

Abstract
Co-expression of auxiliary β subunits with the α1B Ca2+ channel subunit in COS-7 cells resulted in an increase in current density and a hyperpolarising shift in the mid-point of activation. Amongst the β subunits, β2a in particular, but also β4 and β1b caused a significant retardation of the voltage-dependent inactivation compared to currents with α1B alone, whilst no significant changes in inactivation properties were seen for the β3 subunit in this system.
Prevention of β2a palmitoylation, by introducing cysteine to serine mutations (β2a(C3,4S)), greatly reduced the ability of β2a to retard voltage-dependent inactivation.
Deletion of the proximal half of the α1B cytoplasmic amino terminus (α1BΔ1-55) differentially affected β subunit-mediated voltage-dependent inactivation properties. These effects were prominent with the β2a subunit and, to a lesser extent, with β1b. For β2a, the major effects of this deletion were a partial reversal of β2a-mediated retardation of inactivation and the introduction of a fast component of inactivation, not seen with full-length α1B. Deletion of the amino terminus had no other major effects on the measured biophysical properties of α1B when co-expressed with β subunits.
Transfer of the whole α1B amino terminus into α1C (α1bCCCC) conferred a similar retardation of inactivation on α1C when co-expressed with β2a to that seen in parental α1B.
Individual (α1B(Q47A) and α1B(R52A)) and double (α1B(R52,54A)) point mutations within the amino terminus of α1B also opposed the β2a-mediated retardation of α1B inactivation kinetics.
These results indicate that the α1B amino terminus contains determinants for β subunit-mediated voltage-dependent inactivation properties. Furthermore, effects were β subunit selective. As deletion of the α1B amino terminus only partially opposed β subunit-mediated changes in inactivation properties, the amino terminus is likely to contribute to a complex site necessary for complete β subunit function.
The auxiliary β subunit forms part of the functional multimeric neuronal voltage-dependent Ca2+ channel (VDCC) protein, together with pore-forming α1, extracellular α2-δ and, possibly, γ subunits (Hofmann et al. 1994; Letts et al. 1998). A major, high-affinity β subunit-binding site has been described on the intracellular loop connecting domains I and II of the α1 subunit (Pragnell et al. 1994). In addition, lower affinity sites have been described on the carboxyl termini of α1E (Tareilus et al. 1997; Qin et al. 1997) and α1A (Walker et al. 1998), and also on the amino terminus of α1A (Walker et al. 1999). Unlike the I-II loop β subunit-binding domain, the α1A amino- and carboxyl-terminal sites are β subunit selective (Walker et al. 1999). Furthermore, binding to these sites occurs independently of interaction with the I-II loop and it has been shown that the β4 subunit can bind to both the I-II loop and one secondary site (either the amino- or the carboxyl-terminal site, but not both) (Walker et al. 1999). In addition, β subunits contain three different putative binding domains (Hanlon et al. 1999). These subtleties may be important in determining the precise biophysical properties of the VDCC complex, and permit functional modulation of the extensive regulatory pathways that converge at the α1 subunit.
Functionally, the presence of VDCC β subunits in expression system studies has revealed a repertoire of effects on the major α1 subunit, including changes both in current amplitude and kinetics and in the current-voltage relationship (Birnbaumer et al. 1998; Walker & De Waard, 1998). Whilst VDCC α1 subunits contain inherent determinants of voltage-dependent inactivation (Zhang et al. 1994; Herlitze et al. 1997; Hering et al. 1998; Cens et al. 1999; Spaetgens & Zamponi, 1999), association with β subunit isoforms dictates their overall inactivation rate (Olcese et al. 1994). VDCC inactivation at presynaptic termini may contribute to short-term synaptic depression (Forsythe et al. 1998). Similarly, trains of action potentials induce cumulative VDCC inactivation and a depression of Ca2+ entry in expression system studies (Patil et al. 1998). Although the precise mechanism regarding inactivation of N-type (α1B) currents has been the subject of recent controversy (Shirokov, 1999; Jones et al. 1999), it is clear that β subunit composition differentially affects inactivation properties (Patil et al. 1998). The mechanism of another major function of the β subunit, that of increasing Ca2+ current amplitude, is also controversial. The β subunit is thought to act as a chaperone to traffic Ca2+ channels to the cell membrane (Chien et al. 1995; Brice et al. 1997) and also acts on the α1 subunit directly to alter the gating properties (Neely et al. 1993; Kamp et al. 1996). Recent studies provide evidence that multiple β subunit modulatory pathways may co-exist. Injection of a β3 fusion protein into Xenopus oocytes expressing α1C subunits provided the first temporal resolution of allosteric and trafficking effects (Yamaguchi et al. 1998). A recent study has also shown that a point mutation in the major β subunit-binding site on the α1 I-II loop prevented β subunit-mediated chaperoning of α1C to the cell membrane, but had no effect on the allosteric properties (Gerster et al. 1999). Such studies implicate independent β subunit functions and may complement data identifying multiple β subunit-binding sites.
We have recently demonstrated that the amino terminus of the VDCC α1 subunit plays an essential functional role, containing important determinants for Gβγ modulation (Page et al. 1998; Canti et al. 1999). Here, we demonstrate that deletions or mutations within the α1B amino terminus partially oppose β subunit-mediated effects on voltage-dependent inactivation kinetics in a β subunit-selective manner. Transfer of the whole α1B amino terminus into an α1C backbone also transfers β subunit-mediated inactivation properties. As a full reversal of effects does not occur, the results are consistent with the α1B amino terminus contributing determinants to a global site necessary for complete β subunit function.
METHODS
Materials
The following cDNAs were used: rabbit α1B (GenBank accession number D14157); rat α2-δ (neuronal splice variant, M86621); rat β1b (X11394); rat β2a (M80545); rat β3 (M88751); rat β4 (LO2315); rat α1C (isoform CII M67515); the carboxyl-terminal minigene of βARK (M34019); and mut-3 green fluorescent protein (mut-3 GFP) (U73901).
Construction of amino-terminal deletion and point mutations and α1bCCCC chimera
Constructs were created using PCR as described previously (Page et al. 1998; Canti et al. 1999). All constructs were subcloned into the pMT2 vector (Swick et al. 1992).
Deletions
The α1B subunit was truncated at the 5′ end to make the α1BΔ1-55 and α1BΔ2-50 constructs using the forward primers 5′-CGC ACT AGT ACC ATG GCG CTG TAC AA-3′ (α1BΔ1-55) and 5′-CAG ACT AGT ATG CAG CGC GCG CGG ACC AT-3′ (α1BΔ2-50) with the reverse primer 5′-GTC GCT TCT GCT CTT CTT GG-3′. A start codon (methionine) was incorporated into the forward primer before amino acid Q51 to make α1BΔ2-50. The PCR products were digested with the enzymes Spe I and Kpn I and subcloned into α1B-pMT2, which had also been digested with Spe I (polylinker cloning site) and Kpn I (1285 bp position in α1B).
Mutations
The β2a(C3,4S) subunit in which cysteines at positions 3 and 4 were mutated to serines, was made using the forward primer 5′-TTC ATG CAG TCC TCC GGG CT-3′, along with the reverse primer 5′-TG ACA GGT CAG GTA TCT GG-3′. The resultant β2a mutant was identical to that used by Chien et al. (1996), which was shown to prevent palmitoylation of β2a.
For all of the α1B amino-terminal point mutations, primers were designed so that the specific residues were mutated to alanines. The following primers were used: α1B(Q47A), 5′-GTC CTC TAC AAA GCG TCG ATC GCG CAG-3′; α1B(R52A), 5′-TCG ATC GCG CAG GCC GCG CGG ACC ATG-3′; and α1B(R52,54A), 5′-TCG ATC GCG CAG GCC GCG GCG ACC ATG GCG CT-3′. The reverse primer used in each case was 5′-GTC GCT TCT GCT CTT CTT GG-3′. For the PCR extension reactions, the forward primer used was 5′-AGC ACT AGT ATG GTC CGC TTC GGG GAC-3′.
α1bCCCC chimera
The α1bCCCC (where lower case denotes the amino terminus and upper case the transmembrane domain; see Canti et al. 1999) chimera was made by PCR using the chimeric primer 5′-CAC CGA GTG GCC TCC ATT TGA AAT AAT T-3′, the reverse primer 5′-CCA CCA GCA GGT CCA GGA TAT TGA-3′ and α1C-pMT2 template as detailed previously (Canti et al. 1999). The resulting PCR product was extended against the α1B-pMT2 template using a forward primer directed against the vector: 5′-TCT CCA CAG GTG TCC ACT-3′. This PCR product was digested with Kpn I, situated in the cloning site, and Mfe I and subcloned into a Kpn I-Mfe I-digested α1C-pMT2. The construct is made up of amino acid residues α1B1-95, followed by the α1C125-2143 sequence.
