch pipettes were used and the experiments were carried out at room temperature. The pipette solution contained (mmol l−1): NaCl, 5; CsCl, 118; TEA-Cl, 10; glucose, 10; EGTA, 14; CaCl2, 1; Hepes, 10; MgATP, 5 (pH 7.2) and the external solution contained (mmol l−1): NaCl, 5; TEA-Cl, 124; CsCl, 5; glucose, 10; MgCl2, 3; Hepes, 10; pH 7.4. Capacity transients and leak currents were subtracted using the P/5 method. To minimize the steady-state inactivation of Na+ channels, INa densities were measured from a holding potential of −110 mV using a 500 ms long conditioning pulse. Steady-state inactivation was measured by varying the conditioning holding potential from −130 to −40 mV, followed by a test pulse to −30 mV.
The voltage dependence of activation and slope conductance were estimated by fitting the current-voltage relation to the following function:
where I is current density, Gmax is the maximal slope conductance, V is membrane potential, V½ is the potential for half-activation of the Na+ channel, B is the Boltzmann slope factor and VNa is the reversal potential of the Na+ current. Parameters were estimated for each cell using non-linear fitting software (WinNonlin, Scientific Consulting, Inc.).
Ca2+ currents (ICa) were measured in K+-free solution containing (mmol l−1): NaCl, 137; MgCl2, 1; glucose, 10; CaCl2, 2; Hepes, 10; pH 7.4. The pipette solution contained (mmol l−1): CsCl, 111; TEA-Cl, 20; glucose, 10; EGTA, 14; Hepes, 10; MgATP, 5; pH 7.2. In some experiments, cyclic adenosine monophosphate (cAMP) or 8-bromo-cAMP (8-Br-cAMP) were added to the internal solution, as indicated in the text. The membrane potential was either held at −50 mV for 100 ms to inactivate Na+ channels, or NaCl was replaced with 137 mmol l−1 TEA-Cl, when more negative holding potentials (-70 to −90 mV) were used. Peak ICa was measured in response to 50 ms depolarization steps ranging from −50 to 50 mV. Where indicated, isoproterenol was used at a concentration of 5 μmol l−1.
The Na+-Ca2+ exchange current was estimated as the nickel-sensitive outward current generated in response to rapid replacement of Na+ by TEA+. The pipette solution contained (mmol l−1): NaCl, 15; CsCl, 118; glucose, 5.5; EGTA, 14; CaCl2, 3.92; Hepes, 10; MgATP, 3; MgCl2, 0.5; pH 7.1 (free Ca2+∼100 nmol l−1). After breakthrough was achieved, cells were superfused with K+-free Tyrode solution to block the Na+ pump, in part to allow more rapid accumulation of internal Na+ and to eliminate possible Na+ pump currents in response to changes in the Na+ gradient. Since the exchange current shows significant ‘run-up’, solution switches were performed every 2 min until the outward current stabilized within 4–15 min, according to a protocol described previously by Litwin & Bridge (1997).
Statistical analysis
Differences between control and CSQ mice were assessed using a one-way analysis of variance (ANOVA), followed by Student's two-sided t test whenever appropriate. Results were considered statistically significant if the P value was less than 0.05. The increase in heart weight over time was estimated with linear regression analysis and the regression line and the 95 % confidence band plotted (Origin 5.0, Microcal Software, Inc., Northampton, MA, USA). Kaplan-Meier survival plots were generated using the Statview software package, SAS Institute Inc., San Francisco, CA, USA. Age-dependent changes in cellular electrophysiology were either examined by linear regression analysis, or by comparing mean values from mice aged 60 days or less (hypertrophy stage) with those from mice older than 60 days (heart failure stage).
RESULTS
Phenotype of CSQ mice
Time course of cardiac hypertrophy and failure
The heart weight of transgenic mice increased progressively with age (Fig. 1A). Echocardiography was used to monitor cardiac morphology and function in vivo. Progression in cardiac hypertrophy was associated with increased dilatation of the left ventricular chamber with age (Fig. 1B). Contractile function, estimated by fractional shortening, was not significantly different between control and CSQ mice during the first 60 days of life, but deteriorated progressively in older animals (Fig. 1C). Approximately 50 % of the animals older than 120 days showed evidence of overt heart failure (ascites, pleural effusions or generalized oedema) at the time of killing. The 2-D echo image in Fig. 2A illustrates the characteristic biventricular chamber dilatation, pleural and pericardial effusions and ascites of a 150-day-old CSQ mouse, which died shortly after these images were obtained. Left ventricular dimensions and fractional wall shortening were measured from M-mode images (Fig. 2B). The left ventricles of transgenic animals were dilated, with increased ventricular wall thickness. The right ventricle also appeared to be dilated, but this was not consistently quantified.
Figure 1. Time course of cardiac hypertrophy and failure.
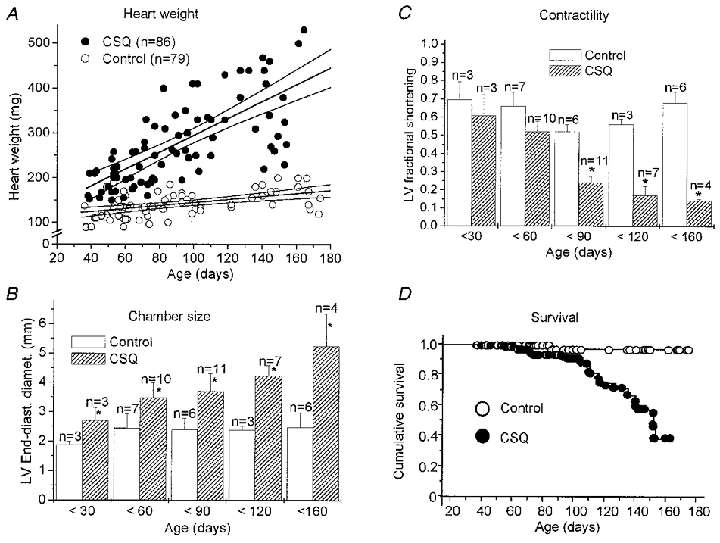
A, heart weight (wet) of CSQ-overexpressing mice (CSQ) and non-transgenic littermates (Control) increased at a different rate with age ( P < 0.001, ANOVA). B and C, left ventricular (LV) end-diastolic diameter (B) and fractional shortening (C) were measured as a function of age by sequential echocardiography. D, Kaplan-Meier plot of survival probability. Survival decreased significantly after about 3 months. In B and C, values are means ±s.e.m.* P < 0.05 vs. control; n, number of animals studied.
Figure 2. Echocardiographic evidence for heart failure.
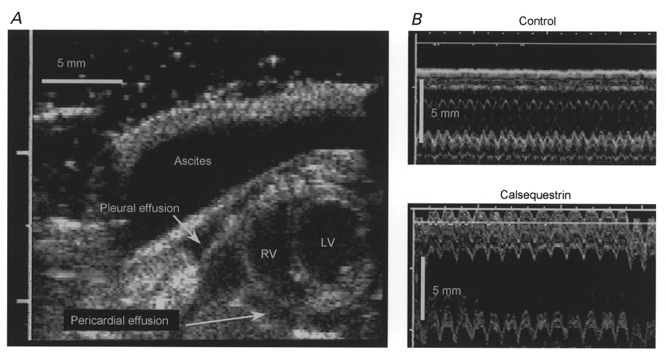
A, 2-D echocardiograph (modified short axis view) of a 5-month-old CSQ mouse, with biventricular dilatation, prominent ascites, and pleural and pericardial effusions (LV, left ventricle; RV, right ventricle). B, M-mode recordings (3 s sweep) from a control and a CSQ mouse heart. Data were obtained with a high-resolution 13 MHz linear array transducer.
Sixteen out of 86 transgenic animals died spontaneously, eight of which had clear evidence of pump failure (pleural effusions, liver congestion or ascites) on necropsy. Survival of transgenic mice was estimated by Kaplan-Meier statistics to be 40 % at 6 months of age (Fig. 1D). Note that 15 of the 16 deaths occurred in CSQ mice older than 60 days, which also showed significant contractile dysfunction by echocardiography (see Fig. 1C).
