

Abstract
The present study investigated the regulatory mechanism of the Na+,K+-ATPase and the level of internal Na+ and Ca2+ in response to persistent Na+ influx in acutely dissociated rat thalamic neurones.
Whole-cell patch-clamp recordings and Na+ imaging revealed a stable [Na+]i and low background pump activity. Exposure to veratridine (50 μm) for 1 h resulted in a progressive rise in [Na+]i (ΔFNa= 64 ± 22 %) and [Ca2+]i (ΔFCa= 44 ± 14%) over 3 h. Increases in [Na+]i and [Ca2+]i were also observed during neuronal exposure to the Na+ ionophore monensin (50 μm).
Subcellular confocal immunofluorescence quantification of α3 catalytic Na+-K+ pump subunits showed that a veratridine-induced rise in [Na+]i was accompanied by a significant increase in pump density in both membrane and cytoplasmic compartments, by 39 and 54%, respectively. Similar results were also obtained in experiments when neurones were treated with monensin.
A fluorescent 9-anthroylouabain binding assay detected a 60 and 110% increase in phosphorylated (active) pumps after veratridine and monensin exposure, respectively.
During the entire experiment, application of ouabain or veratridine alone induced little cell swelling and death, but pump inhibition in cells pre-loaded with Na+ led to rapid cell swelling and necrosis.
The above results indicate that a persistent influx of Na+ may trigger rapid enhancement of pump synthesis, membrane redistribution and functional activity. However, these compensatory mechanisms failed to prevent persistent Na+ accumulation.
Together with K+, Na+ is the predominant ion responsible for electrogenesis in mammalian central neurones. The entry of Na+ from the extracellular space into the cell takes place through multiple routes consisting of voltage-sensitive and ligand-gated ion channels as well as various transporters. Despite large fluctuations in neuronal excitatory activity and persistent entry of Na+, brain cells are capable of maintaining a relatively constant physiological level of internal Na+. Central to this Na+-regulating capacity is the Na+,K+-ATPase, which can rapidly activate or deactivate in response to changes in [Na+]i (Nakao & Gadsby, 1989; Inoue & Matsui, 1990).
Under physiological conditions, regulation of Na+-K+ pump activity takes place in two distinct modes: a relatively rapid modulation of pump kinetics and a relatively slow modification of pump protein expression. In the former, Na+ binds to the catalytic α subunit, which triggers a phosphorylation-induced conformational change leading to increased pump turnover rate and ion extrusion (Haber et al. 1987; Nakao & Gadsby, 1989; Lingrel & Kuntzweiler, 1994; Senatorov et al. 1997a). The latter modulation mode is less well understood. Recent studies show that Na+ may stimulate pump mRNA expression and protein subunit synthesis, especially following persistent activation of Na+ channels. For example, the mRNAs of α and β pump subunits in cultured muscle cells increase severalfold within a few hours following application of veratridine, a Na+ channel activator (Yamamoto et al. 1994). In the skeletal muscle system, such pump upregulation can be utilized to reduce the Na+ overload that occurs during muscle differentiation and following intense contraction, thus protecting cells against potential damage (Clausen, 1986).
The expression-based slow pump regulatory mechanism has not been investigated in adult mammalian central neurones. Compared with muscle fibres, brain tissue appears to be particularly vulnerable to a breakdown of Na+ homeostasis, especially that mediated by non-inactivating conductance pathways (Stys et al. 1992; Taylor & Meldrum, 1995; Agrawal & Fehlings, 1996; Urenjak & Obrenovitch, 1996). It is possible that Na+-pumping mechanisms under these conditions may not be sufficient to deal with such a mode of Na+ entry.
The purpose of the present study was to investigate the cellular responses to persistent Na+ accumulation and the efficacy of upregulation of the Na+ pump as a mechanism against Na+-induced cell injury in mature central neurones. To this end, we utilized isolated neurones from adult rat thalamus in which persistent Na+ channels and pump activities have recently been characterized (Senatorov et al. 1997a; Parri & Crunelli, 1998). Our results showed that the Na+ channel activator veratridine and the Na+ ionophore monensin elicited a sustained but moderate rise in [Na+]i in thalamic neurones, which significantly enhanced pump synthesis, membrane redistribution and phosphorylation activity. Reversal of Na+ overload was not observed, suggesting that in mammalian central neurones pathological Na+ influx may rapidly surpass Na+ extrusion capacity despite these compensatory efforts. Preliminary results of this work have been published in abstract form (Senatorov et al. 1997b).
METHODS
Preparation of dissociated cells
Adult male Long-Evans rats (100-150 g) were decapitated and the brain removed according to a protocol approved by the Hospital Ethics Committee. Tissue containing the bilateral diencephalon was dissected and placed in ice-cold oxygenated (100 % O2) modified Krebs solution (mM): 118 NaCl, 4.7 KCl, 1.2 KH2PO4, 2 CaCl2, 1.2 MgSO4, 20 Na-Hepes and 25 glucose, pH adjusted to 7.4 with HCl. Osmolality was 300 mosmol kg−1. Under a stereomicroscope, the tissues of both medial geniculate bodies were removed, cut into 8–12 small pieces and taken for enzymatic digestion according to procedures described earlier (Senatorov & Hu, 1997). Digestion was performed at room temperature for 40 min with a mixture of pronase (protease type XIV, Sigma-Aldrich Canada Ltd, Oakville, Ontario, Canada) and thermolysin (protease type X, Sigma-Aldrich Canada Ltd) – 0.5 mg ml−1 each. Neurones were mechanically dissociated by gentle pipetting, then allowed to adhere to the bottom of a tissue culture dish (Easy Grip, Becton Dickinson and Company, NJ, USA) in a plastic box with constant circulation of 100 % O2, in the presence of fluorescence probes for 40 min at room temperature. During the experiment, the dish was constantly perfused with warm (30°C) oxygenated (100 % O2) Krebs solution of the above composition (with or without addition of drugs) at a flow of 2 ml min−1. For immunocytochemistry, dissociated neurones were fixed 2 h after the onset of veratridine or monensin exposure with periodate lysine paraformaldehyde (McLean & Nakane, 1974) for 30 min at room temperature, and washed two times (5 min each) in 0.1 M phosphate buffer (pH 7.4) and three times (5 min each) in Tris-buffered saline (TBS, pH 7.2) with 0.5 % Triton X-100.
The average life span of acutely dissociated mammalian central neurones is usually limited to 12–16 h. In control experiments we found that almost all neurones that survived the dissociation showed no detectable pathological changes during the first 4 h. Therefore this time frame was used for all the experiments.