All PCRs were performed using the proof-reading enzyme Pfu (Stratagene). The sequences of the subcloned PCR products were verified by cycle-sequencing using SequiTherm EXCEL II (Epicentre Technologies, Madison, WI, USA).
Expression of constructs
COS-7 cells were transfected by electroporation as described previously (Campbell et al. 1995), using 15, 5, 5 and 1 μg of the α1-, α2-δ-, β- and mut-3 GFP-pMT2 constructs, respectively. In order to limit the effects of endogenous Gβγ in constructs sensitive to G protein modulation (Stephens et al. 1998) the βARK minigene-pMT2 construct (5 μg) was also co-transfected. In experiments in the absence of α2-δ and/or β subunits, blank pMT2 vector was transfected to maintain a total cDNA of 31 μg. Cells were maintained at 37°C for 36–48 h, and prior to recording were replated using a non-enzymatic cell dissociation medium (Sigma) and maintained at 25°C for between 1 and 9 h.
Electrophysiology
Recordings were made from fluorescent COS-7 cells expressing the mut-3 GFP reporter gene. Borosilicate glass electrodes of resistance 2–5 MΩ were filled with a solution containing (mM): caesium aspartate, 140; EGTA, 5; MgCl2, 2; CaCl2, 0.1; K2ATP, 2; and Hepes, 10; pH 7.2, 310 mosmol l−1 with sucrose. The external solution contained (mM): TEA-Br, 160; KCl, 3; NaHCO3, 1.0; MgCl2, 1.0; Hepes, 10; glucose, 4; and BaCl2, 1 or 10 as stated; pH 7.4, 320 mosmol l−1 with sucrose. For all experiments in the presence of β2a, current density was sufficiently large to allow use of 1 mM Ba2+ as a charge carrier; for α1B in the absence of any β subunit it was necessary to use 10 mM Ba2+ to obtain robust, measurable currents; subsequent experiments with β subunits used 10 mM Ba2+ to permit direct comparisons.
Whole-cell currents were recorded using an Axopatch-1D amplifier. Data were filtered at 1–2 kHz, digitised at 5–10 kHz and analysed using pCLAMP 6 and Origin 3.5 and 5.0. The junction potential between external and internal solutions was −6 mV; values given in the figures and text have not been corrected for this. Current records are shown following leak and capacitance current subtraction (P/4 or P/8 protocol). Series resistance was compensated from a minimum of 70% up to 85%. The voltage errors from the residual uncompensated series resistance were < 2 mV for the largest currents, and no further correction was made.
Experiments were performed at room temperature (20-24°C). Data are expressed as means ±s.e.m. Current decay was fitted with a double exponential function of the form:
where y0 is the non-inactivating current component, A1 is the fast component and A2 is the slow component, τ1 is the fast time constant of inactivation and τ2 is the slow time constant of inactivation. In a very small number of cases, current decay was fitted with a single exponential function of the form:
Statistical analysis was performed using Student's paired or unpaired t test as appropriate.
RESULTS
Biophysical properties of α1B co-expressed with auxiliary α2-δ and/or β2a subunits
Initially, we examined the biophysical properties of α1B subunits expressed in COS-7 cells either alone, or with α2-δ or α2-δ/β2a auxiliary subunits (Fig. 1; Tables 1 and 2). Inward currents were recorded using Ba2+ as the charge carrier as it supports voltage-dependent inactivation of Ca2+ channels (in addition, controversy exists as to the precise mechanism of N-type channel inactivation in the presence of extracellular Ca2+; see Jones, 1999). For α1B expressed alone, whole-cell barium current (IBa) was typically high-voltage activated, peaking between +20 and +30 mV with a mid-point of voltage dependence of activation (V½) of +14.7 ± 2.5 mV (n = 8, Fig. 1A). At peak current levels, inactivation was almost complete for a 1.5 s step depolarisation (Fig. 1B and C). Current decay was well fitted with a double exponential function yielding time constants of inactivation τfast = 77 ± 10 ms (55 ± 8%) and τslow = 413 ± 34 ms (41 ± 7%) and a non-inactivating component of 4 ± 1% (n = 4, Table 2). Co-expression of α2-δ with α1B caused a non-significant shift in V½ to +20.8 ± 1.8 mV (n = 12, Fig. 1A). The inactivation time course of α1B/α2-δ currents over 1.5 s was well fitted with a double exponential function with τfast = 93 ± 6 ms (52 ± 5%) and τslow = 474 ± 31 ms (43 ± 5%) and a non-inactivating component of 5 ± 2% (n = 7, Table 2); these values were not significantly different from those for α1B alone. The additional expression of β2a with α1B/α2-δ caused a number of changes in biophysical properties (Fig 1; Tables 1 and 2). Current density was increased by about 10-fold and there was a hyperpolarising shift in V½ to +4.6 ± 3.3 mV (n = 12, Fig. 1A). There was also an increase in voltage sensitivity as shown by the decrease in value of the slope factor k (Table 1). A major effect of β2a co-expression was to significantly retard the voltage-dependent inactivation of α1B (Fig. 1B and C; Table 2). Current decayed according to a single exponential function that was too slow to fit accurately over 1.5 s and had a large non-inactivating component (Fig. 1B). As current decay in the presence of β2a was incomplete over the time course used, inactivation properties were compared in terms of percentage inactivation (Iend/Ipeak) at maximal values of the current density-voltage relationship for step depolarisations of 1.5 s (Fig. 1C). Mean percentage inactivation after 1.5 s for α1B/α2-δ/β2a was 15 ± 5% with 10 mM Ba2+ (Fig. 1B) and 9 ± 1% with 1 mM Ba2+ (Fig. 2B) (these values were not statistically different from each other). Inactivation with β2a was dramatically slowed in comparison to both α1B alone (95 ± 2%, P < 0.001) and α1B/α2-δ (93 ± 2%, P < 0.001) (Fig. 1C). In addition, current activated in two distinct phases in the presence of β2a, with a fast and a slow component (Fig. 1B). Activation kinetics were fitted to the first 500 ms of the α1B/α2-δ/β2a trace shown in Fig. 1B; a double exponential function gave τfast = 9 ms (83%) and τslow = 75 ms (17%). The second, slower component was unresolvable in the absence of β2a due to the high degree of inactivation.
Figure 1. Biophysical properties of α1B and auxiliary α2-δ and β2a subunits in COS-7 cells.
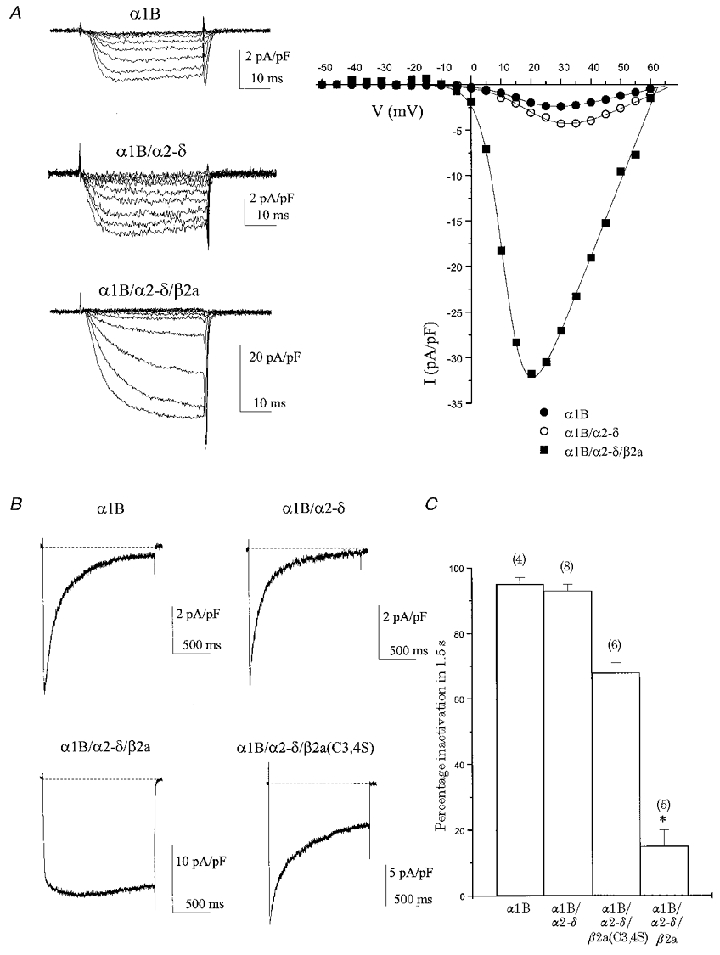
Table 1.
