

Abstract
Adenosine causes voltage- and non-voltage-dependent inhibition of high voltage-activated (HVA) Ca2+ currents in Xenopus laevis embryo spinal neurons.
As this inhibition can be blocked by 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) and mimicked by N6-cyclopentyladenosine (CPA) it appears to be mediated by A1 receptors. Agents active at A2 receptors either were without effect or could be blocked by DPCPX. AMP had no agonist action on these receptors.
By using ω-conotoxin GVIA we found that adenosine inhibited an N-type Ca2+ current as well as a further unidentified HVA current that was insensitive to dihydropyridines, ω-agatoxin TK and ω-conotoxin MVIIC. Both types of current were subject to voltage- and non-voltage-dependent inhibition.
We used CPA and DPCPX to test whether A1 receptors regulated spinal motor pattern generation in spinalized Xenopus embryos. DPCPX caused a near doubling of, while CPA greatly shortened, the length of swimming episodes. In addition, DPCPX slowed, while CPA greatly speeded up, the rate of run-down of motor activity.
Our results demonstrate a novel action of A1 receptors in modulating spinal motor activity. Furthermore they confirm that adenosine is produced continually throughout swimming episodes and acts to cause the eventual termination of activity.
The purinergic transmitters adenosine and ATP regulate the generation of motor patterns in the frog embryo spinal cord (Dale & Gilday, 1996; Dale, 1998). ATP released by spinal neurons during motor activity activates P2Y receptors and reduces voltage-gated K+ currents thereby increasing neuronal excitability. However, ATP is converted to adenosine in the extracellular space. Although adenosine reduces voltage-gated Ca2+ currents, it does not alter synaptic transmission (Dale & Gilday, 1996). This reduction of Ca2+ currents by adenosine reduces neural excitability. As adenosine levels slowly mount (Dale, 1998), the inhibitory action of adenosine gradually overcomes the excitatory action of ATP. This leads to a gradual slowing and weakening of motor activity (termed run-down) and eventually results in its termination.
The mechanism of action of adenosine has not been studied in detail. In particular, the mechanism of block (i.e. whether voltage or non-voltage dependent), the identity of the adenosine receptors and the type of Ca2+ currents involved have not been established. In addition a more subtle but equally important question is whether AMP can act as an agonist for the adenosine receptor (see Ross et al. 1998). This arises because the delayed production of adenosine seems to result from feed-forward inhibition of the ecto-5′-nucleotidase by ADP or ATP. This enzyme produces adenosine from AMP, and numerical simulations suggest that inhibition of this enzyme by ADP (cf. Slakey et al. 1986) not only slows the appearance of adenosine and determines the length of motor activity but also causes a massive accumulation of AMP within the extracellular space of the spinal cord (Dale, 1998). Were AMP to act as an agonist for the adenosine receptor, it would short-circuit this delay.
Adenosine receptors are present in a variety of tissues, including the brain and spinal cord, peripheral sensory and motor neurons, the autonomic nervous system, cardiac tissues and visceral and vascular smooth muscle (Olah & Stiles, 1995; Sebastião & Ribeiro, 1996; Thorne & Housley, 1996; Ralevic & Burnstock, 1998). Four subtypes of the P1 adenosine receptor, which are all G-protein coupled, have now been isolated and cloned. These are termed A1, A2A, A2B and A3 subtypes. Many studies have shown them to be directly involved in the modulation of high voltage-activated (HVA) calcium channels in peripheral and central neurons. The A1 receptor has been shown to reduce N-type (Scholz & Miller, 1991; Mogul et al. 1993; Yawo & Chuhma, 1993; Zhu & Ikeda, 1993; Umemiya & Berger, 1994; Wu & Saggau, 1994) and P/Q-type currents (Ambrosio et al. 1996; 1997). Conversely, the A2 receptor has been reported to enhance P-type calcium currents in the hippocampus (Mogul et al. 1993; Goncalves et al. 1997) and brainstem (Umemiya & Berger, 1994), and to enhance both N- and P/Q- type channels in the striatum (Gubitz et al. 1996). This results in A1 and A2 receptors inhibiting and enhancing calcium-dependent transmitter release, respectively (reviewed by Ribeiro, 1995; Sebastião & Ribeiro, 1996; Henning, 1997; Ralevic & Burnstock, 1998). However, the A2A receptor inhibits L- and N-type currents in PC12 cells (Park et al. 1998), and both A1 and A2 receptors can inhibit calcium currents and transmitter release in hypothalamic neurons (Chen & van den Pol, 1997). The less well studied A3 receptor has been shown to enhance calcium currents in the hippocampus (Fleming & Mogul, 1997).
Spinal neurons of Xenopus larvae (stage 41/42, Nieuwkoop & Faber, 1956) possess N-, L- and P/Q-type HVA Ca2+ currents as well as a small component that defies pharmacological identification (Sun & Dale, 1998). At younger embryonic stages (37/38) spinal neurons have N-type currents (Wall & Dale, 1994), but the other components of the HVA current have not been identified. The purposes of this paper are to characterize in more detail the actions of adenosine on Ca2+ currents; identify the receptor subtype and the types of current involved and to examine the actions of the specific adenosine receptor on motor pattern generation in the frog embryo.
METHODS
Whole cell patch clamp recordings
Stage 37/38 Xenopus embryos (Nieuwkoop & Faber, 1956) were anaesthetized by immersion in MS222 (1 mg ml−1, Sigma, Poole, UK). Their spinal cords were dissected free and, using the methods described by Dale (1991), dissociated to provide acutely isolated neurons. Using a P97 puller (Sutter Instruments, Novato, CA, USA), electrodes were made from thick-walled borosilicate glass (WPI, Sarasota, FL, USA). An Axopatch 200B patch clamp amplifier (Axon Instruments, Foster City, CA, USA), together with a DT31EZ interface (Data Translation, Marlboro, NH, USA), was used to record and digitize current and voltage records, which were stored on the hard drive of a PC. The sampling and analysis software were written by Dale (1995a). Whole cell recordings had access resistance ranging from 5 to 20 MΩ. Between 75 and 85% of this was compensated for electronically.
For recording calcium currents the pipette solution consisted of 100 mM CsOH, 1 mM CaCl2, 100 mM MeSO3H, 10 mM EGTA, 6 mM MgCl2, 5 mM ATPNa2, 20 mM Hepes and 1 mM GTP, adjusted to pH 7.4 and 240 mosmol l−1. The external solution consisted of 57.5 mM NaCl, 57.5 mM tetraethylammonium chloride (TEA-Cl), 2.4 mM NaHCO3, 10 mM Hepes, 5 mM glucose, 3 mM KCl, 1 mM MgCl2, 10 mM CaCl2 and 0.14 μM TTX, adjusted to pH 7.4 and 260 mosmol l−1. Leak subtraction was performed on the recordings by applying a scaled negative version of the test voltage protocol to the same cell immediately after the experimental records were obtained. The resultant current was then scaled up and added to the experimental current record. Drugs were applied through a multi-barrelled microperfusion pipette, which was positioned within 1 mm of the cell. All experiments were performed at room temperature, 18–23°C.