Surface ECG recordings were obtained from sedated animals in two different age groups to examine whether the stage of the disease (compensated hypertrophy vs. failing) had an impact on the ECG morphology (Table 1). We found that most ECG changes were already present during the compensated hypertrophy stage and that the heart rate was even slower in the younger animals (Table 1). Transgenic animals had significantly wider QRS complexes and longer Q-T intervals, independent of age (Table 1). The Q-T interval remained prolonged even after correcting for the slower heart rate (Q-Tc), using the formula Q-Tc = Q-T/(R-R/100)½ (London et al. 1998a). Figure 3A and B illustrates the wide range of ECG abnormalities found in transgenic mice. An alternating T-wave morphology (T-wave alternans) was observed in 2 out of 6 young and in 4 out of 12 old transgenic animals. However, various degrees of atrioventricular conduction abnormalities and widening of the P-wave were present in the older transgenic animals, but not in the younger group or in age-matched non-transgenic littermates. Interestingly, despite grossly enlarged atria, all transgenic animals remained in sinus rhythm with no indication of atrial fibrillation. During the short period of ECG recordings (∼5 min), ventricular tachycardia or ventricular fibrillation were not encountered. The echocardiographic and ECG measurements from young and old mice are summarized in Table 1.
Table 1. Comparison of body weight, heart weight, echocardiography and ECG findings for transgenic (CSQ) mice and age-matched non-transgenic littermates (control).
| Young mice | Old mice | |||
|---|---|---|---|---|
| Control (n = 6) | CSQ (n = 6) | Control (n = 12) | CSQ (n = 12) | |
| Age (days) | 35 ± 4.2 | 38 ± 4.2 | 130 ± 11 | 132 ± 12 |
| Body weight (g) | 13.9 ± 0.8 | 13.8 ± 0.9 | 21.9 ± 1.1 | 22.6 ± 1.6 |
| Heart weight (mg) | 108 ± 5.1 | 162 ± 3.6** | 158 ± 6.8 | 356 ± 25*** |
| LVEDD (mm) | 1.95 ± 0.06 | 3.1 ± 0.23*** | 2.4 ± 0.12 | 4.6 ± 0.3*** |
| LVESD (mm) | 0.48 ± 0.16 | 1.0 ± 0.14** | 0.80 ± 0.13 | 3.6 ± 0.38*** |
| IVSW thickness (mm) | 0.85 ± 0.05 | 1.15 ± 0.09** | 1.01 ± 0.04 | 1.16 ± 0.05* |
| PLVW thickness (mm) | 0.80 ± 0.08 | 1.07 ± 0.05* | 1.02 ± 0.04 | 1.18 ± 0.07* |
| LV FS | 0.75 ± 0.08 | 0.66 ± 0.06 | 0.68 ± 0.05 | 0.17 ± 0.02*** |
| Heart rate (beats min−1) | 564 ± 38 | 374 ± 10*** | 481 ± 76 | 357 ± 50*** |
| P–R interval (ms) | 35 ± 1.2 | 41 ± 3.2 | 37 ± 4.5 | 62 ± 12*** |
| QRS duration (ms) | 16 ± 0.4 | 25 ± 2.3** | 17 ± 3.2 | 28 ± 10* |
| Q–T interval (ms) | 53 ± 2.1 | 98 ± 6.3*** | 65 ± 8.2 | 123 ± 20*** |
| Q–Tc interval (ms) | 51 ± 1.9 | 77 ± 4.3*** | 57 ± 5.7 | 95 ± 20*** |
Data are means ± S.E.M. LV, left ventricle; EDD, end-diastolic internal diameter; ESD, end-systolic internal diameter; IVSW, interventricular septal wall; PLVW, posterior LV wall; FS, fractional shortening.
P < 0.05
P < 0.01
P < 0.001 vs. control.
Figure 3. ECG and action potential measurements.
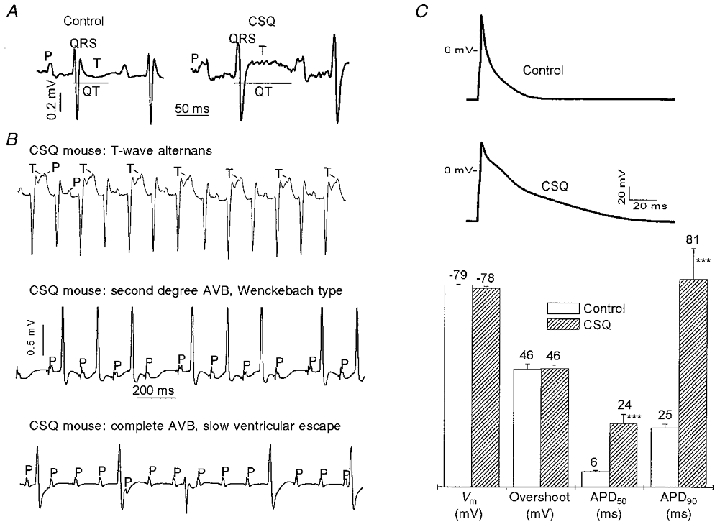
A, representative ECG tracings (lead II) from control and transgenic mice overexpressing CSQ (aged 139 and 134 days, respectively). The various parameters of the ECG are indicated; P represents atrial depolarization (P-wave) and T the ventricular repolarization (T-wave). A wider QRS complex and a prolonged Q-T interval were consistently encountered in CSQ mice. B, approximately 30 % of CSQ mice had an alternating pattern of the T-wave (lead II, T-wave alternans, top panel) in all age groups examined. A long P-R interval (P-R > 45 ms), higher degrees of atrioventricular block (AVB, lead I, middle and bottom panels) or QRS morphologies suggestive of conduction blocks (middle panel) were only present in CSQ mice older than 60 days. C, the cardiac action potential was significantly prolonged in ventricular cells from CSQ mice, both at the 50 % and the 90 % repolarization level (APD50 and APD90, respectively). Resting membrane potential (Vm) and overshoot potential were not significantly different. Mean values are given above each bar; error bars are s.e.m. Control: 19 myocytes from 3 hearts; mean age, 109 days. CSQ: 13 myocytes from 4 hearts; mean age, 139 days. *** P < 0.001 vs. control.
Heart morphology and histopathology
The atrial and ventricular dimensions from control and transgenic animals at 4–6 months of age are compared in Table 2. CSQ mice demonstrated both gross and histological evidence of dilated cardiomyopathy. Cross-sections through the ventricles demonstrated biventricular enlargement with symmetrical increase in wall thickness (Fig. 4A and B). A large proportion of CSQ mice had atrial and ventricular thrombi in various states of organization (Fig. 4B).
Table 2. Cardiac dimensions (in mm) from non-transgenic (control) and transgenic (CSQ) mice, aged 4–6 months.
| RA | LA | RV | LV | RVFW | LVFW | Septum | |
|---|---|---|---|---|---|---|---|
| Control | 3.3 ± 0.2 | 3.0 ± 0.2 | 2.5 ± 0.2 | 1.5 ± 0.5 | 0.43 ± 0.06 | 1.1 ± 0.04 | 0.9 ± 0.17 |
| CSQ | 5.6 ± 0.3** | 6.5 ± 0.6** | 3.9 ± 0.4* | 3.1 ± 0.3* | 0.83 ± 0.05** | 1.4 ± 0.05** | 1.3 ± 0.09** |
Atrial measurements were taken at necropsy (6 control, 12 CSQ mice). Ventricular measurements were obtained from formalin-fixed and haematoxylin-eosin-stained subvalvular transverse sections (3 control, 6 CSQ mice). Values are means ± S.E.M.
P < 0.05
P < 0.01 vs. control. RA and LA, largest external diameter of the right and left atrium, respectively; RV and LV, largest internal diameter of right and left ventricular chamber, respectively; RVFW, right ventricular free wall; LVFW, left ventricular free wall.
Figure 4. Heart morphology.