Biophysical properties of VDCC constructs transiently transfected into COS-7 cells
| Construct | [Ba2+](mM) | Current density (pA pF−1) | Activation V1/2 (mV) | k(mV) | Steady-state inactivation V1/2 (mV) | k(mV) |
|---|---|---|---|---|---|---|
| α1B | 10 | 6 ± 1 (8) | +14.7 ± 2.5 (8) | 5.7 ± 0.2 (8) | — | — |
| α1B/α2-δ | 10 | 3 ± 1 (12) | +20.8 ± 1.8 (12) | 6.8 ± 0.5 (12) | — | — |
| α1B/α2-δ/β2a | 10 | 30 ± 7 (12) | +4.6 ± 3.3 (12) | 3.6 ± 0.3 (12) | — | — |
| α1B/α2-δ/β2a(C3,4S) | 10 | 20 ± 7 (7) | −0.4 ± 4.0 (7) | 4.1 ± 0.4 (7) | −42.4 ± 4.2 (6) | −8.0 ± 0.5 (6) |
| α1BΔ1–55/α2-δ/β2a | 1 | 27 ± 6 (12) | −14.3 ± 1.5 (12) | 3.7 ± 0.2 (12) | — | — |
| α1BΔ2–50/α2-δ/β2a | 1 | 17 ± 6 (8) | −12.6 ± 2.1 (8) | 4.0 ± 0.5 (8) | — | — |
| α1B(R52A)/α2-δ/β2a | 1 | 24 ± 11 (7) | −12.8 ± 2.7 (7) | 3.7 ± 0.3 (7) | — | — |
| α1B(Q47A)/α2-δ/β2a | 1 | 23 ± 5 (9) | −15.3 ± 1.6 (9) | 4.1 ± 0.2 (9) | — | — |
| α1B(R52,54A)/α2-δ/β2a | 1 | 31 ± 13 (13) | −11.2 ± 1.6 (13) | 4.0 ± 0.2 (13) | — | — |
| α1B/α2-δ/β1b | 10 | 46 ± 11 (15) | +4.5 ± 3.2 (15) | 4.0 ± 0.4 (11) | −42.4 ± 3.7 (7) | −9.2 ± 0.6 (7) |
| α1BΔ1–55/α2-δ/β1b | 10 | 42 ± 15 (11) | +7.0 ± 4.8 (10) | 3.4 ± 0.4 (10) | −45.6 ± 7.4 (4) | −7.3 ± 0.6 (4) |
| α1B/α2-δ/β3 | 10 | 86 ± 22 (10) | +5.0 ± 2.0 (10) | 4.0 ± 0.3 (10) | −41.9 ± 1.9 (4) | −8.7 ± 0.9 (4) |
| α1BΔ1–55/α2-δ/β3 | 10 | 89 ± 20 (10) | +3.3 ± 3.5 (10) | 3.2 ± 0.4 (10) | −47.1 ± 7.3 (4) | −8.6 ± 1.4 (4) |
| α1B/α2-δ/β4 | 10 | 50 ± 15 (12) | +4.5 ± 2.5 (12) | 4.7 ± 0.4 (12) | −48.6 ± 2.8 (5) | −9.1 ± 0.8 (5) |
| α1BΔ1–55/α2-δ/β4 | 10 | 59 ± 17 (15) | +7.4 ± 2.4 (14) | 3.5 ± 0.3 (14) | −44.8 ± 5.1 (5) | −7.9 ± 1.1 (5) |
| α1C/α2-δ/β2a | 10 | 16 ± 3 (20) | +9.7 ± 1.9 (20) | 6.1 ± 0.3 (20) | — | — |
| α1bCCCC/α2-δ/β2a | 10 | 7 ± 2 (7) | +8.1 ± 4.0 (7) | 6.5 ± 0.6 (7) | — | — |
Numbers in parentheses are number of experiments.
Table 2.
Voltage-dependent inactivation properties of VDCC constructs for a 1.5 s depolarization to the peak of the current density-voltage relationship
| Construct | n | τfast (MS) | Fast component (%) | τslow (MS) | Slow component (%) | Non-inactivating component (%) |
|---|---|---|---|---|---|---|
| α1B | 4 | 77 ± 10 | 55 ± 8 | 413 ± 34 | 41 ± 7 | 4 ± 1 |
| α1B/α2-δ | 6 | 93 ± 6 | 52 ± 5 | 474 ± 31 | 43 ± 5 | 5 ± 2 |
| α1B/α2 δ/β2a* | — | — | — | — | — | — |
| α1B/α2-δ/β2a(C3,4S) | 4 | 187 ± 38 | 37 ± 8 | 753 ± 55 | 44 ± 4 | 18 ± 6 |
| α1BΔ1–55/α2-δ/β2a | 4 | 167 ± 42 | 11 ± 3 | 4760 ± 857 | 44 ± 4 | 45 ± 7 |
| α1BΔ2–50/α2-δ/β2a | 4 | 168 ± 61 | 19 ± 3 | 1888 ± 202 | 36 ± 4 | 45 ± 4 |
| α1B/α2-δ/β1b | 8 | 155 ± 35 | 48 ± 6 | 704 ± 120 | 40 ± 5 | 12 ± 1 |
| α1BΔ1–55/α2-δ/β1b | 7 | 137 ± 12 | 61 ± 3 | 793 ± 176 | 34 ± 4 | 6 ± 1 |
| α1B/α2-δ/β3 | 8 | 169 ± 23 | 52 ± 7 | 758 ± 79 | 38 ± 6 | 10 ± 2 |
| α1BΔ1–55/α2-δ/β3 | 7 | 130 ± 23 | 42 ± 7 | 506 ± 49 | 47 ± 6 | 10 ± 5 |
| α1B/α2-δ/β4 | 6 | 264 ± 87 | 26 ± 7 | 1003 ± 111 | 51 ± 6 | 23 ± 4 |
| α1BΔ1–55/α2-δ/β4 | 10 | 266 ± 26 | 32 ± 5 | 951 ± 101 | 49 ± 5 | 19 ± 3 |
| α1B(R52A)/α2-δ/β2a | 4 | 259 ± 83 | 9 ± 3 | 1755 ± 209 | 40 ± 5 | 51 ± 8 |
| α1B(Q47A)/α2-δ/β2a | 3 | 63 ± 27 | 9 ± 2 | 1141 ± 51 | 41 ± 3 | 50 ± 6 |
| α1B(R52,54A)/α2-δ/β2a | 3 | 280 ± 178 | 8 ± 4 | 1401 ± 70 | 41 ± 4 | 51 ± 1 |
| α1B(R52,54A) | 5 | 78 ± 9 | 58 ± 6 | 418 ± 69 | 34 ± 4 | 8 ± 3 |
It was not possible to fit parameters to α1B/α2-δ/β2a currents, as described in the text. For all other combinations, current decay was well fitted with a double exponential function (see Methods). In a very small number of cases, current decayed with a single slow exponential function and the fast component was assumed to be zero.
Figure 2. α1B proximal amino-terminal deletion partially opposes β2a-induced slowing of α1B inactivation.
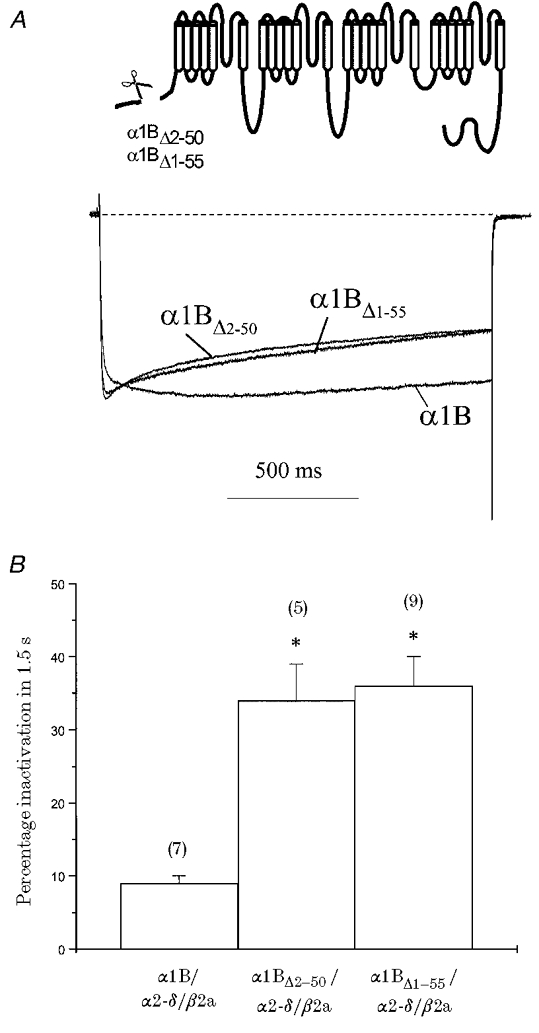
α1B constructs in which the proximal amino-terminal residues (α1BΔ1-55 and α1BΔ2-50) were deleted (see inset) were co-expressed with α2-δ/β2a and compared with α1B/α2-δ/β2a. A, normalised and averaged current traces generated by a 1.5 s step depolarisation eliciting maximal IBa for α1B, α1BΔ2-50 and α1BΔ1-55; VH =−100 mV. Recordings were in 1 mM Ba2+. B, histogram of mean percentage inactivation (Iend/Ipeak) for a 1.5 s step depolarisation taken at maximal IBa; n for each condition is given in parentheses. Deletion of the proximal amino-terminal residues increased β2a-mediated percentage inactivation in comparison with α1B (* P < 0.001).
