

Abstract
It has been proposed that hypoxic pulmonary vasoconstriction (HPV) is mediated via K+ channel inhibition and Ca2+ influx through voltage-gated channels. HPV depends strongly on the degree of preconstriction, and we therefore examined the effect of Ca2+ channel blockade on tension and intracellular [Ca2+] ([Ca2+]i) during HPV in rat intrapulmonary arteries (IPAs), whilst maintaining preconstriction constant. We also investigated the role of intracellular Ca2+ stores.
HPV demonstrated a transient constriction (phase I) superimposed on a sustained constriction (phase II). Nifedipine (1 μm) partially inhibited phase I, but did not affect phase II. In arteries exposed to 80 mm K+ and nifedipine or diltiazem the rises in tension and [Ca2+]i were blunted during phase I, but were unaffected during phase II.
At low concentrations (< 3 μm), La3+ almost abolished the phase I constriction and rise in [Ca2+]i, but had no effect on phase II, or constriction in response to 80 mm K+. Phase II was inhibited by higher concentrations of La3+ (IC50∼50 μm).
IPA treated with thapsigargin (1 μm) in Ca2+-free solution to deplete Ca2+ stores showed sustained constriction upon re-exposure to Ca2+ and an increase in the rate of Mn2+ influx, suggesting capacitative Ca2+ entry. The concentration dependency of the block of constriction by La3+ was similar to that for phase I of HPV. Pretreatment of IPA with 30 μm CPA reduced phase I by > 80%, but had no significant effect on phase II.
We conclude that depolarization-mediated Ca2+ influx plays at best a minor role in the transient phase I constriction of HPV, and is not involved in the sustained phase II constriction. Instead, phase I appears to be mainly dependent on capacitative Ca2+ entry related to release of thapsigargin-sensitive Ca2+ stores, whereas phase II is supported by Ca2+ entry via a separate voltage-independent pathway.
Hypoxic pulmonary vasoconstriction (HPV) is a unique and vital local homeostatic mechanism within the lung. By constricting in response to a hypoxic stimulus, the pulmonary circulation diverts blood flow away from poorly ventilated areas of the lung, thus maintaining ventilation- perfusion matching in the event of localized alveolar hypoxia (von Euler & Liljestrand, 1946).
The mechanisms underlying HPV remain unresolved. However, studies over the past two decades have shown that HPV is associated with membrane depolarization (Madden et al. 1984), and have suggested that HPV requires Ca2+ influx via voltage-gated Ca2+ channels (Vadula et al. 1993; Cornfield et al. 1994). The resting membrane potential in pulmonary artery (PA) smooth muscle cells is set mainly by one or more voltage-gated K+ currents (Smirnov et al. 1994; Osipenko et al. 1997; Yuan et al. 1998), some of which are suppressed by hypoxia, leading to depolarization (Post et al. 1992, 1995; Yuan et al. 1993; Osipenko et al. 1997). These observations support the proposal that K+ channel inhibition in PA smooth muscle cells plays a central role in linking hypoxia to the increase in intracellular Ca2+, and hence HPV (Weir & Archer, 1995; Post et al. 1995; Gelband & Gelband, 1997).
A crucial prediction of this model is that HPV should be prevented by inhibition of voltage-gated Ca2+ channels. In accordance with this, HPV and/or hypoxia-mediated rises in the intracellular Ca2+ concentration ([Ca2+]i) are greatly attenuated by organic Ca2+ channel antagonists in intact dogs (Tucker et al. 1976), blood-perfused rat lung (McMurtry et al. 1976), isolated small PA (Jin et al. 1992; Leach et al. 1994; Savineau et al. 1995; Woodmansey et al. 1995), and single PA cells (Salvaterra & Goldman, 1993; Vadula et al. 1993; Cornfield et al. 1994).
However, one notable property of HPV, which does not appear to have been taken into account in these studies, is that its amplitude is strongly enhanced if PAs are slightly stretched, or if a small degree of pretone is elicited using an agonist (Rodman et al. 1989; Demiryurek et al. 1993; Wadsworth, 1994; Ward & Robertson, 1995; Jabr et al. 1997; Ozaki et al. 1998). A similar phenomenon has long been known to exist in intact animals and isolated lung preparations (Fishman, 1976). Most studies of HPV in isolated PA have therefore examined the effect of hypoxia following preconstriction with agonists such as PGF2α, 5-HT or noradrenaline. However, since preconstriction by these agonists is itself sensitive to Ca2+ channel antagonists (e.g. Jin et al. 1992; Leach et al. 1994), it is possible that inhibition of HPV caused by Ca2+ channel blockade is secondary to the effect on pretone. This is supported by the observation of Rodman et al. (1989) that although nifedipine reduced HPV in rat PA preconstricted with phenylephrine, the response was completely restored if the phenylephrine concentration was raised to bring the level of pretone back to that developed before nifedipine was applied.
Rodman et al. (1989) did not discuss the implications of this observation, and their study focused on the peak response to hypoxia which occurred in the rat main PA within 5 min after hypoxia was imposed. However, we and others have shown that when hypoxia is prolonged for more than 10–15 min, HPV in isolated PA is biphasic (e.g. Bennie et al. 1991; Kovitz et al. 1993; Leach et al. 1994). For example, in intrapulmonary arteries (IPAs) from the rat the response to hypoxia consists of an initial transient constriction (phase I) superimposed on a sustained constriction that is more slowly developing (phase II). The amplitude of both phases is strongly enhanced by a small degree of pretone (Leach et al. 1994; Ward & Robertson, 1995; Ozaki et al. 1998).
In the present study, we have evaluated the effects of inhibitors of both voltage- and non-voltage-gated Ca2+ entry on both phases of HPV in the rat IPA. We have used a protocol in which pretone was kept constant by varying the concentration of PGF2α in order to determine the effects of these inhibitors on HPV per se. Our results suggest that Ca2+ entry through voltage-gated channels plays little part in the response to hypoxia observed in this model of HPV.
METHODS
IPA mounting
Male Wistar rats (250-350 g) were anaesthetized with sodium pentobarbital (55 mg kg−1i.p.) and killed by cervical dislocation. The lungs were excised and placed in a physiological salt solution (PSS) containing (mM): NaCl, 118; NaHCO3, 24; MgSO4, 1; NaH2PO4, 0.435; glucose, 5.56; sodium pyruvate, 5; CaCl2, 1.8; and KCl, 4. Small IPAs (150-550 μm internal diameter, median ∼300 μm) were dissected free of adventitia, mounted in a temperature-controlled myograph at 37°C (Cambustion AM10, Cambustion Ltd, Cambridge, UK), and gassed continuously with 95 % air/5 % CO2 (pH 7.35). They were then stretched to give an equivalent transmural pressure of 30 mmHg as previously described (Leach et al. 1992). All arteries had an intact endothelium, as determined by relaxation in response to 1 μM acetylcholine.
Estimation of [Ca2+]i
IPAs were loaded with the Ca2+-sensitive fluorophore fura PE-3, via incubation of the vessels with the acetoxymethyl ester of fura PE-3 (3 μM) for 2 h at room temperature (Sigma-Aldrich Ltd). The vessels were then washed with PSS, the bath temperature raised to 37°C, and the myograph transferred to the stage of an inverted fluorescence microscope (Olympus IMT2, Olympus Ltd, London UK). Changes in [Ca2+]i were assessed by calculating the ratio of the light emitted through a wide bandpass 500 nm emission filter when the vessel was illuminated at 340 and 380 nm (Cairn spectrophotometer, Cairn Research Ltd, Faversham, Kent, UK).
Hypoxic protocol
IPAs were equilibrated with four exposures to 80 mM KCl-PSS (KPSS) of 2 min duration (isotonic replacement of NaCl by KCl), as previously described (Leach et al. 1992, 1994; Robertson et al. 1995). As we have previously shown, a small degree of agonist-induced tone is required to facilitate the hypoxic response in isolated rat IPA (Leach et al. 1994; Robertson et al. 1995). The vessels were therefore exposed to 3 μM PGF2α for 20 min prior to, and during, the hypoxic challenge, equivalent to ∼12 % of the response to KPSS. This ‘pretone’ was stable over at least the experimental period, as reflected by the developed tension before and after the hypoxic challenge (e.g. Fig. 1; and Robertson et al. 1995).
