

Abstract
Aminoglycoside antibiotics such as gentamicin are known to block the medial olivocochlear efferent system. In order to determine whether this inhibition takes place at the postsynaptic cholinergic receptors in outer hair cells (OHCs), we studied the effects of these polycationic molecules on cholinergic currents evoked in isolated guinea-pig OHCs.
The cholinergic response of OHCs involves nicotinic-like receptors (nAChRs) permeable to Ca2+ ions that activate nearby Ca2+-sensitive K+ channels (KCa(ACh) channels). The extracellular application of gentamicin and neomycin reversibly blocked ACh-evoked K+ current (IK(ACh)) with IC50 values of 5.5 and 3.2 μm, respectively. The results showed that the blocking mechanism of IK(ACh) was due to inhibition of Ca2+ influx via nAChRs.
Our study also provides interesting insights into the functional coupling between nAChRs and KCa(ACh) channels in OHCs. By directly recording the cation current flowing through nAChRs (In(ACh)) using an intracellular solution containing 10 mm BAPTA, we measured an EC50 near 110 μm for ACh-evoked In(ACh). This EC50 for ACh is one order of magnitude higher than that measured indirectly on IK(ACh). This reveals a rather low affinity of ACh for its receptor but a very efficient coupling between nAChRs and KCa(ACh) channels.
We also show that a high external Ca2+ concentration reverts the gentamicin inhibition of IK(ACh) and that gentamicin directly alters the cation current flowing through the nAChRs of OHCs. We propose that gentamicin acts as a non-competitive cholinergic blocker by displacing Ca2+ from specific binding sites at the nAChRs. This block of the nAChRs at the level of the postsynaptic membrane in OHCs could explain the inhibitory effect of gentamicin reported on the crossed medial olivocochlear efferent system in vivo.
Aminoglycoside antibiotics are powerful antimicrobial drugs that bind with high affinity to bacterial ribosomes and inhibit protein synthesis. However, the therapeutic use of these molecules is limited by serious side effects such as nephrotoxicity, ototoxicity and muscle paralysis. While nephro- and ototoxicity seem to depend on intracellular accumulation of these antibiotics (Silverblatt & Kuehn, 1979; Dulon et al. 1993), numerous studies have demonstrated the ability of these polycationic drugs to acutely depress synaptic transmission at the neuromuscular junction, presumably by blocking presynaptic voltage-gated Ca2+ channels (Vital Brazil & Prado-Franceschi, 1969; Prado et al. 1978). These polycationic drugs also block a wide variety of ion channels such as mechanosensitive ion channels (Ohmori, 1985; Kroese et al. 1989; Winegar et al. 1996), purinergic ionotropic channels (Lin et al. 1993) and nicotinic ACh receptors (Okamoto & Sumikawa, 1991; Nishizaki et al. 1994). The molecular mechanisms by which these drugs block these different ion channels in the cell plasma membrane still remain poorly understood.
The aminoglycoside antibiotic gentamicin has also been shown to produce acute side effects in cochlear neurotransmission, characterized by a rapid and reversible block of the medial olivocochlear efferent system (Smith et al. 1994; Lima da Costa et al. 1997, 1998; Yoshida et al. 1999). This efferent system is believed to modulate cochlear micromechanics by regulating the contractile properties of outer hair cells (OHCs) (Brownell et al. 1985; Dallos et al. 1997) and hence to provide a means of adjusting the auditory sensitivity. At doses that do not produce hearing loss, a single injection of gentamicin in the guinea-pig was shown to block the suppression effects of the medial olivocochlear efferent system. The medial efferent fibres form synapses at the basal pole of OHCs and use ACh as the main neurotransmitter (Galambos, 1956; Bobbin & Konishi, 1971; Kujawa et al. 1992). Although the OHC cholinergic synapses have been proposed as the site of the aminoglycoside action (Lima da Costa et al. 1997; Yoshida et al. 1999), we still do not know whether these drugs act presynaptically on voltage-gated Ca2+ channels or postsynaptically at the cholinergic receptors.
In whole-cell patch clamp experiments, ACh has been shown to hyperpolarize OHCs by triggering an outward K+ current (Housley & Ashmore, 1991; Eróstegui et al. 1994; Blanchet et al. 1996; for review see Fuchs, 1996). Similar to the responses of chick cochlear hair cells (Fuchs & Murrow, 1992), the OHC cholinergic responses involve ligand-gated receptors permeable to Ca2+ and co-localized Ca2+-activated K+ channels (Blanchet et al. 1996; Dulon & Lenoir, 1996; Evans, 1996). Strong evidence suggests that the cholinergic ionotropic receptors of OHCs contain nicotinic α9 subunits (Elgoyhen et al. 1994; Glowatzki et al. 1995) while the co-localized Ca2+-activated K+ channels belong to the SK type (Nenov et al. 1996b; Dulon et al. 1998).
The aim of the present study was to determine whether the inhibition of the medial efferent system observed in vivo by gentamicin could be explained by block of the cholinergic receptors at the postsynaptic membrane in OHCs. Using the whole-cell patch clamp recording technique, we studied the effects of gentamicin and two other aminoglycoside antibiotics on ACh-evoked currents in isolated guinea-pig OHCs. Our results indicate that these drugs inhibit the cholinergic response of OHCs in the micromolar range by blocking the Ca2+ influx through the ACh receptor channel.
METHODS
Preparation of isolated cochlear outer hair cells
Guinea-pig outer hair cells were isolated as previously described (Dulon et al. 1990; Blanchet et al. 1996). Briefly, young guinea-pigs (weight 200–300 g) were deeply anaesthetized with an intramuscular injection of 0.3 ml of a mixed solution of 2/3 ketamine hydrochlorate (50 mg ml−1, Ketalar; Parke-Davis, France) and 1/3 xylazine (2 %, Rompum; Bayer, Germany) and decapitated. The bullae were separated and placed in Hanks’ balanced salt solution (HBSS; Sigma), containing (mM): NaCl 136.9, KCl 5.4, MgSO4 0.81, CaCl2 1.26, KH2PO4 0.44, Na2HPO4 0.34, Hepes 5 and glucose 5.5. The HBSS was adjusted to pH 7.35 with 2 mM NaOH and to 300 mosmol (kg H2O)−1 with 6 mM NaCl. The two middle turns of each organ of Corti were dissected and transferred for 10 min to a 40 μl drop of HBSS containing collagenase (type IV, final concentration 0.8-1 mg ml−1, Sigma). The pieces of organ of Corti were transferred to a 50 μl drop of HBSS in the middle of a glass coverslip attached to the perforated bottom of a Petri dish. The cells were then mechanically dissociated with gentle trituration with a Gilson micropipette (Gilson, France), and left to settle for 30 min. The dish was then filled with 3–4 ml of HBSS.
Animals were treated in accordance with French Ministry of Agriculture guidelines in agreement with EEC regulations.
Electrophysiological recordings
Outer hair cells were recorded under voltage clamp configuration with electrodes pulled from borosilicate glass capillaries (GC150TF-10 Clark Electromedical, UK), on a Sachs-Flaming horizontal electrode puller (Sutter Instruments, USA). Recording electrodes were back filled with one of the following internal solutions. Solution 1 (mM): KCl 136, KOH 28, MgCl2 1.5, CaCl2 0.1, EGTA 11, Hepes 5; solution 2 (mM): CsCl 100, KCl 50, KOH 3.5, NaCl 2, MgCl2 2, EGTA 1.1, Hepes 5, glucose 4; solution 3 (mM): KCl 100, KOH 39, NaCl 5.8, MgCl2 2, CaCl2 0.1, BAPTA 10, Hepes 5, glucose 47. All solutions were adjusted to pH 7.20 and to 300 mosmol (kg H2O)−1. Electrode resistance ranged from 3 to 6 MΩ. Patch clamp recordings were performed as previously described in detail (Blanchet et al. 1996) by means of either an Axopatch-1D amplifier (Axon Instruments, Foster City, CA, USA) or a Biologic RK-400 amplifier (Biologic Science Instruments, France). Axotape and pCLAMP software (Axon Instruments) were used for data collection and analysis. Electrode capacitance was compensated as much as possible and series resistance was compensated at 80 %. Voltage errors attributable to residual uncompensated series resistance (less than 5 mV in all recordings) were not corrected for, but those due to liquid junction potentials were corrected for during data analysis (Blanchet et al. 1996). All experiments were performed at room temperature (20-22°C).
Drug application
The test solutions were applied to OHCs by two different drug delivery systems: a U-tubing microperfusion system described elsewhere (Eróstegui et al. 1994) and a Picospritzer puffer system (Picospritzer II, General Valve, Fairfield, NJ, USA) as previously described (Blanchet et al. 1996).
The U-tubing system consisted of a small glass pipette (about 4 mm long and 100 μm internal diameter) inserted in a U-shaped polyethylene tube connected to a valve and to a vacuum pump by one edge and to the test solutions by the other. The tip of the pipette was positioned approximately 200 μm from cells and the response delay, measured by perfusion of a high K+ solution, ranged from 600 to 800 ms. Although presenting a longer delay, the U-tubing system triggered ACh-evoked K+ currents in a similar way to a pressure-puff application. However, because of its rapid desensitization, it was very difficult to record the early cationic current using a slow drug-delivery system such as the U-tubing system. When we used the U-tubing system for drug application, the cells were continuously perfused at about 200 μl min−1 with HBSS (Eróstegui et al. 1994) to effect rapid rinsing of the drugs.