Electrophysiology
Patch electrodes were made from borosilicate glass capillary tubing (1.5 mm o.d., Sutter Instrument Co., Novato, CA, USA) on a Flaming-Brown micropipette puller (model P-87, Sutter Instrument Co.). Electrodes were filled with (mM): 130 potassium gluconate, 10 NaCl, 10 KCl, 10 Na-Hepes, 1 EGTA and 4 Mg-ATP. The solution was buffered to pH 7.4 with 1 M KOH. The DC resistance of the patch electrode was 4–8 MΩ. Signals were recorded using Axopatch 200A amplifiers (Axon Instruments Inc.) and were digitized and stored on videotape with a Vetter PCM Recorder (A. R. Vetter Co. Inc., Reversburg, PA, USA), or digitized with a Digidata 1200 interface (Axon Instruments Inc.) for on-line acquisition using pCLAMP software (Axon Instruments Inc.). Seal resistance was 2–5 GΩ. In all experiments, pipette capacitance was compensated before breaking the cell membrane. Liquid junction potential was measured and subtracted off-line.
Chemicals
The Na+ channel activator veratridine and the Na+ ionophore monensin were used to increase membrane permeability to Na+. The Na+,K+-ATPase inhibitor ouabain was employed to block pump activity. Chemicals (Sigma-Aldrich Canada Ltd) were prepared as stock solutions and added to the standard solution contained in an oxygenated side-bath to produce the final concentrations indicated in the text. Ouabain was first dissolved in DMSO, while veratridine and monensin were dissolved in ethanol. The final concentration of DMSO or ethanol was < 0.1 %, a level generally innocuous to cells. Perfusion medium containing the drugs at their final concentration was delivered to the dish with a perfusion line or, for fast application, directly to the cells with a temperature-regulated cell perfusion system (PTR-200, ALA Scientific Instruments Inc., Wesbury, NY, USA).
Imaging
Healthy neurones in a Petri dish were identified based on previously published morphological criteria (Friedman & Haddad, 1993; Oliet & Bourque, 1993) and their viability was further assessed with a dual-fluorescence cell viability assay consisting of calcein AM (2 μM, green fluorescence) and ethidium homodimer (EthD-1, 4 μM, red fluorescence). Both probes were from Molecular Probes, Inc. Live cells were identified by the presence of ubiquitous intracellular esterase activity, which cleaves cell-permeant calcein AM, giving rise to the intensely green (about 530 nm) fluorescent calcein. Dead neurones were recognized using EthD-1, which enters cells with damaged membranes and undergoes a 40-fold enhancement of fluorescence on binding nucleic acids, thereby producing a bright red (> 600 nm) fluorescence in dead cells. Calcein and EthD-1 were both excited at 485 nm. Complete neuronal disintegration (visible under transmitted light) and/or the conversion from green fluorescence (Sodium Green- or calcein-mediated) to very bright red (EthD-1-mediated) fluorescence in the cell nucleus was taken as indicative of irreversible neuronal cell death. In some ion-imaging experiments all healthy neurones within the visual field of the microscope were monitored. Alternatively, all healthy neurones that passed through the visual field of the microscope during a single scan along the full diameter of the 35 mm Petri dish were surveyed.
To monitor the changes in [Na+]i we used the acetoxymethyl ester (AM) derivative of the fluorescent probe Sodium Green. The cell-permeant AM ester of Calcium Orange was used to monitor the changes in [Ca2+]i. The probes were first dissolved in DMSO, then diluted into the perfusion medium to final concentrations of 8 and 5 μM for Sodium Green and Calcium Orange, respectively. Neurones were incubated with the probes for 1 h at room temperature with a constant flow of oxygen. During and after loading, neurones were kept in the dark to minimize photobleaching. Both probes were from Molecular Probes Inc.
Images of neurones were collected every 30 or 60 min using an inverted epifluorescence microscope (Olympus IX-70) and a dual-colour CCD camera (Sony) linked to a PC computer running image integration software (Empix Imaging, Toronto, Ontario, Canada).
Confocal immunofluorescence microscopy
Fixed cells were incubated for 2 h at room temperature with a monoclonal mouse antibody (3 μg ml−1) against the α3 Na+,K+-ATPase subunit isoform (Affinity Bioreagents, Inc., Golden, CO, USA) followed by three washes of 10 min each. The sensitivity and specificity of these monoclonal antibodies for rat antigens has previously been validated (Senatorov & Hu, 1997). Fluorescence signals were then visualized by 1 h incubation at room temperature with secondary goat anti-mouse antibody (1:250) conjugated to the Cy3 fluorophore (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA). Antibodies were diluted in TBS (pH 7.2) with 0.5 % Triton X-100 and 4 % normal goat serum. The same solution was used for washing. Usually one of two neuronal populations was randomly chosen for 1 h incubation with veratridine or monensin and the other was treated with drug-free standard solution. For negative controls, primary antibody was omitted or replaced by normal mouse serum. Confocal images of acutely dissociated cells labelled with Cy3 were acquired on a Bio-Rad MRC 1024 confocal microscope. Five to six images, 1 μm apart (in the z-axis) were collected from each cell and the largest cross-section was chosen for further analysis. Confocal scanning of the treated and untreated samples was conducted by a technician who had no prior knowledge of the samples. In one experiment, both treated and control neurones were stained with DiI (0.05 %; Molecular Probes) for 40 min at room temperature to study the possible interaction between cell shape and probe fluorescence.
Fluorescent 9-anthroylouabain assay
Na+,K+-ATPase activity was assessed using a fluorescent (excitation wavelength, 365–368 nm; emission wavelength, 480 nm) derivative of ouabain. 9-Anthroylouabain is known to increase in fluorescence upon binding to phosphorylated (active) Na+,K+-ATPase (Fortes, 1977; McCormick, 1990; Li et al. 1995). In these experiments one of two neuronal populations was randomly chosen for 1 h incubation with veratridine or monensin and the other was treated with drug-free standard solution. Both groups were then incubated with 9-anthroylouabain (1 h, 20 μM; Molecular Probes). In control experiments we showed that pre-incubation with 100 μM ouabain blocked 9-anthroylouabain binding. A stock solution (20 mM) was prepared by dissolving 9-anthroylouabain in DMSO. For image collection under low magnification (× 10 objective, 1.5 × magnification) we used a 365 nm excitation filter, a 395 nm chromatic beam splitter and a 420 nm long-pass filter.