Figure 1. Repeatability of hypoxic response in rat IPA, and dependence on external Ca2+.
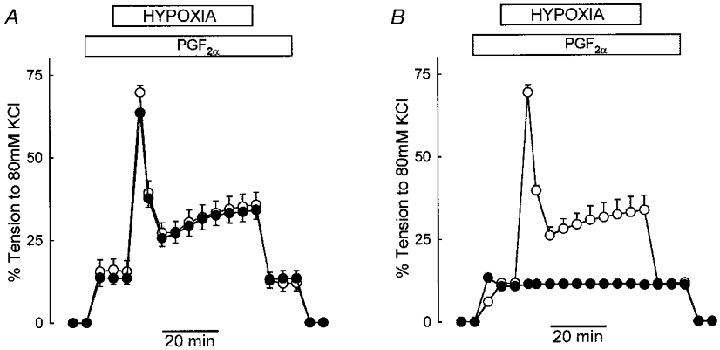
A, IPAs were preconstricted with 3 μM PGF2α and challenged with hypoxia for 45 min before being washed (○). After 60 min the process was repeated (•). The value shown for phase I after 5 min hypoxia is the mean of the peak tension obtained during this phase. It can be seen that the second (experimental) response was identical to the first (control) response. The symbols represent the mean ±s.e.m. of five arteries. Where error bars are not shown, the error is smaller than the symbol. B, HPV in IPAs under control conditions (○, 3 μM PGF2α), and in 0 Ca2+ PSS containing 1 mM EGTA (•, 100 μM PGF2α). Removal of extracellular Ca2+ completely inhibited HPV (n= 3, P < 0.001 for every point).
Hypoxia was induced by continuous gassing with 1 % O2/5 % CO2/balance N2 for 45 min, after which time the vessels were reoxygenated and washed with PSS. There were no changes in PSS pH or osmolarity over this period. Oxygen tension in the myograph chamber was continually monitored via a dissolved oxygen meter (Diamond General electrode, Ann Arbor, MI, US; Strathkelvin oxygen meter, Glasgow, UK). During the hypoxic challenge the chamber PO2 was typically 15–18 mmHg, compared to the control PO2 of 135–145 mmHg. We have previously demonstrated that HPV in IPA of the rat is reproducible providing the vessels are allowed at least 1 h to recover between hypoxic challenges (Fig. 1A; Leach et al. 1994; Robertson et al. 1995). A recovery period of 60–90 min was therefore used between the control and experimental exposures in all experiments.
Protocols for experiments using 0 Ca2+ or La3+
The effect of 0 Ca2+ was examined by omission of Ca2+ from the PSS and addition of 1 mM EGTA. To prevent precipitation, experiments involving La3+ were performed with a Hepes-buffered PSS gassed with air (control) or 1 % O2 in N2 (hypoxia), containing (mM): NaCl, 130; MgCl2, 1; glucose, 5.56; CaCl2, 1.8; KCl, 4; and Hepes, 10, with pH adjusted to 7.4 with NaOH. La3+ is avidly bound by EGTA, and EGTA was therefore not used in 0 Ca2+ solutions for these experiments. The preparations were returned to standard HCO3− PSS gassed with 5 % CO2 in air for 60 min between hypoxic challenges.
Tension is presented as a percentage of the maximum tension obtained in reponse to the final exposure to KPSS during the equilibration procedure. Changes in [Ca2+]i are represented in terms of the change in the fura PE-3 ratio. Mean changes are expressed as a percentage of the maximum ratio change seen during the final KPSS exposure (F340/380); although not linearly related to [Ca2+]i this provides a reliable qualitative index of changes in [Ca2+]i. Results are expressed as means ±s.e.m., and means were compared using ANOVA for repeated measures, or Student's paired or unpaired t test as appropriate (SigmaStat, SPSS Inc., Chicago, IL, USA). A difference was deemed significant if P < 0.05.
RESULTS
Effects of Ca2+ removal and antagonists of voltage-gated Ca2+ channels on IPA force development during hypoxia
Removal of extracellular Ca2+ abolished the response to hypoxia, as shown in Fig. 1B. Following a control hypoxic response and a subsequent 1 h recovery period, arteries were exposed to a Ca2+-free medium containing 1 mM EGTA. A level of pretone equivalent to that used for the control response was evoked with PGF2α (100 μM, as compared to 3 μM used for the control response), and then hypoxia was imposed.
The effect of nifedipine on HPV is shown in Fig. 2. Arteries were subjected to hypoxia under control conditions, allowed to recover for 1 h, and then exposed to nifedipine before a second hypoxic challenge. It was necessary to raise the PGF2α concentration slightly, to 3.5-4 μM, to achieve a level of pretone in the presence of nifedipine which matched that used for the control response. Under these conditions, 1 μM (Fig. 2A) and 10 μM (Fig. 2B) nifedipine caused a similar partial inhibition of phase I (to 48 and 52 %, respectively, both P < 0.05). Neither concentration of nifedipine significantly diminished the amplitude of the phase II constriction measured after 45 min. By comparison, nifedipine at 1 and 10 μM reduced the KPSS-induced constriction in these arteries by 98 and 99 %, respectively (not shown).
Figure 2. Nifedipine suppresses phase I but not phase II of HPV in either polarized or depolarized IPA.
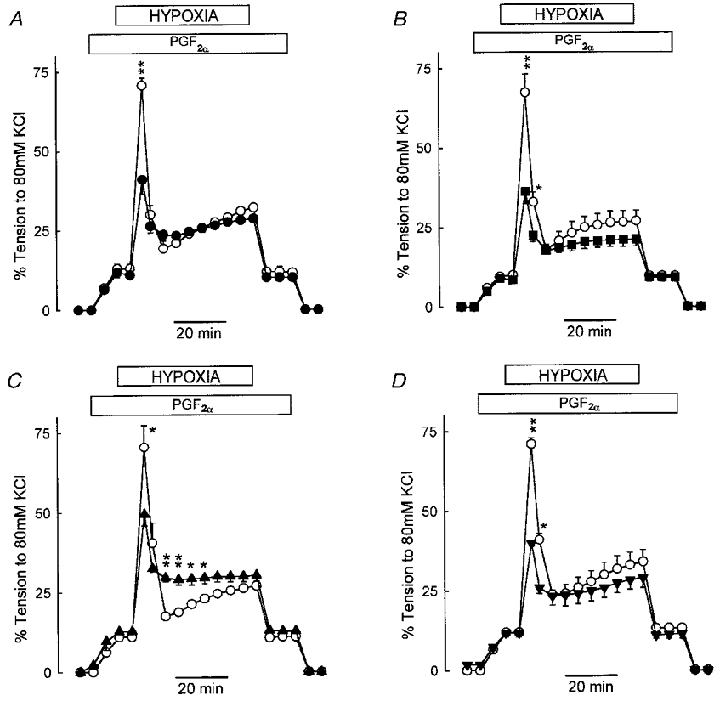
A, HPV in IPA under control conditions (○) and following preincubation with 1 μM nifedipine (•, n= 4, ** P < 0.001). The PGF2α concentration was raised from 3 μM (control) to 3.5 or 4 μM so as to ensure that the pretone level matched that in control conditions. B, similar experiment to that in A, but with 10 μM nifedipine (▪, n= 4, * P < 0.05, ** P < 0.001). C, HPV in depolarized IPA; arteries were exposed to KPSS, and then relaxed to the baseline with 1 μM nifedipine. Pretone was raised to the same level as in controls by addition of 1 μM PGF2α and the hypoxic challenge was performed as before (▴, n= 4, * P < 0.05, ** P < 0.001). D, similar to C, except that 1 μM nifedipine was added before rather than after KPSS (▾, n= 5, * P < 0.05, ** P < 0.001).
The response to hypoxia was also recorded in IPAs that were simultaneously treated with a voltage-gated Ca2+ channel blocker, and KPSS to depolarize the artery. Two procedures were used with 1 μM nifedipine. In the first, a control response to HPV was evoked utilizing 3 μM PGF2α to elicit pretone. Following a 1 h recovery period, IPAs were contracted with KPSS. Nifedipine was then added, causing a rapid relaxation to baseline. In the continuing presence of KPSS and nifedipine, PGF2α (1 μM) was added to elicit a level of pretone similar to that used for the control response, and hypoxia was then imposed. Under these conditions, the phase I constriction was significantly attenuated by 38 % compared to control (P < 0.05); the fall in tension which terminated phase I was also significantly blunted (Fig. 2C). However, the amplitude of the phase II constriction measured after 45 min was not different from that of the control response to hypoxia.