It has been shown that the β2a subunit is unique amongst β subtypes in that its properties are dependent on the palmitoylation of two amino-terminal cysteine residues not found on other β subunits (Chien et al. 1996). Functionally, mutation of the cysteines to serines prevents palmitoylation of these sites and partially reverses the distinguishing features of β2a regulation (Qin et al. 1998). Therefore, we also compared the properties of α1B expressed alone and with either β2a or the mutant β2a(C3,4S) subunit. Prevention of β2a palmitoylation had no significant effects on the density and voltage dependence of activation of α1B current (Table 1), but it greatly reduced the ability of β2a to retard voltage-dependent inactivation (Fig. 1B and C; Table 2). The percentage inactivation after 1.5 s was significantly greater than that seen with β2a (P < 0.001). The α1B current in the presence of β2a(C3,4S) inactivated according to a double exponential function. The C→S mutations introduced a resolvable fast component of inactivation not seen with β2a, with τfast = 187 ± 38 ms (37 ± 8%) and τslow = 753 ± 55 ms (44 ± 4%), and a non-inactivating component of 18 ± 6% (n = 4, Table 2). However, voltage-dependent inactivation properties were not fully reversed in the β2a(C3,4S) mutant. The non-inactivating component for β2a(C3,4S) (18 ± 6%) was more than that in the absence of β subunits (5 ± 2%, P < 0.05) and both the fast (P < 0.05) and slow (P < 0.01) time constants of inactivation were slower than for α1B/α2-δ (Table 2).
Involvement of the α1B amino terminus in β subunit function
We have recently shown that the α1B amino terminus is essential for certain functional properties, in that it contains determinants for voltage-dependent modulation by Gβγ subunits (Page et al. 1998; Canti et al. 1999). We examined the potential role of the α1B amino terminus as a similar determinant of β subunit function (Figs 2 and 3).
Figure 3. Effects of β subunit co-expression and deletion of the proximal 55 amino-terminal residues on α1B voltage-dependent inactivation.
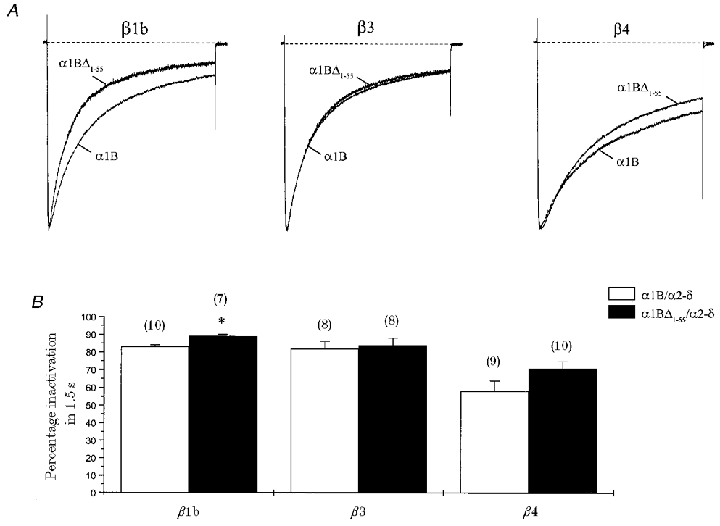
α1B or α1BΔ1-55 (together with α2-δ) was co-expressed with β1b, β3 or β4 subunits. A, normalised and averaged current traces generated by a 1.5 s step depolarisation eliciting maximal IBa for the conditions stated; VH =−100 mV. Recordings were in 10 mM Ba2+. B, histogram of percentage inactivation (Iend/Ipeak) for a 1.5 s step depolarisation taken at maximal IBa. n for each condition is given in parentheses. Deletion of proximal α1B amino-terminal residues increased β1b-mediated percentage inactivation in comparison with α1B (* P < 0.05). No statistically significant effects were seen for β3 or β4.
Initially, we examined responses with the β2a subunit. Constructs in which proximal residues of the α1B amino terminus were deleted (α1BΔ2-50 and α1BΔ1-55) showed no differences in current density and voltage dependence of activation in comparison with parental α1B when co-expressed with β2a (Table 1). However, significant differences in voltage-dependent inactivation properties were seen (Fig. 2; Table 2). The inactivation of currents in response to 1.5 s step depolarisations was significantly greater for α1BΔ1-55 than for parental α1B with β2a (Fig. 2A and B). A similar effect was shown with the α1BΔ2-50 construct (in which amino acids 2–50 were deleted, but the start codon, methionine, was retained; see Methods). Current decay was well fitted with a double exponential function for both α1BΔ2-50 and α1BΔ1-55, with a fast component of 19 ± 3 and 11 ± 3%, respectively (Fig. 2A; Table 2). In addition, the β2a-mediated non-inactivating current component was clearly reduced for both α1BΔ2-50 (45 ± 4%) and α1BΔ1-55 (45 ± 7%). The only major difference in β2a-mediated current between α1BΔ2-50 and α1BΔ1-55 was an approximately 2.5-fold lengthening of the slow time constant of inactivation for α1BΔ1-55 in comparison with α1BΔ2-50 (Table 2). Overall, deletion of proximal α1B amino-terminal residues introduced a resolvable small, fast component of inactivation not seen for parental α1B in the presence of β2a. These data suggest that the ability of β2a to retard inactivation was compromised by the lack of the proximal half of the amino terminus. In addition, parental α1B currents in the presence of β2a showed a second, slower activation phase not resolvable with the more inactivating α1BΔ1-55 and α1BΔ2-50 currents (Fig. 2A). Activation kinetics were fitted to the first 500 ms of the α1B/α2-δ/β2a trace recorded in 1 mM Ba2+ shown in Fig. 2A; a double exponential function gave τfast = 7 ms (87%) and τslow = 116 ms (13%). These values are similar to those given above for α1B/α2-δ/β2a currents recorded in 10 mM Ba2+.
We extended our investigation of the potential role of the α1B amino terminus in β subunit function to include three other major β subunits, β1b, β3 and β4 (Fig. 3). Each subunit was co-expressed with either α1B or α1BΔ1-55 (together with α2-δ). All β subunits caused significant increases in α1B current density and a hyperpolarising shift in V½, similar to results with β2a (Table 1). For all these β subunits, peak current in response to a 1.5 s step depolarisation decayed according to a double exponential function (Fig. 3A; Table 2). The major effect of β subunit co-expression was an increase in the non-inactivating current component for both β1b (12 ± 1%, P < 0.01) and β4 (23 ± 4%, P < 0.01), in comparison with values for α1B/α2-δ (5 ± 2%) (Table 2). For the β4 subunit, the increase in the non-inactivating component was accompanied by a significant decrease in the fast component of inactivation (26 ± 7%, P < 0.05), in comparison with values for α1B/α2-δ (52 ± 5%) (Table 2).
Deletion of the proximal amino terminus had no major effects on the current density and activation properties of α1B measured in the presence of each of the different β subunits (Table 1), but did have subtle effects on voltage-dependent inactivation (Fig. 3A and B; Table 2). Decay of current over 1.5 s was well fitted with a double exponential function for each β subunit. Effects on α1B inactivation were dependent on the subfamily of β subunit co-expressed. Whilst deletion of the proximal α1B amino terminus had no statistically significant effects on β3- or β4-mediated inactivation, small, but significant changes were seen with β1b. The percentage inactivation after 1.5 s for α1BΔ1-55 co-expressed with β1b was slightly increased in comparison with α1B (Fig. 3B). There was also a decrease in the proportion of the non-inactivating current component for α1BΔ1-55 with β1b (6 ± 1%, P < 0.01) in comparison with values for α1B (12 ± 1%) (Table 2). In fact, the values for α1BΔ1-55 were no different from the non-inactivating component in the absence of β subunits (5 ± 2%) (Table 2). For all of these β subunits, deletion of the α1B amino terminus had no significant effect on the fast and slow time constants of inactivation (Table 2) or the mid-point of voltage dependence of inactivation determined from steady-state curves measured at peak current following a 5 s prepulse (Table 1).