Pipettes for the Picospritzer system were pulled similarly to the recording patch clamp pipettes and were placed about 10–20 μm from the basal pole of OHCs. The delay for drugs to reach the cell ranged from 20 to 50 ms as estimated by the junction potential change induced by high K+ solutions.
In experiments with high (10 mM) or low (0.126 mM) Ca2+-containing solutions, only the calcium chloride concentration was altered. Acetylcholine chloride (ACh), neomycin sulphate (NM), streptomycin sulphate (SM) and gentamicin sulphate (GM) were obtained from Sigma and dissolved at the desired concentration on the day of the experiment in HBSS. When repetitive applications of ACh on the same OHC were done, we waited 30 s to 2 min between each application in order to reduce the desensitization of the cholinergic response. Dose-response curves were analysed using GraphPad Prism software (GraphPad Software Inc, San Diego, CA, USA).
RESULTS
The ACh-evoked K+ current depends on a Ca2+ influx
A pressure-puff application of 100 μM ACh (ACh100) typically triggered a rapidly activating outward current when OHCs were voltage clamped at -40 mV (Fig. 1A). This hyperpolarizing current started with a mean latency of 30 ms with respect to the onset of the ACh puff and usually peaked in less than 200 ms when the puff pipette was positioned 10–20 μm away from the base of the cell. Following the peak, the ACh-induced outward current presented a slowly decaying plateau that ended rapidly after the end of the puff. We have previously shown that this outward current is essentially carried by K+ ions and is triggered secondarily to Ca2+ influx through nicotinic-like receptors (nAChRs; Blanchet et al. 1996). Another strong indication of the involvement of Ca2+ influx in ACh-activated K+ currents (IK(ACh)) is given in Fig. 1. Lowering the Ca2+ concentration from 1.26 mM to 0.126 mM in the ACh puff-applied solution (ACh100Ca/10), while OHCs were maintained in 1.26 mM Ca2+ HBSS, reversibly reduced the peak amplitude and the plateau phase of IK(ACh) (Fig. 1A). The plateau phase of IK(ACh) was more affected than the initial peak, suggesting that the decrease in the Ca2+ concentration in the vicinity of the nAChRs during the puff was relatively slow. Indeed, when the same experiment was performed with OHCs directly bathed in a low Ca2+ solution (0.126 mM Ca2+), IK(ACh) was completely abolished when applying ACh100Ca/10, probably because the extracellular Ca2+ concentration in the vicinity of the nAChRs was already low at the beginning of the puff (n= 5; data not shown).
Figure 1. IK(ACh) depends on a Ca2+ influx.
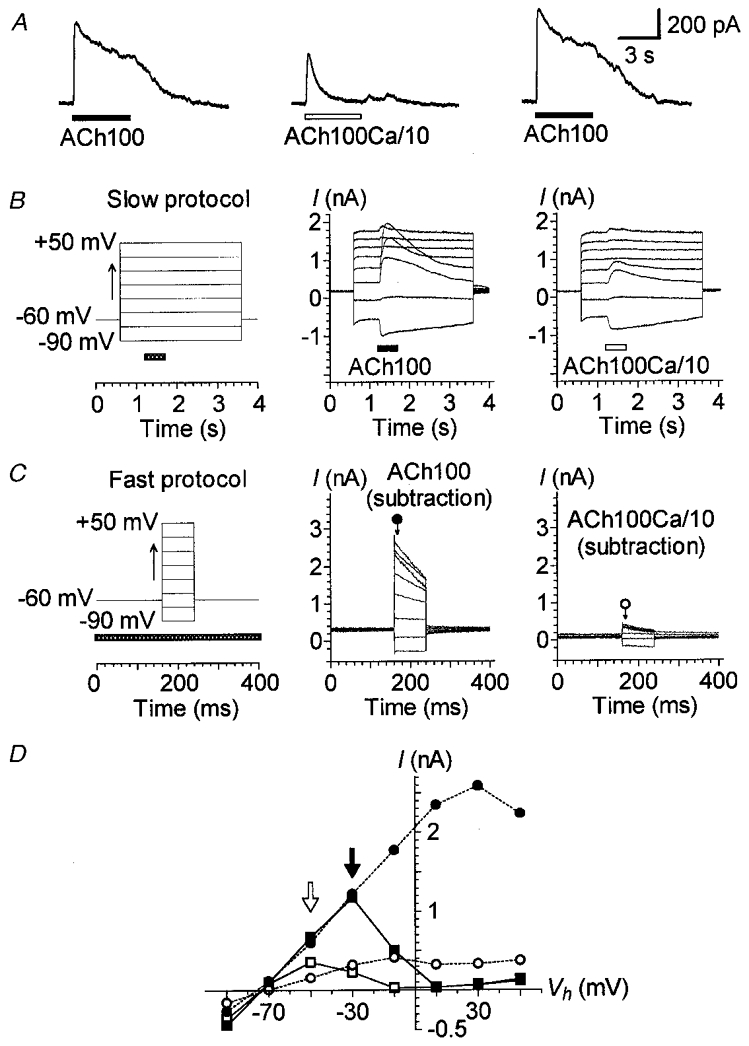
A, effect of lowering extracellular Ca2+ concentration on IK(ACh). ACh (100 μM) was sequentially applied with 1 min intervals between applications. Stimulation was via two puff pipettes containing either a normal carrier solution (1.26 mM CaCl2: ACh100) or a carrier solution with a tenfold lower concentration of Ca2+ (0.126 mM CaCl2: ACh100Ca/10). Vh was set at -40 mV. B, effect of ACh100 and ACh100Ca/10 at different membrane potentials. The cell was stimulated by a slow protocol (left) consisting of 3 s voltage steps to between -90 and +50 mV in 20 mV increments triggered from a Vh of -60 mV. Voltage steps were spaced 30 s apart. ACh100 (middle) or ACh100Ca/10 (right) was applied (horizontal bar) for 500 ms at each voltage step, 600 ms after its onset. C, effect of varying the Vh during an ACh100 or ACh100Ca/10 application. This fast protocol (left) consisted of 80 ms voltage steps to between -90 and +50 mV in 20 mV increments from a Vh of -60 mV spaced 400 ms apart. The fast protocol was performed without ACh application (control), during a continuous 5 s application of ACh100, again in the absence of ACh application (control) and finally during a continuous 5 s application of ACh100Ca/10. The whole sequence was performed twice with a 1 min interval between applications. The pressure-puff application (horizontal bar) of ACh100 or ACh100Ca/10 started 1 s before the voltage-step protocol. The traces are net currents obtained after subtraction of control (leak) currents from those obtained during an ACh100 (middle) or ACh100Ca/10 (right) application. Current amplitudes reported in D were measured 8 ms after the onset of each voltage step as indicated by the arrows under the corresponding symbols. D, I-V relationships of the responses presented in B and C. Currents triggered by ACh100 (filled symbols) and ACh100Ca/10 (open symbols) were measured at their maximum amplitude for the slow protocol (squares) and as described in C for the fast protocol (circles). Filled and open arrows point to the maximum amplitude of the I–V relationships for ACh100- and ACh100Ca/10-evoked currents during the slow protocol, respectively. Recordings in A, B and C are from the same cell dialysed with internal solution 1. All these effects were reversible and repeated several times.
Further evidence for a Ca2+ influx was also visible in the shape of the current-voltage (I–V) relationship of IK(ACh) and its modifications upon different voltage-step protocols with normal or low Ca2+ concentration in the ACh carrier solution (Fig. 1B–D). Although similar experiments on isolated OHCs have been reported (Eróstegui et al. 1994; Evans, 1996; Nenov et al. 1996a,b), we have repeated and extended these experiments in order to determine the effect of Ca2+ depletion on IK(ACh) under our own recording conditions. Two voltage-step protocols were used: first, a slow protocol that allowed 30 s recovery between each ACh application at different holding potentials; second, a fast protocol that consisted of brief voltage steps during a single ACh application. The slow protocol consisted of 3 s voltage steps to between -90 and +50 mV triggered every 30 s from a holding potential (Vh) of -60 mV. ACh100 was sequentially applied for 0.5 s at each voltage step (Fig. 1B). The I–V relationship of IK(ACh) was N-shaped, with a maximum around -30 mV (Fig. 1D, filled arrow) and a minimum near +10 mV. As we have previously shown (Blanchet et al. 1996), the decrease of the amplitude of IK(ACh) between -30 and +10 mV is due to a decrease in the driving force for Ca2+ ions, and therefore a decrease in the Ca2+ influx through the nAChRs. The increase in the current recorded above +10 mV could be explained by the outwardly rectifying nicotinic cation current (In(ACh)). From -90 to -30 mV, but not above, the Ca2+ influx, although decreasing, should have been sufficient to fully activate the Ca2+-dependent K+ current, as suggested by the linearity of the I–V relationship. When we used the same slow voltage-step protocol but applied ACh100Ca/10 instead of ACh100, the peak amplitude of IK(ACh) was reduced at all negative membrane potentials. The I–V relationship displayed a flattened N-shape with a maximum shifted toward hyperpolarized potentials (-50 instead of -30 mV, see arrows in Fig. 1D) and a somewhat inward rectification below. These observations indicated that, from -90 to -10 mV, the Ca2+ influx decreased and as a result activated fewer Ca2+-sensitive K+ (KCa(ACh)) channels. For instance, the effect of low Ca2+ was much less obvious at -90 mV than at -10 mV because at -90 mV the driving force for Ca2+ ions was much larger. This allowed a Ca2+ influx which was nearly sufficient to activate as many KCa(ACh) channels as in normal conditions despite the large reduction of external Ca2+. Otherwise, at positive potentials, the ACh-induced current In(ACh) was apparently not affected by the low extracellular Ca2+ concentration.