The second procedure was identical to the first, except that 1 μM nifedipine was added prior to, rather than subsequent to KPSS, in an effort to prevent any loading of intracellular Ca2+ stores which might occur as a result of the KPSS challenge. Figure 2D shows that under these conditions phase I was reduced by 52 %. In this case, neither the relaxation from phase I nor the amplitude of phase II was different from control.
Simultaneous measurement of tension and [Ca2+]i
The effect of calcium channel blockade (10 μM verapamil) upon tension and [Ca2+]i during HPV in normally polarized IPA is shown in Fig. 3A. Pretone in the presence of verapamil was maintained constant by raising the concentration of PGF2α to 3.5-4 μM. Under these conditions the increase in [Ca2+]i associated with pretone was identical in control (11.3 ± 0.6 % response to KPSS) and verapamil-treated arteries (11.2 ± 0.6 % response to KPSS) suggesting that the relationship between [Ca2+]i and tension was not altered at the slightly higher PGF2α concentration. As with nifedipine, phase I tension was partially inhibited by verapamil, in this case by 40 %, and the phase I Ca2+ transient was substantially reduced. However, verapamil had no significant effect on the magnitude of either the phase II constriction or the associated rise in [Ca2+]i. This concentration of verapamil reduced the KPSS-induced constriction by > 95 %.
Figure 3. Verapamil and diltiazem have no effect upon either the sustained increase in tension or [Ca2+]i during HPV in polarized or depolarized arteries.
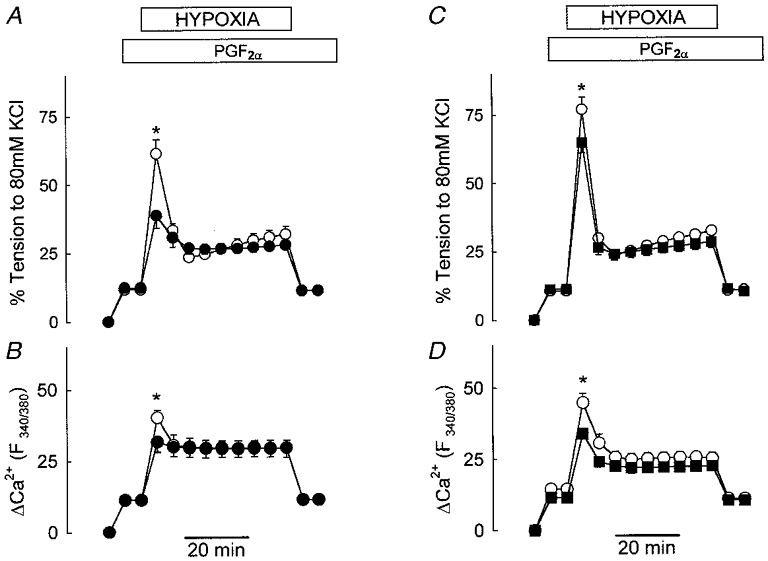
A and B, IPAs were exposed to hypoxia in the presence of 3 μM PGF2α (○) and following preincubation with 10 μM verapamil (•); the PGF2α concentration was raised to 3.5 or 4 μM in the presence of verapamil to match pretone to the control level. A shows tension development in eight IPAs; B depicts fura PE-3 F340/380 fluorescence ratio, an indication of [Ca2+]i, measured simultaneously in four of these arteries (* P < 0.05). C and D, HPV under control conditions (○) and in the same IPA exposed sequentially to diltiazem (10 μM, 20 min), KPSS containing diltiazem (10 min), and KPSS containing diltiazem and sufficient PGF2α (1 or 1.5 μM) to evoke the same pretone as in control conditions (▪). C shows mean tension development in 17 IPAs; D depicts the fura PE-3 F340/380 fluorescence ratio measured simultaneously in seven of these arteries (* P < 0.05).
Figure 3B compares force development and [Ca2+]i during the control hypoxic challenge with the response evoked by hypoxia after arteries were treated with the Ca2+ channel blocker diltiazem (10 μM) and then depolarized with KPSS. As previously described, the PGF2α-induced pretone in the presence of KPSS and diltiazem was set to the same level as that for the control HPV response. Under these conditions, the increase in both [Ca2+]i and tension during phase I was significantly inhibited, by 30 and 19 %, respectively. However, treatment with KPSS and diltiazem did not affect either parameter measured during phase II.
Effects of La3+ and SKF 96365 on HPV
The limited effect of antagonists of voltage-gated Ca2+ entry on HPV in both normally polarized and depolarized arteries implies either that Ca2+ entry during contraction occurs through one or more non-voltage-dependent pathways, or that the rise in [Ca2+]i illustrated in Fig. 3B and D is due to release from intracellular stores. We used the non-selective Ca2+ entry blockers La3+ and SKF 96365 to explore the former possibility. As described above, the concentration of PGF2α was raised in order to maintain the level of pretone (3.5-4 μM PGF2α for 1 μM La3+; 50–100 μM PGF2α for 100 μM La3+; 10 μM PGF2α for 10 μM SKF 96365; 15 μM PGF2α for 100 μM SKF 96365). A Hepes-buffered solution was used with La3+ to avoid precipitation.
Figure 4A and B shows example traces of the effect of two concentrations of La3+ on tension and [Ca2+]i during HPV. The mean response of a number of IPAs is illustrated in Fig. 4C and D. A low concentration of La3+ (1 μM) greatly inhibited the phase I contraction, but did not suppress the phase II contraction. In contrast to the organic Ca2+ channel blockers, La3+ at a higher concentration (100 μM) not only eliminated phase I contraction completely, but also strongly reduced the phase II contraction. The effects of La3+ on [Ca2+]i closely paralleled those on tension; the lower concentration of La3+ selectively suppressed the transient rise in [Ca2+]i associated with phase I, and the higher concentration abolished the transient rise and greatly diminished the sustained increase in [Ca2+]i coinciding with the phase II contraction (Fig. 4B and D).
Figure 4. Effect of La3+ on HPV in IPA.
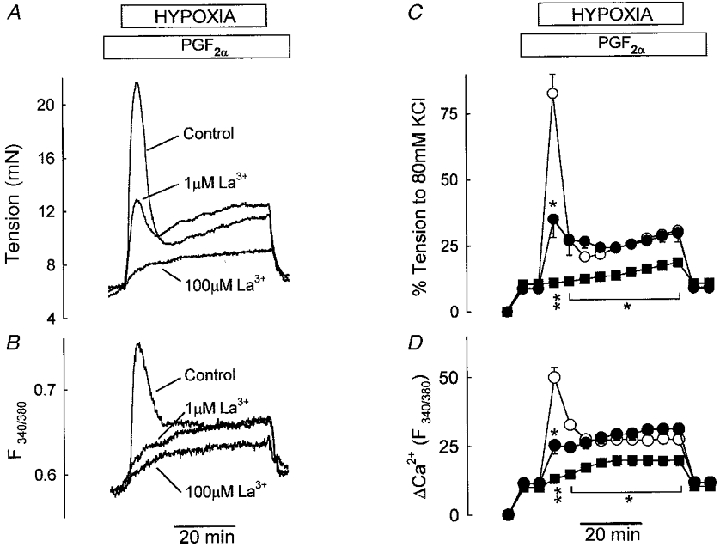
A and B, tension and fura PE-3 F340/380 fluorescence ratio, respectively, measured simultaneously in a single IPA under control conditions and in the presence of 1 and 100 μM La3+. C and D, mean results for five IPAs: control, ○; 1 μM La3+, •; and 100 μM La3+, ▪. * P < 0.05, ** P < 0.001 compared to control.
