

Abstract
We have used an enzyme-based, twin-barrelled sensor to measure adenosine release during hypoxia in the CA1 region of rat hippocampal slices in conjunction with simultaneous extracellular field recordings of excitatory synaptic transmission.
When loaded with a combination of adenosine deaminase, nucleoside phosphorylase and xanthine oxidase, the sensor responded linearly to exogenous adenosine over the concentration range 10 nM to 20 μM.
Without enzymes, the sensor when placed on the surface of hippocampal slices recorded a very small net signal during hypoxia of 40 ± 43 pA (mean ±s.e.m.; n = 7). Only when one barrel was loaded with the complete sequence of enzymes and the other with the last two in the cascade did the sensor record a large net difference signal during hypoxia (1226 ± 423 pA; n = 7).
This signal increased progressively during the hypoxic episode, scaled with the hypoxic depression of the simultaneously recorded field excitatory postsynaptic potential and was greatly reduced (67 ± 6.5 %; n = 9) by coformycin (0.5-2 μM), a selective inhibitor of adenosine deaminase, the first enzyme in the enzymic cascade within the sensor.
For 5 min hypoxic episodes, the sensor recorded a peak concentration of adenosine of 5.6 ± 1.2 μM (n = 16) with an IC50 for the depression of transmission of approximately 3 μM.
In slices pre-incubated for 3-6 h in nominally Ca2+-free artificial cerebrospinal fluid, 5 min of hypoxia resulted in an approximately 9-fold greater release of adenosine (48.9 ± 17.7 μM; n = 6).
High extracellular Ca2+ (4 mM) both reduced the adenosine signal recorded by the sensor during hypoxia (3.5 ± 0.6 μM; n = 4) and delayed the hypoxic depression of excitatory synaptic transmission.
Adenosine plays a well established and important modulatory role in a wide variety of physiological and pathophysiological processes in the central nervous syem. All four adenosine receptor subtypes (A1, A2A, A2B and A3) are expressed in brain (Ralevic & Burnstock, 1998) and are responsible for the diverse actions of adenosine on neuronal function including control of transmitter release (Brundege & Dunwiddie, 1997), spinal motor pattern generation (Dale & Kuenzi, 1997), induction of sleep (Porkka-Heiskanen, 1999) and modulation of synaptic plasticity (De Mendonça & Ribeiro, 1997).
Extracellular levels of adenosine are known to increase during a variety of pathological processes including epileptiform activity (Dunwiddie, 1999) and hypoxia/ischaemia (Schubert et al. 1994). Furthermore, the depression of excitatory synaptic transmission observed in the CA1 region of the hippocampus in vitro during hypoxia (Fowler, 1989; Katchman & Hershkowitz, 1993a), ischaemia (Fowler, 1990), hypoglycaemia (Zhu & Krnjevic, 1993) or cyanide poisoning (Zhu & Krnjevic, 1997) is mediated by the activation of presynaptic adenosine A1 receptors. Thus, the activation of A1 receptors, by both reducing glutamatergic excitation and causing postsynaptic hyperpolarisation (Greene & Haas, 1991; Brundege & Dunwiddie, 1997), prevents excessive activation of the N-methyl-D-aspartate (NMDA) receptor. Adenosine may therefore be an endogenous neuroprotectant. Indeed, in several models of cerebral hypoxia/ischaemia adenosine A1 agonists have been shown to be protective whereas adenosine receptor antagonists exacerbate neuropathology (Rudolphi et al. 1992; Bischofberger et al. 1997). Thus, an understanding of the release of adenosine during hypoxia/ischaemia may underpin the development of novel therapeutic strategies in disorders such as stroke or cardiac arrest.
To facilitate such an understanding a method is required which allows continuous, on-line measurement of extracellular adenosine to be combined with simultaneous measurement of electrophysiological activity from defined neuronal populations. Recently, a novel adenosine sensor has been developed (Dale, 1998) which satisfies these criteria. The probe is sensitive to adenosine levels as low as 10 nM and maintains a linear response into the micromolar range. The sensor has been used to measure adenosine release during episodes of fictive locomotion in Xenopus embryo spinal cord where it has been possible to resolve changes in adenosine levels varying from 20 to 600 nM during single episodes of swimming on a time scale of seconds (Dale, 1998). Given the success of the sensor in measuring continuously, for the first time, adenosine release during physiological activity, we have applied this technique to investigate adenosine production in the hippocampus during hypoxia.