Involvement of the α1B amino terminus in β2a subunit function
The above data suggest that the α1B amino terminus plays a role in β subunit function and that these effects are largely restricted to voltage-dependent inactivation properties. Such effects were β subunit specific, and may be ranked in the order β2a > β1b ⋙ β4 > β3. Subsequent experiments were performed with β2a, the subunit exhibiting the most significant effects.
We sought to demonstrate that the transfer of the α1B amino terminus alone could confer β subunit-mediated voltage-dependent inactivation properties on another VDCC α1 subunit. We took advantage of the fact that the α1C subunit, whose amino terminus lacks any significant homology to that of α1B (see Canti et al. 1999), showed significantly stronger voltage-dependent inactivation than α1B when co-expressed with β2a (Fig. 4A and B). We investigated the potential role of the amino terminus in this difference, by examining the inactivation properties of a construct in which the entire α1C amino terminus was replaced with that of α1B (to give α1bCCCC, where lower case denotes amino terminus and upper case transmembrane domains) (see Fig. 4A, inset). The α1bCCCC construct had properties generally similar to those of α1C when co-expressed with β2a (Table 1); in particular, the reduced voltage sensitivity (increased slope factor, k), a characteristic difference between α1B and α1C in this expression system, was retained in α1bCCCC. The only notable exception was in the inactivation properties (Fig. 4A and B). For the α1bCCCC construct, the percentage inactivation after 1.5 s was no different from that of parental α1B and in both cases a significantly smaller percentage inactivation was seen than for α1C co-expressed with β2a (Fig. 4B). This gain-of-function chimeric construct further suggests that the presence of the α1B amino terminus is in part responsible for the functional properties of the β2a subunit. The α1bCCCC construct also more closely resembled α1B than α1C in that it had two components of activation; as discussed above, the second, slower component is unresolvable in faster inactivating constructs (such as α1C).
Figure 4. Substitution of the α1B amino terminus reduces the β2a-mediated inactivation of α1C.
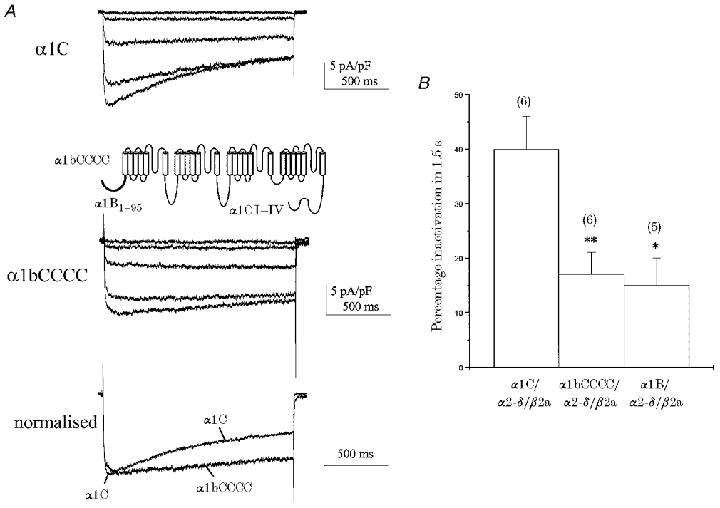
Parental α1C or α1bCCCC constructs (in which the amino terminus of α1C was exchanged for that of α1B; see inset) were co-expressed with α2-δ/β2a. A, example I–V profiles elicited by a family of 1.5 s step depolarisations to the levels described in 10 mV increments: α1C, −20 to +20 mV; α1bCCCC, −30 to +10 mV; VH =−100 mV. Recordings were in 10 mM Ba2+. Normalised peak IBa traces are shown for direct comparison. α1C inactivation meant that any slower activation phase seen with α1bCCCC and β2a was not resolvable. B, histogram of percentage inactivation (Iend/Ipeak) for a 1.5 s step depolarisation taken at maximal IBa; n for each condition is given in parentheses. Significantly less inactivation was seen for both α1bCCCC and α1B compared to α1C (* P < 0.05, ** P < 0.01). Furthermore, there was no difference between percentage inactivation for α1bCCCC and α1B under identical conditions.
Within the α1B amino-terminal sequence, we previously identified an 11 amino acid region (α1B45-55) whose deletion renders the α1B subunit unresponsive to G protein modulation (Canti et al. 1999). We examined the inactivation of α1B constructs containing mutations within this amino-terminal sequence (see Fig. 5A, inset), both as determinants for β subunit function, and also for possible overlap with the Gβγ-interaction sites, as reported for identified β subunit-binding sites (De Waard et al. 1997; Zamponi et al. 1997; Qin et al. 1997). We selected two point mutations in this region, α1B(Q47A) (normal modulation by co-expressed Gβγ) and α1B(R52A) (compromised Gβγ modulation) and the double mutant α1B(R52,54A) construct (no Gβγ modulation) (Canti et al. 1999). For these constructs, there were no differences in current density and activation properties from those seen for α1B when co-expressed with β2a (Table 1). However, Fig. 5 illustrates that all of these constructs demonstrated an increase in percentage inactivation after 1.5 s in comparison with parental α1B. The current in the presence of these mutants inactivated according to a double exponential function (Fig. 5A). Thus, a resolvable small fast component of β2a-mediated inactivation, not apparent with parental α1B, was introduced in the α1B(Q47A) (9 ± 2%), α1B(R52A) (9 ± 3%) and α1B(R52,54A) (8 ± 4%) constructs (Table 2). These constructs showed similar percentage inactivation and exponential decay properties to those seen for both α1BΔ1-55 and α1BΔ2-50 under the same conditions (with the exception of a lengthening in the slow time constant of inactivation for α1BΔ1-55). Figure 5 also shows that the second, slower component of activation seen in parental α1B with β2a was not resolvable in faster inactivating mutant α1B constructs. Slowing of current activation may be due in part to the interaction of endogenous Gβγ subunits with α1B (Stephens et al. 1998). However, the Gβγ-binding protein βARK was present here to limit Gβγ availability; in addition, no differences were seen between the α1B(Q47A) construct (which retains similar Gβγ modulation properties to parental α1B) and the other mutants with compromised Gβγ modulation, suggesting that this effect is negligible. The finding that Q47 of α1B is a determinant for β subunit-mediated inactivation properties in the present study, coupled with the observation that it is not essential for Gβγ function (Canti et al. 1999), differentiates regions contributing to these modulation pathways.
Figure 5. α1B amino-terminal sequence mutations partially oppose β2a-mediated slowing of inactivation.
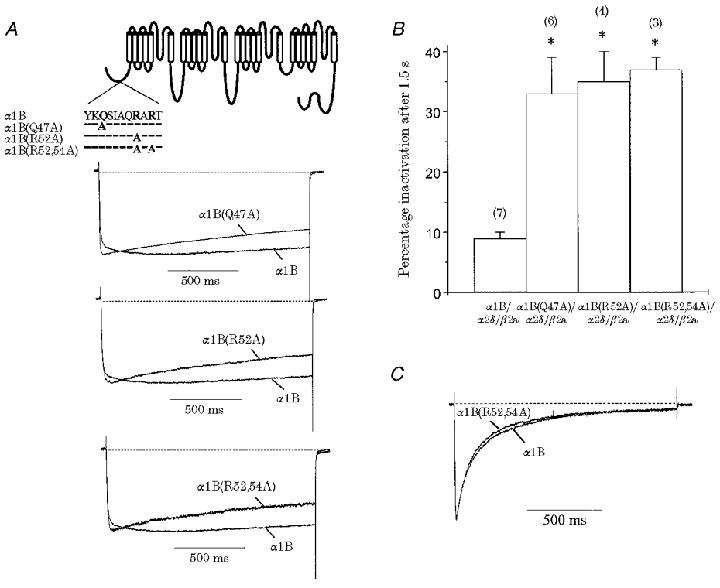
Constructs in which single (α1B(Q47A) and α1B(R52A)) and double (α1B(R52,54A)) point mutations within the proximal α1B amino-terminal sequence (see inset) were co-expressed with α2-δ/β2a and compared with α1B/α2-δ/β2a. A, normalised and averaged current traces generated by a 1.5 s step depolarisation eliciting maximal IBa for α1B(R52A), α1B(Q47A) and α1B(R52,54A); VH =−100 mV. Recordings were in 1 mM Ba2+. Also included is the corresponding α1B/α2-δ/β2a trace from Fig. 2A for comparison. B, histogram of percentage inactivation (Iend/Ipeak) for a 1.5 s step depolarisation taken at maximal IBa; n for each condition is given in parentheses. Mutations within the α1B proximal amino terminus increase β2a-mediated percentage inactivation in comparison with α1B (* P < 0.001). C, normalised and averaged current traces generated by a 1.5 s step depolarisation eliciting maximal IBa for α1B alone (n = 7) and α1B(R52,54A) (n = 5); VH =−100 mV. Recordings were in 10 mM Ba2+. α1B(R52,54A) channels expressed alone showed no inherent differences in voltage-dependent inactivation properties from those seen for α1B.