In the light of previous evidence that release of Ca2+ from intracellular stores is an early consequence of hypoxia in pulmonary artery smooth muscle (Jabr et al. 1997; Gelband & Gelband, 1997), it seemed possible that micromolar concentrations of La3+ might be acting to block Ca2+ entry via a store-operated Ca2+ channel or ‘capacitative’ Ca2+ entry mechanism (CCE), analogous to that existing in other types of cell (Putney, 1986; Broad et al. 1999). In this case, it would be predicted (1) that agents which selectively deplete intracellular Ca2+ stores should cause a sustained increase in Ca2+ influx which would also be sensitive to low concentrations of La3+, and (2) that activation of CCE in the presence of extracellular Mn2+ should cause an influx of this cation which would be demonstrable as a quenching of the fura signal at its isosbestic wavelength (∼360 nm) (Morgan & Jacob, 1994).
In order to investigate the first prediction, IPAs were placed into Ca2+-free solution, and then treated for 10 min with thapsigargin (1 μM), an agent that blocks the smooth endoplasmic reticulum Ca2+-ATPase (SERCA). Thapsigargin did not cause a contraction under these conditions. However, subsequent restoration of the normal concentration of Ca2+ caused a sustained contraction (Fig. 5A). This contraction was likely to be due to Ca2+ entry via CCE, since it was not observed if thapsigargin treatment during the Ca2+-free period was omitted (Fig. 5B). As predicted, application of 1 μM La3+ prior to the re-admission of Ca2+ abolished the contraction seen in thapsigargin-treated IPAs (Fig. 5A). Similar results were obtained with 10 μM cyclopiazonic acid (CPA, n= 5, data not shown).
Figure 5. Thapsigargin-induced tension in IPA.
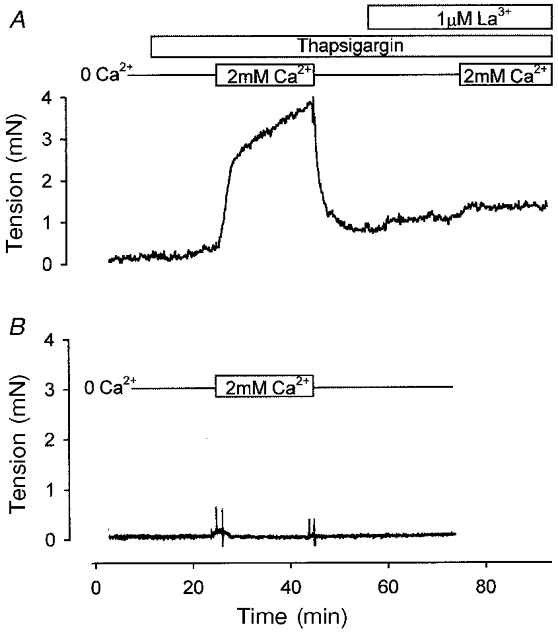
A, the trace shows tension developed by an IPA incubated in Ca2+-free solution (no EGTA). Thapsigargin (1 μM) was applied after 10 min, and 2 mM Ca2+ was added to the bath 10 min later. The tension developed on re-admission of Ca2+ was completely ablated when the procedure was repeated in the presence of 1 μM La3+. B, re-admission of Ca2+ without prior application of thapsigargin did not cause constriction, suggesting that the contraction was due to a capacitative Ca2+ entry mechanism. Results are typical for at least four independent experiments for the two conditions.
The second prediction was evaluated as follows. IPAs were loaded with fura-2 AM, which we found to be more effectively quenched by Mn2+ than fura PE-3. The 360 nm signal was monitored in normal PSS, and during an 80 s exposure to PSS containing 50 μM Mn2+. Addition of Mn2+ led to an acceleration of the slow rate of decay of the signal (Fig. 6A, control trace; Fig. 6B, open circles, mean data). Mn2+ was then removed, causing an immediate return of the rate of decay of the signal to the pre-Mn2+ level (not shown). IPAs were then treated for 10 min with 1 μM thapsigargin, which itself did not affect the 360 nm signal. Thapsigargin was then removed, and Mn2+ was reapplied. Figure 6A (see also Fig. 6B, filled circles) illustrates that the resulting acceleration of the decay of the fura-2 signal, indicative of Mn2+ entry, was now greatly enhanced. Results are expressed in terms of the total quenchable 360 nm signal; maximum quench was ascertained by treating the IPA at the end of the experiment with ionomycin in the presence of Mn2+, and allowing the signal to decay completely.
Figure 6. Thapsigargin-induced Mn2+ influx in IPA; evidence for CCE.
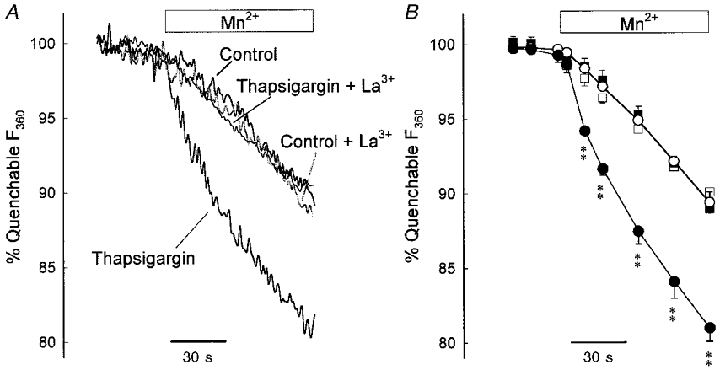
A, typical traces of the change in fluorescence at 360 nm of IPA loaded with fura-2 AM due to addition of 50 μM Mn2+, under control conditions and following treatment with thapsigargin, and in the presence of 1 μM La3+. Values are expressed as a proportion of the 360 nm signal quenchable by Mn2+. B, mean response for control IVA (○, n= 8), thapsigargin-treated IVA (•, n= 5), control IVA in the presence of 1 μM La3+ (□, n= 3), and thapsigargin-treated IVA in the presence of 1 μM La3+ (▪, n= 3). Thapsigargin treatment substantially increased the rate of Mn2+ influx; this increase was abolished by 1 μM La3+, which did not affect the control rate. Each symbol is the mean of rolling averages (6 s) derived from the individual experiments. ** P < 0.001 compared to control.
Similar experiments were carried out in IPA exposed to 1 μM La3+ before (for 10 min) and during Mn2+ application. La3+ had no effect on the rate of decay of the 360 nm signal in control IPA either before or during the application of Mn2+ (Fig. 6A; see also Fig. 6B, open squares). However, La3+ abolished the increased rate of signal decay evident when Mn2+ was added after thapsigargin pretreatment (Fig. 6A; see also Fig. 6B, filled squares). These results show that thapsigargin pretreatment activated a Mn2+-permeable influx pathway which was completely blocked by 1 μM La3+.
Figure 7 presents the concentration dependencies of the inhibition by La3+ of both phases of HPV, as well as the contractions evoked by KPSS, and following store depletion by thapsigargin (1 μM). For the hypoxic challenges the level of pretone was maintained in the presence of La3+ by increasing the concentration of PGF2α (to 3.5-4 μM for 1 μM La3+; 5–6 μM for 3 μM La3+; 5–10 μM for 10 μM La3+; 20–30 μM for 30 μM La3+; and 50–100 μM for 100 μM La3+). Pretone was not diminished by concentrations of La3+ below 1 μM. La3+ inhibited store release-associated contraction over a concentration range approximately 500-fold lower than that required to block depolarization-mediated contraction (Fig. 7). The potency of La3+ in inhibiting the thapsigargin-induced contraction was slightly greater than that for the phase I contraction (IC50∼200 vs.∼100 nM). The phase II contraction was also suppressed by La3+, though with a substantially greater IC50 than that for phase I (∼50 μM). At 100 μM La3+ a significant fraction of the phase II contraction remained (see also Fig. 4), although contraction in response to high K+ solution was suppressed to a significantly greater extent (KPSS: 88 ± 6 %, n= 7; phase II: 60 ± 5 %, n= 4; P < 0.01).
Figure 7. Dose dependency of La3+ block in IPA.
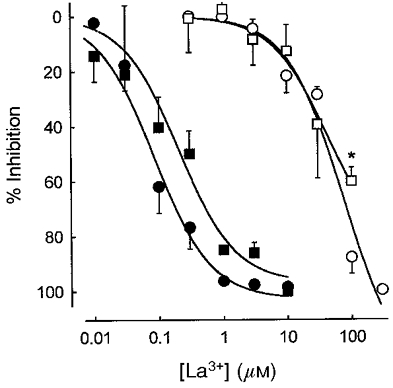
Effect of La3+ on phase I (▪) and phase II (□) of HPV, and thapsigargin- (•) and 80 mM KCl-induced tension (○). Tension is expressed in terms of inhibition from control. Each point is the mean of three to eight experiments. * P < 0.01, 80 mM KCl compared to phase II.