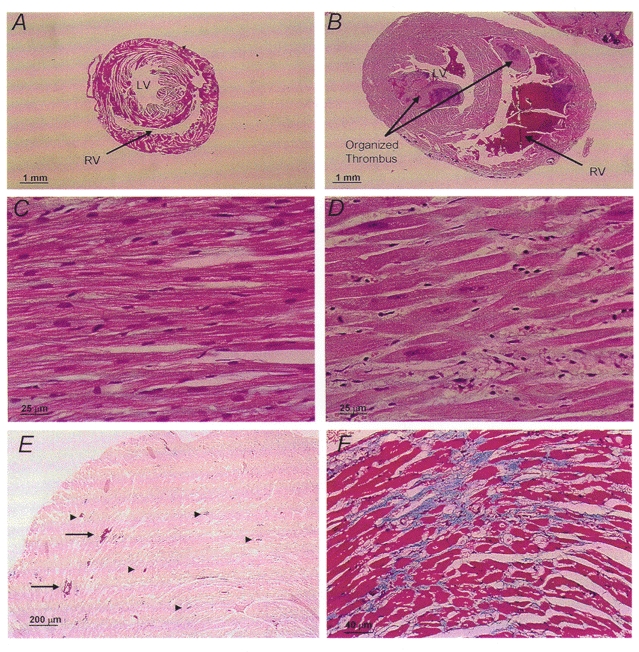
Subvalvular transverse cross-sections from control (A) and CSQ-overexpressing (B) mouse hearts. Biventricular chamber dilatation, wall thickening and an organized ventricular thrombus (arrow) were present in the CSQ heart. Haematoxylin-eosin (H-E)-stained sections from control (C) and CSQ (D) hearts demonstrate individual myocyte hypertrophy with enlarged nuclei, prominent nucleoli and a mild lymphohistocytic infiltrate. Tissue calcifications (E; H-E stain) involving single myofibrils (arrowheads) and clusters (arrows) and areas of interstitial fibrosis (F; trichrome stain) were only present in CSQ hearts.
Histological examination of control (n = 3) and transgenic mice (n = 6) revealed striking differences. CSQ myocytes were hypertrophied, showing consistently enlarged vesiculated nuclei, prominent nucleoli (Fig. 4C and D) and occasionally multiple nuclei. These hearts contained patchy mononuclear inflammatory infiltrates in both atrial and ventricular walls (Fig. 4D). There were scattered biventricular and atrial dystrophic calcifications, involving both single myocytes and larger wall areas (Fig. 4E). Calcifications did not appear to be linked to inflammatory infiltrates. Trichrome staining showed mild diffuse biventricular fibrosis with focal areas of increased intramural collagen deposits (Fig. 4F).
Lung sections from three out of six transgenic animals examined showed evidence of pulmonary congestion and haemosiderin-laden macrophages (heart failure cells) in the alveolar spaces. Liver sections from these animals had significant congestion with sinusoidal dilatation, diffuse mononuclear parenchymal infiltrates, with scattered micro-abscesses and degenerating hepatocytes. Kidney sections from transgenic animals showed no significant histopathological abnormalities (data not shown).
Cellular electrophysiological remodelling in transgenic mice with dilated cardiomyopathy
Only 50% of transgenic mice that died spontaneously had clear evidence of pump failure (ascites, pleural effusion or oedema) on necropsy, suggesting that cardiac arrhythmias could have contributed to the high mortality of CSQ mice. Since 15 of 16 deaths occurred in CSQ mice older than 60 days, we examined the cellular electrophysiology of this age group first.
Cell capacitance and action potential duration
Ventricular myocytes from transgenic animals (18 mice; mean age, 116 days) had significantly larger cell capacitance (357 ± 13 pF, n = 79 myocytes) compared with myocytes obtained from age-matched non-transgenic littermates (20 mice; mean age, 124 days; 207 ± 7 pF, n = 78 myocytes, P < 0.001). This finding is consistent with our previous reports of cellular hypertrophy and suggests a 50–100 % increase in myocyte size (Jones et al. 1998).
At 36°C, the cellular action potential was significantly longer in CSQ myocytes (Fig. 3C). APD50 and APD90, respectively, were 6 ± 0.4 and 25 ± 1.3 ms in control (n = 19 myocytes) and 24 ± 3.6 and 81 ± 12 ms in CSQ myocytes (n = 13 myocytes, P < 0.001 for each set). Resting and overshoot potentials were not statistically different between CSQ and control myocytes.
Ionic currents underlying the action potential
Outward K+ current
Mouse ventricular myocytes express large depolarization-activated and Ca2+-independent K+ currents. Figure 5A (upper panel) shows representative outward K+ currents elicited from a control and a CSQ myocyte. Cells were dialysed with internal solution containing 14 mmol l−1 EGTA to suppress both contraction and the Ca2+-activated component of the outward current. The depolarization-activated outward K+ currents were significantly suppressed in CSQ myocytes. Peak current densities at +40 mV were 12.2 ± 0.7 pA pF−1 in CSQ and 44 ± 5.6 pA pF−1 in control cells (Fig. 5A, middle panel, P < 0.01). The sustained K+ currents at the end of the 400 ms depolarization pulses were also smaller (current density at +40 mV: 7.2 ± 0.6 pA pF−1 in CSQ and 16.5 ± 1.8 pA pF−1 in control cells; Fig. 5A, lower panel, P < 0.01).
Figure 5. Ito and IK1 are suppressed in CSQ mice.
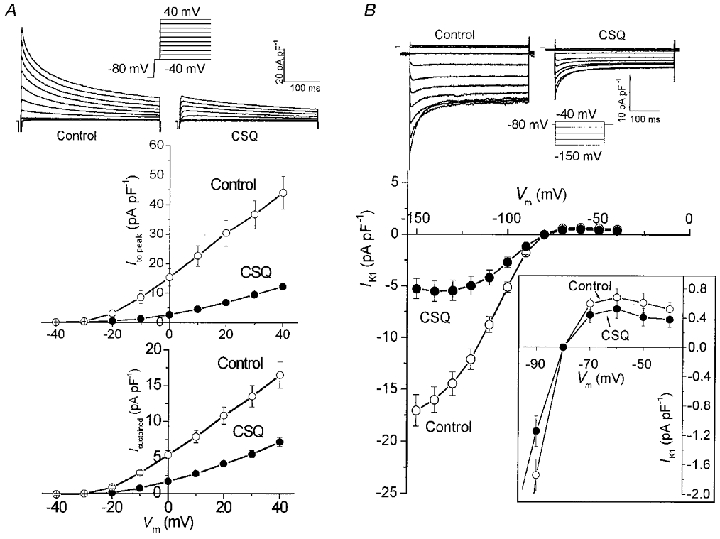
A, transient outward K+ currents (Ito) were significantly decreased in CSQ myocytes ( P < 0.01). Top panel, superimposed current traces in response to 10 mV step depolarizations from −40 to 40 mV applied from a holding potential of −80 mV in control and CSQ myocytes. Na+ current was inactivated by a 5 ms voltage step to −40 mV. Peak Ito (middle panel) and sustained K+ currents measured at the end of the 400 ms steps (bottom panel; Isustained) were markedly suppressed at all voltages in CSQ myocytes. Values are means ±s.e.m.○, ten control myocytes from 4 hearts; mean age, 145 days. •, eleven CSQ myocytes from 4 hearts; mean age, 132 days. B, top panel: representative traces of IK1 from control and CSQ myocytes in response to 10 mV voltage steps from −150 to −40 mV. Bottom panel: inward rectifier K+ currents (IK1) were significantly decreased in CSQ myocytes ( P < 0.01). Inset: magnified segment of current-voltage relationship from −90 to −40 mV. Outward IK1 was not significantly different between control and CSQ myocytes. Values are means ±s.e.m.○, twelve control myocytes from 4 hearts; mean age, 132 days. •, ten CSQ myocytes from 4 hearts; mean age, 136 days.
Inward rectifier K+ current
IK1 is thought to regulate the resting potential as well as the rapid repolarization phase of the action potential. Ventricular myocytes from non-transgenic mice developed a large IK1 in response to hyperpolarizing voltage steps (Fig. 5B). In CSQ myocytes, the density of hyperpolarization-induced IK1 was significantly smaller. Figure 5B (lower panel) shows that currents measured negative to −90 mV were significantly smaller in CSQ compared with control myocytes (IK1 at −100 mV was −2.7 ± 0.5 pA pF−1 in CSQ vs. −5.1 ± 0.5 pA pF−1 in control cells, P < 0.01). The outward portion of IK1 also appeared to be somewhat smaller, but was not statistically different from that of control myocytes (Fig. 5B, inset).
Na+ current
Large Na+ currents were measured in both control and transgenic ventricular myocytes (Fig. 6A). Figure 6B shows the pooled current-voltage relations for control and CSQ cells measured in extracellular and intracellular solutions containing symmetrical 5 mmol l−1 Na+. Peak INa, when plotted at different potentials, was significantly smaller in CSQ myocytes, but its voltage dependence was shifted towards more positive membrane potentials. The maximal slope conductance of INa was significantly reduced in CSQ myocytes ( 0.28 ± 0.02 vs. 0.48 ± 0.02 pA pF−1 mV−1, n = 31 and 24 myocytes, respectively, P < 0.001), with the half-activation potential shifted by 4 mV towards more positive potentials (-44 ± 0.8 vs.−48 ± 0.9 mV, P < 0.01). The slope factor for activation of INa (CSQ, 4.9 ± 0.15; control, 5.0 ± 0.15) and the estimated VNa (CSQ, −2.5 ± 1.4 mV; control, −1.0 ± 1.4 mV) were not significantly different between CSQ and control myocytes.