Image analysis
To calculate cell volume, the cross-sectional area was first obtained by tracing images of neuronal soma recorded under bright-field optics (Friedman & Haddad, 1993; Oliet & Bourque, 1993; Churchwell et al. 1996). The final value of the cross-sectional area was obtained after averaging results of two tracings. Cell volume changes (ΔV) in each neurone were calculated as a percentage of its baseline volume recorded during control imaging at the beginning of each experiment using the following equation:
where A0 and Ai are cross-sectional areas recorded before and at any given time point after drug application, respectively. This approach assumes that the soma swells and shrinks in a symmetrical manner as if it were a sphere, and has been successfully used to measure volume in dissociated CA1 hippocampal (Friedman & Haddad, 1993) and supraoptic neurones (Oliet & Bourque, 1993). Churchwell and co-workers (1996) compared the volume calculation using cross-sectional area in a single focal plane with that using optical-sectioning techniques and found no significant difference when measuring veratridine-induced swelling in rat cortical cell cultures. In pilot experiments, we also validated this assumption by comparing cell volume calculated based on cross-sectional area with that calculated by the summation of confocal optical sections. Fixed neurones of different sizes immunostained with Cy3 (n = 9) or live neurones stained with DiI (0.05 %) and treated with veratridine plus ouabain (n = 6) were used for this purpose. The difference in measurements was statistically insignificant.
Fluorescence image analysis was performed after background fluorescence was subtracted from all measurements. Mean (per pixel) and integrated (per area) fluorescence intensity was calculated. For [Na+]i and [Ca2+]i measurements ΔFi (%) was determined according to the following relationship:
where ΔFi is the normalized percentage change in probe fluorescence corrected for cell volume, and Fi and Vi are the mean fluorescence intensity and cell volume, respectively, for any given image i. Normalization was performed with respect to the baseline fluorescence, F0, measured during control imaging at the beginning of each experiment. Correction for cell volume was performed based on two assumptions. The first assumption was that if the intracellular content of the probe is constant, changes in its concentration reflect changes in cell water volume. The second assumption was that if the concentration of the fluorophore is not excessive, the emitted fluorescence is directly proportional to the concentration of the fluorophore. These assumptions were previously validated and utilized in electrophysiological measurements of cell volume changes using tetramethylammonium as an impermeant probe (Alvarez-Leefmans et al. 1992), and in optical measurements of cell volume changes using fluorescent dyes such as BCECF, fura-2 and calcein (Tauc et al. 1990; Muallem et al. 1992; Crowe et al. 1995). Working concentrations of 8 μM for Sodium Green and 5 μM for Calcium Orange were chosen empirically following a series of pilot experiments which showed that these were the minimal concentrations giving stable fluorescence. All measurements were performed using NIH Image (version 1.62; http://rsb.info.nih.gov/nih-image). Parametric statistical tests were used to examine differences among cells, including Student's t test and ANOVA. Values are presented as means ±s.e.m.
RESULTS
Pump activity under control conditions
The great majority of freshly dissociated neurones chosen for surveillance in our experiments remained viable and maintained their basic morphological features for at least 4 h (Fig. 1B and C). Furthermore, they had a relatively stable level of internal Na+ (ΔFNa not significant, Fig. 1A and C) and an apparent high input resistance (between 0.5 and 1.0 GΩ). They also lacked a Na+-permeable hyperpolarization-activated inwardly rectifying conductance (Ih) (n = 11, Fig. 2A–C). A 1 h exposure to ouabain (250 μM) failed to produce statistically significant changes in internal Na+ levels during 3 h of surveillance (Fig. 2D). We also found that ouabain (100 μM, 20 s) induced a significantly smaller inward current (23 ± 4 pA, n = 3, data not shown) recorded in voltage-clamp experiments in dissociated cells as compared with that in a thalamic explant (Senatorov et al. 1997a). This low and stable background Na+ permeability and resting Na+,K+-ATPase activity provided a ‘low noise’ baseline suitable for studying pump upregulation induced by Na+ influx.
Figure 1. Under control conditions, acutely dissociated thalamic neurones stayed viable, retained their basic morphology and maintained a stable [Na+]i.

A, results of sodium imaging from a group of neurones in control experiments. B, cell volume changes under control conditions. Cont, control values at the beginning of the incubation. C, images of a representative neurone collected in a control experiment. Top row, phase-contrast images. Note that the cell retained its shape and volume during the experiment; minor rounding and swelling were noticeable only at the end of the experiment. Bottom row, fluorescence images of the same neurone. The cell was incubated with Sodium Green. The absence of an increase in green fluorescence provides evidence of a relatively stable [Na+]i.
Figure 2. Electrophysiological properties of isolated thalamic neurones and their responses to drug applications.

A, phase-contrast image of a representative neurone during patch-clamp recording. B, voltage traces induced by current pulses injected into the neurone in current-clamp mode. Note the absence of inwardly rectifying current and the high amplitude of the action potentials with zero overshoot. C, current traces recorded in voltage-clamp mode in an isolated neurone in response to hyperpolarizing and depolarizing voltage pulses. Note the absence of inwardly rectifying current and the high amplitude of the fast depolarization-activated transient inward current. D, results of sodium imaging from a group of neurones before (Cont), during and after a 1 h exposure to 250 μM ouabain (indicated by the filled bar). The absence of an increase in sodium fluorescence indicates a relatively stable [Na+]i. E, the inward current shift recorded in an isolated thalamic neurone in voltage-clamp mode after a 1 min application of veratridine, providing evidence of functioning Na+ channels.
Changes in [Na+]i and [Ca2+]i following veratridine and monensin application
To examine whether veratridine could increase membrane permeability to Na+ in such a low background conductance state, we recorded membrane current under voltage clamp after a 1 min application of 50 μM veratridine. At this concentration veratridine consistently induced a slowly increasing inward current with a mean peak amplitude of 41 ± 13 pA (n = 4, Fig. 2E).
In the next series of experiments we examined the changes in Na+ concentration after a 1 h application of veratridine. It was found that over a 3 h time period veratridine induced a progressive increase in [Na+]i with maximal ΔFNa= 64 ± 22 % (Fig. 3A and C). Similar changes in [Na+]i were observed in neurones during a 3 h exposure to 50 μM monensin, a Na+ ionophore, with maximal ΔFNa= 46 ± 16 % (P < 0.05, n = 5, data not shown). During and after veratridine application we also measured changes in [Ca2+]i. A moderate increase in [Ca2+]i was recorded during the course of the experiment, with maximal ΔFCa= 44 ± 14 %, observed 3 h after the onset of drug application (Fig. 3B and C). Incubation with monensin produced only a small rise in [Ca2+]i, with maximal ΔFCa= 17 ± 6 % (P < 0.05, n = 6, data not shown).