The fast protocol consisted of 80 ms voltage steps to between -90 and +50 mV triggered every 400 ms from a Vh of -60 mV. This fast protocol was performed before, during and after a single 5 s ACh application. ACh-evoked currents (Fig. 1C) were obtained after subtraction of the leak currents measured with the fast protocol before the ACh application from the current recorded during the ACh application (full return to control leak currents was assessed by fast protocols performed after the ACh application). In these conditions, ACh100 triggered a large current whose amplitude and speed of decline increased with depolarization. When the measurements of the current amplitude were made immediately after the onset of each voltage step (as indicated in Fig. 1C), the I–V relationship of IK(ACh) displayed a different profile from the one recorded with I–V relationships deduced from slow voltage-step protocols (Fig. 1D). With a standard concentration of Ca2+ (1.26 mM) in the ACh carrier solution (ACh100), the I–V curve from -90 to -30 mV followed a similar pattern to that obtained with the slow protocol. However, above -30 and up to +10 mV, the I–V curve obtained with the fast protocol flattened out. This could be due to the fact that when using a fast voltage-step protocol, the amplitude of IK(ACh) at the beginning of each step potential depends on the Ca2+ entering at the initial holding potential (i.e. -60 mV), this Ca2+ influx being sufficient to activate all KCa(ACh) channels.
With a low Ca2+ concentration (0.126 mM) in the carrier solution (ACh100Ca/10), the corresponding I–V curve obtained with the fast protocol was also strongly flattened, presumably because the Ca2+ influx at -60 mV was much smaller, activating in consequence only a fraction of the KCa(ACh) channels. Furthermore, it is interesting to note that the decline in IK(ACh) increased during the most depolarized step potentials in both conditions (ACh100 and ACh100Ca/10; Fig. 1C). This current decline was probably dependent on the decrease in the driving force for Ca2+ ions during depolarized step potentials, which was stronger with larger depolarization, and on the speed of intracellular Ca2+ buffering near KCa(ACh) channels.
Aminoglycoside antibiotics reversibly blocked IK(ACh)
The effect of gentamicin was first assessed by comparing the responses of OHCs to pressure-puff applications of ACh with or without the antibiotic. All tested OHCs showed a reversible decrease in their IK(ACh) when 100 μM ACh was co-applied with 50 μM GM (ACh100GM50, Fig. 2A; representative of 6 cells). In the presence of GM, the maximum amplitude of IK(ACh) was decreased by 40–90 % and at the end of a 2 s application by 70–100 %, thus mimicking the effect of a low Ca2+ concentration (compare with Fig. 1A). The dose dependency of the effect of GM was measured by means of the U-tubing system in order to test several aminoglycoside concentrations on each OHC. Since the effect of GM was progressive and stable only after more than 5 s (not shown), IK(ACh) amplitudes were measured at the end of each 10 s application. In these conditions, the dose-inhibition curve of IK(ACh) indicated 50 % inhibition (IC50) at a concentration of 5.5 μM GM and a Hill coefficient (nH) of 0.95 (Fig. 2B). Similarly, neomycin (NM) also blocked IK(ACh) of five OHCs with an IC50 of 3.2 μM and a nH of 0.38 (Fig. 2B). The Hill coefficients for NM and GM were significantly different (P < 0.05; Student's unpaired t test). Compared with GM or NM, streptomycin (SM) was a much less potent inhibitor of IK(ACh) as tested in five other OHCs; at the highest concentration tested (i.e. 100 μM), the response was inhibited by only 33 % (Fig. 2B). In the rest of the study, we focused our experiments on the characterization of the block by GM, the only aminoglycoside antibiotic together with netilmicin which has been shown to block the medial efferent system in vivo (Lima da Costa et al. 1998).
Figure 2. Aminoglycoside antibiotics reversibly block IK(ACh) in a dose-dependent manner.
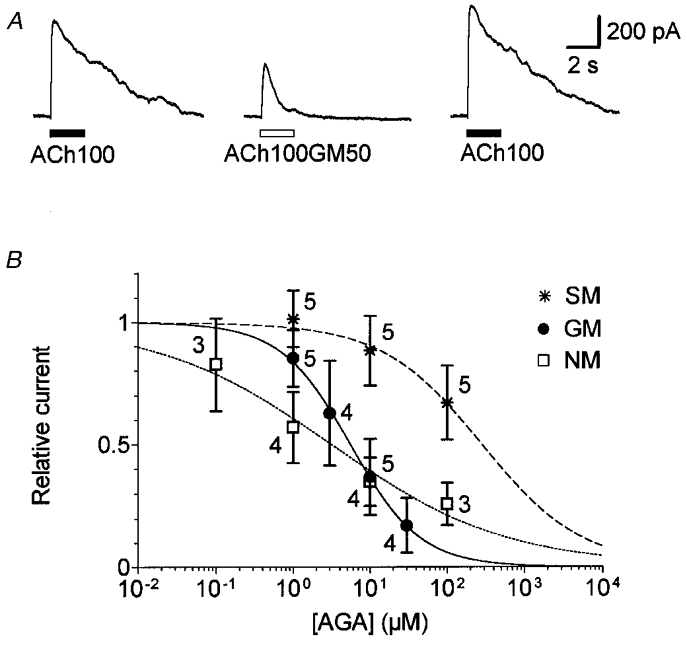
A, effect of 50 μM gentamicin on IK(ACh). Cholinergic responses were evoked by sequential pressure-puff applications of 100 μM ACh alone (ACh100) or in combination with 50 μM gentamicin (ACh100GM50) with 1 min intervals between applications. Vh was set at -40 mV. B, relative amplitude of IK(ACh) as a function of aminoglycoside antibiotic (AGA) concentration. ACh was applied at 100 μM for 10 s alone and with increasing concentrations of gentamicin (•), neomycin (□) or streptomycin (*) by means of the U-tubing system. The amplitude of IK(ACh) was measured at the steady state (i.e. at the end of each 10 s application). To maximize current sizes, measurements were taken at 0 mV, 20 ms after a voltage step from -60 mV, as previously described (Eróstegui et al. 1994). Current amplitudes were expressed relative to the initial control value for each cell. Data represent the mean value (±s.d.) with the number of cells tested indicated beside each symbol. Dose-inhibition curves are the best fits calculated from the empirical Hill equation Y= 1/(1 + (X/IC50)nH) where X is the AGA concentration. For GM, IC50= 5.5 μM and nH= 0.95. For NM, IC50= 3.2 μM and nH= 0.38. For SM, IC50 and nH values were extrapolated to 282.6 μM and 0.66, respectively.
Gentamicin impaired the Ca2+ influx necessary to activate IK(ACh)
The effects of GM on IK(ACh) were studied as a function of membrane potential. When we used a slow protocol identical to that described in Fig. 1, ACh-induced currents were reduced at all membrane potentials when 50 μM GM was co-applied with 100 μM ACh (Fig. 3A and B). The peak IK(ACh) amplitude in the presence of GM displayed a flattened N-shape when plotted as a function of membrane potential (Fig. 3B). Moreover, the maximum amplitude of IK(ACh) in the presence of GM (open arrow) shifted toward more negative potentials with respect to the I–V relationship obtained with 100 μM ACh alone. These GM effects were observed on the two OHCs tested and were remarkably similar at negative potentials to the effects obtained with a low Ca2+ concentration in the carrier solution instead of GM (compare with Fig. 1B and D). This suggested that a similar mechanism might block IK(ACh), i.e. a decrease in the Ca2+ influx flowing through nAChRs. Interestingly, at positive potentials, the ACh-induced current (which here corresponded to the ionotropic current In(ACh)) also appeared to be inhibited by GM, suggesting that GM might directly affect the nAChRs of OHCs.
Figure 3. Block of IK(ACh) by gentamicin at different membrane potentials.
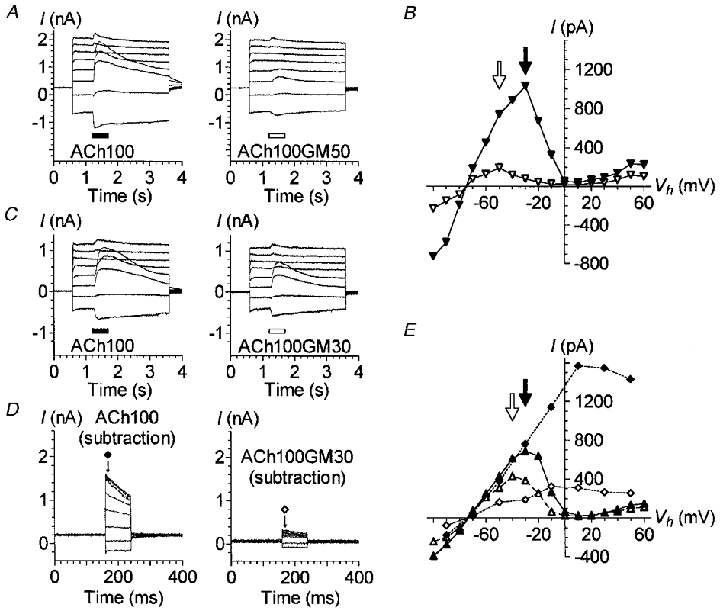
A, effect of 100 μM ACh alone or in combination with 50 μM GM at different membrane potentials. The cell was stimulated with a slow protocol as described in Fig. 1B. ACh100 (left) or ACh100GM50 (right) was applied for 500 ms at each voltage step, 600 ms after its onset. B, I-V relationships of the responses displayed in A, plus others obtained subsequently with a similar slow protocol but with voltage steps to between -100 and +60 mV. Currents triggered by ACh100 (▾) and ACh100GM50 (▿) were measured at their maximum amplitude (filled and open arrows, respectively). C, same experiment as in A, except that 30 μM GM, rather than 50 μM, was co-applied with ACh (ACh100GM30). D, effect of varying the Vh during an ACh100 or ACh100GM30 application using the same fast protocol as described in Fig. 1C. The traces shown are the net currents obtained after subtraction of the control (leak) currents recorded with the fast protocol in between ACh100 and ACh100GM30 applications. Current amplitudes reported in E were measured 8 ms after the onset of each voltage step as indicated by the arrows under the corresponding symbols. E, I-V relationships of the responses presented in D and C plus others as described in B. Currents triggered by ACh100 (filled symbols) and ACh100GM30 (open symbols) were measured at their maximum amplitude for the slow protocol (triangles) and as described in D for the fast protocol (diamonds). Filled and open arrows indicate the maximum amplitude of the I–V relationships for ACh100- and ACh100GM30-evoked currents during the slow protocol, respectively. A and B are from the same cell, C–E are from another cell. Both cells were dialysed with internal solution 1. All these effects were reversible and repeated several times.