Figure 6. INa is decreased, but its availability is shifted in CSQ mice.
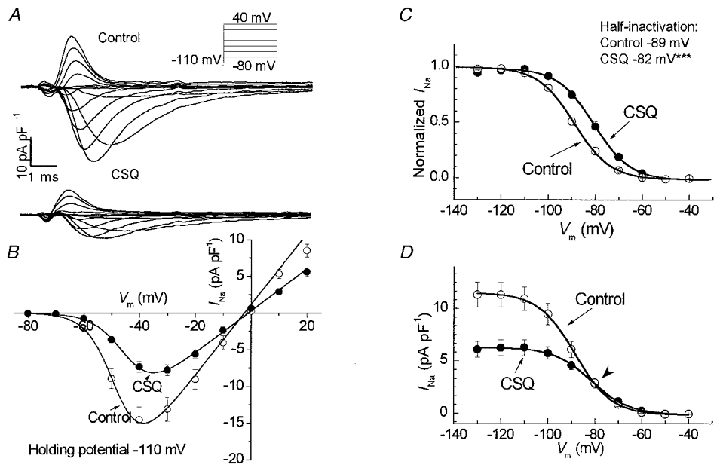
A, superimposed current traces of INa from control and CSQ myocytes in response to 10 mV voltage steps from −80 to 40 mV. B and C, peak INa density of CSQ cells was about 50 % smaller (B) and both its activation (B) and steady-state inactivation (C) were shifted towards more positive membrane potentials. *** P < 0.001 vs. control. D shows that this results in no change in absolute INa density at a resting membrane potential of −80 mV (arrowhead). Measurements were obtained at 22°C in symmetrical Na+ concentrations of 5 mmol l−1. Values are means ±s.e.m.○, twenty-four control myocytes from 5 hearts; mean age, 123 days. •, thirty-one CSQ myocytes from 6 hearts; mean age, 87 days.
The steady-state half-inactivation potential for INa was significantly shifted towards more positive potentials in CSQ myocytes (-82 ± 1.2 vs.−89 ± 1.3 mV, n = 23 and 29 myocytes, P < 0.001) without a change in the slope factor (CSQ, 6.4 ± 0.26; control, 6.7 ± 0.17). Normalized values of INa in control and transgenic myocytes are shown in Fig. 6C. Thus for any given membrane potential, the availability of Na+ channels appeared to be higher in CSQ myocytes, which may, in part, compensate for the decrease in the density of INa. In fact, at potentials positive to −80 mV, there was little or no difference in the density of INa between the control and CSQ-overexpressing myocytes (Fig. 6D). This observation is consistent with the finding that there were no significant differences between the overshoot potentials of the control and transgenic myocytes.
L-type Ca2+ current
Figure 7A (upper panels) shows representative Ca2+ current tracings from control and CSQ-overexpressing myocytes. Peak ICa was significantly smaller (Fig. 7A, lower panels) and its rate of inactivation was markedly slower in CSQ myocytes (time constant: 28 ± 4 vs. 7 ± 1 ms, n = 10 and 9 myocytes, P < 0.01).
Figure 7. Basal and isoproterenol-stimulated ICa are decreased in CSQ mice, but partly recover following dialysis with 8-Br-cAMP or exposure to IBMX.
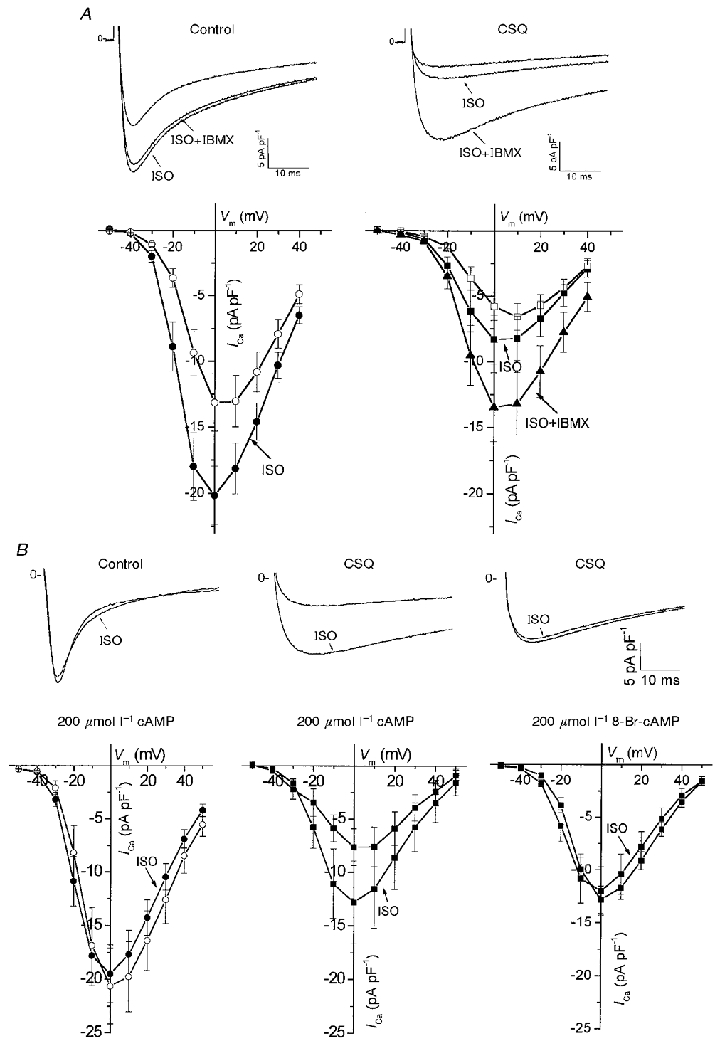
A, top panels: representative current traces from a control (left) and CSQ (right) myocyte in response to a voltage step from −50 to 0 mV in the absence and presence of 5 μmol l−1 isoproterenol (ISO) and/or 200 μmol l−1 IBMX. Bottom panels: respective voltage-dependent effects of ISO and IBMX on averaged peak currents. B, similar recordings of ICa were made in myocytes dialysed with 200 μmol l−1 cAMP or its non-hydrolysable analogue 8-Br-cAMP, as indicated. Note the strong enhancement of ICa with ISO only in CSQ myocytes, which was abolished by cell dialysis with 8-Br-cAMP. Values are means ±s.e.m. Open symbols, 7–10 control myocytes from 3–4 hearts; mean age, 151 days. Closed symbols, 5–8 CSQ myocytes from 4 hearts; mean age, 141 days.
There were also major differences between CSQ-overexpressing and control myocytes in the β-adrenergic/cAMP-dependent regulation of ICa. While isoproterenol enhanced ICa in control myocytes, the effect was significantly blunted in CSQ myocytes (Fig. 7A). Interestingly, the combination of isoproterenol and the phosphodiesterase inhibitor isobutylmethylxanthine (IBMX) further enhanced ICa in CSQ but not in control myocytes (Fig. 7A). These observations suggest either an increased phosphodiesterase activity or an impaired isoproterenol-dependent cAMP synthesis in CSQ myocytes. In the next set of experiments we dialysed the myocytes with saturating concentrations (200 μmol l−1) of cAMP and found that ICa remained smaller in CSQ compared with control myocytes (Fig. 7B, left and middle panels). Application of 5 μmol l−1 isoproterenol surprisingly enhanced ICa in CSQ but not in control myocytes. To determine whether increased cAMP hydrolysis was responsible for the continued effect of isoproterenol in the presence of high concentrations of cAMP, we used a non-hydrolysable analogue of cAMP, 8-Br-cAMP. In CSQ myocytes dialysed with 200 μmol l−1 8-Br-cAMP, the density of ICa was not only larger than that in myocytes dialysed with cAMP, but also isoproterenol failed to further enhance ICa in a manner similar to that in control cells (Fig. 7B, right panel). Taken together these results suggest that CSQ myocytes may have had higher phosphodiesterase activity.