Here we provide a detailed description and careful validation of the use of this adenosine sensor in the mammalian hippocampus during hypoxia. Such an approach is an important prerequisite for the general acceptance and adoption of a novel technology which offers several advantages over more traditional measures of purine release. We have been careful to establish the specificity of the sensor for adenosine. This is indicated by the dependence of the signal recorded by the sensor during hypoxia on the presence of the full enzyme cascade within the sensor and its reduction by the selective adenosine deaminase inhibitor coformycin. We show that the increase in extracellular adenosine parallels the depression of excitatory synaptic transmission in area CA1 and that the release of adenosine is independent of extracellular Ca2+. Indeed, external Ca2+ exerts an inhibitory influence on adenosine production, with approximately 9-fold greater levels of adenosine being released in nominally Ca2+-free medium compared to physiological 2 mM extracellular Ca2+. In addition elevated extracellular Ca2+ further inhibited the release of adenosine and retarded the hypoxic depression of the fEPSP. Our use of the sensor and simultaneous recording of synaptic transmission has therefore revealed novel aspects of adenosine release.
Some of this work has appeared in abstract form (Frenguelli & Dale, 1999).
METHODS
Preparation of hippocampal slices
Twelve- to 24-day-old Sprague-Dawley rats of either sex were killed by cervical dislocation in accordance with Schedule 1 of the UK Government Animals (Scientific Procedures) Act 1986. Following decapitation, the brain was rapidly removed and immersed in high-Mg2+, ice-cold artificial cerebrospinal fluid (ACSF) of the following composition (mM): NaCl, 124; NaHCO3, 26; KCl, 3; CaCl2, 2; MgSO4, 11; NaH2PO4, 1.25; D-glucose, 10; pH 7.4; bubbled with 95 % O2-5 % CO2. Each hemisphere was sub-dissected to yield a tissue block containing the hippocampus, which was glued (cyanoacrylate) to a perspex block such that the mid-line of the brain was uppermost and horizontal. In this orientation, 400 μm horizontal slices were cut with a Vibratome whilst being continuously immersed in ice-cold ACSF. The sections containing transverse hippocampal slices were transferred to an incubation chamber similar to that described by Edwards et al. (1989) and immersed in standard ACSF (1 mM MgSO4) at room temperature for at least 1 h. In some experiments, after this 1 h incubation, slices were transferred (for between 3 and 6 h) to a similar chamber in which the Ca2+ in the ACSF had been replaced with 2 mM MgSO4 (nominally Ca2+-free ACSF).
Electrophysiology
Individual slices were placed in a recording chamber where they were completely submerged by oxygenated ACSF flowing at ∼6 ml min−1 (32-34°C). Field excitatory postsynaptic potentials (fEPSPs) were recorded with an ACSF-filled glass microelectrode from stratum radiatum in response to stimulation of the Schaffer collateral-commissural pathway, at 15 s intervals, with either a similar ACSF-filled glass microelectrode or a twisted bipolar tungsten electrode (50 μm Teflon-coated tungsten wire). Stimulation and recording parameters were under the control of ‘LTP’ software (W. W. Anderson, University of Bristol; http://www.ltp-program.com; Anderson & Collingridge, 1997). Individual fEPSPs were displayed on-line on a PC which calculated their amplitude or rising slope and either individual fEPSPs or the average of four sequential fEPSPs (1 min) were stored to disk. Hypoxia was induced by perfusing slices with identical ACSF pre-equilibrated with 95 % N2-5 % CO2 as previously described (Frenguelli, 1997). This results in a change in bath oxygen tension from approximately 80-90 % saturation to <10 % as measured by a Diamond General oxygen microelectrode.
Adenosine sensor
A development of the sensor technique originally devised by Dale (1998) has been used to measure adenosine release during hypoxia in rat hippocampal slices. The sensor (Sycopel International Ltd, Jarrow, Tyne & Wear, UK) has an overall width of approximately 500 μm and comprises two identical parallel barrels (Fig. 1A). Each barrel contained a 50 μm platinum wire polarised to +650 mV. The platinum wires were located on the bottom of the barrels such that they were in closest proximity to the hippocampal slice. One barrel (the adenosine barrel) was filled with adenosine deaminase (AD; EC 3.5.4.4), nucleoside phosphorylase (NP; EC 2.4.2.1) and xanthine oxidase (XO; EC 1.1.3.22). This enzyme combination results in the sequential breakdown of adenosine to inosine to hypoxanthine and hence to uric acid and hydrogen peroxide (Fig. 1A). The hydrogen peroxide is oxidised on the platinum wire to yield a current proportional to the concentration of hydrogen peroxide. In the second barrel, the enzyme combination lacked AD and is thus referred to as the reference barrel. A differential recording between the two barrels yields a current that is linearly related to the concentration of adenosine. This procedure helps to subtract out the effects of any non-specific interferents and the contribution of inosine (which can be released from hippocampal slices) thereby increasing the specificity of the measured signal.