Two other cells were challenged with 100 μM ACh with or without 30 μM GM (ACh100GM30) at various membrane potentials by means of slow voltage-step protocols. This lower GM concentration (30 instead of 50 μM) also reduced ACh-evoked currents at all potentials (Fig. 3C and E). The effects were, however, less pronounced, the I–V relationship plotted from ACh100GM30-induced peak currents being much less flattened and its maximum (open arrow in Fig. 3E) shifted only 10 mV toward more negative potentials with respect to the I–V relationship obtained with ACh100 alone.
The same two OHCs were also submitted to fast voltage-step protocols before, during and after a single 5 s application of ACh100 or ACh100GM30. The recordings obtained with ACh100 or ACh100GM30 after subtraction of the leak currents are displayed in Fig. 3D. The GM effects on the I–V relationships of IK(ACh) were again remarkably similar to those observed with a low Ca2+ concentration in the carrier solution, i.e. a flattening of the I–V curve (compare Fig. 3D and E with Fig. 1C and D). As explained above, this might be due to the fact that with the fast protocol, the IK(ACh) amplitude at the beginning of each step potential depended on the amount of Ca2+ entering at the holding potential (i.e. -60 mV), and that this amount was largely reduced in the presence of GM.
All these observations suggested that GM decreased the Ca2+ influx flowing through the nAChRs in a dose-dependent manner. Interestingly, Fig. 3 shows that GM also reduced the ACh-induced current at positive potentials above +20 mV. At these potentials, IK(ACh) should no longer be activated because the Ca2+ influx is drastically reduced. The current remaining at these positive potentials should in fact correspond only to the ionotropic current, i.e. the current flowing through the nAChRs (Blanchet et al. 1996 and see below).
To further ensure that GM effectively impaired Ca2+ influx through nAChRs, we studied the effects of 30 μM GM with a modified fast protocol, called the tail protocol. This protocol was identical to the fast protocol of Figs 1 and 3, except that the voltage steps started from a holding potential of -10 instead of -60 mV (Fig. 4A). With such a protocol, voltage steps were triggered about 1 s after the beginning of the ACh application, i.e. during the plateau phase of IK(ACh). At -10 mV and in the absence of GM, the amplitude of the ACh-activated current at the plateau phase was close to zero before the voltage steps (about 40 pA in Fig. 4A, bottom right panel). This indicates that the Ca2+ influx at this potential was probably just at the threshold to allow activation of only a few KCa(ACh) channels. During the brief 80 ms voltage steps ranging from -90 to -30 mV, but not above, the IK(ACh) amplitude increased slowly. However, large tail currents were observed immediately after the return to -10 mV. The more hyperpolarized the cell during the preceding step potential, the larger the tail current. These observations may be easily explained if we consider that the driving force for Ca2+ was enhanced during voltage steps to potentials below -10 mV. The enhanced Ca2+ influx (through the cholinergic receptor channels) therefore allowed intracellular accumulation of Ca2+ ions near the KCa(ACh) channels and their progressive activation. Shifting the membrane potential back to -10 mV suddenly enhanced the driving force for K+ ions and thus triggered large tail currents. Examining the tail current amplitudes of IK(ACh) as a function of the preceding step-potential value thus provided an indirect quantification of the Ca2+ influx during each step potential (Fig. 4B). In the presence of 30 μM GM, IK(ACh) was not apparently activated at -10 mV before or after the voltage steps (Fig. 4A, bottom left panel). Nor was it activated during the voltage steps, with the exception of some transitory activation at -90 mV. It is to be noted that the five OHCs tested with this tail protocol displayed reduced but still large IK(ACh) with ACh100GM30 when the membrane potential was maintained at -40 mV or stepped from -60 mV with a normal fast protocol, as shown in Fig. 3D. It is reasonable to assume that if the inhibition by GM had taken place directly at the KCa(ACh) channels, proportionally reduced but sizeable ACh-activated tail currents would have been triggered with the tail protocol. These results therefore confirm that GM blocks IK(ACh) essentially by impairing Ca2+ entry at the nAChRs of OHCs.
Figure 4. Gentamicin impairs the Ca2+ influx necessary for IK(ACh) activation.
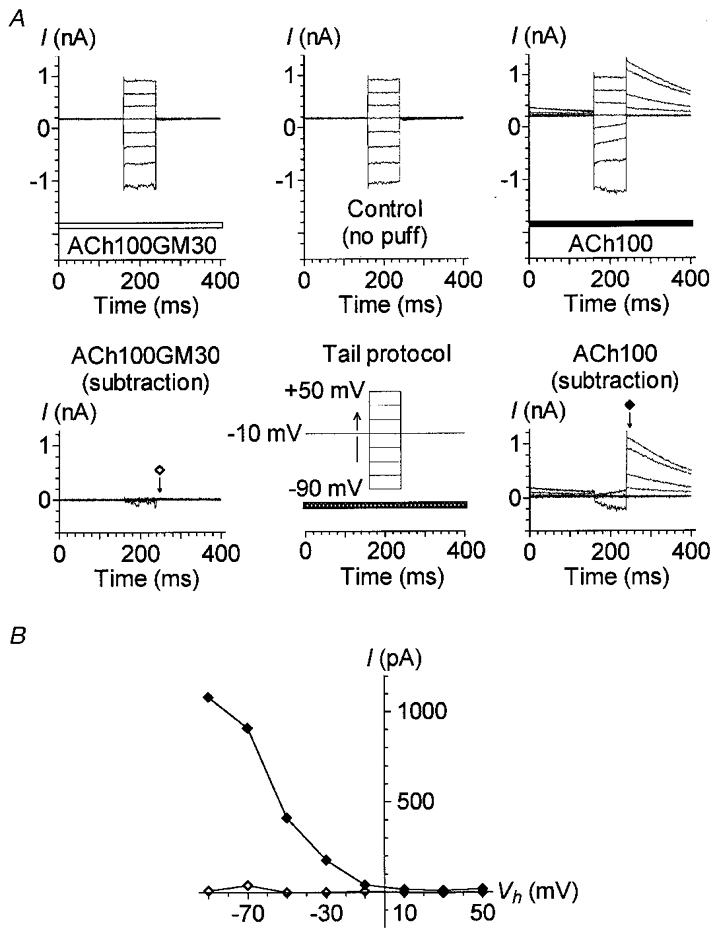
A, currents recorded using a tail protocol (bottom middle) in control conditions (top middle) or during an ACh100 (top right, filled bar) or ACh100GM30 (top left, open bar) application. The tail protocol is identical to the fast protocol described in Fig. 1C, except that voltage steps were triggered from a Vh of -10 mV; the horizontal bar indicates the timing of pressure-puff application. Net currents triggered by ACh100 (bottom right) or ACh100GM30 (bottom left) during the tail protocol were obtained after subtraction of the control leak currents recorded in between. Current amplitudes reported in B were measured 8 ms after the offset of each voltage step as indicated by the arrows under the corresponding symbols. B, I-V relationships of the tail currents presented in A. Tail current amplitudes triggered during ACh100 (♦) or ACh100GM30 (⋄) application were measured as described in A. Data presented in this figure are from the same cell as illustrated in Fig. 3C–E recorded with internal solution 1. All these effects were reversible and repeated several times.
Increasing extracellular Ca2+ or ACh concentration suppressed gentamicin inhibition of IK(ACh)
Since GM interferes with the Ca2+ influx necessary for IK(ACh) activation, we wonder whether raising the concentration of extracellular Ca2+ from 1.26 mM to 10 mM could affect GM inhibition of the cholinergic response. Here ACh and GM were applied using the U-tubing system, thus explaining the somewhat longer delay of activation of the outward K+ current in Fig. 5. In these conditions, co-applying 50 μM GM with 300 μM ACh in a carrier solution with 10 mM Ca2+ suppressed the block exerted by GM on IK(ACh) in normal extracellular Ca2+ conditions. This effect of 10 mM Ca2+ was observed with the three OHCs tested and was confirmed in two other OHCs challenged with 100 μM ACh instead of 300 μM. This indicated that raising the concentration of extracellular Ca2+ could displace GM from Ca2+ binding sites and/or restore a Ca2+ influx at the nAChRs similar to that occurring in control conditions (1.26 mM Ca2+, no GM), owing to the increased driving force for Ca2+ ions (about 26 mV here). However, the increase of the Ca2+ driving force may only be part of the explanation, because as shown in Fig. 3B, shifting the membrane potential from -40 mV (the same membrane potential as in Fig. 5A) to -100 mV did not allow a complete recovery of the response despite a 60 mV increase of the driving force for Ca2+ ions.