Na+-Ca2+ exchange current
It is postulated that the Na+-Ca2+ exchanger contributes to both the influx of Ca2+ early during depolarization and to the efflux of Ca2 later during the action potential. The activity of the Na+-Ca2+ exchanger is reported to increase during heart failure in humans and in animal models of heart failure (Hatem et al. 1994; Hasenfuss et al. 1997). Figure 8A illustrates an attempt to quantify the maximal activity of the exchanger in its Ca2+-influx mode (outward current) using the procedure of rapid removal of external Na+ (Litwin & Bridge, 1997). Step removal of extracellular Na+ generated slowly developing but large (1-3 pA pF−1) currents, which decayed back to resting values upon replacement of extracellular Na+ (Fig. 8A). When extracellular Na+ is replaced, the exchanger switches again into forward mode and starts extruding the Ca2+ that has accumulated in the cell at the end of the Na+-free period. This process is most probably responsible for the transient Ni2+-sensitive inward current following the step removal of extracellular Na+. Since 5 mmol l−1 Ni2+ effectively and reversibly blocks the exchanger, only the Ni2+-sensitive outward currents were used to quantify the exchanger activity. This analysis showed that, on average, Na+-Ca2+ exchange current density increased by 33 % in CSQ compared with control myocytes (Fig. 8B; P < 0.01).
Figure 8. Na+-Ca2+ exchange current is increased in CSQ mice.
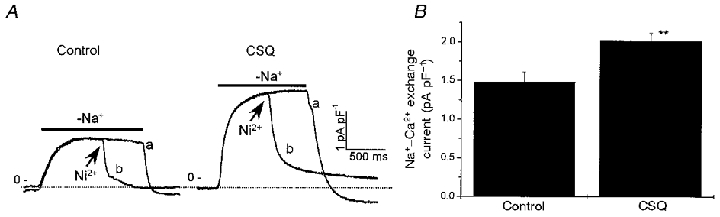
A, Na+-Ca2+ exchange current was quantified as the Ni2+-blockable component (trace a – b) of the outward current generated on rapid removal of external Na+. Arrows indicate addition of 5 mmol l−1 Ni2+. The holding potential was −40 mV. Note the current recovers to levels negative to resting values, as expected from higher cellular levels of Ca2+ following the large influx of Ca2+. B, mean densities of Na+-Ca2+ exchange currents measured as described in A were significantly higher in CSQ compared with control myocytes (** P < 0.01). Error bars are s.e.m. Control: 6 myocytes from 3 hearts; mean age, 138 days. CSQ: 6–10 myocytes from 3 hearts; mean age, 150 days.
Remodelling of ionic currents during progression from hypertrophy to heart failure
Echocardiography showed that the cardiac function of CSQ mice is normal during the first 60 days of life (hypertrophy stage). As the CSQ mice get older, contractile function deteriorates and dilated cardiomyopathy with overt heart failure develops (see Fig. 1C). We examined the possible age-dependent alterations in electrophysiological properties of ventricular myocytes during the progression from compensated hypertrophy to heart failure. Figure 9 shows plots of cell capacitance, action potential duration and densities of several ionic currents as a function of age, using linear regression analysis. In general, cell capacitance and action potential duration increased significantly with age in CSQ myocytes, whereas all examined current densities declined. We then compared the data from younger mice with compensated hypertrophy (< 60 days) with those of older animals with heart failure. The analysis showed that cell capacitance had already increased during the hypertrophy stage (CSQ: 201 ± 11 pF, n = 30 cells from 4 mice, vs. control: 159 ± 8 pF, n = 23 cells from 6 mice, P < 0.01) and continued to increase progressively with age during the failure stage (Fig. 9A).
Figure 9. Cellular electrophysiological parameters as a function of age.
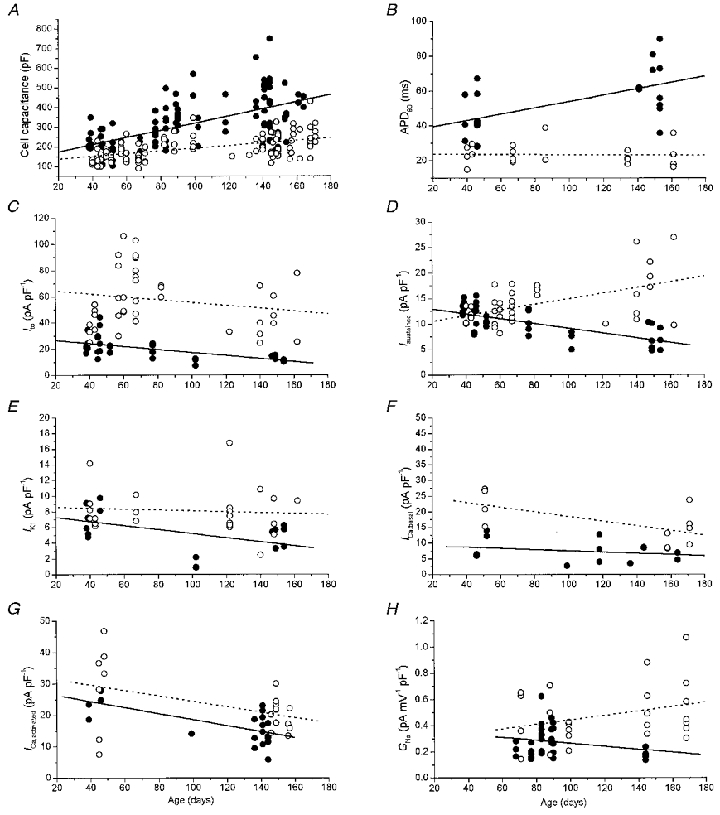
Parameter values of control (○) and CSQ (•) myocytes and the respective linear regression lines for control (dashed) and CSQ (continuous) myocytes. A, cell capacitance increased significantly with age both in control and CSQ myocytes. Control: r = 0.51, P < 0.0001, 101 myocytes from 26 hearts vs. CSQ: r = 0.60, P < 0.0001, 109 myocytes from 22 hearts. B, action potential duration (APD90) increased only in CSQ myocytes. Control: r =−0.02, P = 0.89, 24 myocytes from 9 hearts vs. CSQ: r = 0.57, P < 0.01, 23 myocytes from 6 hearts. C, Ito was downregulated in young CSQ mice and decreased further with age. Control: r =−0.19, P = 0.25, 39 myocytes from 10 hearts vs. CSQ: r =−0.62, P < 0.0001, 36 myocytes from 9 hearts. D, the sustained outward K+ current (Isustained) increased significantly with age in control myocytes, but decreased significantly in CSQ myocytes. Control: r = 0.51, P < 0.001, 39 myocytes from 10 hearts vs. CSQ: r =−0.67, P < 0.0001, 36 myocytes from 9 hearts. E, IK1 remained unchanged in control myocytes, but decreased in CSQ myocytes. Control: r =−0.08, P = 0.69, 23 myocytes from 9 hearts vs. CSQ: r =−0.50, P < 0.05, 18 myocytes from 6 hearts. F, basal ICa (ICa,basal) decreased significantly with age in control myocytes, but remained unchanged in CSQ myocytes. Control: r =−0.62, P < 0.05, 11 myocytes from 3 hearts vs. CSQ: r =−0.26, P = 0.37, 14 myocytes from 7 hearts. G, maximally activated ICa (ICa,activated; 200 μmol l−1 8-Br-cAMPi) decreased significantly with age in both control and CSQ myocytes. Control: r =−0.45, P < 0.05, 21 myocytes from 6 hearts vs. CSQ: r =−0.67, P < 0.001, 20 myocytes from 6 hearts. H, no significant age-dependent changes in INa were present (GNa, Na+ channel conductance). Control: r = 0.29, P = 0.15, 24 myocytes from 5 hearts vs. CSQ: r =−0.22, P = 0.22, 31 myocytes from 6 hearts. In C and D, currents were measured at 40 mV; in E, currents were measured at −100 mV.