Figure 3. Cellular response to veratridine-induced Na+ influx.
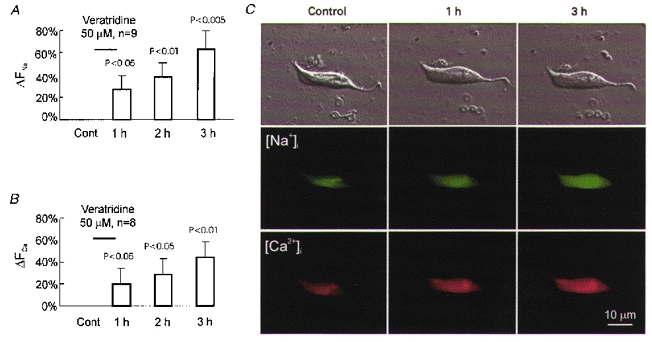
A, results of Na+ imaging obtained from a group of neurones before (Cont), and after a 1 h exposure to 50 μM veratridine. B, results of Ca2+ imaging before (Cont), and after a 1 h exposure to 50 μM veratridine. C, simultaneous increase in [Na+]i and [Ca2+]i in representative neurones after veratridine exposure. Cells were preincubated with Sodium Green and Calcium Orange. Top row, phase-contrast images. Middle row, Na+ fluorescence images of the same neurone. Bottom row, Ca2+ fluorescence images of the same neurone. Note the increase in green and red fluorescence indicating a simultaneous rise in [Na+]i and [Ca2+]i.
Effects of Na+-K+ pump inhibition
The capacity of the Na+,K+-ATPase to restore the Na+ gradient was assessed by adding 250 μM ouabain with 50 μM veratridine and comparing the internal Na+ level before and after pump inhibition at each time point. The data showed that addition of ouabain resulted in an even higher elevation of [Na+]i than with veratridine alone (ΔFNa= 85 ± 29 %, Fig. 4A). Although the additional increase in the Na+ fluorescence level (about 20 %) with ouabain was statistically insignificant, the increase in the total internal Na+ content of the cell could be much higher considering the 59 ± 24 % increase in cell volume with combined veratridine and ouabain exposure (Fig. 4B). In contrast, veratridine application alone produced insignificant volume changes (Fig. 4B).
Figure 4. Cellular Na+ and volume responses to combined application of veratridine and ouabain.
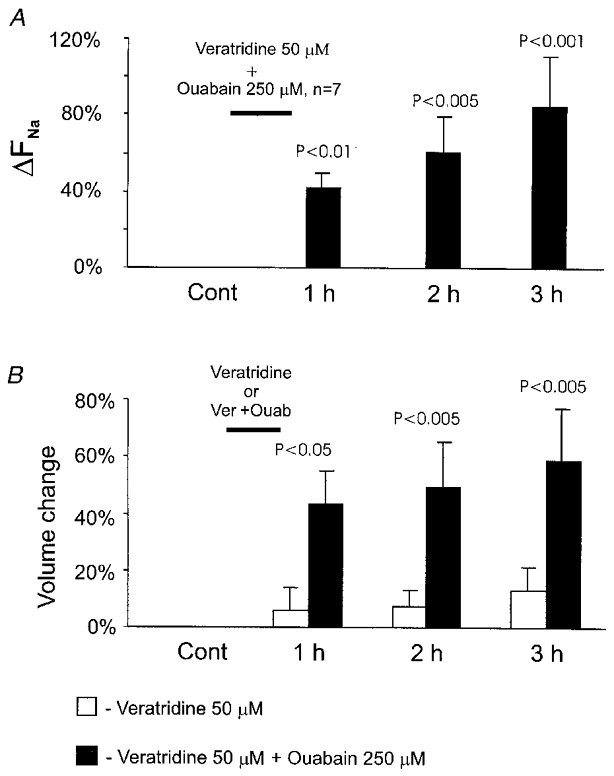
A, results of Na+ imaging obtained from a group of neurones before (Cont) and after a 1 h exposure to 50 μM veratridine together with 250 μM ouabain. B, results of volume measurements obtained from the same groups of neurones as in A (veratridine plus ouabain) and Fig. 3A (veratradine alone).
The physiological role of the Na+,K+-ATPase in restoring the Na+ gradient was assessed by comparing the cytotoxic effect of veratridine alone versus simultaneous application of veratridine and ouabain. A 1 h application of 50 μM veratridine alone resulted in the death of only one out of 22 neurones (< 5 %) after 3 h and did not produce significant neuronal swelling during the first 3 h. In contrast, even a 1 h application of 50 μM veratridine together with 250 μM ouabain produced rapid morphological deterioration with cell rounding and swelling. Combined application of veratridine and ouabain resulted in the death of five out of 18 observed neurones (27 %) after 3 h.
Veratridine-induced upregulation of Na+,K+-ATPase α3 subunit expression
To examine whether upregulation of pump expression indeed took place, we measured the density of α3 pump subunits immunostained in veratridine-treated neurones. Fluorescence intensity measurements from optical sections of the neurone showed that veratridine-treated (50 μM) cells had 48 % higher fluorescence expressed as mean pixel intensity (in arbitrary units) compared with controls (85 ± 9 versus 57 ± 3, Fig. 5A, C and D). In the cytoplasmic domain, the mean pixel intensity was 54 % higher than the control (78 ± 8 versus 51 ± 4, Fig. 5A), whereas fluorescence from the membrane domain was 39 % higher in veratridine-treated neurones (98 ± 8 versus 70 ± 4, Fig. 5A). Incubation with monensin (50 μM) produced similar increases in pump density: 28 % in membrane and 93 % in cytoplasm (Fig. 5B). These data clearly showed that a significantly higher number of pump molecules were being produced and recruited to the cell membrane after a persistent increase in [Na+]i. Comparison of fluorescence measurements between cytoplasmic and membrane domains demonstrated that a large proportion of the increase in fluorescence originated from the former. Furthermore, we showed that the increase in membrane immunofluorescence of veratridine-treated neurones was not associated with alterations in somatic shape. In some experiments, we stained veratridine-treated (n = 19) and non-treated (n = 24) neurones with DiI. Confocal immunofluorescence scanning and image analysis did not reveal significant differences in fluorescence intensity between the two groups.