Figure 5. Increasing the extracellular concentration of Ca2+ or ACh reverts the block of IK(ACh) by gentamicin.
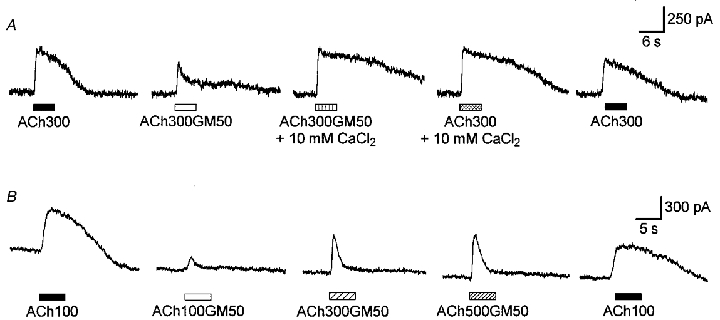
A, example of sequential current traces evoked by 300 μM ACh alone (ACh300) or with 50 μM GM (ACh300GM50). The carrier solution was either standard (1.26 mM Ca2+) or supplemented with 10 mM CaCl2 as indicated. Recording traces are separated by 1 min intervals. B, example of sequential current traces evoked by 100 μM ACh (ACh100) and by increasing concentrations of ACh (100, 300 and 500 μM) supplemented with 50 μM GM (ACh100GM50, ACh300GM50 and ACh500GM50). Recording traces are separated by 1 min intervals. A and B are from two different OHCs voltage clamped at -40 mV and dialysed with internal solution 1. Drugs were applied to both cells using the U-tubing system described in Methods, thus explaining the longer delay of the responses.
To determine whether GM could compete with ACh at its binding site on the nAChRs, we tested the effect of increasing concentrations of ACh for a fixed concentration of GM (Fig. 5B). The various solutions containing ACh (100, 300 or 500 μM) with or without 50 μM GM were again applied with the U-tubing system. As shown above, GM largely inhibited the cholinergic current activated by 100 μM ACh. Increasing the ACh concentration to 300 and 500 μM reverted the block exerted by GM on IK(ACh) at the peak, but not (or very weakly) at the plateau phase (n= 6). The recovery of the peak IK(ACh) response in the presence of ACh300GM50 or ACh500GM50 suggested that GM could compete with ACh on the nAChRs. However, the absence of recovery of the response at the plateau phase does not indicate competition between ACh and GM at the receptor sites because the overall shape of the response should have been unchanged with such an interaction. This may indicate another mechanism of inhibition.
Although 100 μM ACh has been shown to fully activate IK(ACh) in guinea-pig OHCs (Housley & Ashmore, 1991; Kakehata et al. 1993; Eróstegui et al. 1994) and to lead to maximum current activation in Xenopus oocytes expressing rat homomeric α9 nAChRs (Elgoyhen et al. 1994), it might be that the nAChRs of guinea-pig OHCs are not saturated at such an ACh concentration. On this assumption, increasing the ACh concentration above 100 μM would reduce the GM inhibition of IK(ACh), even if it is non-competitive, by activating more nAChRs and thus allowing a larger Ca2+ influx to trigger the activation of more KCa(ACh) channels. To test this hypothesis, we examined and compared in detail the dose-response relationships of IK(ACh) and In(ACh), the ionotropic nicotinic-like current of OHCs.
Comparison of the ACh dose-response relationships of the early nicotinic cation current In(ACh) and the secondary activated K+ current IK(ACh)
The dose-response relationships of IK(ACh) and In(ACh) were examined by measuring the response to a test ACh concentration relative to that obtained at a concentration of 100 μM (see Figs 6A and 7A) by means of two pressure-puff pipettes (the delivery time of the U-tubing system was not fast enough to allow the observation of In(ACh)). To compare the dose-response relationships of IK(ACh) and In(ACh) under the same conditions, and, when possible, on the same cell, we used intracellular solution 2 (see Methods) which allowed extended recording at positive potentials. Dose-response relationships were thus established from measurements performed at three membrane potentials: -65 and -20 mV for IK(ACh) and +70 mV for In(ACh) (Fig. 6). For each potential, peak amplitude measurements relative to the reference response to 100 μM ACh were plotted as a function of the ACh test concentration on a semi-logarithmic scale and fitted with the empirical Hill equation. Best fits yielded apparent half-effective concentrations (EC50 values) of 21, 33 and 122 μM and nH values of 1.9, 2.3 and 1.7 at -65, -20 and +70 mV, respectively. The EC50 values determined at -65 and -20 mV reflecting the efficiency of the combination In(ACh) plus IK(ACh) were close to previously published values (Housley & Ashmore, 1991; Kakehata et al. 1993; Eróstegui et al. 1994). The small discrepancies in the values may be due to differences in the holding potentials at which the measurements were made and/or the extracellular Ca2+ concentrations used in these studies (both conditions affecting the amount of Ca2+ entry and therefore the proportion of KCa(ACh) channels activated). On the other hand, the EC50 measured at +70 mV, a membrane potential at which only the nicotinic cation current should be activated, was found to be 4–6 times larger than the EC50 values measured at negative potentials. The half-effective ACh concentration (122 μM) for activation of the nAChRs in OHCs appeared sufficient to activate IK(ACh) fully at negative potentials (Fig. 6B). This indicated that, under our experimental conditions, the activation of half the number of nAChRs present in OHCs was sufficient to allow an influx of Ca2+ large enough to trigger the opening of all KCa(ACh) channels.
Figure 6. The dose-response relationship of the OHC cholinergic response shifts with membrane potential.
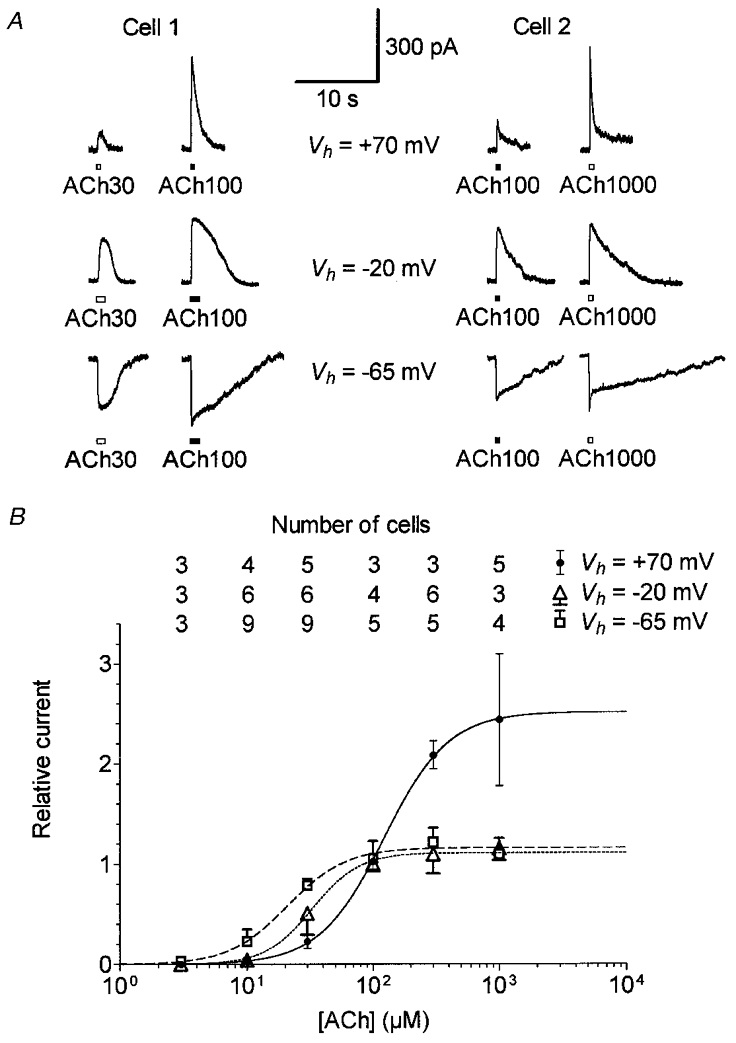
A, example of cholinergic currents from two OHCs evoked by pressure-puff application of 30 μM ACh (Cell 1) or 1000 μM ACh (Cell 2) compared with 100 μM ACh (used as reference) at Vh=−65, -20 and +70 mV. Internal solution 2, containing 100 mM CsCl and 1.1 mM EGTA, was used because it allowed extended recordings at positive potentials. B, dose-response relationships of the ACh-evoked currents at three Vh values. Peak amplitudes of ACh-evoked currents were expressed relative to the corresponding reference response obtained with 100 μM ACh. Each symbol is the mean relative response from the number of cells reported vertically for each Vh. For clarity, only +s.d., –s.d. and ±s.d. are displayed (unless masked by symbols) for measurements performed at -65, -20 and +70 mV, respectively. Some cells could be successively challenged at the three Vh values as exemplified in A. For each Vh, dose-response curves were fitted with the empirical Hill equation Y= 1/(1 + (EC50/X)nH). Best fits yield EC50 values of 21, 33 and 122 μM and nH values of 1.9, 2.3 and 1.7 at Vh=−65 mV (dashed line), -20 mV (dotted line) and +70 mV (continuous line), respectively.