The ventricular action potential was significantly longer in the hypertrophy stage (CSQ: APD50, 14.4 ± 2.7 ms; APD90, 44 ± 4.2 ms, n = 10 cells from 3 mice vs. control: APD50, 5.7 ± 0.5 ms; APD90, 24 ± 2.6 ms, n = 5 cells from 2 mice, P < 0.01). Resting and overshoot potentials, on the other hand, were not significantly different. The action potential was prolonged significantly with age in CSQ but not in control myocytes (Fig. 9B), suggesting differential regulation of the underlying ionic currents. Ito was unique in that both its absolute magnitude (CSQ: 4655 ± 295 pA, vs. control: 9328 ± 1282 pA, P < 0.001) and its density (CSQ: 24 ± 1.7 pA pF−1, n = 20 cells from 3 mice vs. control: 53 ± 5.7 pA pF−1, n = 16 cells from 3 mice, P < 0.001) were significantly smaller during the hypertrophy stage of the disease. The density of Ito, as well as its absolute magnitude, decreased further with age in the CSQ myocytes (Fig. 9C). In contrast, the sustained K+ current appeared to increase during the hypertrophy stage (CSQ: 2281 ± 157 pA, vs. control: 2069 ± 205 pA, P = 0.38), therefore its current density remained the same as in control myocytes (CSQ: 11.8 ± 0.5 pA pF−1, n = 20 cells from 3 mice vs. control: 12.0 ± 0.6 pA pF−1, n = 16 cells from 3 mice, P = 0.80). With progression to heart failure, a profound decrease in the density of the sustained K+ current occurred (Fig. 9D), probably reflecting the failure of de novo protein synthesis to keep up with cell hypertrophy. Similar changes were also observed in IK1 measured at −100 mV, i.e. increased absolute currents during the hypertrophy stage (CSQ: −1510 ± 136 pA, vs. control: −1135 ± 84 pA, P < 0.05) and no difference in current density (CSQ: 7.4 ± 0.7 pA pF−1, n = 8 cells from 3 mice, vs. control: 8.3 ± 1.0 pA pF−1, n = 7 cells from 2 mice, P = 0.45). With progression to heart failure, absolute values of IK1 also failed to increase appropriately with cell hypertrophy and IK1 density decreased significantly (Fig. 9E).
Longitudinal analysis of basal ICa showed that this current was already suppressed during the hypertrophy stage (CSQ: 8.9 ± 1.7 pA pF−1, n = 5 cells from 3 mice vs. control: 22 ± 2.8 pA pF−1, n = 4 cells from 1 mouse, P < 0.05) and continued to remain suppressed in older animals (Fig. 9F). However, ICa maximally activated by isoproterenol (CSQ: 25 ± 7.8 pA pF−1, n = 5 cells from 2 mice vs. control: 26 ± 4.6 pA pF−1, n = 5 cells from 2 mice, n.s.) or by intracellular dialysis with 8-Br-cAMP (CSQ: 24 ± 1.7 pA pF−1, n = 6 cells from 2 mice vs. control: 29 ± 5.4 pA pF−1, n = 7 cells from 2 mice, n.s.) was not significantly different from that of control myocytes during the hypertrophy stage. The absolute values of maximally activated ICa were higher at any age in CSQ myocytes, even though ICa density declined with a similar slope both in control and CSQ myocytes (Fig. 9G).
Similar observations were made for the maximally available Na+ channel conductance. Although absolute Na+ channel conductance was not significantly different in control and CSQ myocytes (CSQ: 86 ± 7.7 pA mV−1, n = 31 cells from 6 mice vs. control: 103 ± 12 pA mV−1, n = 24 cells from 5 mice, n.s.), the Na+ channel conductance corrected for cell capacitance showed a trend towards an age-dependent decrease in CSQ myocytes, but an increase in control myocytes (Fig. 9H). This difference may be responsible for the observation that INa density was significantly smaller in CSQ myocytes during the heart failure stage (see Fig. 6).
DISCUSSION
The major finding of this report is that overexpression of cardiac calsequestrin causes progressive cardiac hypertrophy, leading to a form of dilated cardiomyopathy with severe contractile impairment and extensive electrical remodelling, resulting in increased mortality (Fig. 1). The electrophysiological changes are accompanied by Q-T interval and action potential prolongation (Fig. 3), caused most probably by downregulation in Ito and IK1 (Fig. 5), the marked slowing in the inactivation kinetics of ICa (Fig. 7A) and an increase in Na+-Ca2+ exchanger activity (Fig. 8). In addition, Ca2+ channel currents appear to be less responsive to intracellular cAMP, most probably caused by an enhanced phosphodiesterase activity in CSQ myocytes of older animals (Fig. 7). The remodelling of ionic currents occurred in an age-dependent manner during the progression from compensated hypertrophy to heart failure (see Figs 1 and 9).
Electrophysiological remodelling
Comprehensive cellular electrophysiological studies in transgenic mice developing heart failure are as yet to be reported. In a murine model of familial hypertrophic cardiomyopathy induced by α-myosin heavy chain Arg403Gln missense mutation, Berul and colleagues (Berul et al. 1997) showed Q-T prolongation, ventricular arrhythmias and premature deaths. The Q-T prolongation in that study was much smaller (Q-Tc: 54 vs. 63 ms, control vs. transgenic, rate correction according to Mitchell et al. 1998) compared with our findings (Fig. 3) and no cellular electrophysiological parameters were reported. Although other murine models of cardiac hypertrophy or failure have not as yet been examined, action potential prolongation and a downregulation of the transient outward current (Ito) have been consistently observed in spontaneously hypertensive rats with cardiac hypertrophy (Cerbai et al. 1994). In mice, Q-T prolongation was observed when Ito was decreased by functional knockout of the genes encoding either Kv1.5 or Kv4.3 (London et al. 1998b; Barry et al. 1998). Interestingly, athough the action potential was prolonged the functional knockout of these genes did not cause cardiac hypertrophy and increased mortality.
It is likely that the impaired Ca2+ signalling typically found in CSQ mice (Jones et al. 1998; Sato et al. 1998) may also contribute significantly to the Q-T prolongation of this model. Winslow and coworkers recently modelled the mechanism of altered excitation-contraction coupling in canine tachycardia-induced heart failure and showed that the reduction of Ito had only a minor effect on the action potential duration, whereas the decrease in the sarcoplasmic reticulum (SR) Ca2+ release significantly prolonged the action potential (Winslow et al. 1999). These investigators attributed the action potential prolongation to the slower kinetics of inactivation of the L-type Ca2+ current. Our finding that the inactivation kinetics of ICa are markedly slowed in CSQ mice supports this idea (Fig. 7).
Heart rate regulation in CSQ-overexpressing mice
One of our surprising findings was that CSQ mice had lower heart rates compared with control mice (Table 1). This finding may be secondary to a higher sensitivity of mice with heart failure to sedation. However, younger CSQ mice without evidence of heart failure showed even more profound bradycardia (Table 1), which does not support this idea. In our initial study of CSQ mice we reported a faster heart rate in CSQ mice based on qualitative examinations of the pulse rate (Jones et al. 1998). Given the wide variability in heart rate and the possible artifacts of sedation, we believe the earlier qualitative measurements to be less trustworthy. Others have also reported lower heart rates in younger CSQ mice without heart failure (Cho et al. 1999). It is unlikely that suppression of Ito alone was responsible for the decreased heart rate, because the functional knockout of Ito had no effect on the heart rate (London et al. 1998b; Barry et al. 1998). It is more likely that the impaired Ca2+ release and slowing of the inactivation kinetics of ICa contribute significantly to the observed bradycardia. In this respect, interventions that reduce SR Ca2+ release in sino-atrial cells have been reported to decrease the spontaneous beating of guinea-pig sino-atrial nodal preparations (Rigg & Terrar, 1996).
Age- and disease dependence of the changes in ionic currents
One of the novel aspects of this study is the finding that there are differential age- and disease-dependent changes in ionic currents of both control and transgenic myocytes. For instance, the absolute magnitude of IK1, the sustained K+ current (Isustained) and INa increased significantly with age in control myocytes. This appears to be an appropriate compensation for the observed increase in heart weight and cellular surface area during the first 180 days of life (see Figs 1A and 9), contributing to keeping the action potential duration (Fig. 9B) and current densities (Fig. 9C–H) relatively constant. Similar age-dependent changes in IK1, Ito and Isustained have been observed in normal rat ventricular myocytes (Leblanc et al. 1998). A much more profound increase in heart weight and cell size appears to occur in CSQ mice (see Figs 1A and 9A). The CSQ-induced hypertrophy was compensated in the first 2 months of life, but then progressed into a severe dilated cardiomyopathy (Fig. 1C), as also recently reported by Cho et al. (1999). Ito appeared to have already decreased during the compensated hypertrophy stage, but other ionic currents were relatively preserved. With age, as CSQ mice progressed into the heart failure stage, the myocyte surface area increased further, but the absolute ionic currents remained relatively unchanged, resulting in a decline of all examined current densities (Fig. 9C–H). For example, the density of IK1 was not significantly different from that of control in the smaller myocytes of younger mice, but thereafter absolute IK1 failed to increase further and the density of the current declined with age (Fig. 9E). Similar results were obtained for Isustained and INa. This suggests a dynamic regulation of these ion channels during the hypertrophic process, i.e. initially, the number of functional channels may have increased appropriately with cell growth, but during the heart failure stage the hypertrophied cells seem not to have been able to keep up with channel production.