In order to discount the possibility that mutations in the α1B subunit itself are responsible for the recorded differences in inactivation properties, we expressed the mutant α1B(R52,54A) construct alone and examined the inactivation properties in detail (Fig. 5C; Table 2). Normalised averaged traces from a 1.5 s depolarisation for α1B and α1B(R52,54A) alone showed very similar inactivation time courses (Fig. 5C). The α1B(R52,54A) current decay was well fitted with a double exponential function, with τfast = 78 ± 9 ms (58 ± 6%) and τslow = 418 ± 69 ms (34 ± 4%) and a non-inactivating component of 8 ± 3% (n = 5, Table 2); these values were not significantly different from those of α1B alone. Therefore, it is unlikely that intrinsic α1 properties were responsible for the differences in inactivation kinetics.
Taken together, these data are consistent with the α1B amino terminus contributing determinants for β subunit-mediated voltage-dependent inactivation properties in a β subunit-selective manner. The results identify this region as a requirement for normal β subunit function.
DISCUSSION
The present study has investigated the role of the α1B amino terminus in the function of auxiliary VDCC β subunits. The findings suggest that this region selectively affects β subunit-mediated voltage-dependent inactivation properties, whilst having little effect on other biophysical properties. Effects were β subunit selective and may be ranked in the order β2a > β1b ⋙ β4 > β3. Such selectivity may shape Ca2+-mediated responses dependent on differential VDCC phenotype expression.
Effects of β subunit expression on α1B in COS-7 cells
When co-expressed with α1B, members of each of the β1-β4 subfamilies all caused increases in current density and a hyperpolarising shift in the current-voltage relationship. On the other hand, β subunit-selective effects were seen for voltage-dependent inactivation properties. When expressed alone, α1B currents inactivated almost completely during a 1.5 s step depolarisation. Whilst the auxiliary α2-δ subunit had no clear effects on α1B voltage-dependent inactivation, co-expression of β2a, β4 or β1b subunits decreased the percentage inactivation measured over the same time course. Effects were particularly dramatic for the β2a subunit. The functional properties of β2a are dependent on the palmitoylation of two cysteine residues not found in other β subunits (Chien et al. 1996; Chien & Hosey, 1998). Palmitoylation was prevented by mutating these cysteines to serines. These mutations caused a significant opposition to β2a-mediated retardation of inactivation kinetics and introduced a resolvable fast component of inactivation that was not apparent with parental β2a. Inactivation properties of β2a(C3,4S) were closer to those seen with the more inactivating β1b, β3 and β4 isoforms rather than β2a, suggesting that these two cysteine residues play an important role in the characteristically slow β2a-mediated inactivation kinetics and may contribute to a β subunit domain important for functional interaction with the α1 subunit (see Walker & De Waard, 1998). These findings are in agreement with voltage-dependent inactivation results obtained in Xenopus oocytes for both α1B (C. Canti, Y. Bogdanov & A. C. Dolphin, manuscript in preparation) and α1E (Qin et al. 1998) when co-expressed with mutant β2a subunits in which palmitoylation was prevented. The present study substantiates such electrophysiological results in a mammalian expression system where endogenous palmitoylation pathways may differ.
We also show that the mid-point of the voltage dependence of inactivation (V½) was no different when α1B was co-expressed with β1b, β3, β4 or β2a(C3,4S) subunits. The lack of inactivation of α1B in the presence of β2a in this system meant that accurate values could not be measured for β2a. However, these results are in agreement with α1B data obtained in Xenopus oocytes (C. Canti, Y. Bogdanov & A. C. Dolphin, unpublished data), which further show that β2a is unique in causing a depolarising shift in α1B steady-state inactivation in contrast to the uniform hyperpolarisation seen with other β subunits. A similar differentiation has been reported for α1E (Jones et al. 1998). α1E V½ was hyperpolarised by co-expression of β1b (∼10 mV), β3 (∼15 mV) and β4 (∼10 mV); in contrast, β2a depolarised V½ (∼15 mV), in comparison to α1E alone. Jones et al. (1998) also examined the effects of β2a and β3 on steady-state inactivation of α1C; with the α1C subunit, there was no difference in V½ for β3 and β2a co-expression. Taken together these data indicate that β subunit-induced shifts in steady-state inactivation are dependent on specific α1 subunit association.
Role of α1B amino terminus in β subunit-mediated properties
We examined the role of the α1B amino terminus in β subunit function using a series of deletions, mutations and a gain-of-function chimeric construct. The effects were β subunit dependent and were largely confined to changes in the kinetics of voltage-dependent inactivation. The most striking effects were seen with β2a, where the deletion of the α1B amino terminus markedly opposed the β subunit-mediated retardation of inactivation. Similar effects were seen for the β1b subunit; the amino-terminal deletion reduced the non-inactivating current component to levels similar to those recorded in the absence of β subunits. In contrast, deletion of the α1B amino terminus had little effect on β4- or β3-mediated inactivation properties.
It was possible to confer similar β2a-mediated inactivation properties of α1B onto the α1C subunit by exchanging the amino-terminal sequences of these subunits. Using a similar approach, Walker et al. (1999) used a loss-of-function construct, replacing the α1A amino terminus with the corresponding α1C region, to demonstrate a functional interaction between β subunits and the α1A amino terminus.
We have previously identified an 11 amino acid region (α1B45-55) as an essential determinant for Gβγ modulation of α1B (Canti et al. 1999). We tested the effects of a number of mutations to investigate whether this region also contained determinants for β subunit function. For both single (α1B(Q47A) and α1B(R52A)) and double (α1B(R52,54A)) point mutations, significant opposition to β2a-mediated retardation of inactivation was seen. In all cases, these effects were not significantly different from those seen when the proximal half of the amino terminus was deleted, indicating that individual mutations are equally disruptive to β subunit function. These findings are consistent with the existence of multiple interdependent determinants in the amino terminus, possibly contributing to a highly structured region, necessary for β subunit function.
VDCC α1 subunits contain inherent determinants of voltage-dependent inactivation (see Hering et al. 1998). The differentiating effects of β subunits on the inactivation properties of full-length and amino-terminal truncated α1B suggest that disruption of the amino terminus alone is not responsible for the changes in voltage-dependent inactivation. If this was an intrinsic property of the α1 subunit then allβ subunits would be expected to show a difference when co-expressed with α1B amino-terminal deletion constructs. We have been unable to achieve sufficiently robust expression of amino-terminal deletion constructs in the absence of β subunits to accurately measure baseline inactivation. This may be due in part to the amino terminus playing a role in channel expression levels, as also suggested for an α1A amino-terminal chimeric construct (Walker et al. 1999). Importantly, however, we were able to express the α1B(R52,54A) construct, containing a less-disruptive double point mutation, in the absence of β subunits. This construct showed similar differences in β2a-induced inactivation properties to the amino-terminal deletion constructs; however, when expressed alone, α1B(R52,54A) showed no differences in inactivation properties from those of α1B alone (see Table 2). Together with the differentiating effects of the β subunit isoforms, these results confirm that amino-terminal disruption per se did not affect inactivation.
Potential overlap of determinants for VDCC β subunit and Gβγ subunit function
Within the α1B45-55 sequence, the combined mutation of two arginines to alanines (R52A, R54A) was shown previously to prevent modulation of the subunit by G proteins; furthermore, four individual point mutations (S48A, I49A, R52A and R54A) caused G protein modulation to be compromised (Canti et al. 1999). Here we have shown that both the α1B(R52,54A) and α1B(R52A) constructs also had compromised β2a function, as did α1B(Q47A), which was shown previously to undergo normal Gβγ modulation (Canti et al. 1999). A Gβγ-binding site on the Ca2+ channel α1 subunit intracellular I-II loop (De Waard et al. 1997; Zamponi et al. 1997) partially coincides with binding sites for auxiliary β subunits (Pragnell et al. 1994). However, further studies showed that the three amino acids critical for β subunit interaction are not within, but adjacent to, the QxxER consensus sequence implicated in Gβγ binding (De Waard et al. 1996). A partial overlap in VDCC β subunit- and Gβγ-binding sites has been proposed for the α1E carboxyl-terminal site (Qin et al. 1997). However, deletion of the majority of this α1E site prevented Gβγ modulation, but full sensitivity to β2a was retained, suggesting that another binding site is the prime mediator of the β subunit response (see also Jones et al. 1998). Taken together with our previous study (Canti et al. 1999), the results indicate that the α1B amino terminus contributes determinants for both VDCC β subunit and Gβγ subunit function. However, the differential effects of mutating Q47 indicate that although the overall region involved may partially coincide, the determinants are not identical.