Figure 5. Veratridine- and monensin-induced overexpression of the Na+,K+-ATPase α3 subunit.

A, quantification of the subcellular distribution of the Na+,K+-ATPase α3 subunit after a 1 h exposure to 50 μM veratridine using confocal immunofluorescence microscopy. B, subcellular distribution of the Na+,K+-ATPase α3 subunit after a 1 h exposure to 50 μM monensin as determined with confocal immunofluorescence scanning. C, fluorescence images of two representative neurones – control (left) and exposed to veratridine (right). D, 3D intensity profiles of the same neurones as in C. Note the higher content of the Na+,K+-ATPase α3 subunit in the veratridine-treated neurone.
Veratridine-induced changes in membrane density of phosphorylated Na+-K+ pump molecules
We employed a fluorescent 9-anthroylouabain assay to assess the relative changes in the amount of phosphorylated (active) pump molecules. 9-Anthroylouabain, a fluorescent derivative of ouabain, has been shown to selectively bind to phosphorylated (active) pump molecules in live cells (Fortes, 1977; McCormick, 1990; Li et al. 1995). As shown in Fig. 6, in the control group, in which neurones were incubated with 9-anthroylouabain without previous exposure to veratridine or monensin, the mean pixel fluorescence of the anthroylouabain signal was 6 ± 1. This baseline level of fluorescence was very stable and showed practically no variations during surveillance. Two hours after the onset of a 1 h veratridine (50 μM) application, 9-anthroylouabain-mediated fluorescence increased by 60 % to a mean pixel intensity of 10 ± 1 (Fig. 6A, C and D). A more than 2-fold (110 %) increase in 9-anthroylouabain fluorescence (from 6 ± 1 to 13 ± 2) was found when monensin (50 μM) was used instead of veratridine (Fig. 6B).
Figure 6. Veratridine- and monensin-induced changes in the amount of phosphorylated (active) pump molecules measured with 9-anthroylouabain.
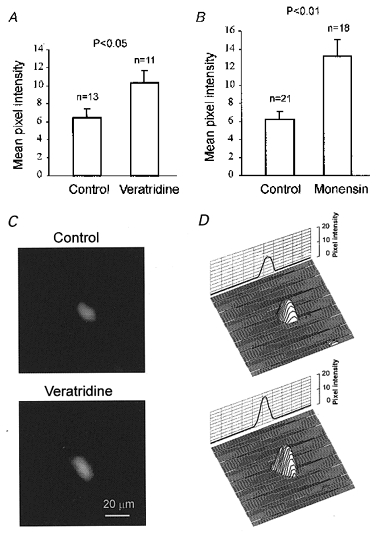
Exposure to veratridine (50 μM, A) or monensin (50 μM, B) results in a higher membrane fluorescence of phosphorylated (active) Na+,K+-ATPase. C, fluorescence images of two representative neurones labelled with 9-anthroylouabain. D, 3D fluorescence intensity profiles of the same neurones as in C.
DISCUSSION
Methodological considerations
Isolated neurones from different brain regions have been shown to retain their basic morphological and physiological characteristics in vitro (Friedman & Haddad, 1993, 1994; Oliet & Bourque, 1993). In the present study, the dissociated cell preparation allowed electrophysiological recording techniques to be used in conjunction with fluorescence microscopy for measuring pump activity along with the changes in ionic gradients and cell viability. Dissociated cells are also devoid of synaptic contacts and glia which permitted the study of veratridine-induced Na+ accumulation in isolation from excitotoxicity. In addition, single dissociated neurones provided better resolution for fluorescence quantification and morphometric measurements, which were essential for the present study. There is a distinct possibility that cells maintained at 30°C will have a lower level of ATP production than at physiological temperature, which could influence the activity of pumps. However, it was observed that the anthroylouabain binding following monensin treatment nearly doubled suggesting that a lack of ATP supply was not a factor limiting pump phosphorylation activity.
We used the cell-permeant Sodium Green tetraacetate as an indicator of internal Na+ activity (Friedman & Haddad, 1994; Amorino & Fox, 1995; Housley et al. 1998). With the ionophore gramicidin, Sodium Green gives calibration curves similar to the ratiometric Na+ probe SBFI (sodium-binding benzofuran isophthalate). The dissociation constant (Kd) of Sodium Green is about 21 mM at 22°C in solutions with a combined Na+ and K+ concentration of 135 mM, approximating physiological ionic strength. This Kd value is close to that of SBFI, which is 11.3 mM under the same conditions. In general, intracellular ion indicators have a detectable response in the concentration range from approximately 0.1 ×Kd to 10 ×Kd, with a maximal optical response near Kd (Haugland, 1993). The Kd of Sodium Green is compatible with the [Na+] range in mammalian central neurones, which is reportedly from about 10 to about 30 mM depending upon the cell type and the method of measurement (Erecinska et al. 1991; Rose & Ransom, 1997; Silver et al. 1997). Although Sodium Green is not ratiometric, it has a number of advantages including greater selectivity for Na+ than K+ (41-fold versus 18-fold for SBFI), higher quantum yield, and low energy requirement for excitation. Calcium Orange is a fluorescent indicator previously used to monitor [Ca2+]i changes in brain slices (Duffy & MacVicar, 1996). The spectral characteristics of Calcium Orange allow dual-colour fluorescence analysis to be performed together with Sodium Green for simultaneous measurement of [Ca2+]i and [Na+]i.
Regulation of Na+,K+-ATPase activity
The purified pump contains α and β chains in a 1:1 ratio (Horisberger et al. 1991; Lingrel & Kuntzweiler, 1994). Since both the ATP-binding site and the phosphorylation site are located on the α chain, the α subunit is thought to be responsible for catalytic activity (Horisberger et al. 1991; Lingrel & Kuntzweiler, 1994). We have previously shown that the auditory thalamus neurones have dominant expression of the α3 isoform which may be specific to central neurones, but only very weak expression of the α1 isoform and almost no expression of the α2 isoform (Senatorov & Hu, 1997). Therefore expression of the α3 isoform was specifically chosen for analysis in the current paper. Under normal physiological conditions, when Na+ is present at non-saturating concentrations, [Na+]i predominantly regulates Na+,K+-ATPase activity (Haber et al. 1987; Nakao & Gadsby, 1989; Inoue & Matsui, 1990). However, alternative regulatory mechanisms may be required during a persistent increase of [Na+]i. One of these may be post-translational regulation via accelerated membrane insertion of pre-existing pump molecules and/or their assembly from α and β subunits (Blot-Chabaud et al. 1990; Fambrough et al. 1991; Coutry et al. 1992). In cortical collecting tubules of rabbit kidney a moderate rise in [Na+]i induces a 3-fold increase in the number of pumps within 1–2 min, suggesting the presence of a pre-existing pool of pump molecules for rapid recruitment (Blot-Chabaud et al. 1990). Such pump recruitment is Ca2+ independent (Coutry et al. 1992).