Since Ca2+ channels had lower basal activity at any age (Fig. 9F), it is likely that possible channel dephosphorylation secondary to high levels of phosphodiesterase activity also contributes to the quantification of ICa density. Consistent with this idea, stimulation with isoproterenol was able to overcome this apparent inhibition of ICa by higher phosphodiesterase activity in the younger but not older CSQ mice. These results indicate that the universal suppression of ionic currents in the failing myocytes is multifactorial; functional ion channels may be downregulated (Ito), or fail to increase with cell hypertrophy (IK1, Isustained, INa) or may be dephosphorylated (ICa). It is possible that ionic current may behave differently in vivo, as most of the measurements in this study were made in isolated myocytes under highly artificial conditions.
Arrhythmogenesis and mortality
The decrease in Ito and IK1 in dog models (Kaab et al. 1996) and human heart failure (Beuckelmann et al. 1993; Koumi et al. 1995) has been implicated in the development of ventricular arrhythmias. It is tempting to speculate that the profound decrease in Ito and IK1 (Fig. 5) could have contributed to arrhythmogenesis and the increased mortality of CSQ mice. Several findings argue against a single cause for arrhythmias in the CSQ model: (1) Ito was already decreased during the hypertrophy stage not associated with increased mortality; and (2) the outward component of IK1 was not significantly decreased in failing CSQ myocytes. Although there was a trend towards decreased outward IK1 in CSQ myocytes, it did not reach statistical significance because of the large variations in IK1 between different cells.
In the short periods of ECG recordings, we were able to measure conduction blocks, but no ventricular tachyarrhythmias. The frequent conduction blocks of older CSQ mice may be related to the fibrotic changes and tissue calcifications observed in CSQ hearts (Fig. 4E and F). It is likely that other mechanisms (e.g. pulmonary emboli) may also contribute to the deaths of animals, especially since atrial thrombi were present in almost every transgenic animal examined (Fig. 4). Telemetric studies on conscious mice might be helpful in determining the incidence of ventricular arrhythmias in the CSQ mouse model.
The decreased Na+ current density in CSQ myocytes (Fig. 6) could have also contributed to the conduction blocks (Fig. 3) and the resulting increased mortality in a manner similar to that encountered with the use of Na+ channel blocking antiarrhythmic agents (The Cardiac Arrhythmia Suppression Trial II Investigators, 1992). On the other hand, INa was found not to be altered in a dog heart failure model (Kaab et al. 1996). We also did not find any differences in the overshoot potential (Fig. 3C), an indicator of Na+ channel function. This discrepancy may result from the compensatory shift in the steady-state inactivation of INa (Fig. 6C). The shift in steady-state inactivation of INa towards more positive membrane potentials is also consistent with decreased phosphorylation of the Na+ channels (Murphy et al. 1996), caused by the increased phosphodiesterase activity observed in CSQ myocytes (e.g. Fig. 7).
Na+-Ca2+ exchanger function has been shown to increase in end-stage heart failure (Studer et al. 1994; Hatem et al. 1994) and could contribute to the action potential prolongation (Winslow et al. 1999). Although the level of SR Ca2+-ATPase does not appear to be decreased in the CSQ model (Jones et al. 1998), Ca2+ transients did decay slowly in CSQ myocytes, thus allowing the exchanger to play a greater role in Ca2+ removal. In older CSQ-overexpressing myocytes, we found Na+-Ca2+ exchange current density to be increased by 35% (Fig. 8), with a greater than 2-fold increase in the absolute current magnitude. The increased exchanger activity is thereby likely to contribute to the action potential prolongation of failing CSQ myocytes. Linck et al. (1998) reported that exchanger protein levels and activity were not changed in younger CSQ mice, which has recently been confirmed in another CSQ-overexpressing mouse model (Sato et al. 1998). This may suggest that increased exchanger activity develops in response to the heart failure rather than being directly regulated by CSQ.
Significance of the CSQ mouse model
Despite the fact that CSQ does not appear to be upregulated in human heart failure, its overexpression produces hypertrophy and eventually heart failure, which was not the case for transgenic mice overexpressing any other protein of the Ca2+-signalling cascade (Ca2+ channel, Na+-Ca2+ exchanger, triadin, phospholamban, Ca2+-ATPase). The CSQ mouse is also the only transgenic model with significantly impaired cardiac Ca2+ signalling. The fact that this Ca2+-signalling impairment appears to occur before all the other molecular changes in the membrane points to the extreme importance of intact cardiac Ca2+ homeostasis. It is well recognized that cardiac Ca2+ homeostasis is defective in heart failure. As such, the CSQ model could be very valuable in the study of interventions aimed at improving cardiac Ca2+ metabolism as a potential target for the treatment of heart failure. We have fully characterized the time course of contractile dysfunction and electrical remodelling in this model, which may provide the basis for further studies of modifiers of Ca2+ signalling.
Acknowledgments
This work was supported by NIH Clinical Pharmacology Training Grant GM08386 and an AHA fellowship no. 9920387U (to B.C.K.), RO1-HL-16152 (to M.M.) and RO1-HL-28556 (to L.R.J.).
References
- Barry DM, Xu H, Schuessler RB, Nerbonne JM. Functional knockout of the transient outward current, long-QT syndrome and cardiac remodeling in mice expressing a dominant-negative Kv4 α subunit. Circulation Research. 1998;83:560–567. doi: 10.1161/01.res.83.5.560. [DOI] [PubMed] [Google Scholar]
- Berul CI, Christe ME, Aronovitz MJ, Seidman CE, Seidman JG, Mendelsohn ME. Electrophysiological abnormalities and arrhythmias in α MHC mutant familial hypertrophic cardiomyopathy mice. Journal of Clinical Investigation. 1997;99:570–576. doi: 10.1172/JCI119197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuckelmann DJ, Nabauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circulation Research. 1993;73:379–385. doi: 10.1161/01.res.73.2.379. [DOI] [PubMed] [Google Scholar]
- Cerbai E, Barbieri M, Li Q, Mugelli A. Ionic basis of action potential prolongation of hypertrophied cardiac myocytes isolated from hypertensive rats of different ages. Cardiovascular Research. 1994;28:1180–1187. doi: 10.1093/cvr/28.8.1180. [DOI] [PubMed] [Google Scholar]
- Cho MC, Rapacciuolo A, Koch WJ, Kobayashi Y, Jones LR, Rockman HA. Defective β-adrenergic receptor signaling precedes the development of dilated cardiomyopathy in transgenic mice with calsequestrin overexpression. Journal of Biological Chemistry. 1999;274:22251–22256. doi: 10.1074/jbc.274.32.22251. [DOI] [PubMed] [Google Scholar]
- Cleemann L, Morad M. Role of Ca2+ channel in cardiac excitation-contraction coupling in the rat: evidence from Ca2+ transients and contraction. The Journal of Physiology. 1991;432:283–312. doi: 10.1113/jphysiol.1991.sp018385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbert MC, Hall DG, Kimball TR, Witt SA, Lorenz JN, Kirby ML, Hewett TE, Klevitsky R, Robbins J. Cardiac compartment-specific overexpression of a modified retinoic acid receptor produces dilated cardiomyopathy and congestive heart failure in transgenic mice. Journal of Clinical Investigation. 1997;100:1958–1968. doi: 10.1172/JCI119727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, Dorn GW., II Transgenic Gαq overexpression induces cardiac contractile failure in mice. Proceedings of the National Academy of Sciences of the USA. 1997;94:8121–8126. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fentzke RC, Korcarz CE, Lang RM, Lin H, Leiden JM. Dilated cardiomyopathy in transgenic mice expressing a dominant-negative CREB transcription factor in the heart. Journal of Clinical Investigation. 1998;101:2415–2426. doi: 10.1172/JCI2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenfuss G, Meyer M, Schillinger W, Preuss M, Pieske B, Just H. Calcium handling proteins in the failing human heart. Basic Research in Cardiology. 1997;92(suppl. 1):87–93. doi: 10.1007/BF00794072. [DOI] [PubMed] [Google Scholar]