Although the concentration dependency of SKF 96365 was not studied in detail, its effect on HPV resembled that of La3+. At a concentration of 10 μM, SKF 96365 markedly inhibited the HPV phase I contraction (control, 59.6 ± 2.3 % response to KPSS; SKF 96365 (10 μM), 24 ± 4.6 %; n= 4, P < 0.001) but had no effect on phase II (control at 45 min, 22.3 ± 6.6 %; SKF 96365, 19.3 ± 0.8 %). However, at 100 μM SKF 96365 abolished both phases of contraction (phase I, 3.0 ± 3.3 %; phase II, 1.6 ± 2.6 %; n= 4, P < 0.001). Notably, 100 μM SKF 96365 required only 15 μM PGF2α to maintain pretone, whereas 100 μM La3+ required 50–100 μM PGF2α.
Effect on HPV of CPA, thapsigargin, caffeine and ryanodine, and preconstriction with 20 mM K+
The apparent dependency of the HPV phase I contraction on a CCE mechanism implies that Ca2+ release must be occurring in these arteries before or during this transient contraction. In accord with this prediction, pretreatment of IPA with CPA (30 μM) greatly reduced the phase I contraction (Fig. 8A). An apparent suppression of the phase II contraction by CPA did not reach significance. In separate experiments, 30 nM thapsigargin was applied for 10 min, and then removed from the bath 10 min before pretone was evoked with 0.5-1 μM PGF2α. Under these conditions phase I of HPV was also strongly depressed (control, 62.7 ± 2.7 % response to KPSS; thapsigargin, 6.6 ± 1.7 %; n= 7, P < 0.001).
Figure 8. Effect of CPA, caffeine and ryanodine, and preconstriction with 20 mM K+ on HPV in IPA.
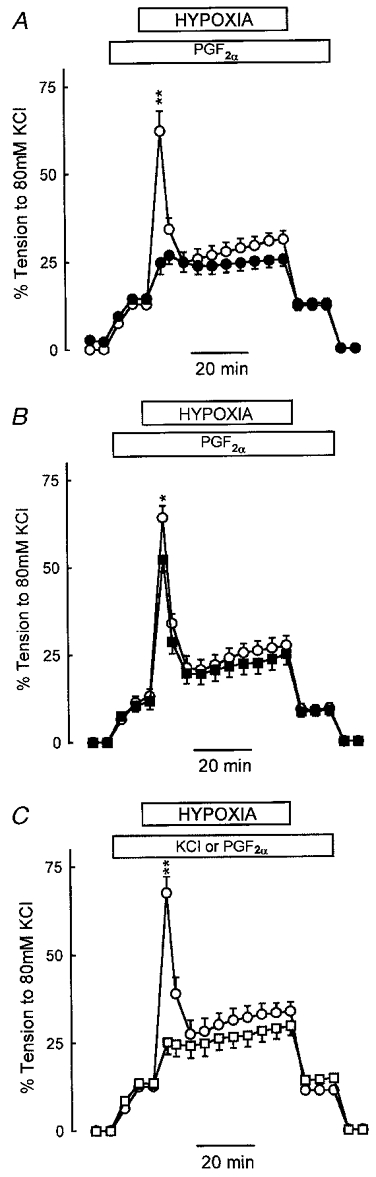
A and B, IPAs were exposed to hypoxia in the presence of 3 μM PGF2α (○) and following preincubation with 30 μM CPA (A, •, n= 5), or 10 μM ryanodine and 10 mM caffeine (B, ▪, n= 7). For CPA only, PGF2α had to be reduced to 1 μM to maintain the same level of pretone as control. For ryanodine and caffeine no alteration from 3 μM PGF2α was required. C, IPAs were exposed to hypoxia following preconstriction with 3 μM PGF2α (○) or 20 mM K+ (□, n= 5). * P < 0.05, ** P < 0.001 compared to control.
To examine the role of caffeine- and ryanodine-sensitive stores, IPAs were treated with 10 μM ryanodine for 20 min prior to and during the hypoxic challenge. The IPAs were then challenged with caffeine (10 mM) for two periods of 2 min each at 10 and 5 min before preconstriction with PGF2α. The first addition of caffeine caused a small transient constriction that returned to baseline; there was no response to the second addition. Ryanodine and caffeine had no effect on the level of preconstriction elicited by 3 μM PGF2α. In contrast to the effect of CPA, treatment with caffeine and ryanodine had only a small effect on HPV (Fig. 8B), with a reduction in phase I of < 20 %.
In separate experiments we examined the possibility that hypoxia-mediated Ca2+ store release, and therefore phase I, might be facilitated by the presence of an agonist. We therefore used PSS containing 20 mM K+ to set pretone rather than PGF2α. Under these conditions the response to hypoxia was almost identical to that following treatment with CPA (Fig. 8C), in that phase I was much less pronounced, whereas phase II was similar to that observed with PGF2α.
DISCUSSION
We have previously suggested that the mechanism(s) underlying HPV is multifactorial (Ward & Aaronson, 1999). Although hypoxia is generally reported to elicit a sustained monophasic rise in PA pressure in vivo and in blood-perfused lungs, isolated PA preparations often exhibit a biphasic response when hypoxia is maintained for more than ∼10-15 min. This is more apparent in some species (e.g. rat, pig) than others, and is enhanced in the presence of agonist-induced pretone (see Ozaki et al. 1998; Ward & Aaronson, 1999). The biphasic response usually consists of a transient phase I constriction superimposed on a sustained phase II constriction. The sustained component has been shown to be endothelium dependent in rat (Leach et al. 1994), rabbit (Johns et al. 1989), pig (Kovitz et al. 1993) and human (Demiryurek et al. 1993), although in the latter a transient phase I was not apparent. In contrast, it has been reported that the phase II constriction in rat IPA is not dependent on the endothelium (Bennie et al. 1991; Jin et al. 1992), although these studies were difficult to interpret since very high levels of preconstriction were used, and hypoxia actually caused a net relaxation.
Phase I may have less relevance to sustained HPV in vivo, since it is by nature transient (discussed in Ward & Robertson, 1995; Ward & Aaronson, 1999), although it could be argued that phase I may have a role in the initial rapid response to hypoxia seen in vivo, or for moment-to-moment regulation of the ventilation-perfusion ratio. Whatever the relative importance of the two phases, it is clear from this study that they involve different Ca2+ entry mechanisms.
Effects of selective antagonists of voltage-gated Ca2+ channels
Our results show that when the level of pretone is kept constant, both phases of HPV in rat IPA are clearly apparent in the presence of organic Ca2+ channel blockers. Moreover, HPV persists even when these blockers are applied to IPA in which the membrane potential has been strongly depolarized by high K+. Similar responses were obtained with all three organic Ca2+ channel blockers tested. In each case, phase I was only partially suppressed by Ca2+ channel blockade with, or without, depolarization. However, both the rise in [Ca2+]i and the development of tension during phase II were essentially independent of Ca2+ influx via voltage-gated Ca2+ channels. These results demonstrate that depolarization and Ca2+ influx through such channels are not required for either the plateau elevation of [Ca2+]i or the progressive rise in tension which characterize phase II in this model of HPV (Robertson et al. 1995).
We were careful to maintain the same level of pretone during the various interventions that were used to block depolarization-induced Ca2+ influx as that for the control hypoxic challenge. This approach was based on the dependency of HPV on the level of pretone which has been consistently demonstrated in vivo, in perfused lung preparations and in isolated pulmonary arteries (e.g. Fishman, 1976; Ward & Robertson, 1995; Ozaki et al. 1998). It is of interest that Rodman et al. (1989) also demonstrated that nifedipine did not suppress the early peak contraction in response to hypoxia in rat PA if pretone was maintained at control levels by elevating the concentration of phenylephrine, although this contraction was reduced if pretone was not maintained in this way. This previous report, together with the present more systematic study of both phases of HPV, suggests strongly that the reported inhibitory action of Ca2+ channel blockers on HPV in several models (Tucker et al. 1976; McMurtry et al. 1976; Jin et al. 1992; Leach et al. 1994; Savineau et al. 1995; Woodmansey et al. 1995) could have been due to a non-specific decrease in initial pulmonary vascular tone, in other words pretone (Ward & Aaronson, 1999).