Extracellular recordings
Xenopus embryos were prepared for extracellular ventral root recordings in accordance with the UK Animals (Scientific Procedures) Act (1986) using previously described methods (Dale, 1995b). Drug access was facilitated by bilateral removal of the rostral myotomes and loosening the dorsal attachment of the remaining myotomes. The spinal cord was transected at the first or second post-otic myotome. The preparation was placed in a small bath (0.2 ml volume) and continually superfused with saline containing 115 mM NaCl, 3 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 2.4 mM NaHCO3, 10 mM Hepes, adjusted to pH 7.4 at room temperature. Drugs were applied from a multi-barrelled device which directed a continuous stream of saline over the exposed spinal cord. A DT31EZ interface and Neurolog stimulus isolator were used to generate electrical stimuli to the skin to trigger swimming with an interval of 3 min between the beginning and end of consecutive episodes. A WPI Iso-dam amplifier was used to amplify the ventral root activity and swimming episodes were digitized on-line by means of the DT31EZ interface and stored on computer.
Analysis of swimming episodes and cycle periods
For analysis, ventral root activity was digitally rectified and integrated with a 2 ms time window. Cycle period was measured by an automated threshold-crossing method in which a ventral root burst was defined as the period from the first crossing above threshold to the last crossing below threshold. If a further threshold crossing occurred within 10 ms from the last threshold crossing it was considered to be part of one ventral root burst. Cycle period was measured between peak amplitudes of the successive rectified and integrated ventral root bursts. Gradients of run-down were obtained by numerically fitting a linear regression line to the cycle periods plotted against time. In some embryos, run-down was much more rapid over the first second of activity. Consequently, in these cases, the first second of cycle period data was excluded from all analysis.
Because both episode lengths and the gradients of run-down varied greatly between different embryos (more than an order of magnitude of difference), these measures were not normally distributed and their variance correlated with their mean. To facilitate the use of parametric statistical tests, the data were log-transformed before analysis. This gave much better distributions and removed the correlation between the variance and the mean. Student's paired sample t tests were used on the transformed data to compare the control and drug treatments. As an extra level of analysis, the non-parametric Friedman two-way analysis of variance was used with non-transformed data to compare control, drug and wash. This confirmed and supported the conclusions obtained from analysis of the log-transformed data and both sets of probabilities are reported in the text.
HPLC analysis
AMP solutions were derivatized with chloro-acetaldehyde at 80°C to form the fluorescent etheno-derivatives. Reverse phase HPLC analysis was performed using a LUNA C8 (2) column (Phenomenex, Macclesfield, UK), a Thermo Separation Products (San Jose, CA, USA) HPLC gradient pump and fluorescence detector. The mobile phase consisted of the following two solutions, made according to the methods of Schweinsberg & Loo (1980): solution A, 20 mM potassium phosphate (pH 6.0); and solution B, 75% 20 mM potassium phosphate (pH 6.0) and 25% methanol. All reagents were HPLC grade. A concave gradient was run, going from 100% A to 100% B in 10 min. Typical elution times were around 5 min for AMP and 9 min for adenosine. Between runs the column was re-equilibrated for 10 min with solution A.
Chemicals
The adenosine receptor agonists and antagonists used were as follows. Adenosine (RBI, Poole, UK), adenosine monophosphate (AMP, Sigma, Poole, UK), N6-cyclopentyladenosine (CPA, RBI), 5′-(N-cyclopropyl)-carboxamidoadenosine (CPCA, RBI), 2-p-(2-carboxethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine hydrochloride (CGS 21680, Tocris Cookson, Bristol, UK), 1-deoxy-1-[6-[[(3-iodophenyl)methyl]amino]-9H-purin-9-yl]-N-methyl-β-D-ribofuranuronamide (IB-MECA, Tocris Cookson), 8-cyclopentyl-1,3-dipropylxanthine (DPCPX, RBI), 4-(2-[7-amino-2-(2-furyl)[1,2, 4]triazolo[2,3-a][1,3,5]triazin-5-ylamino]ethyl)phenol (ZM241385, Tocris Cookson), alloxazine (benzo[g]pteridine-2,4(1H,3H)-dione, RBI), 8-(3-chlorostyrl)caffeine (CSC, RBI). The selective calcium channel blockers used were as follows. ω-Conotoxin GVIA (CgTx GVIA, Allomone Labs, Jerusalem, Israel), ω-conotoxin MVIIC (CgTx MVIIC, Allomone Labs), ω-agatoxin TK (AgTx, Sigma), nifedipine (Sigma). Adenosine deaminase was obtained from Sigma.
RESULTS
Adenosine causes voltage- and non-voltage-dependent inhibition of Ca2+ currents
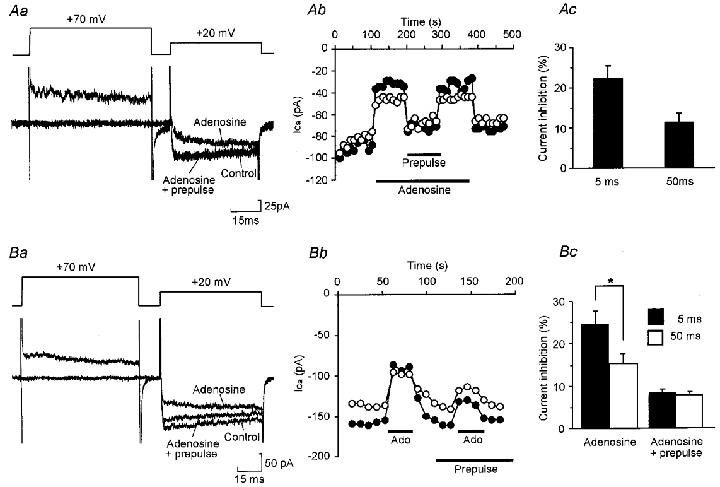
Whole cell HVA calcium currents were evoked by stepping from a holding potential of -50 mV to a test potential of +20 mV. Application of adenosine at concentrations of 0.1-100 μM caused dose-dependent inhibition of the HVA Ca2+ currents (Fig. 1). The IC50 for inhibition was 1.2 ± 0.63 μM (mean ±s.e.m.), with significant inhibition occurring at adenosine levels as low as 0.3 μM and saturating at 10 μM. The mean inhibition evoked by 10 μM adenosine was 24 ± 1.7% (n= 76), measured 5 ms into the test voltage step (Table 1). Adenosine produced modulation in all classes of neuron examined, including monopolar, commissural-like and multipolar neurons (Dale, 1991). Overall around 75% of neurons tested responded to adenosine. This suggests that adenosine receptors are common on most, if not all, classes of spinal neuron.
Figure 1. Adenosine causes dose-dependent inhibition of HVA Ca2+ currents.
A, graph showing the effect of application of adenosine at doses between 100 nM and 100 μM on the amplitude of HVA currents, measured 5 ms after activation of the current, evoked by a series of voltage steps from -50 to +20 mV delivered once every 10 s. B, representative current records from times a and b in A. C, logdose-response curve for adenosine. Points represent mean current inhibition (±s.e.m.) normalized to the inhibition obtained at the highest dose of adenosine. The curve was fitted using the Hill equation (Hill coefficient = 1.51, IC50= 1.2 μM).
Table 1.
Summary of the effects on modulation of HVA currents of the various agonists and antagonists used
| Percentage block of agonist | ||||||
|---|---|---|---|---|---|---|
| Agonist | Inhibition (%) | IC50(nM) | DPCPX | Alloxazine | CSC | ZM241385 |
| Adenosine | 24 ± 11.7 (76) | 1202 ± 630 | 59 ± 6.5 (27) | 15 ± 10.7 (19) | −19 ± 5.3 (8)* | −38 ± 17.6 (5)* |
| CPA | 20 ± 3.8 (25) | 6.9 ± 12 | 79 ± 13.2 (3) | 67 ± 22.7 (4) | — | — |
| CPCA | 20 ± 3.8 (14) | 892 ± 710 | 66 ± 9.9 (5) | — | — | — |
| CGS21680 | 2 ± 1.2 (7) | — | 17 ± 38.9 (2) | — | — | — |
| IB-MECA | 14 ± 2.4 (16) | — | 43 ± 24.9 (5) | — | — | — |
Numbers of cells are given in parentheses.