- Hatem SN, Sham JS, Morad M. Enhanced Na+-Ca2+ exchange activity in cardiomyopathic Syrian hamster. Circulation Research. 1994;74:253–261. doi: 10.1161/01.res.74.2.253. [DOI] [PubMed] [Google Scholar]
- Jones LR, Suzuki YJ, Wang W, Kobayashi YM, Ramesh V, Franzini-Armstrong C, Cleemann L, Morad M. Regulation of Ca2+ signaling in transgenic mouse cardiac myocytes overexpressing calsequestrin. Journal of Clinical Investigation. 1998;101:1385–1393. doi: 10.1172/JCI1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaab S, Nuss HB, Chiamvimonvat N, O'Rourke B, Pak PH, Kass DA, Marban E, Tomaselli GF. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circulation Research. 1996;78:262–273. doi: 10.1161/01.res.78.2.262. [DOI] [PubMed] [Google Scholar]
- Knollmann BC, Duc J, Groth A, Weissmann NJ, Cleemann L, Morad M. Cardiac phenotype of transgenic mice overexpressing calsequestrin (abstract) Circulation. 1998a;98(suppl. 1):I–490. [Google Scholar]
- Knollmann BC, Jones LR, Morad M. Electrophysiological properties of transgenic myocytes overexpressing cardiac calsequestrin (abstract) Circulation. 1998b;98(suppl. 1):I–187. [Google Scholar]
- Koumi S, Backer CL, Arentzen CE. Characterization of inwardly rectifying K+ channel in human cardiac myocytes. Alterations in channel behavior in myocytes isolated from patients with idiopathic dilated cardiomyopathy. Circulation. 1995;92:164–174. doi: 10.1161/01.cir.92.2.164. [DOI] [PubMed] [Google Scholar]
- Kubota T, McTiernan CF, Frye CS, Slawson SE, Lemster BH, Koretsky AP, Demetris AJ, Feldman AM. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-α. Circulation Research. 1997;81:627–635. doi: 10.1161/01.res.81.4.627. [DOI] [PubMed] [Google Scholar]
- Leblanc N, Chartier D, Gosselin H, Rouleau JL. Age and gender differences in excitation-contraction coupling of the rat ventricle. The Journal of Physiology. 1998;511:533–548. doi: 10.1111/j.1469-7793.1998.533bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linck B, Neumann J, Huke S, Kirchhefer U, Schmitz W, Volkery F, Jones LR. Functional alterations in mice overexpressing calsequestrin in the heart (abstract) Circulation. 1998;98(suppl. 1):I–490. [Google Scholar]
- Litwin SE, Bridge JH. Enhanced Na+-Ca2+ exchange in the infarcted heart. Implications for excitation-contraction coupling. Circulation Research. 1997;81:1083–1093. doi: 10.1161/01.res.81.6.1083. [DOI] [PubMed] [Google Scholar]
- London B, Jeron A, Zhou J, Buckett P, Han X, Mitchell GF, Koren G. Long QT and ventricular arrhythmias in transgenic mice expressing the N terminus and first transmembrane segment of a voltage-gated potassium channel. Proceedings of the National Academy of Sciences of the USA. 1998a;95:2926–2931. doi: 10.1073/pnas.95.6.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London B, Wang DW, Hill JA, Bennett PB. The transient outward current in mice lacking the potassium channel gene Kv1.4. The Journal of Physiology. 1998b;509:171–182. doi: 10.1111/j.1469-7793.1998.171bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mende U, Kagen A, Cohen A, Aramburu J, Schoen FJ, Neer EJ. Transient cardiac expression of constitutively active Gαq leads to hypertrophy and dilated cardiomyopathy by calcineurin-dependent and independent pathways. Proceedings of the National Academy of Sciences of the USA. 1998;95:13893–13898. doi: 10.1073/pnas.95.23.13893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q-T interval in the conscious mouse. American Journal of Physiology. 1998;274:H747–751. doi: 10.1152/ajpheart.1998.274.3.H747. [DOI] [PubMed] [Google Scholar]
- Mitra R, Morad M. A uniform enzymatic method for dissociation of myocytes from hearts and stomachs of vertebrates. American Journal of Physiology. 1985;249:H1056–1060. doi: 10.1152/ajpheart.1985.249.5.H1056. [DOI] [PubMed] [Google Scholar]
- Momtaz A, Coulombe A, Richer P, Mercadier JJ, Coraboeuf E. Action potential and plateau ionic currents in moderately and severely DOCA-salt hypertrophied rat hearts. Journal of Molecular and Cellular Cardiology. 1996;28:2511–2522. doi: 10.1006/jmcc.1996.0243. [DOI] [PubMed] [Google Scholar]
- Murphy BJ, Rogers J, Perdichizzi AP, Colvin AA, Catterall WA. cAMP-dependent phosphorylation of two sites in the α subunit of the cardiac sodium channel. Journal of Biological Chemistry. 1996;271:28837–28843. doi: 10.1074/jbc.271.46.28837. [DOI] [PubMed] [Google Scholar]
- Nabauer M, Kaab S. Potassium channel downregulation in heart failure. Cardiovascular Research. 1998;37:324–334. doi: 10.1016/s0008-6363(97)00274-5. [DOI] [PubMed] [Google Scholar]
- O'Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R, Marban E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circulation Research. 1999;84:562–570. doi: 10.1161/01.res.84.5.562. [DOI] [PubMed] [Google Scholar]
- Pan X-H, Baker L, Lee JS, Kubota T, Feldman AM, Salama G, London B. Atrial and ventricular arrhythmias in transgenic mice with myocardial expression of TNF-α (abstract) Circulation. 1998;98(suppl. 1):I–745. [Google Scholar]
- Rigg L, Terrar DA. Possible role of calcium release from the sarcoplasmic reticulum in pacemaking in guinea-pig sino-atrial node. Experimental Physiology. 1996;81:877–880. doi: 10.1113/expphysiol.1996.sp003983. [DOI] [PubMed] [Google Scholar]
- Sato Y, Ferguson DG, Sako H, Dorn GW, II, Kadambi VJ, Yatani A, Hoit BD, Walsh RA, Kranias EG. Cardiac-specific overexpression of mouse cardiac calsequestrin is associated with depressed cardiovascular function and hypertrophy in transgenic mice. Journal of Biological Chemistry. 1998;273:28470–28477. doi: 10.1074/jbc.273.43.28470. [DOI] [PubMed] [Google Scholar]
- Scott BT, Simmerman HK, Collins JH, Nadal-Ginard B, Jones LR. Complete amino acid sequence of canine cardiac calsequestrin deduced by cDNA cloning. Journal of Biological Chemistry. 1988;263:8958–8964. [PubMed] [Google Scholar]
- Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, Just H, Holtz J, Drexler H. Gene expression of the cardiac Na+-Ca2+ exchanger in end-stage human heart failure. Circulation Research. 1994;75:443–453. doi: 10.1161/01.res.75.3.443. [DOI] [PubMed] [Google Scholar]
- The Cardiac Arrhythmia Suppression Trial II Investigators. Effect of the antiarrhythmic agent moricizine on survival after myocardial infarction. New England Journal of Medicine. 1992;327:227–233. doi: 10.1056/NEJM199207233270403. [DOI] [PubMed] [Google Scholar]
- Thuringer D, Deroubaix E, Coulombe A, Coraboeuf E, Mercadier JJ. Ionic basis of the action potential prolongation in ventricular myocytes from Syrian hamsters with dilated cardiomyopathy. Cardiovascular Research. 1996;31:747–757. doi: 10.1016/0008-6363(96)00018-1. [DOI] [PubMed] [Google Scholar]
- Tomaselli GF, Beuckelmann DJ, Calkins HG, Berger RD, Kessler PD, Lawrence JH, Kass D, Feldman AM, Marban E. Sudden cardiac death in heart failure. The role of abnormal repolarization. Circulation. 1994;90:2534–2539. doi: 10.1161/01.cir.90.5.2534. [DOI] [PubMed] [Google Scholar]
- Winslow RL, Rice J, Jafri S, Marban E, O'Rourke B. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, II: model studies. Circulation Research. 1999;84:571–586. doi: 10.1161/01.res.84.5.571. [DOI] [PubMed] [Google Scholar]