Potential mechanisms for α1B amino terminus effects on β subunit function
The functional contribution of the α1B amino terminus to β subunit-mediated inactivation properties may be due to the presence of a direct β subunit-selective binding site in this region. Alternatively, an allosteric effect may be translated to the amino terminus when β subunits occupy a different binding site.
In addition to the α1 I-II loop and carboxyl-terminal β subunit-binding sites discussed above, an amino-terminal site has been identified recently (Walker et al. 1999). Amongst neuronal α1A, α1B and α1C subunits, only the α1A amino terminus was found to bind β subunits to any significant extent. The α1A-binding site was β subunit selective; importantly, it was shown that β subunits may occupy both the I-II loop and the amino-terminal β subunit-binding sites simultaneously (Walker et al. 1999). GST-α1B amino-terminal fusion proteins did not show any specific β subunit binding (Walker et al. 1999). The present data clearly demonstrate that the α1B amino-terminal region does contribute to β subunit function and so may appear to contradict the binding data. However, β subunit-binding affinity may be below the limits of detection in vitro, possibly compounded by the lack of secondary or tertiary structure. Interestingly, GST fusion proteins of specific regions of α1A amino terminus highlighted the role of residues 42–52 in β subunit binding, a very similar region to the α1B45-55 sequence which contains determinants for β subunit function and is important for Gβγ modulation (Canti et al. 1999).
The present study suggests that an amino-, rather than a carboxyl-, terminal site is required for the functional effects of β2a. The α1bCCCC carboxyl terminus is derived from α1C, therefore it does not contain a β subunit-binding sequence (Qin et al. 1997), and the different α1bCCCC properties can only be conferred by the amino terminus. In contrast, all constructs used in the present study do contain the high affinity I-II loop site (Pragnell et al. 1994). The relative reported affinities for different β subunit-binding sites would suggest that the I-II loop represents the primary β subunit interaction site in these constructs. It has been suggested that the VDCC I-II loop itself may dictate voltage-dependent inactivation properties, acting as a blocking particle analogous to the amino-terminal inactivation ball in voltage-dependent potassium channels (Cens et al. 1999). In this scenario, the I-II loop is stabilised by β subunit interaction. In the present study, the lack of the proximal amino terminus may somehow destabilise the β subunit interaction with the I-II loop to increase inactivation; as yet such a mechanism is still highly speculative.
We cannot discount the possibility that β subunit binding to the I-II loop transmits a conformational change to the amino terminus. However, given the caveats mentioned above, the demonstration of simultaneous β subunit binding to the α1A I-II loop and the amino terminus (Walker et al. 1999) makes a mechanism whereby the β subunit interacts directly with both the I-II loop and the amino terminus an attractive candidate to explain the functional data presented here for α1B. Assuming that the β subunit binds with high affinity to the α1B I-II loop site, we can speculate on the relative contribution of the amino terminus to β subunit function. The fact that deletion of the proximal half of the α1B amino terminus reduced β subunit-mediated inactivation, but did not fully reverse it to levels seen without β subunits, is consistent with this region contributing determinants to, but not being fully responsible for, β subunit-mediated voltage-dependent inactivation properties. The α1bCCCC construct retains the α1C I-II loop sequence, which would be expected to bind β2a with an affinity similar to α1B (De Waard et al. 1995). If we discount effects of the carboxyl-terminal β subunit-binding site as discussed above, this means that the differences in the β2a-mediated inactivation properties between α1bCCCC and α1C (illustrated in the normalised inactivation trace shown in Fig. 4A) represent the relative contribution of the α1B amino terminus. On these criteria the amino terminus contributes ∼25% of the α1 determinants for voltage-dependent inactivation. A similar percentage is obtained when comparing the values of β2a-mediated inactivation for α1B with the values for α1BΔ2-50 (∼26%) and α1BΔ1-55 (∼25%) (Fig. 2).
In summary, we have shown that the α1B amino terminus contributes determinants for β subunit function with β subunit-selective effects on voltage-dependent inactivation properties. Several lines of evidence support this conclusion. Firstly, deletion of the proximal half of the amino terminus partially opposes β subunit-mediated slowing of inactivation in a β subunit-selective manner. Secondly, transfer of the α1B amino terminus to an α1C backbone results in a gain-of-function chimera with inactivation properties no different from those of parental α1B in the presence of β2a. Thirdly, mutations within the α1B45-55 amino-terminal sequence also oppose the β2a-mediated slowing of inactivation. This work extends the hypothesis that the α1 subunit contains multiple sites required for complete β subunit function and highlights the role of β subunits in dictating α1 subunit voltage-dependent inactivation properties.
Acknowledgments
We thank the following for generous gifts of cDNAs: Dr Y. Mori (Seiriken, Okazaki, Japan), rabbit α1B; Dr T. Snutch (UBC, Vancouver, Canada), rat α1C, rat β1b; Dr H. Chin (NIH, Bethesda, MD, USA), rat α2-δ-1; Dr E. Perez-Reyes (Loyola University, Chicago, IL, USA), rat β2a, β3, β4; R. Lefkowitz (Duke University, Durham, NC, USA), β-ARK1; T. Hughes (Yale University, New Haven, CT, USA), mut-3 GFP; Genetics Institute (CA, USA), pMT2. We also gratefully acknowledge financial support from the Wellcome Trust, and thank N. Balaguero and M. Li for technical assistance and F. Bertaso for help in manuscript preparation.