These antagonists appeared to increase modulation, hence the negative values. However, this was found to be due to a direct partial block of HVA currents by the antagonists themselves.
We examined whether this modulation was voltage dependent. This was suggested by the observation that adenosine caused kinetic slowing of the current as seen by comparing modulation of the current at 5 ms into the test pulse with that evoked after 50 ms into the test pulse (Fig. 2). By using 70 ms prepulses to +70 mV ending 10 ms prior to the start of the test pulse, we measured the magnitude of the voltage- and non-voltage-dependent components of modulation in 39 neurons. The modulatory responses evoked by adenosine fell into two categories, which are illustrated in Fig. 2.
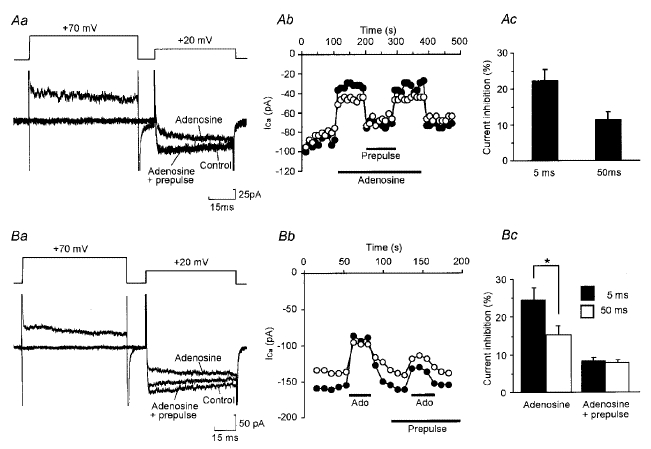
Figure 2. Inhibition of HVA Ca2+ currents occurs through voltage- and non-voltage-dependent mechanisms.
Aa, current record from a cell in which a prepulse totally relieved modulation by 10 μM adenosine of HVA currents evoked by a voltage step from -50 to +20 mV. Ab, time series showing the effect of adenosine and a prepulse on HVA currents measured every 10 s in the same cell. •, current measured 5 ms into the test pulse; ○, current measured 50 ms into the test pulse. Ac, summary histogram of 21 cells in which a prepulse totally abolished modulation. Modulation 5 ms into the test voltage pulse was significantly greater than that at 50 ms (P < 0.01, paired t test). B, as for panels in A except records are from a cell in which a prepulse only partially relieved modulation by 10 μM adenosine (Ado). For c, 18 cells. Total modulation 5 ms into the test voltage pulse was significantly greater than that at 50 ms (*P < 0.01, paired t test). However, for the ‘steady state’ voltage-independent modulation not relieved by a prepulse, there was no significant difference between that measured at 5 ms and that measured at 50 ms.
Neurons in the first category (21/39 neurons) showed kinetic slowing. Modulation by 10 μM adenosine of the early current was 22 ± 3.3%, significantly greater than modulation of the sustained current which was 11 ± 2.3% (P < 0.01, paired t test). However, in these neurons the prepulse relieved all of the inhibition (Fig. 2A). This suggests that in the first category of neurons all of the inhibition was voltage dependent.
The second category consisted of 18/39 neurons which also showed kinetic slowing, but in which the prepulse only partially relieved modulation. Prepulses removed the kinetic slowing but left a ‘steady state’ non-voltage-dependent modulation, which was sustained through the step and accounted for an 8 ± 1.1% reduction of the total current (Fig. 2B). Interestingly, the mean total inhibition by 10 μM adenosine in this second category of neuron was 25 ± 3.4% and 15 ± 2.5% for the current at 5 and 50 ms, respectively. This was not significantly different from the respective values for the first class of cells which exhibited only voltage-dependent modulation. This suggests that the two classes of neurons may have a very similar amount of modulatable current and that the two forms of modulation, voltage and non-voltage dependent, may not be completely independent.
Adenosine acts through A1 receptors
We next used selective agonists and antagonists to classify the adenosine receptor involved in the inhibition of Ca2+ currents. The A1 agonist CPA, at concentrations between 10 nM and 1 μM caused dose-dependent inhibition (Fig. 3). By contrast, the selective A2 agonist CPCA at 100 nM failed to produce modulation in any cell tested (n= 15), but produced inhibition at higher, non-selective doses (1-100 μM, 12 cells, Fig. 3). A second selective A2 agonist CGS 21680, also failed to evoke inhibition at concentrations below 10 μM in 12 cells. The selective A3 agonist IB-MECA would only produce modulation at high, non-selective concentrations of 1–10 μM (n= 19). These results are summarized in Table 1. Given the potent action of CPA and the much less potent actions of the other agonists, our results strongly suggest that modulation of Ca2+ currents was mediated entirely by the A1 receptor subtype.
Figure 3. A1 receptors mediate inhibition of HVA currents.
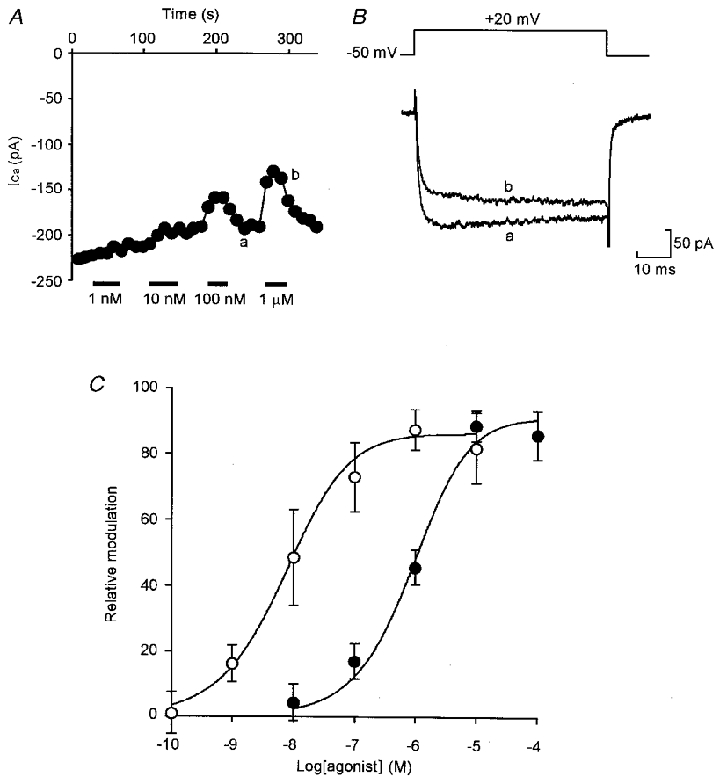
A, graph showing the dose-dependent effect of the A1-selective agonist CPA on the amplitude of HVA currents evoked by a series of steps from -50 to +20 mV delivered once every 20 s. The points represent the current measured 5 ms into the voltage step. B, representative current records from times a and b in A showing the effect of 1 μM CPA. C, logdose-response curve for CPA (○) and the A2-selective agonist CPCA (•). Error bars represent s.e.m. Curves were fitted using the Hill equation (Hill coefficient = 0.79 and IC50= 6.9 nM for CPA; Hill coefficient = 0.9 and IC50= 896 nM for CPCA).