Figure 1. Schematic representation of adenosine sensor and recording arrangement.
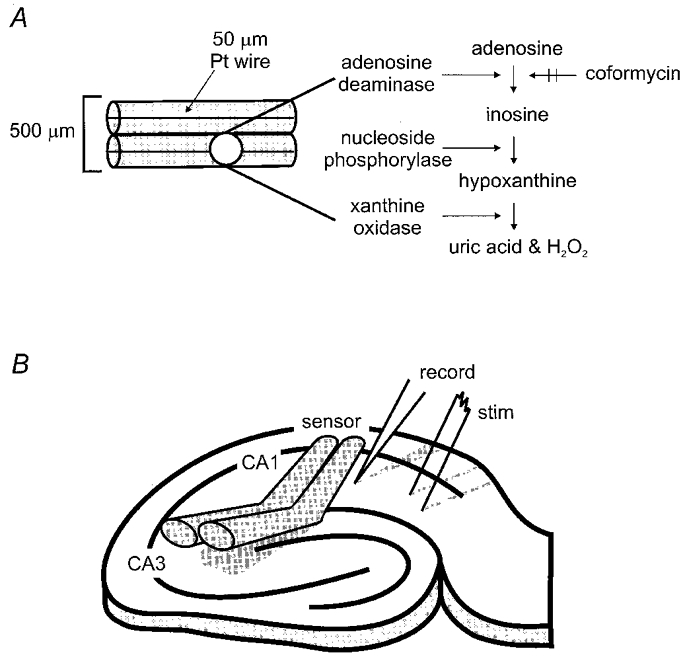
A, the adenosine sensor consisted of two parallel tubes of semi-permeable glass (overall width ≈500 μm) each housing a 50 μm platinum (Pt) wire held at a potential of +650 mV. B, placement of stimulating and recording electrodes and adenosine sensor on the CA1 region of the hippocampal slice.
Balancing of the adenosine sensor
Accurate differential measurements between the adenosine and reference barrels require both compensation for differences in the intrinsic sensitivity of the two barrels and also orientation of the probe so that both sensing elements (internal platinum wires) are placed in equal proximity to the tissue. The outputs of the two sensor barrels were fed to a laboratory-built differential amplifier in which the relative gain of the two input channels could be adjusted to compensate for slight differences (<25 %) in sensitivity between the two barrels. To determine the correct balance, the following manipulations were performed. Firstly, each output was tested with either hydrogen peroxide (0.03 %) or inosine (2 μM) and the balance control was adjusted to minimize the net difference signal. In a further refinement, the sensor was placed upon an agar ‘slice’ that had been made up with a solution containing 20 μM inosine. Once again, balancing was done to minimise the net signal when the sensor was placed on or removed from the agar slice. This latter method was designed to facilitate optimal orientation of the sensor with the hippocampal slice.
Sensor placement
The sensor was gently placed on the surface of the hippocampal slice under visual guidance such that it lightly deformed the surface of the slice thereby minimising diffusion distance from slice to sensor. A shallow (30 deg) bend in the sensor (Fig. 1B) facilitated its placement parallel to the surface of the slice. The tip of the sensor was aligned so as to impinge slightly into stratum oriens whereas the greater part of contact with the tissue was on stratum pyramidale and stratum radiatum. The probe was inclined such as to make no contact with the dentate gyrus region of the slice. With the sensor in position, the stimulating and recording electrodes were placed on either side, or on one side of the sensor with the recording electrode proximal to the sensor (within approximately 200 μm), as depicted schematically in Fig. 1B. This allowed simultaneous monitoring of synaptic transmission which acted as an independent measure of experimental manipulations. With this arrangement experiments measuring both adenosine release and synaptic transmission routinely lasted in excess of 6 h. The outputs from both the sensor and extracellular recording amplifier were simultaneously displayed on a two-channel chart recorder.
Calibration of sensor and determination of released adenosine concentration
Exogenous adenosine (most commonly 2 μM) was bath applied until the peak current recorded by the sensor had achieved an asymptote (usually 2-3 min). In order to assess any possible probe rundown over time (observed in early experiments and subsequently reduced by the inclusion of NaHCO3 (26 mM) in the saline used to dissolve and load the enzymes into the probe) we tested probe sensitivity to exogenous adenosine between hypoxic episodes. Since the dose- response relationship between adenosine and probe polarisation was linear (see below), all subsequent hypoxia-induced adenosine release was measured relative to this single concentration of exogenous standard. Calibrations were performed prior to or shortly after episodes of hypoxia and under identical ionic conditions. This experimental design excluded the (unlikely) possibility that changes in extracellular ion concentration could influence the calibration of adenosine signals since both were recorded under identical conditions.