While our data do not exclude the existence of similar mechanisms of pump upregulation in central neurones, our findings cannot be fully explained by simple recruitment of α subunits from a pre-existing cytoplasmic pool. Subcellular quantification of fluorescence signals in both veratridine- and monensin-treated neurones demonstrated that not only does the cytoplasmic content of the catalytic ATPase subunit increase, but also this increase is greater in the cytoplasmic than in the membrane domain. This was especially evident in experiments with monensin, where the increase in the cytoplasmic enzyme content was 3-fold higher than that in the membrane, suggesting significant upregulation of pump synthesis. This observation is consistent with previous studies in non-neuronal tissues where persistent Na+ channel activation leads to an increase in the levels of both pump mRNAs and α/β subunits (Fambrough et al. 1991; Yamamoto et al. 1994).
Mechanisms of Na+,K+-ATPase upregulation
Regulation of pump synthesis and membrane redistribution may involve multiple cellular signalling mechanisms. Furthermore, both veratridine and monensin were shown to stimulate breakdown (hydrolysis) of phosphoinositides by phospholipase C to yield inositol phosphates and diacylglycerides (Gusovski & Daly, 1988). Alternatively, Na+ channel activation may also trigger Ca2+ influx through Ca2+-permeable channels and/or through reduced operation or reversal of the Na+-Ca2+ exchanger (Choi, 1992; Stys et al. 1992; Friedman & Haddad, 1993; Agrawal & Fehlings, 1996; Churchwell et al. 1996). Elevation of [Ca2+] may cause an increase in the rate of transcription of α1 subunit mRNA and slow the rate of degradation of β1 subunit mRNA (Rayson, 1991). Finally, Na+ can directly modify gene expression (Yamamoto et al. 1993, 1994; Murata et al. 1996; Ruiz-Opazo et al. 1997). Yamamoto et al. (1993) reported that in Ca2+-free medium, ouabain induced a significant increase in [Na+]i and an accumulation of α1 Na+,K+-ATPase subunit mRNA in cultured neonatal rat cardiocytes. Similarly, veratridine induced α1 and β1 subunit protein accumulation in the absence of extracellular Ca2+ in vascular smooth muscle cells (Yamamoto et al. 1994). In this last study, transfection with chimeric plasmids containing 5′ flanking sequences of the α1 pump isoform gene and a luciferase reporter gene revealed a 3-fold increase in luciferase activity, suggesting the presence of Na+-responsive elements (Yamamoto et al. 1994). A transcriptional Na+-response mechanism has been recently characterized in rat A10 embryonic aortic smooth muscle cells (Ruiz-Opazo et al. 1997). Data from the analysis of α1 (position −1288) and α2 (position −798) Na+,K+-ATPase promoter regions identified a positive Na+-response regulatory region within −358 bp from the transcription start site in both 5′ flanking regions. The authors detected monensin-induced upregulation of a 95 kDa nuclear DNA-binding protein which may represent a putative Na+-response transcription factor. A sustained rise in [Na+]i was also implicated in the induction of DNA synthesis and regulation of cell proliferation in CNS neurones and oligodendrocyte progenitor cells (Cone & Cone, 1976; Cone, 1980; Knutson et al. 1997). It is noteworthy that the effect of veratridine on cell proliferation was independent of extracellular calcium ions (Knutson et al. 1997). In the present study, we observed a moderate rise in both [Na+]i and [Ca2+]i in veratridine-treated neurones. In monensin-treated neurones, the difference in the magnitude of [Ca2+]i and [Na+]i increases was greater (17versus 66 %). A similar small rise in [Ca2+]iversus a considerable increase in [Na+]i was observed in cultured cortical neurones treated with monensin (Erecinska et al. 1991). Unfortunately, because of the non-ratiometric technique used, we cannot reliably compare the changes in [Na+]i and [Ca2+]i. However, it is noteworthy that while a rise in [Ca2+]i is triggered by Na+ overload, veratridine-induced [Na+]i elevation is a Ca2+-independent process, which can be observed in Ca2+-free medium (V. V. Senatorov, P. K. Stys & B. Hu, manuscript in preparation).
Persistent Na+ accumulation and neuronal injury
Persistent Na+ accumulation and in particular that mediated by non-inactivating Na+ channels plays an important role in brain injury (Stys et al. 1992; Fried et al. 1995; Taylor & Meldrum, 1995; Urenjak & Obrenovich, 1996; Barzo et al. 1997). For example, a recent magnetic resonance imaging study has shown that Na+-driven cellular oedema is a major contributor to brain swelling after traumatic brain injury (Barzo et al. 1997). A similar mechanism was also observed for ischaemic injury (for review see Odland & Sutton, 1999). It was suggested that Na+-induced intracellular hyperosmolarity occurring minutes after ischaemia triggers an extracellular-to-intracellular fluid shift which further results in vasogenic oedema acting together with inflammatory agents. The main factor implicated in the intracellular hyperosmolarity is the loss of the Na+,K+-ATPase (Odland & Sutton, 1999). In our study, we found that the Na+,K+-ATPase plays a central role in maintaining the viability of neurones subject to an increased membrane permeability to Na+. Such a role was demonstrated by a 5-fold (from 5 to 27 %) increase in the rate of neuronal death as a result of pump inhibition. This process may be essential in the light of the finding that under the same conditions, removing external calcium and the consequent drop in [Ca2+]i neither prevents the rise in [Na+]i nor results in significant cell death reduction (V. V. Senatorov, P. K. Stys & B. Hu, manuscript in preparation). Furthermore, despite the fact that Na+ influx induced rapid upregulation of the Na+,K+-ATPase, the trend for a rise in [Na+]i continued (up to 66 % from the baseline), apparently surpassing the Na+ extrusion capacity of the cell. This observation may partially explain why under normoxic conditions with pumps fully functioning, sustained Na+ influx, such as that induced by intense epileptic activity or overactivation of glutamate receptors, can cause neuronal injury (Choi, 1988).
Acknowledgments
This work was supported by the Medical Research Council and the Heart and Stroke Foundation of Canada. We are indebted to Dr Kathleen J. Sweadner for her advice on using monoclonal antibodies.