To provide further evidence, we used selective antagonists to block modulation. The classic A1 antagonist DPCPX (100 nM) totally abolished the response to CPA (n= 3) (Fig. 4B) and to adenosine (n= 22) (Fig. 4A) in all cells tested. DPCPX also abolished the responses to CPCA (n= 5) (Fig. 4C) and IB-MECA (n= 3) again indicating that their ability to evoke inhibition was almost certainly due to cross-activation of the A1 receptor subtype at non-selective concentrations (Fig. 4). In contrast, the A2 selective antagonists ZM241385 (100-200 nM, n= 8) and CSC (100-200 nM, n= 5) would not produce any block of adenosine-evoked modulation (Fig. 4Aa). A third A2 antagonist, alloxazine (100-200 nM), produced a moderate block of modulation in 6 of 19 cells tested, but was found to effectively block the action of CPA (n= 4), and therefore was antagonizing the A1 receptor at non-selective concentrations. These results are summarized in Table 1.
Figure 4. Modulation of Ca2+ currents by adenosine is blocked by A1 but not A2 antagonists.
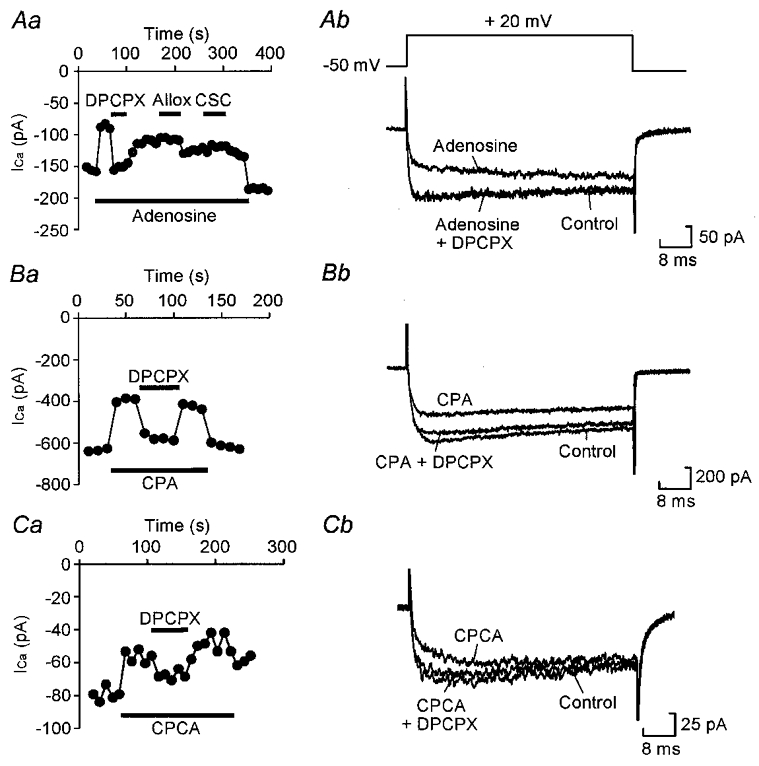
Aa, graph showing the effect of the A1 antagonist DPCPX (100 nM) and the A2 antagonists alloxazine (Allox, 100 nM) and CSC (100 nM) on inhibition by 10 μM adenosine of current evoked by a series of steps from -50 to +20 mV once every 10 s. Current was measured 5 ms into the voltage step. DPCPX totally abolished the modulation, whereas alloxazine (Allox) and CSC were without effect. Ab, representative current records from the same cell showing total block of modulation by DPCPX. Ba, modulation by 100 nM CPA is also blocked by 100 nM DPCPX. Bb, representative current records from the same cell. Ca, modulation by 10 μM CPCA is also blocked by 100 nM DPCPX, indicating that this agonist cross-activates the A1 receptor. Cb, representative current records from the same cell.
AMP has no agonist action
AMP has been reported to activate adenosine receptors (Ross et al. 1998). Any such activation would short-circuit the purinergic control system within the spinal cord because the large pool of AMP which accumulates during activity, and from which adenosine is produced, would have an agonist action of its own. We therefore tested the actions of AMP.
Commercial AMP is contaminated by adenosine. We quantified this by fluorescence HPLC and found that even a freshly made solution of 100 μM AMP contained around 1 μM adenosine (Fig. 5A). This contaminating level of adenosine (already sufficient to activate A1 receptors) rapidly increased over time to reach just over 20 μM after 4 h (Fig. 5A). We therefore treated our AMP solutions with adenosine deaminase (0.2 U ml−1) which converts adenosine to inosine but has no effect on AMP itself (Agarwhal & Parks, 1978). Although untreated solutions of AMP reduced Ca2+ currents, those treated with adenosine deaminase had no effect (Fig. 5B). We therefore conclude that AMP itself has no action on A1 receptors.
Figure 5. AMP is unstable but has no agonist action.
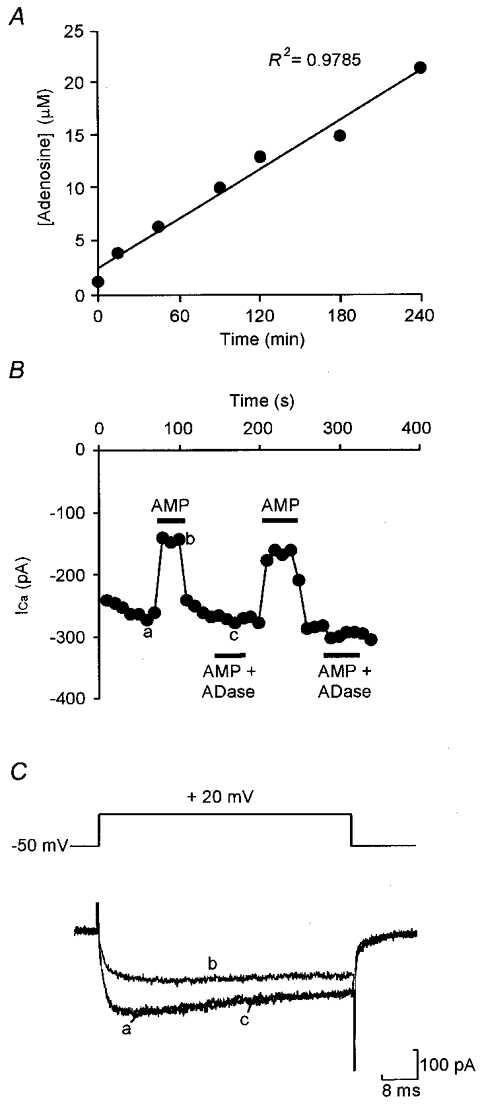
A, a 100 μM solution of commercial AMP in recording saline was initially contaminated with 1.3 μM adenosine, measured by reverse phase HPLC analysis, which increased to 21.4 μM over 4 h when kept under experimental conditions. B, a solution of 100 μM AMP caused modulation of HVA currents evoked by a series of steps from -50 to +20 mV at 0.1 Hz, but treatment with adenosine deaminase (ADase) to remove the contaminating adenosine abolished modulation. Current was measured 5 ms into the voltage step. C, representative current traces from the same cell, times a-c in B showing that the inibition of Ca2+ currents by untreated AMP solutions looks very similar to the inhibition evoked by adenosine.