We observed that the initial relaxation following phase I was diminished in arteries which had been first depolarized and then exposed to nifedipine (Fig. 2C). This did not occur if nifedipine was applied before KPSS (Fig. 2D). Although we did not further explore this dichotomy, we speculate that the loading of intracellular Ca2+ stores that occurred when KPSS was applied prior to nifedipine may have caused a more prolonged release of Ca2+ during phase I, thus offsetting the subsequent relaxation. The mechanism of this relaxation, which is especially prominent if pretone is marked (e.g. Bennie et al. 1991) remains unclear. However, its persistence in depolarized arteries suggests that it is not caused by hyperpolarization or the offset of depolarization following phase I.
A possible criticism of the present experiments is that the changes in PGF2α concentration needed to maintain a constant level of pretone during voltage-gated Ca2+ channel blockade may have biased the subsequent response to hypoxia, because the nature of the transduction mechanisms activated by different concentrations of PGF2α may have varied. For example, higher concentrations of PGF2α might cause increased sensitivity of the artery to hypoxia, or of the contractile apparatus to Ca2+. This seems unlikely for two reasons. Firstly, in experiments with normally polarized arteries, the PGF2α concentration had to be increased only from 3 to a maximum of 4 μM in order to maintain pretone in the presence of organic Ca2+ channel blockers (e.g. Fig. 2). Secondly, in experiments with depolarized arteries the concentration of PGF2α was not increased, but instead decreased to 1 μM (e.g. Fig. 3) in order to maintain a constant pretone. The former effect is likely to be due to a partial dependency of PGF2α constriction on Ca2+ influx via voltage-gated channels, while the latter is consistent with the possibility that depolarization favours constriction via other mechanisms, for example enhancement of IP3 production (Ganitkevich & Isenberg, 1993). In both cases, however, the relationship between tension and [Ca2+]i during pretone was not altered from control, which was perhaps not surprising since the range of PGF2α concentrations used (1-4 μM) was narrow. This suggests that over this range any existing level of Ca2+ sensitization is constant. Taken together, these data imply that varying the concentration of PGF2α within this range has no additional effects on HPV, other than those related to the level of pretone.
Partial inhibition by organic Ca2+ channel blockers of the rapid hypoxia-induced rise in [Ca2+]i has previously been described in isolated IPA cells (Vadula et al. 1993; Cornfield et al. 1994). In the present study all three organic Ca2+ channel blockers tested caused partial inhibition of phase I, at concentrations sufficient to cause complete inhibition of high K+-induced contraction. Therefore, whereas the phase II contraction in rat IPA did not require voltage-gated Ca2+ influx, phase I may be partly dependent on this pathway. With regard to the remainder of the phase I contraction, past reports have suggested that HPV is also dependent to a variable degree on the release of intracellular Ca2+ stores (Vadula et al. 1993; Gelband & Gelband, 1997; Jabr et al. 1997). Our demonstration that CPA greatly diminished the amplitude of the phase I contraction in rat IPA is in agreement with the earlier finding of Gelband & Gelband (1997) that thapsigargin eliminated the rapid hypoxia-mediated contraction and rise in [Ca2+]i in these arteries.
Effects of La3+
It is established that the release of intracellular Ca2+ stores is in many cell types associated with activation of a Ca2+ influx pathway mediated by store-operated Ca2+ channels (e.g. Putney, 1986; Berridge, 1995; Broad et al. 1999). It is proposed that in mammals this ‘capacitative’ Ca2+ entry (CCE) is mediated by one or more of a family of seven proteins homologous to the transient receptor potential (Trp) protein of Drosophila (Hardie & Minke, 1993; Birnbaumer et al. 1996). In addition to store release, CCE can be activated by a number of other stimuli related to G protein activation (Boulay et al. 1997; Kiselyov et al. 1998; Hofmann et al. 1999).
CCE has been shown to occur in rat main pulmonary artery following treatment with thapsigargin, CPA or noradrenaline, and was blocked by 50 μM La3+ (Gonzalez De La Fuente et al. 1995). We have confirmed that CCE also exists in these smaller rat IPAs by showing both that thapsigargin treatment under Ca2+-free conditions causes a contraction upon Ca2+ repletion (Fig. 5; analogous to Kwan et al. 1990), and that thapsigargin treatment causes an increased entry of Mn2+, manifested as quenching of the fura-2 isosbestic signal, which is abolished by 1 μM La3+ (Fig. 6; see Morgan & Jacob, 1994). Although the former observation could also be accounted for if thapsigargin prevented the functioning of a superficial buffer barrier (Loutzenhizer & van Breemen, 1983), the selective effect of La3+ on thapsigargin-stimulated Mn2+ influx is difficult to explain in this way. Apart from the known ability of La3+ to block a release-activated Mn2+ influx pathway (i.e. CCE; Kwan et al. 1990), this observation could only be explained if micromolar La3+ was blocking Ca2+ release, which is extremely unlikely considering that La3+ was first introduced as a tool precisely because it blocks transmembrane Ca2+ fluxes but not Ca2+ release (van Breemen, 1969).
In the light of previous evidence that hypoxia evokes the release of intracellular Ca2+ stores in pulmonary arteries, we used La3+ to investigate whether CCE might underlie the organic Ca2+ blocker-insensitive response to hypoxia. The experiments described in Figs 5 and 7 revealed that thapsigargin-mediated CCE was exquisitely sensitive to blockade by La3+ (IC50∼100 nM). La3+ suppressed the hypoxia-induced phase I contraction over a similar, albeit slightly higher, concentration range. Consistent with this, 1 μM La3+ profoundly reduced the rise in [Ca2+]i associated with the phase I contraction (Fig. 4), but had no effect on the high K+-induced constriction (Fig. 7). These results, as well as the observation that phase I is greatly suppressed by prior treatment of IPA with CPA or thapsigargin, suggest that the transient phase I constriction in response to hypoxia in isolated rat IPA is mostly due to CCE triggered by release of intracellular Ca2+ stores.
Whereas the 1 μM La3+ and CPA/thapsigargin data suggest that phase I is almost entirely mediated via CCE, the organic Ca2+ channel blocker data suggest that voltage-gated Ca2+ influx contributes up to 50 % of this phase (see above, and Figs 2 and 3). However, there is evidence that these agents may directly reduce store release, as Kanaide et al. (1988) have shown that verapamil and diltiazem inhibit noradrenaline-induced Ca2+ store release in rat vascular smooth muscle cells (IC50∼4 and ∼25 μM, respectively). Saida & van Breemen (1983) also showed that 10 μM diltiazem and 1 μM nisoldipine inhibited store release-mediated contractions by 30–40 % in membrane-skinned rabbit mesenteric artery. It is therefore possible that the effect of these drugs on phase I overestimates the contribution of depolarization-mediated Ca2+ entry.
Jabr et al. (1997) reported that the response to hypoxia in canine small PA was almost abolished by ryanodine and caffeine, but was potentiated by CPA or thapsigargin. They concluded that HPV was due to release of a ryanodine-sensitive Ca2+ store, which was buffered by a separate thapsigargin/CPA-sensitive store. They also found that although nisoldipine abolished HPV under control conditions, it caused only a partial inhibition after pretreatment with CPA or thapsigargin, and therefore suggested that hypoxia might activate a nisoldipine-insensitive Ca2+ entry. Conversely, in rat IPA preconstricted with PGF2α, CPA and thapsigargin almost abolished phase I, whereas caffeine and ryanodine had only a small effect. This difference may arise because separate ryanodine- and thapsigargin-sensitive Ca2+ pools respond differentially to hypoxia and store depletion in various species. For example, the results of Jabr et al. (1997) suggest that in canine IPA, hypoxia releases a ryanodine-sensitive Ca2+ store, but that a separate thapsigargin/CPA-sensitive Ca2+ store controls CCE. Conversely, our results indicate that in rat IPA hypoxia acts directly on the thapsigargin/CPA-sensitive store.
A large transient phase I constriction would appear to be associated with pretone induced by a receptor-coupled agonist (i.e. PGF2α), since phase I was much less pronounced when pretone was set with 20 mM K+. One possible explanation for this effect is that G protein stimulation may promote CCE, as the opening of certain Trp channels has been shown to be activated by diacylglycerol and IP3 (Kiselyov et al. 1998; Hofmann et al. 1999).