References
- Agrawal SK, Fehlings MG. Mechanisms of secondary injury to spinal cord axons in vitro: role of Na+, Na+-K+-ATPase, the Na+-H+ exchanger, and the Na+-Ca2+ exchanger. Journal of Neuroscience. 1996;16:545–552. doi: 10.1523/JNEUROSCI.16-02-00545.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Leefmans FJ, Gamiño SM, Reuss L. Cell volume changes upon sodium pump inhibition in Helix aspersa neurones. The Journal of Physiology. 1992;458:603–619. doi: 10.1113/jphysiol.1992.sp019436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amorino GP, Fox MH. Intracellular Na+ measurements using sodium green tetraacetate with flow cytometry. Cytometry. 1995;21:248–256. doi: 10.1002/cyto.990210305. [DOI] [PubMed] [Google Scholar]
- Barzo OP, Marmarou A, Fatouros P, Hayasaki K, Corwin F. Contribution of vasogenic and cellular edema to traumatic brain swelling measured by diffusion-weighted imaging. Journal of Neurosurgery. 1997;87:900–907. doi: 10.3171/jns.1997.87.6.0900. [DOI] [PubMed] [Google Scholar]
- Blot-Chabaud M, Wanstok F, Bonvalet JP, Farman N. Cell sodium-induced recruitment of Na+-K+-ATPase pumps in rabbit cortical collecting tubules is aldosterone-dependent. Journal of Biological Chemistry. 1990;265:11676–11681. [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Choi DW. Excitotoxic cell death. Journal of Neurobiology. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- Churchwell KB, Wright SH, Emma F, Rosenberg PA, Strange K. NMDA receptor activation inhibits neuronal volume regulation after swelling induced by veratridine-stimulated Na+ influx in rat cortical cultures. Journal of Neuroscience. 1996;16:7447–7457. doi: 10.1523/JNEUROSCI.16-23-07447.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen T. Regulation of active Na+-K+ transport in skeletal muscle. Physiological Reviews. 1986;66:542–580. doi: 10.1152/physrev.1986.66.3.542. [DOI] [PubMed] [Google Scholar]
- Cone CD., Jr Ionically mediated induction of mitogenesis in CNS neurons. Annals of the New York Academy of Sciences. 1980;339:115–131. doi: 10.1111/j.1749-6632.1980.tb15973.x. [DOI] [PubMed] [Google Scholar]
- Cone CD, Jr, Cone CM. Induction of mitosis in mature neurons in central nervous system by sustained depolarization. Science. 1976;192:155–158. doi: 10.1126/science.56781. [DOI] [PubMed] [Google Scholar]
- Coutry N, Blot-Chabaud M, Mateo P, Bonvalet JP, Farman N. Time course of sodium-induced Na+-K+-ATPase recruitment in rabbit cortical collecting tubule. American Journal of Physiology. 1992;263:C61–68. doi: 10.1152/ajpcell.1992.263.1.C61. [DOI] [PubMed] [Google Scholar]
- Crowe WE, Altamirano J, Huerto L, Alvarez-Leefmans FJ. Volume changes in single N1E-115 neuroblastoma cells measured with a fluorescent probe. Neuroscience. 1995;69:283–296. doi: 10.1016/0306-4522(95)00219-9. [DOI] [PubMed] [Google Scholar]
- Duffy S, MacVicar BA. In vitro ischemia promotes calcium influx and intracellular calcium release in hippocampal astrocytes. Journal of Neuroscience. 1996;16:71–81. doi: 10.1523/JNEUROSCI.16-01-00071.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erecinska M, Dagani F, Nelson D, Deas J, Silver IA. Relations between intracellular ions and energy metabolism: a study with monensin in synaptosomes, neurones, and C6 glioma cells. Journal of Neuroscience. 1991;11:2410–2421. doi: 10.1523/JNEUROSCI.11-08-02410.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fambrough DM, Wolitzky BA, Taormino JP, Tamkun MM, Takeyasu K, Somerville D, Renaud KJ, Lemas MV, Lebovitz RM, Kone BC. A cell biologist's perspective on sites of Na,K-ATPase regulation. Society of General Physiologists Series. 1991;46:17–30. [PubMed] [Google Scholar]
- Fortes PA. Anthroylouabain: a specific fluorescent probe for the cardiac glycoside receptor of the Na-K ATPase. Biochemistry. 1977;16:531–540. doi: 10.1021/bi00622a030. [DOI] [PubMed] [Google Scholar]
- Fried E, Amorim P, Chambers G, Cottrell JE, Kass IS. The importance of sodium for anoxic transmission damage in rat hippocampal slices: mechanisms of protection by lidocaine. The Journal of Physiology. 1995;489:557–565. doi: 10.1113/jphysiol.1995.sp021072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JE, Haddad GG. Major differences in response to anoxia between neonatal and adult rat CA1 neurones: role of and Journal of Neuroscience. 1993;13:63–72. doi: 10.1523/JNEUROSCI.13-01-00063.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JE, Haddad GG. Anoxia induces an increase in intracellular sodium in rat central neurones in vitro. Brain Research. 1994;663:329–334. doi: 10.1016/0006-8993(94)91281-5. [DOI] [PubMed] [Google Scholar]
- Gusovsky F, Daly JW. Formation of second messengers in response to activation of ion channels in excitable cells. Cellular and Molecular Neurobiology. 1988;8:157–169. doi: 10.1007/BF00711242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber RS, Pressley TA, Loeb JN, Ismail-Beigi F. Ionic dependence of active Na-K transport: ‘clamping’ of cellular Na+ with monensin. American Journal of Physiology. 1987;253:F26–33. doi: 10.1152/ajprenal.1987.253.1.F26. [DOI] [PubMed] [Google Scholar]
- Haugland R. Intracellular ion indicators. In: Mason WT, editor. Fluorescent and Luminescent Probes for Biological Activity. London: Academic Press; 1993. pp. 34–43. [Google Scholar]
- Horisberger JD, Lemas V, Kraehenbuhl JP, Rossier BC. Structure-function relationship of Na,K-ATPase. Annual Review of Physiology. 1991;53:565–584. doi: 10.1146/annurev.ph.53.030191.003025. [DOI] [PubMed] [Google Scholar]
- Housley GD, Raybould NP, Thorne PR. Fluorescence imaging of Na+ influx via P2X receptors in cochlear hair cells. Hearing Research. 1998;119:1–13. doi: 10.1016/s0378-5955(97)00206-2. [DOI] [PubMed] [Google Scholar]