Adenosine inhibits an N-type and an unidentified HVA Ca2+ current
We first characterized the HVA Ca2+ currents present in Xenopus embryo neurons. As expected from previous work (Wall & Dale, 1994) the selective N-type channel blocker ω-conotoxin GVIA at 1 μM blocked 53 ± 2.1% (n= 31, range 22 to 77.4%) of the total HVA current (Fig. 6). The dihydropyridine nifedipine at 10 μM blocked only 8.8 ± 1.4% (n= 9, range 4 to 17%) of the total HVA current suggesting that a small amount of L-type current is present. The remaining current (30-40%) defied pharmacological characterization. In particular the P/Q-type channel blockers ω-agatoxin TK at 100–200 nM and ω-conotoxin MVIIC at 1 μM had no effect, suggesting that these channels are absent from the embryonic neurons. This suggests that ‘R-type’ channels may be present. However, it has been shown that the possible R-type channel blocker SNX-482 has no effect on the HVA currents in Xenopus neurons (G. McLaren & N. Dale, unpublished observations).
Figure 6. Adenosine inhibits N-type and unidentified HVA currents.
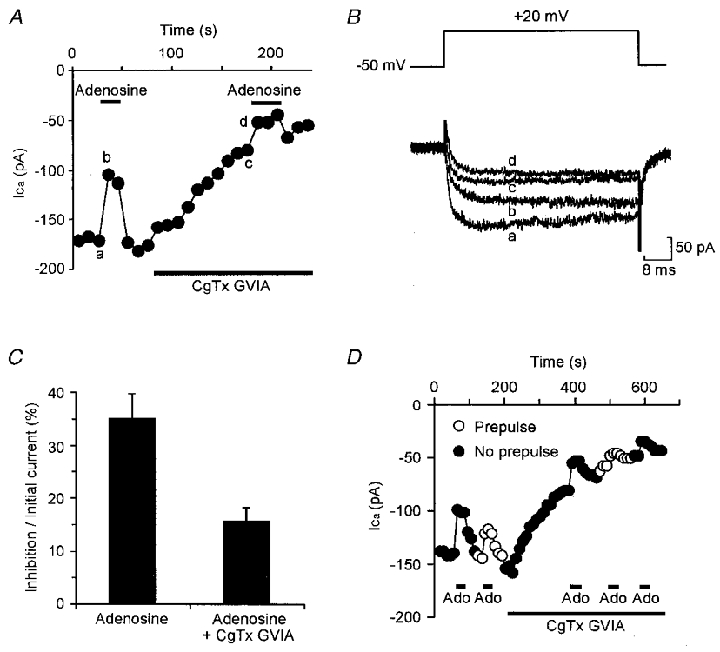
A, graph showing the amplitude of Ca2+ currents evoked by a series of steps from -50 to +20 mV once every 10 s. The inhibition evoked by 10 μM adenosine was partially occluded once 1 μM CgTx GVIA had been added to block the N-type current. B, representative current records from the same cell times a-d in A. C, summary histogram of 10 cells in which modulation before block was compared with modulation after block, expressed as a percentage of the unblocked current. Modulation was significantly reduced (P < 0.01, paired t test). D, prepulses partially relieved inhibition by adenosine (Ado) both before and after current block by CgTx GVIA, indicating that both voltage-dependent and -independent modulation are partially occluded by block of the N-type current.
By comparing the effects of adenosine before and after application of ω-conotoxin GVIA, we found that modulation was occluded by around half, from 35 ± 4.8% to 16 ± 2.6% of the initial unblocked current (Fig. 6). N-type channels are therefore modulated by adenosine. However, the partial rather than total occlusion by ω-conotoxin GVIA suggests that other channel types are modulated by adenosine too. Although there is a small L-type current these channels are not subject to modulation by adenosine because application of nifedipine caused no occlusion of inhibition (inhibition 30 ± 6.8% and 26 ± 7.1% before and after nifedipine, respectively, n= 5, paired t test, P > 0.1). We therefore conclude that an unknown, possibly R-type, HVA channel is also subject to modulation by adenosine.
As many cells show two distinct components of modulation, voltage dependent and non-voltage dependent, we tested whether the two currents, N-type and non-N, non-P/Q-type, are differentially subject to the different forms of modulation. We therefore measured the effect of a prepulse on modulation both before and after block by ω-conotoxin GVIA. This could only be achieved successfully in three cells. Figure 6C illustrates that both the voltage- and non-voltage-dependent components of modulation were occluded equally by ω-conotoxin GVIA. Total modulation was reduced from 37 ± 9.4 to 15 ± 5.4% of the initial current and non-voltage-dependent modulation was reduced from 12 ± 2.4 to 4.6 ± 1.6% of the initial current. This represents a reduction of 63 ± 5.9 and 64 ± 8.7%, respectively.
A1 receptors modulate motor pattern generation
Our previous work with Xenopus embryos has established that adenosine appears only to modulate Ca2+ channels. Adenosine has no effect on the leak or voltage-gated K+ conductances (Dale & Gilday, 1996) and does not alter the magnitude of synaptic transmission either during swimming activity or at rest (Dale & Gilday, 1996). Furthermore, computer simulations using an experimentally determined mathematical model of the Xenopus spinal network (Dale, 1995b) suggest that modest modulation of Ca2+ currents is sufficient to cause run-down and termination of motor activity (Dale & Gilday 1996; Dale, 1998). This evidence therefore strongly suports a causal link between the modulation of Ca2+ currents in single neurons and run-down of circuit activity in the intact spinal cord.
As our work with isolated neurons indicates that the A1 receptor subtype modulates Ca2+ channels, an examination of whether these same receptors also regulate motor pattern generation in the intact spinal cord constitutes an additional test of the causal link between channel modulation and circuit function. The length of swimming episodes in Xenopus embryos was therefore monitored before, during and after the application of DPCPX and CPA.
DPCPX (500 nM) caused a substantial lengthening of swimming episodes from a mean value in control of 54.4 ± 23.3 to 72.7 ± 29.4 s (P < 0.01, paired t test; P < 0.03, Friedman, n= 6) with recovery on wash to 46.3 ± 19.9 s (Figs 7A, 7C and 8Aa). Both the onset of action and washout of DPCPX were slow, possibly reflecting rather poor access of this agent into the spinal cord. In contrast to DPCPX, the A1 agonist CPA at 100 nM greatly shortened swimming episodes from a mean value in control of 38.3 ± 19.4 to 7.3 ± 4.0 s (P < 0.01, paired t test; P < 0.001, Friedman, n= 5) with recovery on wash to 30.1 ± 15.2 s (Figs 6A, 6C and 7Ba).
Figure 7. A1 receptors modulate swimming episodes.
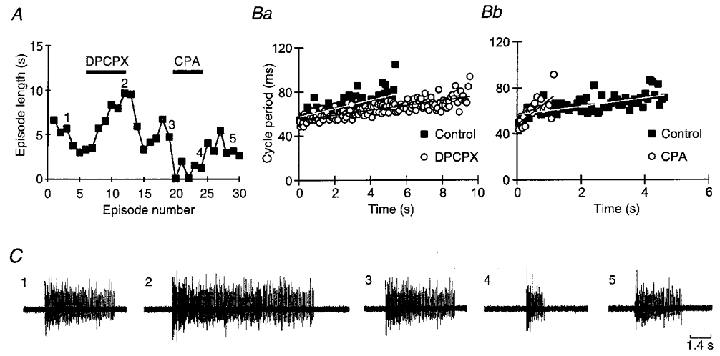
A, graph showing the length of all swimming episodes in an experiment where 500 nM DPCPX and 100 nM CPA were applied. DPCPX resulted in a doubling of episode length whereas CPA greatly shortened episodes. The numbers on the graph refer to the sample records in control, DPCPX, wash, CPA and wash shown in C. Ba and b illustrate how DPCPX and CPA, respectively, alter the time-dependent lengthening of cycle periods during swimming (run-down). Each graph shows a plot of cycle period versus time for control and drug (episodes 1 and 2, 3 and 4, respectively, from C). Linear regression lines fitted to the data show how the rate of run-down is altered by the treatments. Wash caused complete reversal of these effects but is not shown for clarity.