Figures 1–3 demonstrate that voltage-dependent Ca2+ influx played almost no part in mediating the phase II response to hypoxia in these arteries. In addition, phase II was not abolished by CPA, or caffeine and ryanodine, suggesting that it was not dependent on Ca2+ release. However, phase II was abolished by removal of extracellular Ca2+ or addition of 100 μM SKF 96365, and was substantially reduced by 100 μM La3+, suggesting that it does require Ca2+ influx. Taken together, these data imply that phase II involves a voltage-independent Ca2+ influx that is not dependent on store release and CCE.
The concentration-dependent effect of La3+ on the thapsigargin-induced contraction, and phase I and phase II of HPV in these experiments is similar to that of the trivalent cation Gd3+ reported by Broad et al. (1999) in rat A7r5 vascular smooth muscle cells stimulated with vasopressin. In the latter study, CCE was blocked by 1 μM Gd3+, whereas block of a voltage-independent but non-capacitative Ca2+ entry mechanism required least 100 μM Gd3+. Broad et al. (1999) suggested that this non-capacitative mechanism was directly activated by arachidonic acid. It remains to be seen whether this or a related pathway could be responsible for the sustained rise in [Ca2+]i during phase II of HPV in rat IPA.
We have previously shown that removal of the endothelium has little effect on the phase I increase in tension but abolishes phase II, although it does not alter the associated changes in [Ca2+]i during either phase (see Ward & Aaronson, 1999). This suggests not only that the endothelium does not affect Ca2+ influx or handling in the smooth muscle during HPV, but also that the rise in [Ca2+]i on its own is insufficient to cause contraction during phase II of HPV. We have previously speculated that full development of phase II requires both a rise in [Ca2+]i and an increase in Ca2+ sensitivity caused by an as yet unidentified endothelium-derived mediator (Ward & Aaronson, 1999). It is conceivable that release of such a mediator might be dependent on endothelial cell [Ca2+]i and therefore also affected by, for example, the higher concentrations of La3+ required to inhibit phase II. Although such an effect might contribute to the suppression of phase II tension under such conditions, it does not alter our conclusions regarding [Ca2+]i in the smooth muscle during HPV, as this is apparently independent of the endothelium (Ward & Aaronson, 1999).
In summary, our results demonstrate that at least in this model of HPV, depolarization and Ca2+ entry via voltage-dependent channels play at best a supporting role in the hypoxia-induced rise in [Ca2+]i. Instead, voltage-independent capacitative and non-capacitative Ca2+ entry pathways may provide the major sources of Ca2+ during hypoxia, and support the transient and sustained phases of HPV, respectively.
Acknowledgments
We are grateful to the Wellcome Trust (grant number 043357), the British Heart Foundation (grant number PG96044) and the Special Trustees of St Thomas’ Hospital for their support of this work.
References
- Bennie RE, Packer CS, Powell DR, Jin N, Rhoades RA. Biphasic contractile response of pulmonary artery to hypoxia. American Journal of Physiology. 1991;261:L156–163. doi: 10.1152/ajplung.1991.261.2.L156. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Capacitative calcium entry. Biochemical Journal. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaumer L, Zhu X, Jiang M, Boulay G, Peyton M, Vannier B, Brown D, Platano D, Sadeghi H, Stefani E, Birnbaumer M. On the molecular basis and regulation of cellular capacitative calcium entry: roles for Trp proteins. Proceedings of the National Academy of Sciences of the USA. 1996;93:15195–15202. doi: 10.1073/pnas.93.26.15195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay G, Zhu X, Peyton M, Jiang M, Hurst R, Stefani E, Birnbaumer L. Cloning and expression of a novel mammalian homolog of Drosophila transient receptor potential (Trp) involved in calcium entry secondary to activation of receptors coupled by the Gq class of G protein. Journal of Biological Chemistry. 1997;272:29672–29680. doi: 10.1074/jbc.272.47.29672. [DOI] [PubMed] [Google Scholar]
- Broad LM, Cannon TR, Taylor CW. A non-capacitative pathway activated by arachidonic acid is the major Ca2+ entry mechanism in rat A7r5 smooth muscle cells stimulated with low concentrations of vasopressin. The Journal of Physiology. 1999;517:121–134. doi: 10.1111/j.1469-7793.1999.0121z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornfield DN, Stevens T, McMurtry IF, Abman SH, Rodman DM. Acute hypoxia causes membrane depolarization and calcium influx in fetal pulmonary artery smooth muscle cells. American Journal of Physiology. 1994;266:L469–475. doi: 10.1152/ajplung.1994.266.4.L469. [DOI] [PubMed] [Google Scholar]
- Demiryurek AT, Wadsworth RM, Kane KA, Peacock AJ. The role of endothelium in hypoxic constriction of human pulmonary artery rings. American Review of Respiratory Diseases. 1993;147:283–290. doi: 10.1164/ajrccm/147.2.283. [DOI] [PubMed] [Google Scholar]
- Fishman AP. Hypoxia on the pulmonary circulation. How and where it acts. Circulation Research. 1976;38:221–231. doi: 10.1161/01.res.38.4.221. [DOI] [PubMed] [Google Scholar]
- Ganitkevich VY, Isenberg G. Membrane potential modulates inositol 1,4,5-trisphosphate-mediated Ca2+ transients in guinea-pig coronary myocytes. The Journal of Physiology. 1993;470:35–44. doi: 10.1113/jphysiol.1993.sp019845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelband CH, Gelband H. Ca2+ release from intracellular stores is an initial step in hypoxic pulmonary vasoconstriction of rat pulmonary artery resistance vessels. Circulation. 1997;96:3647–3654. doi: 10.1161/01.cir.96.10.3647. [DOI] [PubMed] [Google Scholar]
- Gonzalez De La Fuente P, Savineau JP, Marthan R. Control of pulmonary vascular smooth muscle tone by sarcoplasmic reticulum Ca2+ pump blockers: thapsigargin and cyclopiazonic acid. Pflügers Archiv. 1995;429:617–624. doi: 10.1007/BF00373982. [DOI] [PubMed] [Google Scholar]
- Hardie RC, Minke B. Novel Ca2+ channels underlying transduction in Drosophila photoreceptors: implications for phosphoinositide-mediated Ca2+ mobilization. Trends in Neurosciences. 1993;16:371–376. doi: 10.1016/0166-2236(93)90095-4. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- Jabr RI, Toland H, Gelband CH, Wang XX, Hume JR. Prominent role of intracellular Ca2+ release in hypoxic vasoconstriction of canine pulmonary artery. British Journal of Pharmacology. 1997;122:21–30. doi: 10.1038/sj.bjp.0701326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin N, Packer CS, Rhoades RA. Pulmonary arterial hypoxic contraction: Signal transduction. American Journal of Physiology. 1992;263:L73–78. doi: 10.1152/ajplung.1992.263.1.L73. [DOI] [PubMed] [Google Scholar]
- Johns RA, Linden JM, Peach MJ. Endothelium-dependent relaxation and cyclic GMP accumulation in rabbit pulmonary artery are selectively impaired by moderate hypoxia. Circulation Research. 1989;65:1508–1515. doi: 10.1161/01.res.65.6.1508. [DOI] [PubMed] [Google Scholar]
- Kanaide H, Kobayashi S, Nishimura J, Hasegawa M, Shogakiuchi Y, Matsumoto T, Nakamura M. Quin2 microfluorometry and effects of verapamil and diltiazem on calcium release from rat aorta smooth muscle cells in primary culture. Circulation Research. 1988;63:16–26. doi: 10.1161/01.res.63.1.16. [DOI] [PubMed] [Google Scholar]