G. J. Stephens and K. M. Page contributed equally to this work.
References
- Birnbaumer L, Qin N, Olcese R, Tareilus E, Platano D, Costantin J, Stefani E. Structures and functions of calcium channel β subunits. Journal of Bioenergetics and Biomembranes. 1998;30:357–375. doi: 10.1023/a:1021989622656. [DOI] [PubMed] [Google Scholar]
- Brice NL, Berrow NS, Campbell V, Page KM, Brickley K, Tedder I, Dolphin AC. Importance of the different β subunits in the membrane expression of the α1A and α2 calcium channel subunits: studies using a depolarization-sensitive α1A antibody. European Journal of Neuroscience. 1997;9:749–759. doi: 10.1111/j.1460-9568.1997.tb01423.x. [DOI] [PubMed] [Google Scholar]
- Campbell V, Berrow N, Brickley K, Page K, Wade R, Dolphin AC. Voltage-dependent calcium channel β-subunits in combination with α1 subunits have a GTPase activating effect to promote hydrolysis of GTP by Gαo in rat frontal cortex. FEBS Letters. 1995;370:135–140. doi: 10.1016/0014-5793(95)00813-o. [DOI] [PubMed] [Google Scholar]
- Canti C, Page KM, Stephens GJ, Dolphin AC. Identification of residues in the N-terminus of α1B critical for inhibition of the voltage-dependent calcium channel by Gβγ. Journal of Neuroscience. 1999;19:6855–6864. doi: 10.1523/JNEUROSCI.19-16-06855.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cens T, Restituito S, Galas S, Charnet P. Voltage and calcium use the same molecular determinants to inactivate calcium channels. Journal of Biological Chemistry. 1999;274:5483–5490. doi: 10.1074/jbc.274.9.5483. [DOI] [PubMed] [Google Scholar]
- Chien AJ, Carr KM, Shirokov RE, Rios E, Hosey MM. Roles of a membrane-localized β subunit in the formation and targeting of functional L-type Ca2+-channels. Journal of Biological Chemistry. 1995;270:30036–30044. doi: 10.1074/jbc.270.50.30036. [DOI] [PubMed] [Google Scholar]
- Chien AJ, Carr KM, Shirokov RE, Rios E, Hosey MM. Identification of palmitoylation sites within the L-type calcium channel β2a subunit and effects on channel function. Journal of Biological Chemistry. 1996;271:26465–26468. doi: 10.1074/jbc.271.43.26465. [DOI] [PubMed] [Google Scholar]
- Chien AJ, Hosey MM. Post-translational modifications of β subunits of voltage-dependent calcium channels. Journal of Bioenergetics and Biomembranes. 1998;30:377–386. doi: 10.1023/a:1021941706726. [DOI] [PubMed] [Google Scholar]
- De Waard M, Liu H, Walker D, Scott VE, Gurnett CA, Campbell KP. Direct binding of G-protein βγ complex to voltage-dependent calcium channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- De Waard M, Scott VE, Pragnell M, Campbell KP. Identification of critical amino acids involved in α1-β interaction in voltage-dependent Ca2+ channels. FEBS Letters. 1996;380:272–276. doi: 10.1016/0014-5793(96)00007-5. [DOI] [PubMed] [Google Scholar]
- De Waard M, Witcher DR, Pragnell M, Liu H, Campbell KP. Properties of the α1-β anchoring site in voltage-dependent Ca2+ channels. Journal of Biological Chemistry. 1995;27:12056–12064. doi: 10.1074/jbc.270.20.12056. [DOI] [PubMed] [Google Scholar]
- Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. [DOI] [PubMed] [Google Scholar]
- Gerster U, Neuhuber B, Groschner K, Striessnig J, Flucher BE. Current modulation and membrane targeting of the calcium channel α1C subunit are independent functions of the β subunit. The Journal of Physiology. 1999;517:353–368. doi: 10.1111/j.1469-7793.1999.0353t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon MR, Berrow NS, Dolphin AC, Wallace BA. Modelling of a voltage-dependent Ca2+ channel β subunit as a basis for understanding its functional properties. FEBS Letters. 1999;445:366–370. doi: 10.1016/s0014-5793(99)00156-8. [DOI] [PubMed] [Google Scholar]
- Hering S, Berjukow S, Aczel S, Timin EN. Ca2+ channel block and inactivation: common molecular determinants. Trends in Pharmacological Sciences. 1998;19:439–443. doi: 10.1016/s0165-6147(98)01258-9. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Hockerman GH, Scheuer T, Catterall WA. Molecular determinants of inactivation and G protein modulation in the intracellular loop connecting domains I and II of the calcium channel α1A subunit. Proceedings of the National Academy of Sciences of the USA. 1997;94:1512–1516. doi: 10.1073/pnas.94.4.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann F, Biel M, Flockerzi V. Molecular basis for Ca2+ channel diversity. Annual Review of Neuroscience. 1994;17:399–418. doi: 10.1146/annurev.ne.17.030194.002151. [DOI] [PubMed] [Google Scholar]
- Jones LP, DeMaria CD, Yue DT. N-type calcium channel inactivation probed by gating-current analysis. Biophysical Journal. 1999;76:2530–2552. doi: 10.1016/S0006-3495(99)77407-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LP, Wei SK, Yue DT. Mechanism of auxiliary subunit modulation of neuronal α1E calcium channels. Journal of General Physiology. 1998;112:125–143. doi: 10.1085/jgp.112.2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SW. Inactivation of N-type Ca2+ channels: Ca2+vs. voltage. The Journal of Physiology. 1999;518:630. doi: 10.1111/j.1469-7793.1999.0630p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamp TJ, Pérez-García MT, Marban E. Enhancement of ionic current and charge movement by coexpression of calcium channel β1A subunit with α1C subunit in a human embryonic kidney cell line. The Journal of Physiology. 1996;492:89–96. doi: 10.1113/jphysiol.1996.sp021291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letts VA, Felix R, Biddlecome GH, Arikkath J, Mahaffey CL, Valenzuela A, Bartlett FS, Mori Y, Campbell KP, Frankel WN. The mouse stargazer gene encodes a neuronal Ca2+-channel gamma subunit. Nature Genetics. 1998;19:340–347. doi: 10.1038/1228. [DOI] [PubMed] [Google Scholar]
- Neely A, Wei X, Olcese R, Birnbaumer L, Stefani E. Potentiation by the β subunit of the ratio of the ionic current to the charge movement in the cardiac calcium channel. Science. 1993;262:575–578. doi: 10.1126/science.8211185. [DOI] [PubMed] [Google Scholar]
- Olcese R, Qin N, Schneider T, Neely A, Wei X, Stefani E, Birnbaumer L. The amino terminus of a calcium channel β subunit sets rates of channel inactivation independently of the subunit's effect on activation. Neuron. 1994;13:1433–1438. doi: 10.1016/0896-6273(94)90428-6. [DOI] [PubMed] [Google Scholar]
- Page KM, Canti C, Stephens GJ, Berrow NS, Dolphin AC. Identification of the amino terminus of neuronal Ca2+ channel α1 subunits α1B and α1E as an essential determinant of G-protein modulation. Journal of Neuroscience. 1998;18:4815–4824. doi: 10.1523/JNEUROSCI.18-13-04815.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil PG, Brody DL, Yue DT. Preferential closed-state inactivation of neuronal calcium channels. Neuron. 1998;20:1027–1038. doi: 10.1016/s0896-6273(00)80483-3. [DOI] [PubMed] [Google Scholar]
- Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP, Campbell KP. Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature. 1994;368:67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- Qin N, Platano D, Olcese R, Costantin JL, Stefani E, Birnbaumer L. Unique regulatory properties of the type 2a Ca2+ channel β subunit caused by palmitoylation. Proceedings of the National Academy of Sciences of the USA. 1998;95:4690–4695. doi: 10.1073/pnas.95.8.4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin N, Platano D, Olcese R, Stefani E, Birnbaumer L. Direct interaction of Gβγ with a C-terminal Gβγ-binding domain of the Ca2+ channel α1 subunit is responsible for channel inhibition by G protein-coupled receptors. Proceedings of the National Academy of Sciences of the USA. 1997;94:8866–8871. doi: 10.1073/pnas.94.16.8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirokov R. Interaction between permeant ions and voltage sensor during inactivation of N-type Ca2+ channels. The Journal of Physiology. 1999;518:697–703. doi: 10.1111/j.1469-7793.1999.0697p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaetgens RL, Zamponi GW. Multiple structural domains contribute to voltage-dependent inactivation of rat brain α1E calcium channels. Journal of Biological Chemistry. 1999;274:22428–22436. doi: 10.1074/jbc.274.32.22428. [DOI] [PubMed] [Google Scholar]
- Stephens GJ, Brice NL, Berrow NS, Dolphin AC. Facilitation of rabbit α1B calcium channels: involvement of endogenous Gβγ subunits. The Journal of Physiology. 1998;509:15–27. doi: 10.1111/j.1469-7793.1998.015bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swick AG, Janicot M, Cheneval-Kastelic T, McLenithan JC, Lane MD. Promoter-cDNA-directed heterologous protein expression in Xenopus laevis oocytes. Proceedings of the National Academy of Sciences of the USA. 1992;89:1812–1816. doi: 10.1073/pnas.89.5.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tareilus E, Roux M, Qin N, Olcese R, Zhou J, Stefani E, Birnbaumer L. A Xenopus oocyte β subunit: evidence for a role in the assembly/expression of voltage-gated calcium channels that is separate from its role as a regulatory subunit. Proceedings of the National Academy of Sciences of the USA. 1997;94:1703–1708. doi: 10.1073/pnas.94.5.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker D, Bichet D, Campbell KP, De Waard M. A β4 isoform-specific interaction site in the carboxyl-terminal region of the voltage-dependent Ca2+ channel α1A subunit. Journal of Biological Chemistry. 1998;273:2361–2367. doi: 10.1074/jbc.273.4.2361. [DOI] [PubMed] [Google Scholar]
- Walker D, Bichet D, Geib S, Mori E, Cornet V, Snutch TP, Mori Y, De Waard M. A new β subtype-specific interaction in α1A subunit controls P/Q-type Ca2+ channel activation. Journal of Biological Chemistry. 1999;274:12383–12390. doi: 10.1074/jbc.274.18.12383. [DOI] [PubMed] [Google Scholar]
- Walker D, De Waard M. Subunit interaction sites in voltage-dependent Ca2+ channels: role in channel function. Trends in Neurosciences. 1998;21:148–154. doi: 10.1016/s0166-2236(97)01200-9. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Hara M, Strobeck M, Fukasawa K, Schwartz A, Varadi G. Multiple modulation pathways of calcium channel activity by a β subunit. Direct evidence of β subunit participation in membrane trafficking of the α1C subunit. Journal of Biological Chemistry. 1998;273:19348–19356. doi: 10.1074/jbc.273.30.19348. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C is mediated by the calcium channel α1 subunit I-II linker. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- Zhang J-F, Ellinor PT, Aldrich RW, Tsien RW. Molecular determinants of voltage-dependent inactivation in calcium channels. Nature. 1994;372:97–100. doi: 10.1038/372097a0. [DOI] [PubMed] [Google Scholar]