Figure 8. Both episode length and the rate of run-down (as measured by the time-dependent lengthening of cycle period during swimming) are controlled by A1 receptors.
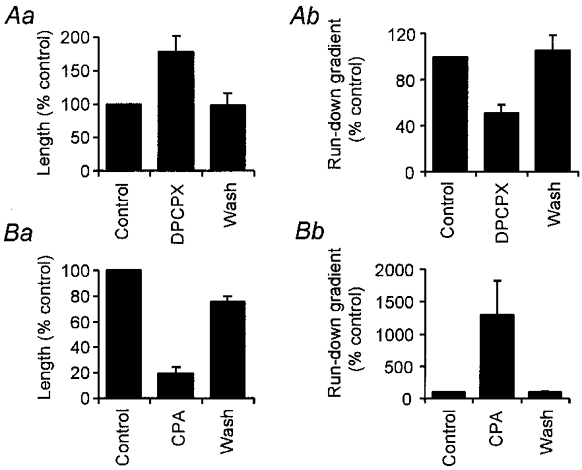
Summary histograms showing the effects of DPCPX and CPA on episode length (Aa and Ba, respectively) and run-down (Ab and Bb, respectively).
We next measured the changes in cycle period throughout the swimming episodes. This time-dependent lengthening of cycle period is a measure of run-down of the swimming pattern. To analyse the effects of DPCPX and CPA, we measured the cycle periods of the episode just before drug application, the final episode in the presence of the drug, and the first episode where maximal wash-out of drug had been obtained for each embryo. By fitting regression lines to the cycle period data plotted against time we were able to estimate the rate or gradient of run-down for all the treatments. In addition, we tested whether DPCPX or CPA altered the cycle period at which circuit activity ceased, by calculating the mean of the last five cycle periods for each swimming episode.
In six of six embryos DPCPX reduced the gradient of run-down (Fig. 7Ba). Overall the mean gradient of run-down decreased from 2.1 ± 1.2 ms s−1 in the control to 0.75 ± 0.34 ms s−1 in the presence of DPCPX (P < 0.01, paired t test; P < 0.029, Friedman, n= 6, and Fig. 8Ab). On washout of DPCPX the gradient of run-down returned to 2.6 ± 1.7 ms s−1. Despite the lengthening of the swimming episodes, the cycle period at which swimming activity ceased was not significantly altered by DPCPX (92 ± 7 ms in control, 88 ± 5 ms in DPCPX, n= 6). We conclude that the longer duration of swimming activity in the presence of DPCPX is due to a slowing of the rate of run-down rather than an extension of the ability of the spinal motor circuit to operate at longer cycle periods.
In five of five embryos CPA greatly increased the gradient of run-down (Fig. 7Bb). Overall the mean gradient of run-down changed from 2.3 ± 0.8 ms s−1 in the control to 31.1 ± 22.1 ms s−1 in the presence of CPA (P < 0.01, paired t test; P < 0.024, Friedman, n= 5, Fig. 8Bb). On washout of CPA the gradient of run-down returned to 2.6 ± 1.0 ms s−1. However, just like DPCPX, CPA had no significant effect on the final cycle period at which swimming episodes stopped (87 ± 6 ms in control, 88 ± 7 ms in CPA, n= 5). We therefore conclude that the shortening of swimming epsiodes by CPA occurs because run-down has been accelerated by the agonist.
DISCUSSION
Inhibition of Ca2+ currents
Our results have extended those of Dale & Gilday (1996) by identifying both the receptor and the HVA channels involved in the actions of adenosine. Our pharmacological results strongly suggest that the actions of adenosine are mediated only through A1 receptors: the classic A1 agonist, CPA, potently mimicked the effects of adenosine which was completely blocked by the classic A1 antagonist DPCPX. The agonists for other receptor subtypes were either without effect or blocked by DPCPX.
About half of the current modulated by adenosine was of the N-type. However, we were unable to identify the remainder of the modulated current since other agents used to classify HVA Ca2+ currents were without effect. Thus adenosine does not modulate either P/Q-type or L-type currents in Xenopus spinal neurons. Our results are similar to those of others, where N-type channels have been inhibited by adenosine acting via the A1 receptor subtype (Scholz & Miller, 1991; Mogul et al. 1993; Yawo & Chuhma, 1993; Zhu & Ikeda, 1993; Umemiya & Berger, 1994; Wu & Saggau, 1994). Modulation of a non-N-, non-P/Q-type calcium current by A1 receptors has also been reported in the striatum and hippocampus (Ambrosio et al. 1996; 1997), and in sympathetic neurons of the superior cervical ganglion (Zhu & Ikeda, 1993).
Although N-type currents are involved in transmitter release in Xenopus (Wall & Dale, 1994), adenosine itself does not mediate presynaptic inhibition in this system (Dale & Gilday, 1996). The extent of modulation of the N-type current, or the location of the modulated channels may be such that transmitter release remains unaffected but that neural excitability is reduced thus mediating the inhibitory action of adenosine. We do not know whether the unidentified HVA current also contributes to synaptic transmission, but it is tempting to speculate that this current also makes a large contribution to neural excitability (see Wall & Dale, 1994).
Mechanisms of inhibition
The HVA currents undergo both voltage- and non-voltage-dependent inhibition. Some neurons showing either one type, or both. The voltage-dependent inhibition almost certainly is mediated by direct binding of the G protein βγ subunits to the HVA channel. Non-voltage-dependent inhibition is generally thought to involve a diffusible messenger. This has been demonstrated for the modulation of HVA currents by 5-HT in Xenopus neurons (Sun & Dale, 1998, 1999). Like adenosine, 5-HT also mediates both voltage- and non-voltage-dependent inhibition of HVA currents. However, for 5-HT more than one receptor is involved. The 5-HT1A receptor predominantly causes non-voltage-dependent inhibition of P/Q-type channels, while the 5-HT1D receptor causes mainly voltage-dependent inhibition of N-type channels (Sun & Dale, 1998). However, for adenosine, the A1 receptor appears to mediate both types of inhibition on both types of current (N- and non-N-, non-P/Q-type channels). This may suggest that the A1 receptor is less selective at coupling to G proteins than the 5-HT receptors. Furthermore the total inhibition in those neurons that were subject to both voltage- and non-voltage-dependent inhibition was no greater than in those cells which were subject only to the voltage-dependent modulation. This could be explained by the observation of Zamponi et al. (1997) that PKC and Gβγ target the same site on the α1 subunit, and that channel phosphorylation inhibits Gβγ binding. Therefore voltage-independent modulation in Xenopus neurons, if mediated by channel phosphorylation, would occur at the expense of some of the voltage-dependent modulation.
As the voltage-dependent inhibition can be relieved by prior depolarization, the functional effect of this inhibition during motor activity might differ from that of the non-voltage-dependent inhibition. In particular, a burst of spikes might be expected to cause progressive relief from voltage-dependent inhibition. During swimming in the embryo, neurons fire only single spikes on each cycle suggesting that such relief is unlikely to occur. However, swimming in older larvae involves bursts of action potentials (Sillar et al. 1991) raising the possibility that relief of voltage-dependent inhibition during a motor burst might be significant. This has been studied for the actions of 5-HT where the two types of inhibition are predominantly mediated by separate receptor subtypes. Surprisingly, there appears to be no difference in the efficacy of voltage- and non-voltage-dependent inhibition in modulating motor activity in Xenopus larvae (Q.-Q. Sun & N. Dale, unpublished observations).