- Kiselyov K, Xu X, Mozhayeva G, Kuo T, Pessah I, Mignery G, Zhu X, Birnbaumer L, Muallem S. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature. 1998;396:478–482. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- Kovitz KL, Aleskowitch TD, Sylvester JT, Flavahan NA. Endothelium-derived contracting and relaxing factors contribute to hypoxic responses of pulmonary arteries. American Journal of Physiology. 1993;265:H1139–1148. doi: 10.1152/ajpheart.1993.265.4.H1139. [DOI] [PubMed] [Google Scholar]
- Kwan CY, Takemura H, Obie JF, Thastrup O, Putney JW., Jr Effects of MeCh, thapsigargin, and La3+ on plasmalemmal and intracellular Ca2+ transport in lacrimal acinar cells. American Journal of Physiology. 1990;258:C1006–1015. doi: 10.1152/ajpcell.1990.258.6.C1006. [DOI] [PubMed] [Google Scholar]
- Leach RM, Robertson TP, Twort CH, Ward JPT. Hypoxic vasoconstriction in rat pulmonary and mesenteric arteries. American Journal of Physiology. 1994;266:L223–231. doi: 10.1152/ajplung.1994.266.3.L223. [DOI] [PubMed] [Google Scholar]
- Leach RM, Twort CHC, Cameron IC, Ward JPT. A comparison of the pharmacological and mechanical properties in vitro of large and small pulmonary arteries of the rat. Clinical Science. 1992;82:55–62. doi: 10.1042/cs0820055. [DOI] [PubMed] [Google Scholar]
- Loutzenhiser R, Van Breemen C. The influence of receptor occupation on Ca2+ influx-mediated vascular smooth muscle contraction. Circulation Research. 1983;52:I97–103. [PubMed] [Google Scholar]
- McMurtry IF, Davidson AB, Reeves JT, Grover RF. Inhibition of hypoxic pulmonary vasoconstriction by calcium antagonists in isolated rat lungs. Circulation Research. 1976;38:99–104. doi: 10.1161/01.res.38.2.99. [DOI] [PubMed] [Google Scholar]
- Madden JA, Harder DR, Dawson C. Hypoxia-induced activation in small isolated pulmonary arteries from the cat. Journal of Applied Physiology. 1984;59:113–118. doi: 10.1152/jappl.1985.59.1.113. [DOI] [PubMed] [Google Scholar]
- Morgan AJ, Jacob R. Ionomycin enhances Ca2+ influx by stimulating store-regulated cation entry and not by a direct action at the plasma membrane. Biochemical Journal. 1994;300:665–672. doi: 10.1042/bj3000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipenko ON, Evans AM, Gurney AM. Regulation of the resting potential of rabbit pulmonary artery myocytes by a low threshold, O2-sensing potassium current. British Journal of Pharmacology. 1997;120:1461–1470. doi: 10.1038/sj.bjp.0701075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki M, Marshall C, Amaki Y, Marshall BE. Role of wall tension in hypoxic responses of isolated rat pulmonary arteries. American Journal of Physiology. 1998;275:L1069–1077. doi: 10.1152/ajplung.1998.275.6.L1069. [DOI] [PubMed] [Google Scholar]
- Post JM, Gelband CH, Hume JR. [Ca2+]i inhibition of K+ channels in canine pulmonary artery: Novel mechanism for hypoxia-induced membrane depolarization. Circulation Reserach. 1995;77:131–139. doi: 10.1161/01.res.77.1.131. [DOI] [PubMed] [Google Scholar]
- Post JM, Hume JR, Archer SL, Weir EK. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. American Journal of Physiology. 1992;262:C882–890. doi: 10.1152/ajpcell.1992.262.4.C882. [DOI] [PubMed] [Google Scholar]
- Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Robertson TP, Aaronson PI, Ward JPT. Hypoxic vasoconstriction and intracellular Ca2+ in pulmonary arteries: Evidence for PKC-independent Ca2+ sensitization. American Journal of Physiology. 1995;268:H301–307. doi: 10.1152/ajpheart.1995.268.1.H301. [DOI] [PubMed] [Google Scholar]
- Rodman DM, Yamaguch T, O'Brien RF, McMurtry IF. Hypoxic contraction of isolated rat pulmonary artery. Journal of Pharmacology and Experimental Therapeutics. 1989;248:952–959. [PubMed] [Google Scholar]
- Saida K, van Breemen C. Mechanism of Ca2+ antagonist induced vasodilation; Intracellular actions. Circulation Research. 1983;52:137–142. doi: 10.1161/01.res.52.2.137. [DOI] [PubMed] [Google Scholar]
- Salvaterra CG, Goldman WF. Acute hypoxia increases cytosolic calcium in cultured pulmonary arterial myocytes. American Journal of Physiology. 1993;264:L323–328. doi: 10.1152/ajplung.1993.264.3.L323. [DOI] [PubMed] [Google Scholar]
- Savineau JP, Gonzalez De La Fuente P, Marthan R. Cellular mechanisms of hypoxia-induced contraction in human and rat pulmonary arteries. Respiration Physiology. 1995;99:91–98. doi: 10.1016/0034-5687(94)00091-d. [DOI] [PubMed] [Google Scholar]
- Smirnov SV, Robertson TP, Ward JPT, Aaronson PI. Chronic hypoxia is associated with reduced delayed rectifier K+ current in rat pulmonary artery muscle cells. American Journal of Physiology. 1994;266:H365–370. doi: 10.1152/ajpheart.1994.266.1.H365. [DOI] [PubMed] [Google Scholar]
- Tucker A, McMurtry IF, Grover RF, Reeves JT. Attenuation of hypoxic pulmonary vasoconstriction by verapamil in intact dogs. Proceedings of the Society for Experimental Biology and Medicine. 1976;151:611–614. doi: 10.3181/00379727-151-39271. [DOI] [PubMed] [Google Scholar]
- Vadula MS, Kleinman JG, Madden JA. Effect of hypoxia and norepinephrine on cytoplasmic free Ca2+ in pulmonary and cerebral arterial myocytes. American Journal of Physiology. 1993;265:L591–597. doi: 10.1152/ajplung.1993.265.6.L591. [DOI] [PubMed] [Google Scholar]
- Van Breemen C. Blockade of membrane calcium fluxes by lanthanum in relation to vascular smooth muscle contractility. Archives Internationales de Physiologie et de Biochimie. 1969;77:710–716. doi: 10.3109/13813456909059783. [DOI] [PubMed] [Google Scholar]
- Von Euler US, Liljestrand G. Observations on the pulmonary arterial blood pressure in the cat. Acta Physiologica Scandinavica. 1946;12:301–320. [Google Scholar]
- Wadsworth RM. Vasoconstrictor and vasodilator effects of hypoxia. Trends in Pharmacological Sciences. 1994;15:47–53. doi: 10.1016/0165-6147(94)90109-0. [DOI] [PubMed] [Google Scholar]
- Ward JPT, Aaronson PI. Mechanisms of hypoxic pulmonary vasoconstriction: Can anyone be right? Respiration Physiology. 1999;115:261–271. doi: 10.1016/s0034-5687(99)00025-0. [DOI] [PubMed] [Google Scholar]
- Ward JPT, Robertson TP. The role of the endothelium in hypoxic pulmonary vasoconstriction. Experimental Physiology. 1995;80:793–801. doi: 10.1113/expphysiol.1995.sp003887. [DOI] [PubMed] [Google Scholar]
- Weir EK, Archer SL. The mechanism of acute hypoxic pulmonary vasoconstriction: the tale of two channels. Federation of American Sciences and Experimental Biology Journal. 1995;9:183–189. doi: 10.1096/fasebj.9.2.7781921. [DOI] [PubMed] [Google Scholar]
- Woodmansey PA, Zhang F, Channer KS, Morice AH. Effect of the calcium antagonist amlodipine on the two phases of hypoxic pulmonary vasoconstriction in rat large and small isolated pulmonary arteries. Journal of Cardiovascular Pharmacology. 1995;25:324–329. doi: 10.1097/00005344-199502000-00019. [DOI] [PubMed] [Google Scholar]
- Yuan XJ, Goldman WF, Tod ML, Rubin LJ, Blaustein MP. Hypoxia reduces potassium currents in cultured rat pulmonary but not mesenteric arterial myocytes. American Journal of Physiology. 1993;264:L116–123. doi: 10.1152/ajplung.1993.264.2.L116. [DOI] [PubMed] [Google Scholar]
- Yuan XJ, Wang J, Juhaszova M, Golovina VA, Rubin LJ. Molecular basis and function of voltage-gated K+ channels in pulmonary arterial smooth muscle cells. American Journal of Physiology. 1998;274:L621–635. doi: 10.1152/ajplung.1998.274.4.L621. [DOI] [PubMed] [Google Scholar]