Figure 7. Dose-response relationships of the In(ACh) at two Vh values: voltage-independent low affinity of ACh for nAChRs.
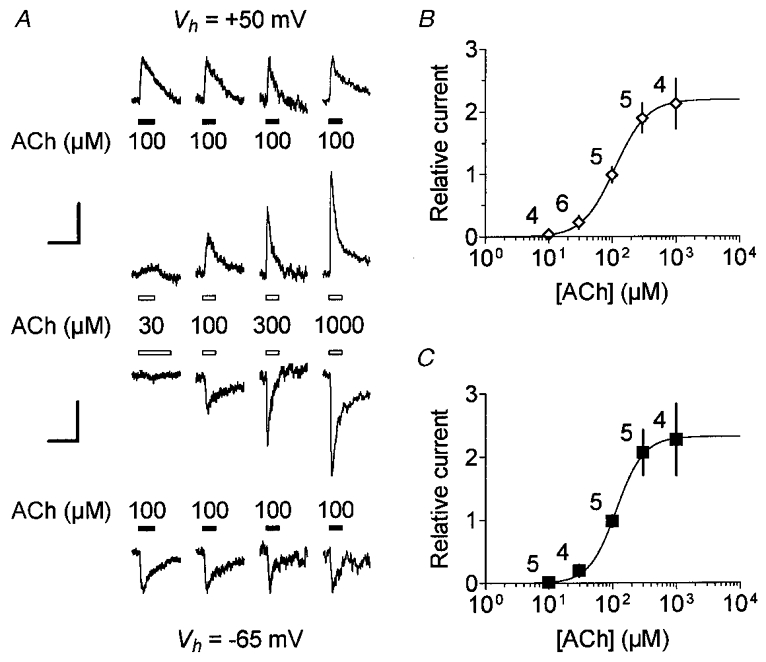
A, example of cholinergic currents from four OHCs evoked by pressure-puff application of ACh at various concentrations compared with 100 μM ACh (used as reference) at Vh=−65 and +50 mV. The 10 mM BAPTA buffered internal solution 3 was used to prevent IK(ACh) activation. The vertically aligned traces were from a single cell first challenged at -65 mV. The current traces of the four OHCs tested for increasing concentrations of ACh (as indicated in the middle) are displayed horizontally. To improve visual comparison, reference responses were scaled to the same peak amplitude both at +50 mV (top) and at -65 mV (bottom). Vertical scale bars therefore represent, from left to right traces, 167, 184, 115 and 280 pA at +50 mV and 88, 51, 70 and 143 pA at -65 mV, while horizontal scale bars represent 1 s for all traces. B, dose-response relationship of the ACh-evoked cation current at +50 mV. Peak amplitudes of ACh-evoked currents were expressed relative to the corresponding reference response at 100 μM ACh. Each symbol is the mean relative response (±s.d.) from the number of cells indicated beside. The dose-response curve was fitted with the empirical Hill equation Y= 1/(1 + (EC50/X)nH). Best fit yields an EC50 of 110 μM and a nH of 1.8. C, the dose-response relationship of the ACh-evoked cation current at -65 mV. Plots and fits were performed as described in B but with current recorded at -65 mV. Some cells could also be challenged at +50 mV as shown in A. Data represent the mean relative response (±s.d.) with the number of cells indicated beside each symbol. Best fit yields an EC50 of 114 μM and a nH of 2.0. For clarity in B and C,±s.d. are displayed as simple bars unless they are masked by symbols.
To ensure that the EC50 measured at +70 mV corresponded solely to In(ACh), we measured dose-response relationships of In(ACh) isolated by means of 10 mM intracellular BAPTA in the recording solution (see solution 3 in Methods). The use of the fast chelating Ca2+ buffer BAPTA prevented the activation of IK(ACh) (Blanchet et al. 1996). Peak amplitudes of In(ACh), measured at -65 or +50 mV, were plotted and fitted as a function of the logarithm of the ACh concentration (Fig. 7). The best fits revealed EC50 values of 114 and 110 μM and nH values of 2.0 and 1.8 at -65 and +50 mV, respectively. These EC50 values were similar to those measured at +70 mV using the EGTA-containing intracellular solution 2 (Fig. 6), thus indicating that they effectively indicated the same current.
Effects of gentamicin on the ACh-evoked ionotropic current
We studied the effects of GM on In(ACh) recorded using internal solution 3 containing 10 mM BAPTA. A concentration of 50 μM GM significantly reduced In(ACh) when co-applied with 100 μM ACh at both negative and positive potentials (Fig. 8). However, the reduction of the peak amplitude was weak or absent at the first co-application of GM with ACh but increased at subsequent co-applications. Furthermore, while the inhibition appeared to be reversible within a few minutes at positive membrane potentials (2 OHCs), full recovery was observed at negative potentials only in one of eight OHCs after 20 min washout (6 of the 8 cells showed partial recovery but could not be recorded for an extended time and GM had no apparent effect on 1 cell). At negative membrane potentials, GM also accelerated the apparent desensitization of In(ACh). Interestingly, GM abolished most of the residual current following the peak. At negative membrane potentials, this residual current is thought to be carried, at least partially, by Ca2+ ions and to be responsible for the sustained plateau phase of IK(ACh) when activation of KCa(ACh) channels is not prevented by intracellular BAPTA. This effect of GM measured directly on In(ACh) fitted well with the blocking pattern observed on IK(ACh) (Fig. 2A).
Figure 8. Effect of gentamicin on In(ACh).
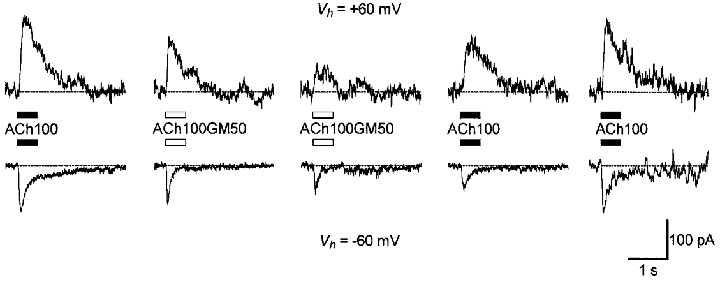
Sequential ACh-evoked cation currents were activated by a pressure puff of 100 μM ACh alone (ACh100) or with 50 μM GM (ACh100GM50) on a single OHC at +60 mV (top) or on another cell at -60 mV (bottom). The 10 mM BAPTA-containing internal solution 3 was used to prevent IK(ACh) activation. Recording traces were separated by 1–2 min intervals except for the last at -60 mV, which was obtained after 20 min washout.
Finally, it is to be remembered that the inhibitory effect of 30 or 50 μM GM on In(ACh) was also observed with intracellular solution 1 at positive membrane potentials on four OHCs (2 cells for each GM concentration; see Fig. 3), thus confirming the apparent absence of voltage-dependent inhibition for GM.
DISCUSSION
Micromolar concentration of gentamicin blocks Ca2+ influx at the level of the nAChRs
The cholinergic efferent synapse of cochlear OHCs is an inhibitory synapse which involves nicotinic-like receptors (nAChRs) permeable to Ca2+ ions and co-localized Ca2+-activated K+ (KCa(ACh)) channels (Blanchet et al. 1996; Dulon & Lenoir, 1996; Evans, 1996). Our study demonstrates for the first time that aminoglycoside antibiotics such as gentamicin and neomycin block ACh-evoked hyperpolarizing K+ currents (IK(ACh)) of OHCs in the micromolar range. In this respect, it is interesting to note that the GM inhibition of the OHC cholinergic response occurred with GM concentrations similar to those measured in guinea-pig perilymph after a single intramuscular injection (Lima da Costa et al. 1998). Since aminoglycosides have been shown to be more than tenfold less potent in affecting Ca2+ channels (Dulon et al. 1989; Nakagawa et al. 1992; Parsons et al. 1992; Haws et al. 1996), our results suggest that the inhibitory effect of GM on the medial olivocochlear function in vivo (Smith et al. 1994; Lima da Costa et al. 1997, 1998) primarily takes place at the postsynaptic level in the nAChRs of OHCs.
Aminoglycoside antibiotics have also been reported to directly block Ca2+-activated K+ channels in cochlear efferent nerve terminals (Takeuchi & Wangemann, 1993) and inner hair cells (Dulon et al. 1995) but at concentrations in the sub-millimolar range, i.e. about two orders of magnitude higher than that found in this study. Although we cannot exclude a direct effect on KCa(ACh) channels at such high concentrations, the present findings indicate that aminoglycosides, or at least GM, primarily impair the Ca2+ influx necessary for IK(ACh) activation by directly inhibiting the cholinergic receptor channels of OHCs. This block of Ca2+ influx at the OHC nAChRs may underlie the suppression by GM of both the fast and the slow component of the olivocochlear activation (Lima da Costa et al. 1997; Yoshida et al. 1999), the fast component being related to a voltage effect on OHC electromotility (Dallos et al. 1997) and the slow component being related to a more slowly developing intracellular Ca2+ increase near the cell contractile apparatus (Dulon et al. 1990).
The blocking mechanism of gentamicin
A screening charge effect?