Linking Ca2+ channel modulation to run-down of motor circuits
There are now several strands of evidence that support our hypothesis that modulation of HVA Ca2+ channels underlies the effects of adenosine on the spinal circuits and contributes to run-down of motor activity. Firstly, both pharmacological experiments (Wall & Dale, 1994) and mathematical modelling (Dale, 1995b) show that the HVA Ca2+ channels help to determine the spike threshold and excitability of Xenopus spinal neurons. Pharmacological blockade of N-type channels severely diminishes the ability of the spinal circuit to operate (Wall & Dale, 1994). However, it is probably the large suppression of synaptic transmission resulting from such a blockade, rather than the more subtle changes in neural excitability, that accounts for most of this effect. Nevertheless in realistic computer simulations where it is possible to reduce HVA Ca2+ currents without altering the magnitude synaptic transmission, we have found that the resulting lowering of excitability severely reduces the ability of the model network to operate (Dale & Gilday, 1996). Secondly, our previous work has shown that adenosine does not: change the resting conductance or membrane potential of neurons; alter voltage-gated K+ currents or affect synaptic transmission (Dale & Gilday, 1996). This leaves inhibition of HVA Ca2+ currents as the only known cellular action of adenosine in this system. Thirdly, computer simulations that incorporate the purinergic modulators ATP and adenosine and their known actions on the voltage-gated currents of spinal neurons, demonstrate that in these models at least, the action of adenosine on Ca2+ currents is sufficient to mediate run-down (Dale, 1998). The results we present here, that only the A1 receptor subtype inhibits Ca2+ currents and that this same receptor also modulates motor activity, further strengthen the causal link between channel modulation and circuit function.
A1 receptors and regulation of motor behaviour
Although A1 receptors have been localized to spinal cord, their functional effects have not been extensively studied. So far A1 receptors have been shown to mediate inhibition of sensory transmission (Burnstock & Wood, 1996; Sawynock, 1998), but there have been no reports for direct effects of A1 receptors on spinal motor circuitry. In the basal ganglia and striatum, A2 receptors alter the propensity for motor activity (Ferre et al. 1997). However, these higher centres are concerned with the initiation rather than the primary generation of motor behaviour (reviewed by Hauber, 1998). Our results are thus novel in demonstrating a direct effect of A1 receptors on the spinal motor circuitry. By identifying the adenosine receptor involved, we have extended previous work on the frog embryo and have shown that just one type of adenosine receptor, rather than several, is involved in regulation of motor activity.
Blockade of A1 receptors lengthened swimming episodes. This could occur in two ways. Firstly, the rate of run-down could remain unaltered, but the effect of receptor blockade could be to allow the spinal network to become capable of generating motor activity at longer cycle periods, hence extending the duration of activity. The SK channel blocker apamin extends swimming episodes in this manner by blocking a slowly activating Ca2+-dependent K+ current (Wall & Dale, 1995). A second way that episodes could be extended is that the rate of run-down itself could be slowed. Measurement of adenosine production (Dale, 1998) suggests that it is slow and continuous through swimming activity. If previous suggestions (Dale & Gilday, 1996) that the production of adenosine underlies this run-down, then blockade or activation of adenosine receptors should alter the rate of run-down itself. The effects of CPA and DPCPX confirm this prediction and show that adenosine is present throughout the swimming episode and plays a major role in run-down of the motor pattern.
Delayed adenosine production and accumulation of AMP
The delay in accumulation of adenosine may arise from the inhibition of the ecto-5′-nucleotidase by either ATP or ADP, a phenomenon that occurs for this enzyme in many tissues including brain (James & Richardson, 1993). Borrowing enzyme kinetics from endothelium (Slakey et al. 1986), Dale (1998) showed, using computer simulations of the spinal locomotor network, that such a mechanism would be a powerful determinant of the rate of adenosine production during swimming and ultimately the rate of run-down. However, the unavoidable concomitant of feed-forward inhibition of ecto-5′-nucleotidase is that its substrate, AMP, will accumulate during motor activity. Simulations suggest that this could easily reach levels of tens of micromolar (Dale, 1998). An important prediction of our work on the purinergic control of motor activity is that AMP should not act as an agonist. Although AMP has been reported as an agonist at adenosine receptors (Ross et al. 1998), were it to do so here, it would short-circuit the delay between the excitatory actions of ATP and the inhibitory actions of adenosine. As AMP could accumulate to quite high levels, even a weak agonist action would be significant. Our evidence that AMP itself has no agonist action even at 100 μM is therefore an important step in establishing that the purines can indeed mediate time-dependent control of motor pattern generation.
Acknowledgments
We thank the Wellcome Trust for generous support.
References
- Agarwhal RP, Parks RE. Adenosine deaminase from human erythrocytes. Methods in Enzymology. 1978;51:502–507. doi: 10.1016/s0076-6879(78)51069-0. [DOI] [PubMed] [Google Scholar]
- Ambrosio AF, Malva JO, Carvalho AP, Carvalho CM. Modulation of Ca2+ channels by activation of adenosine A1 receptors in rat striatal glutamatergic nerve terminals. Neuroscience Letters. 1996;220:163–166. doi: 10.1016/s0304-3940(96)13252-3. [DOI] [PubMed] [Google Scholar]
- Ambrosio AF, Malva JO, Carvalho AP, Carvalho CM. Inhibition of N- and P/Q- and other types of Ca2+ channel in rat hippocampal nerve terminals by the adenosine A1 receptor. European Journal of Pharmacology. 1997;340:301–310. doi: 10.1016/s0014-2999(97)01451-9. [DOI] [PubMed] [Google Scholar]
- Burnstock G, Wood JN. Purinergic receptors – their role in nociception and primary afferent neurotransmission. Current Opinion in Neurobiology. 1996;6:526–532. doi: 10.1016/s0959-4388(96)80060-2. [DOI] [PubMed] [Google Scholar]
- Chen G, Van Den Pol AN. Adenosine modulation of calcium currents and presynaptic inhibition of GABA release in suprachiasmatic and arcuate nucleus neurones. Journal of Neurophysiology. 1997;77:3035–3047. doi: 10.1152/jn.1997.77.6.3035. [DOI] [PubMed] [Google Scholar]
- Dale N. The isolation and identification of spinal neurones that control movement in the Xenopus embryo. European Journal of Neuroscience. 1991;3:1025–1035. doi: 10.1111/j.1460-9568.1991.tb00039.x. [DOI] [PubMed] [Google Scholar]