- Inoue N, Matsui H. Activation of a brain type Na pump after glutamate excitation of cerebral neurones. Brain Research. 1990;534:309–312. doi: 10.1016/0006-8993(90)90146-3. [DOI] [PubMed] [Google Scholar]
- Knutson P, Ghiani CA, Zhou JM, Gallo V, McBain CJ. K+ channel expression and cell proliferation are regulated by intracellular sodium and membrane depolarization in oligodendrocyte progenitor cells. Journal of Neuroscience. 1997;17:2669–2682. doi: 10.1523/JNEUROSCI.17-08-02669.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Eygensteyn J, Lock R, Verbost P, Heijden A, Bonga S, Flik G. Branchial chloride cells in larvae and juveniles of freshwater tilapia Oreochromis mossambicus. Journal of Experimental Biology. 1995;198:2177–2184. doi: 10.1242/jeb.198.10.2177. [DOI] [PubMed] [Google Scholar]
- Lingrel JB, Kuntzweiler T. Na+,K+-ATPase. Journal of Biological Chemistry. 1994;269:19659–19662. [PubMed] [Google Scholar]
- McCormick SD. Fluorescent labelling of Na+, K+-ATPase in intact cells by use of a fluorescent derivative of ouabain: salinity and teleost chloride cells. Cell and Tissue Research. 1990;260:529–533. doi: 10.1007/BF00297233. [DOI] [PubMed] [Google Scholar]
- McLean IW, Nakane PK. Periodate-lysine-paraformaldehyde fixative. A new fixation for immunoelectron microscopy. Journal of Histochemistry and Cytochemistry. 1974;22:1077–1083. doi: 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- Muallem S, Zhang BX, Loessberg PA, Star RA. Simultaneous recording of cell volume changes and intracellular pH or Ca2+ concentration in single osteosarcoma cells UMR-106–01. Journal of Biological Chemistry. 1992;267:17658–17664. [PubMed] [Google Scholar]
- Murata T, Yamato I, Igarashi K, Kakinuma Y. Intracellular Na+ regulates transcription of the ntp operon encoding a vacuolar-type Na+-translocating ATPase in Enterococcus hirae. Journal of Biological Chemistry. 1996;271:23661–23666. doi: 10.1074/jbc.271.39.23661. [DOI] [PubMed] [Google Scholar]
- Nakao M, Gadsby DC. [Na] and [K] dependence of the Na/K pump current-voltage relationship in guinea pig ventricular myocytes. Journal of General Physiology. 1989;94:539–565. doi: 10.1085/jgp.94.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odland RM, Sutton RL. Hyperosmosis of cerebral injury. Neurological Research. 1999;21:500–508. [PubMed] [Google Scholar]
- Oliet SH, Bourque CW. Mechanosensitive channels transduce osmosensitivity in supraoptic neurones. Nature. 1993;364:341–343. doi: 10.1038/364341a0. [DOI] [PubMed] [Google Scholar]
- Parri HR, Crunelli V. Sodium current in rat and cat thalamocortical neurones: role of a non-inactivating component in tonic and burst firing. Journal of Neuroscience. 1998;18:854–867. doi: 10.1523/JNEUROSCI.18-03-00854.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayson BM. [Ca2+]i regulates transcription rate of the Na+/K+-ATPase α1 subunit. Journal of Biological Chemistry. 1991;266:21335–21338. [PubMed] [Google Scholar]
- Rose CR, Ransom BR. Regulation of intracellular sodium in cultured rat hippocampal neurones. The Journal of Physiology. 1997;499:573–587. doi: 10.1113/jphysiol.1997.sp021951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Opazo N, Cloix JF, Melis MG, Xiang XH, Herrera VL. Characterization of a sodium-response transcriptional mechanism. Hypertension. 1997;30:191–198. doi: 10.1161/01.hyp.30.2.191. [DOI] [PubMed] [Google Scholar]
- Senatorov VV, Hu B. Differential Na+-K+-ATPase activity in rat lemniscal and non-lemniscal auditory thalami. The Journal of Physiology. 1997;502:387–395. doi: 10.1111/j.1469-7793.1997.387bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senatorov VV, Mooney D, Hu B. The electrogenic effects of Na+-K+-ATPase in rat auditory thalamus. The Journal of Physiology. 1997a;502:375–385. doi: 10.1111/j.1469-7793.1997.375bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senatorov VV, Stys PK, Hu B. Over-expression of membrane Na,K-ATPase in cell rounding and acute neuronal death in vitro . Society for Neuroscience Abstracts. 1997b;23:1412. [Google Scholar]
- Silver IA, Deas J, Erecinska M. Ion homeostasis in brain cells: differences in intracellular ion responses to energy limitation between cultured neurones and glial cells. Neuroscience. 1997;78:589–601. doi: 10.1016/s0306-4522(96)00600-8. [DOI] [PubMed] [Google Scholar]
- Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na+-Ca2+ exchanger. Journal of Neuroscience. 1992;12:430–439. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauc M, Le Maout S, Poujeol P. Fluorescent video-microscopy study of regulatory volume decrease in primary culture of rabbit proximal convoluted tubule. Biochimica et Biophysica Acta. 1990;1052:278–284. doi: 10.1016/0167-4889(90)90222-y. [DOI] [PubMed] [Google Scholar]
- Taylor CP, Meldrum BS. Na+ channels as targets for neuroprotective drugs. Trends in Pharmacological Sciences. 1995;16:309–316. doi: 10.1016/s0165-6147(00)89060-4. [DOI] [PubMed] [Google Scholar]
- Urenjak J, Obrenovitch TP. Pharmacological modulation of voltage-gated Na+ channels: a rational and effective strategy against ischemic brain damage. Pharmacological Reviews. 1996;48:21–67. [PubMed] [Google Scholar]
- Yamamoto K, Ikeda U, Okada K, Saito T, Kawakami K, Shimada K. Sodium ion mediated regulation of Na/K-ATPase gene expression in vascular smooth muscle cells. Cardiovascular Research. 1994;28:957–962. doi: 10.1093/cvr/28.7.957. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Ikeda U, Seino Y, Tsuruya Y, Oguchi A, Okada K, Ishikawa S, Saito T, Kawakami K, Hara Y. Regulation of Na,K-adenosine triphosphatase gene expression by sodium ions in cultured neonatal rat cardiocytes. Journal of Clinical Investigation. 1993;92:1889–1895. doi: 10.1172/JCI116781. [DOI] [PMC free article] [PubMed] [Google Scholar]