We found that GM had a slightly higher IC50 than NM. This might be due to a difference in valence between these aminoglycosides since at pH 7.40 GM and NM carry 3.46 and 4.37 positive charges, respectively (Josepovitz et al. 1982). Accordingly, the much less potent inhibitory effect of SM might be due to its lower valence (about 2 positive charges at pH 7.40). The effectiveness of block measured on IK(ACh), which increased with the number of charges of the aminoglycosides might therefore be a consequence of a reduction of the permeant ion concentration near the entrance of the receptor channels by screening fixed negative charges, as proposed by Suarez-Kurtz & Reuben (1987). Indeed, these molecules have a strong propensity to associate with negatively charged lipid bilayers (Brasseur et al. 1984) and to compete at Ca2+ binding sites on the plasma membrane of OHCs (Williams et al. 1987). However, contrary to our present study, both association with negatively charged lipid bilayers and competition at Ca2+ binding sites on the OHC plasma membrane were only observed at high concentrations of aminoglycoside antibiotics (above 100 μM), suggesting that such high concentrations are required to screen a significant proportion of fixed negative charges. Furthermore, the dose-inhibition curves for NM and GM significantly differ in their slope (nH of 0.38 and 0.95, respectively). This suggests that the IK(ACh) inhibition mechanisms of these antibiotics differ, at least partially. These differences between aminoglycosides argue against a screening charge effect as the main mechanism of inhibition. For instance, IK(ACh) was more sensitive to 30 μM GM than to 100 μM NM (see Fig. 2B), contrary to what might be expected since the effectiveness in screening surface charge is an exponential function of blocker charge. Much more specific effects of aminoglycosides might therefore be involved.
An open-channel block mechanism?
Aminoglycosides have been reported to act as open-channel blockers on many ion channel types including ATP-ionotropic channels (Lin et al. 1993) and mechanosensitive ion channels (Ohmori, 1985; Kroese et al. 1989; Winegar et al. 1996). In our study, GM enhanced the apparent desensitization of In(ACh) and inhibited this current increasingly upon successive applications. These effects fit well with an open-block mechanism. However, the fact that the polycationic molecule similarly inhibited In(ACh) at positive and negative membrane potentials argues against such a blocking mechanism, since charged open-channel blockers are usually strongly voltage dependent.
An interaction with specific Ca2+ binding sites at the nAChRs?
Our observation that GM blockade of IK(ACh) could be removed by raising the extracellular Ca2+ concentration suggests that GM could interact with specific Ca2+ binding sites at the nAChRs. These sites might be allosteric sites such as the Ca2+ potentiating sites identified at the α7 nAChR subunit (Galzi et al. 1996). The presence of Ca2+ potentiating sites at other nicotinic receptor subunits is strongly suspected since extracellular Ca2+ potentiates current through numerous native and reconstituted neuronal nicotinic receptors (Mulle et al. 1992; Vernino et al. 1992; Liu & Berg, 1999). We have also previously shown that Ca2+ ions are required in the extracellular medium in order to maintain functional ACh-evoked ionotropic responses (In(ACh)) in OHCs, thus suggesting the presence of such regulatory binding sites for Ca2+ at the nAChRs of OHCs (Blanchet et al. 1996). A similar observation was made in chick hair cells where extracellular Ca2+ ions were required as a ‘cofactor’ in the ligand gating of the nAChR (McNiven et al. 1996). Interaction of GM with these specific Ca2+ binding sites of the nAChRs may alter the cation current (especially its Ca2+ component) through the receptor-channel and therefore underlie the inhibition of the cholinergic response of OHCs.
ACh has a low affinity for its receptors but couples very efficiently with KCa(ACh) channels
Our experiments also give interesting insights into the functional coupling of nAChRs and KCa(ACh) channels in cochlear OHCs. We have measured for the first time in mammalian OHCs the real EC50 of ACh for nAChRs by directly recording In(ACh) using an intracellular solution containing 10 mM BAPTA. This EC50 value is one order of magnitude higher than that measured on rat α9 homomeric receptors reconstituted in Xenopus oocytes (Elgoyhen et al. 1994) or on chick hair cell nAChRs when recording the nicotinic cation current in 10 mM BAPTA conditions (McNiven et al. 1996). These discrepancies might be due to the involvement of other nicotinic subunits such α7 in mammalian OHCs as previously suggested (Housley et al. 1994; Morley et al. 1998) or to species differences. Our measured EC50 value for In(ACh) is also one order of magnitude higher than that previously measured for IK(ACh) (Housley & Ashmore, 1991; Kakehata et al. 1993; Eróstegui et al. 1994) and 4–6 times higher than what we found for this IK(ACh). This indicates that in OHCs the coupling between nAChRs and KCa(ACh) channels is efficient enough to allow saturation of the Ca2+-activated K+ current long before the cation current flowing through the nAChRs saturates.
Acknowledgments
This work was supported by the Fondation pour la Recherche Médicale and the Conseil Régional d'Aquitaine.
References
- Blanchet C, Erostegui C, Sugasawa M, Dulon D. Acetylcholine-induced potassium current of guinea-pig outer hair cells: its dependence on a calcium influx through nicotinic-like receptors. Journal of Neuroscience. 1996;16:2574–2584. doi: 10.1523/JNEUROSCI.16-08-02574.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobbin RP, Konishi T. Acetylcholine mimics crossed olivocochlear bundle stimulation. Nature. 1971;231:222–223. doi: 10.1038/newbio231222a0. [DOI] [PubMed] [Google Scholar]
- Brasseur R, Laurent G, Ruysschaert JM, Tulkens P. Interactions of aminoglycoside antibiotics with negatively-charged lipid bilayer: biochemical and conformational studies. Biochemical Pharmacology. 1984;33:629–637. doi: 10.1016/0006-2952(84)90319-8. [DOI] [PubMed] [Google Scholar]
- Brownell WE, Bader CR, Bertrand D, De Ribeaupierre Y. Evoked mechanical responses of isolated cochlear hair cells. Science. 1985;227:194–196. doi: 10.1126/science.3966153. [DOI] [PubMed] [Google Scholar]
- Dallos P, He DZ, Lin X, Sziklai I, Mehta S, Evans BN. Acetylcholine, outer hair cell electromotility, and the cochlear amplifier. Journal of Neuroscience. 1997;17:2212–2226. doi: 10.1523/JNEUROSCI.17-06-02212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulon D, Hiel H, Aurousseau C, Erre JP, Aran JM. Pharmacokinetics of gentamicin in the sensory hair cells of the organ of Corti: rapid uptake and long term persistence. Comptes Rendus de l'Académie des Sciences Paris serie III. 1993;316:682–687. [PubMed] [Google Scholar]
- Dulon D, Lenoir M. Cholinergic responses in developing outer hair cells of the rat cochlea. European Journal of Neuroscience. 1996;8:1945–1952. doi: 10.1111/j.1460-9568.1996.tb01338.x. [DOI] [PubMed] [Google Scholar]
- Dulon D, Luo L, Zhang C, Ryan AF. Expression of small-conductance calcium-activated potassium channels (SK) in outer hair cells of the rat cochlea. European Journal of Neuroscience. 1998;10:907–915. doi: 10.1046/j.1460-9568.1998.00098.x. [DOI] [PubMed] [Google Scholar]
- Dulon D, Sugasawa M, Blanchet C, Erostegui C. Direct measurements of Ca2+ activated K+ currents in inner hair cells of the guinea-pig cochlea using photolabile Ca2+ chelators. Pflügers Archiv. 1995;430:365–373. doi: 10.1007/BF00373911. [DOI] [PubMed] [Google Scholar]
- Dulon D, Zajic G, Aran JM, Schacht J. Aminoglycoside antibiotics impair calcium entry but not viability and motility in isolated cochlear outer hair cells. Journal of Neuroscience Research. 1989;24:338–346. doi: 10.1002/jnr.490240226. [DOI] [PubMed] [Google Scholar]
- Dulon D, Zajic G, Schacht J. Increasing intracellular free calcium induces circumferential contractions in isolated outer hair cells. Journal of Neuroscience. 1990;10:1388–1397. doi: 10.1523/JNEUROSCI.10-04-01388.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S. α9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell. 1994;79:705–715. doi: 10.1016/0092-8674(94)90555-x. [DOI] [PubMed] [Google Scholar]
- Eróstegui C, Norris CH, Bobbin RP. In vitro pharmacologic characterization of a cholinergic receptor on outer hair cells. Hearing Research. 1994;74:135–147. doi: 10.1016/0378-5955(94)90182-1. [DOI] [PubMed] [Google Scholar]
- Evans MG. Acetylcholine activates two currents in guinea-pig outer hair cells. The Journal of Physiology. 1996;491:563–578. doi: 10.1113/jphysiol.1996.sp021240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs PA. Synaptic transmission at vertebrate hair cells. Current Opinion in Neurobiology. 1996;6:514–519. doi: 10.1016/s0959-4388(96)80058-4. [DOI] [PubMed] [Google Scholar]