- Dale N. Kinetic characterization of the voltage-gated currents possessed by Xenopus embryo spinal neurones. The Journal of Physiology. 1995a;489:473–488. doi: 10.1113/jphysiol.1995.sp021066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N. Experimentally derived model for the locomotor pattern generator in the Xenopus embryo. The Journal of Physiology. 1995b;489:489–510. doi: 10.1113/jphysiol.1995.sp021067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N. Delayed production of adenosine underlies temporal modulation of swimming in frog embryo. The Journal of Physiology. 1998;511:265–272. doi: 10.1111/j.1469-7793.1998.265bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N, Gilday D. Regulation of rhythmic movements by purinergic neurotransmitters in frog embryos. Nature. 1996;383:259–263. doi: 10.1038/383259a0. [DOI] [PubMed] [Google Scholar]
- Ferre S, Fredholm BB, Morelli M, Popoli P, Fuxe K. Adenosine-dopamine receptor-receptor interactions as an integrative mechanism in the basal ganglia. Trends in Neurosciences. 1997;20:482–487. doi: 10.1016/s0166-2236(97)01096-5. [DOI] [PubMed] [Google Scholar]
- Fleming KM, Mogul DJ. Adenosine A3 receptors potentiate hippocampal calcium currents by a PKA-dependent/PKC independent pathway. Neuropharmacology. 1997;36:353–362. doi: 10.1016/s0028-3908(97)83762-8. [DOI] [PubMed] [Google Scholar]
- Goncalves ML, Cunha RA, Ribeiro JA. Adenosine A2A receptors facilitate Ca – 452+ uptake through class A calcium channels in rat hippocampal CA3 but not CA1 synaptosomes. Neuroscience Letters. 1997;238:73–77. doi: 10.1016/s0304-3940(97)00803-3. [DOI] [PubMed] [Google Scholar]
- Gubitz AK, Widdowson L, Kurokawa M, Kirkpatrick KA, Richardson PJ. Dual signalling by the adenosine A2a receptor involves activation of both N- and P-type calcium channels by different G-proteins and protein kinases in the same striatal nerve terminals. Journal of Neurochemistry. 1996;67:374–381. doi: 10.1046/j.1471-4159.1996.67010374.x. [DOI] [PubMed] [Google Scholar]
- Hauber W. Involvement of basal ganglia transmitter systems in movement initiation. Progress in Neurobiology. 1998;56:507–540. doi: 10.1016/s0301-0082(98)00041-0. [DOI] [PubMed] [Google Scholar]
- Henning RH. Purinoreceptors in neuromuscular transmission. Pharmacology and Therapeutics. 1997;74:115–128. doi: 10.1016/s0163-7258(97)00015-6. [DOI] [PubMed] [Google Scholar]
- James S, Richardson PJ. Production of adenosine from extracellular ATP at the striatal cholinergic synapse. Journal of Neurochemistry. 1993;60:219–227. doi: 10.1111/j.1471-4159.1993.tb05841.x. [DOI] [PubMed] [Google Scholar]
- Mogul DJ, Adams ME, Fox AP. Differential activation of adenosine receptors decreases N-type but potentiates P-type calcium current in hippocampal CA3 neurones. Neuron. 1993;10:327–334. doi: 10.1016/0896-6273(93)90322-i. [DOI] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J. Normal Tables of Xenopus laevis (Daudin) North Holland, Amsterdam: 1956. [Google Scholar]
- Olah ME, Stiles GL. Adenosine receptor sub-types – Characteristics and therapeutic regulation. Annual Review of Pharmacology and Toxicology. 1995;35:581–606. doi: 10.1146/annurev.pa.35.040195.003053. [DOI] [PubMed] [Google Scholar]
- Park TJ, Chung SK, Han MK, Kim UH, Kim KT. Inhibition of voltage sensitive calcium channels by the A2A adenosine receptor in PC12 cells. Journal of Neurochemistry. 1998;71:1251–1260. doi: 10.1046/j.1471-4159.1998.71031251.x. [DOI] [PubMed] [Google Scholar]
- Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacological Reviews. 1998;50:413–492. [PubMed] [Google Scholar]
- Ribeiro JA. Purinergic inhibition of neurotransmitter release in the central nervous system. Pharmacology and Toxicology. 1995;77:299–305. doi: 10.1111/j.1600-0773.1995.tb01031.x. [DOI] [PubMed] [Google Scholar]
- Ross FM, Brodie MJ, Stone TW. Adenosine monophosphate as a mediator of ATP effects at P1 purinoreceptors. British Journal of Pharmacology. 1998;124:818–824. doi: 10.1038/sj.bjp.0701890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawynok J. Adenosine receptor activation and nociception. European Journal of Pharmacology. 1998;347:1–11. doi: 10.1016/s0014-2999(97)01605-1. [DOI] [PubMed] [Google Scholar]
- Scholz KP, Miller RJ. Analysis of adenosine actions on Ca2+ currents and synaptic transmission in cultured rat hippocampal pyrimidal neurones. The Journal of Physiology. 1991;435:373–393. doi: 10.1113/jphysiol.1991.sp018515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweinsberg PD, Loo TL. Simultaneous analysis of ATP, ADP, AMP, and other purines in human erythrocytes by high-performance liquid chromatography. Journal of Chromatography. 1980;181:103–107. doi: 10.1016/s0378-4347(00)81276-1. [DOI] [PubMed] [Google Scholar]
- Sebastião AM, Ribeiro JA. Adensosine A2 receptor-mediated excitatory actions on the nervous system. Progress in Neurobiology. 1996;48:167–189. doi: 10.1016/0301-0082(95)00035-6. [DOI] [PubMed] [Google Scholar]
- Sillar KT, Wedderburn JFS, Simmers AJ. The development of swimming rhythmicity in post-embryonic Xenopus laevis. Proceedings of the Royal Society B. 1991;246:147–153. doi: 10.1098/rspb.1991.0137. [DOI] [PubMed] [Google Scholar]
- Slakey L, Cosimini K, Earls JP, Thomas C, Gordon EL. Simulation of extracellular nucleotide hydrolysis and determination of kinetic constants for ectonucleotidases. Journal of Biological Chemistry. 1986;261:15505–15507. [PubMed] [Google Scholar]
- Sun QQ, Dale N. Differential inhibition of N and P/Q Ca2+ currents by 5-HT1A and 5-HT1D receptors in spinal neurones of Xenopus larvae. The Journal of Physiology. 1998;510:103–120. doi: 10.1111/j.1469-7793.1998.103bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun QQ, Dale N. G-proteins are involved in 5-HT receptor-mediated modulation of N- and P/Q- but not T-type Ca2+ currents. Journal of Neuroscience. 1999;19:890–899. doi: 10.1523/JNEUROSCI.19-03-00890.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne PR, Housley GD. Purinergic signalling in sensory systems. Seminars in the Neurosciences. 1996;8:233–246. [Google Scholar]
- Umemiya M, Berger AJ. Activation of adenosine A1 and A2 receptors differentially modulates calcium channels and glycinergic synaptic transmission in the rat brain-stem. Neuron. 1994;13:1439–1446. doi: 10.1016/0896-6273(94)90429-4. [DOI] [PubMed] [Google Scholar]
- Wall MJ, Dale N. GABAB receptors modulate an ω-conotoxin-sensitive calcium current that is required for synaptic transmission in the Xenopus embryo spinal cord. Journal of Neuroscience. 1994;14:6248–6255. doi: 10.1523/JNEUROSCI.14-10-06248.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall MJ, Dale N. A slowly activating Ca2+-dependent K+ current that plays a role in termination of swimming in Xenopus embryos. The Journal of Physiology. 1995;487:557–572. doi: 10.1113/jphysiol.1995.sp020900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of the hippocampus. Neuron. 1994;12:1139–1148. doi: 10.1016/0896-6273(94)90321-2. [DOI] [PubMed] [Google Scholar]
- Yawo H, Chuhma N. Preferential inhibition of ω-conotoxin-sensitive presynaptic Ca2+ channels by adenosine autoreceptors. Nature. 1993;365:256–258. doi: 10.1038/365256a0. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G-proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Ikeda SR. Adenosine modulates voltage-gated Ca2+ channels in adult rat sympathetic neurones. Journal of Neurophysiology. 1993;70:610–620. doi: 10.1152/jn.1993.70.2.610. [DOI] [PubMed] [Google Scholar]