The amplitude of signals recorded at the end of the hypoxic episode (usually 5 or 10 min) was measured relative to the preceding baseline. Occasionally, baseline drift required interpolation of the baseline from before to after the hypoxic episode. In these cases, the amplitude was measured relative to the interpolated line. In some instances, a negative shift was seen on the chart trace shortly after the onset of hypoxia (e.g. Fig. 3B). This is likely to reflect the intrinsic sensitivity of the polarised platinum wires to bath oxygen tension. In these cases, the hypoxic adenosine signal was measured relative to the peak of the negative shift. However, the adenosine signals recorded were generally of such amplitude that baseline drift or the small negative shift would not have contributed appreciably to the measurements.
Figure 3. Absolute dependence of hypoxic adenosine signal on presence of enzymes and inhibition by coformycin.
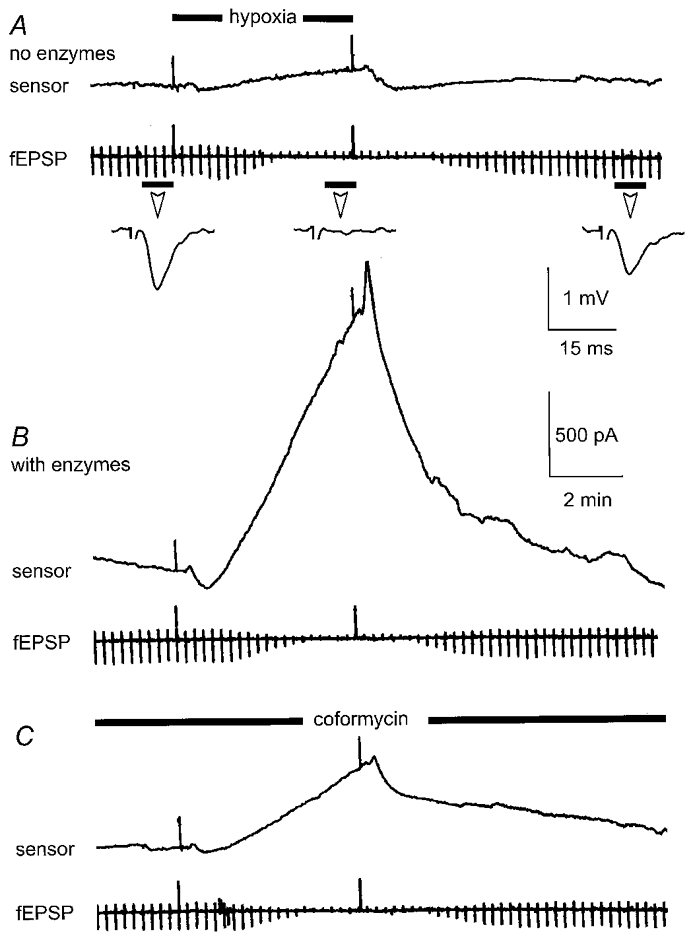
A, the sensor, filled with enzyme-free buffer, was placed on the surface of the slice. Hypoxia was induced for 5 min as indicated, in this and subsequent figures, by the black bar and upward deflections of chart event marker. Note the minimal effect of hypoxia on the sensor trace whilst hypoxia clearly reversibly depressed the simultaneously recorded fEPSP (periodic downward deflections every 15 s and inset traces which are averages of 4 consecutive fEPSPs taken at the times indicated by the short black bar). B, in the same slice, the sensor was loaded with the appropriate enzyme mixture and returned to the same position as that in A. Hypoxia induced a profound polarisation of the sensor which reversed on return to normoxia. The fEPSP was once again reversibly depressed. C, the application of the selective adenosine deaminase inhibitor, coformycin (1 μM), greatly attenuated the hypoxia-induced polarisation of the sensor.
Statistical analysis
Data were pooled into appropriate groups and statistical significance (P = 0.05) was tested with Student's t test, one-way ANOVA or linear regression analysis as indicated in the text. Data are presented as means ±s.e.m.
Drugs
Adenosine deaminase (EC 3.5.4.4), nucleoside phosphorylase (EC 2.4.2.1) and xanthine oxidase (EC 1.1.3.22) were obtained from Sigma. The chemicals used in the ACSF were obtained from BDH. Coformycin was supplied by Calbiochem and adenosine by either RBI or Sigma. 8-Cyclopentyl-1,3-dipropylxanthine (DPCPX) was obtained from RBI.