- Fuchs PA, Murrow BW. Cholinergic inhibition of short (outer) hair cells of the chick's cochlea. Journal of Neuroscience. 1992;12:800–809. doi: 10.1523/JNEUROSCI.12-03-00800.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galambos R. Suppression of auditory nerve activity by stimulation of efferent fibers of the cochlea. Journal of Neurophysiology. 1956;19:424–437. doi: 10.1152/jn.1956.19.5.424. [DOI] [PubMed] [Google Scholar]
- Galzi JL, Bertrand S, Corringer PJ, Changeux JP, Bertrand D. Identification of calcium binding sites that regulate potentiation of a neuronal nicotinic acetylcholine receptor. EMBO Journal. 1996;15:5824–5832. [PMC free article] [PubMed] [Google Scholar]
- Glowatzki E, Wild K, Brändel U, Falker G, Falker B, Zenner HP, Ruppersberg JP. Cell-specific expression of the α9 n-ACh receptor subunit in auditory hair cells revealed by single-cell RT-PCR. Proceedings of the Royal Society. 1995;B262:141–147. doi: 10.1098/rspb.1995.0188. [DOI] [PubMed] [Google Scholar]
- Haws CM, Winegar DD, Lansmann JB. Block of single L-type Ca2+ channels in skeletal muscle fibers by aminoglycoside antibiotics. Journal of General Physiology. 1996;107:421–432. doi: 10.1085/jgp.107.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housley GD, Ashmore JF. Direct measurement of the action of acetylcholine on isolated outer hair cells of the guinea pig cochlea. Proceedings of the Royal Society B. 1991;244:161–167. doi: 10.1098/rspb.1991.0065. [DOI] [PubMed] [Google Scholar]
- Housley GD, Batcher S, Kraft M, Ryan AF. Nicotinic acetylcholine receptor subunits expressed in rat cochlea detected by polymerase chain reaction. Hearing Research. 1994;75:47–53. doi: 10.1016/0378-5955(94)90054-x. [DOI] [PubMed] [Google Scholar]
- Josepovitz C, Pastoriza-Munoz E, Timmerman D, Scott M, Feldman S, Kaloyanides J. Inhibition of gentamicin uptake in rat renal cortex in vivo by aminoglycosides and organic polycations. Journal of Pharmacology and Experimental Therapeutics. 1982;223:314–321. [PubMed] [Google Scholar]
- Kakehata S, Nakagawa T, Takasaka T, Akaike N. Cellular mechanism of acetylcholine-induced response in dissociated outer hair cells of guinea-pig cochlea. The Journal of Physiology. 1993;463:227–244. doi: 10.1113/jphysiol.1993.sp019592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroese ABA, Das A, Hudspeth AJ. Blockage of the transduction channels of hair cells in the bullfrog's sacculus by aminoglycoside antibiotics. Hearing Research. 1989;37:203–218. doi: 10.1016/0378-5955(89)90023-3. [DOI] [PubMed] [Google Scholar]
- Kujawa S, Glattke TJ, Fallon M, Bobbin RP. Intracochlear application of acetylcholine alters sound-induced mechanical events within the cochlear partition. Hearing Research. 1992;61:106–116. doi: 10.1016/0378-5955(92)90041-k. [DOI] [PubMed] [Google Scholar]
- Lima Da Costa D, Chibois A, Erre JP, Blanchet C, Charlet De Sauvage R, Aran JM. The fast, slow and steady state effects of contralateral acoustic activation of the medial olivocochlear efferent system in awake guinea pigs. Action of gentamicin. Journal of Neurophysiology. 1997;78:1826–1836. doi: 10.1152/jn.1997.78.4.1826. [DOI] [PubMed] [Google Scholar]
- Lima Da Costa D, Erre JP, Pehourq F, Aran JM. Aminoglycoside ototoxicity and the medial efferent system: II. Comparison of acute effects of different antibiotics. Audiology. 1998;37:162–173. doi: 10.3109/00206099809072970. [DOI] [PubMed] [Google Scholar]
- Lin X, Hume RI, Nuttall AL. Voltage-dependent block by neomycin of the ATP-induced whole cell current in guinea pig outer hair cells. Journal of Neurophysiology. 1993;70:1593–1605. doi: 10.1152/jn.1993.70.4.1593. [DOI] [PubMed] [Google Scholar]
- Liu QS, Berg DK. Extracellular calcium regulates responses of both α3- and α7-containing nicotinic receptors on chick ciliary ganglion neurons. Journal of Neurophysiology. 1999;82:1124–1132. doi: 10.1152/jn.1999.82.3.1124. [DOI] [PubMed] [Google Scholar]
- McNiven AI, Yuhas WA, Fuchs PA. Ionic dependence and agonist preference of an acetylcholine receptor in hair cells. Auditory Neuroscience. 1996;2:63–79. [Google Scholar]
- Morley BJ, Li HS, Hiel H, Drescher DG, Elgoyhen AB. Identification of the subunits of the nicotinic cholinergic receptors in the rat cochlea using RT-PCR and in situ hybridization. Brain Research. Molecular Brain Research. 1998;53:78–87. doi: 10.1016/s0169-328x(97)00272-6. [DOI] [PubMed] [Google Scholar]
- Mulle C, Léna C, Changeux JP. Potentiation of nicotinic receptor response by external calcium in rat central neurons. Neuron. 1992;8:937–945. doi: 10.1016/0896-6273(92)90208-u. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Kakehata S, Akaike N, Komune S, Takasaka T, Uemura T. Effects of Ca2+ antagonists and aminoglycoside antibiotics on Ca2+ current in isolated outer hair cells of guinea pig cochlea. Brain Research. 1992;580:345–347. doi: 10.1016/0006-8993(92)90966-d. [DOI] [PubMed] [Google Scholar]
- Nenov AP, Norris C, Bobbin RP. Acetylcholine response in guinea pig outer hair cells. I. Properties of the response. Hearing Research. 1996a;101:132–148. doi: 10.1016/s0378-5955(96)00142-6. [DOI] [PubMed] [Google Scholar]
- Nenov AP, Norris C, Bobbin RP. Acetylcholine response in guinea pig outer hair cells. II. Activation of a small conductance Ca2+-activated K+ channel. Hearing Research. 1996b;101:149–172. doi: 10.1016/s0378-5955(96)00143-8. [DOI] [PubMed] [Google Scholar]
- Nishizaki T, Morales A, Gehle VM, Sumikawa K. Differential interactions of gentamicin with mouse junctional and extrajunctional ACh receptors expressed in Xenopus oocytes. Brain Research. Molecular Brain Research. 1994;21:99–106. doi: 10.1016/0169-328x(94)90382-4. [DOI] [PubMed] [Google Scholar]
- Ohmori H. Mechano-electrical transduction currents in isolated vestibular hair cells of the chick. The Journal of Physiology. 1985;359:189–217. doi: 10.1113/jphysiol.1985.sp015581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T, Sumikawa K. Antibiotics cause changes in the desensitization of ACh receptors expressed in Xenopus oocytes. Brain Research. Molecular Brain Research. 1991;9:165–168. doi: 10.1016/0169-328x(91)90144-m. [DOI] [PubMed] [Google Scholar]
- Parsons TD, Obaid AL, Salzberg BM. Aminoglycoside antibiotics block voltage-dependent calcium channels in intact vertebrate nerve terminals. Journal of General Physiology. 1992;99:491–504. doi: 10.1085/jgp.99.4.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado WA, Corrado AP, Marseillan RF. Competitive antagonism between calcium and antibiotics at the neuromuscular junction. Archives Internationales de Phamacodynamie et de Thérapie. 1978;231:297–307. [PubMed] [Google Scholar]
- Silverblatt FJ, Kuehn C. Autoradiography of gentamicin uptake by the rat proximal tubule cell. Kidney International. 1979;15:335–345. doi: 10.1038/ki.1979.45. [DOI] [PubMed] [Google Scholar]
- Smith DW, Erre JP, Aran JM. Rapid, reversible elimination of medial olivocochlear efferent function following single injections of gentamicin in the guinea pig. Brain Research. 1994;652:243–248. doi: 10.1016/0006-8993(94)90233-x. [DOI] [PubMed] [Google Scholar]
- Suarez-Kurtz G, Reuben JP. Effects of neomycin on calcium channel currents on clonal GH3 pituitary cells. Pflügers Archiv. 1987;410:517–523. doi: 10.1007/BF00586535. [DOI] [PubMed] [Google Scholar]
- Takeuchi S, Wangemann P. Aminoglycoside antibiotics inhibit maxi-K+ channels in single isolated cochlear efferent terminals. Hearing Research. 1993;67:13–19. doi: 10.1016/0378-5955(93)90227-r. [DOI] [PubMed] [Google Scholar]
- Vernino S, Amador M, Luetje CW, Patrick J, Dani JA. Calcium modulation and high calcium permeability of neuronal nicotinic acetylcholine receptors. Neuron. 1992;8:127–134. doi: 10.1016/0896-6273(92)90114-s. [DOI] [PubMed] [Google Scholar]
- Vital Brazil O, Prado-Franceschi J. The nature of neuromuscular block produced by neomycin and gentamicin. Archives Internationales de Phamacodynamie et de Thérapie. 1969;179:78–85. [PubMed] [Google Scholar]
- Williams EW, Zenner HP, Schacht J. Three molecular steps of aminoglycosides ototoxicity demonstrated in outer hair cells. Hearing Research. 1987;30:11–18. doi: 10.1016/0378-5955(87)90177-8. [DOI] [PubMed] [Google Scholar]
- Winegar BD, Haws CM, Lansman JB. Subconductance block of single mechanosensitive ion channels in skeletal muscle fibers by aminoglycoside antibiotics. Journal of General Physiology. 1996;107:433–443. doi: 10.1085/jgp.107.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida N, Liberman MC, Brown MC, Sewell WF. Gentamicin blocks both fast and slow effects of olivocochlear activation in anesthetized guinea pigs. Journal of Neurophysiology. 1999;82:3168–3174. doi: 10.1152/jn.1999.82.6.3168. [DOI] [PubMed] [Google Scholar]