RESULTS
Characteristics and specificity of the sensor
Linearity of sensor
In order to test the relationship between adenosine and polarisation of the sensor, we immersed the sensor in the recording chamber used to contain hippocampal slices and perfused the sensor with ACSF containing various concentrations of exogenous adenosine. In differential recording mode, where the signal from the reference barrel is subtracted from the signal obtained from the adenosine barrel, this gave rise to concentration-dependent polarisation of the sensor (Fig. 2). These polarisations were greatly attenuated or abolished by the selective adenosine deaminase inhibitor coformycin (0.5-2 μM) indicative of their origin from the exogenous adenosine. In four separate experiments, each with different probes, the relationship between polarisation of the probe and concentration of adenosine was linear (r = 0.9; P = 0.02; linear regression analysis) over the concentration range 10 nM to 20 μM (the highest concentration tested).
Figure 2. Linearity of sensor to adenosine.
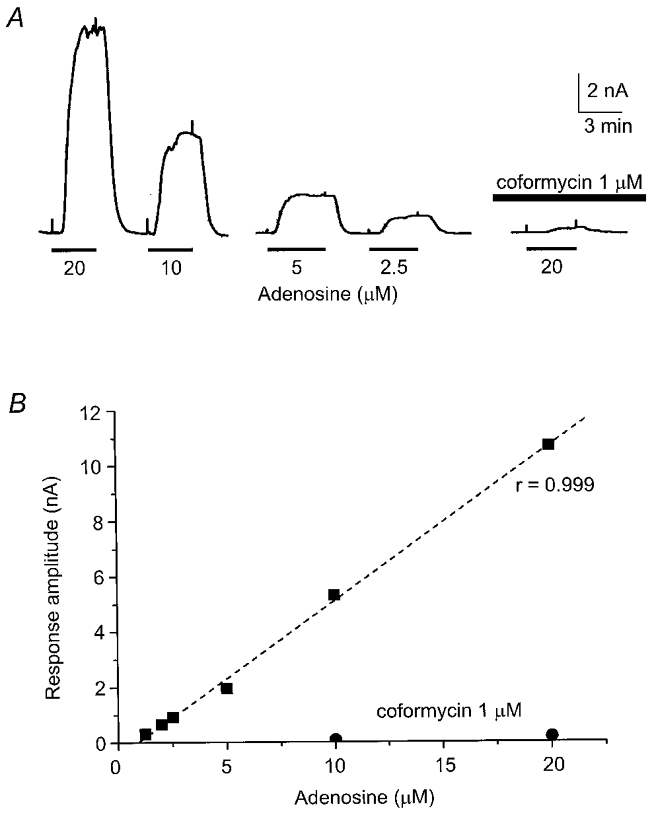
A, concentration-dependent polarisations of sensor in response to exogenous adenosine (1.25-20 μM) applied for the duration shown by the black bar and between upward deflections of chart event marker. The selective adenosine deaminase inhibitor, coformycin (1 μM), greatly attenuated the adenosine-dependent polarisation. B, graph of the experiment depicted in A showing linearity of sensor to adenosine over the concentration range most commonly encountered in this study, with the effects of coformycin on the adenosine signal.
Testing the selectivity of the sensor for adenosine released during hypoxia
The specificity of the probe for adenosine resides in the inclusion of AD in one of the two barrels and accurate subtraction of the inosine signals recorded from both barrels. Therefore, three criteria must be satisfied before the signal recorded can be attributed selectively to adenosine. Firstly, the signal must show an absolute dependence on the presence of the enzymes within the probe. Secondly, application of coformycin, which blocks AD, the first enzyme in the cascade, should greatly reduce the net signal. Thirdly, to confirm the efficacy of the subtraction procedure, when identical complements of enzymes are present in each barrel (i.e. NP and XO) there should be no net difference signal between the two barrels. In our documentation of the control experiments designed to test the compliance of the probe with these three criteria, we report the output of the sensor in units of current. This minimizes any assumptions about the nature of the signal generated by the sensor.
To examine the first of these criteria, we placed the probe, without enzymes, in the recording chamber and changed the perfusing medium to the standard hypoxic ACSF. This resulted in either no change or a very small negative shift in the probe signal (-33 ± 48 pA; n = 3 from 2 differential and 1 single-ended measurements; data not shown) and is likely to reflect the intrinsic sensitivity of the polarised platinum wires to bath oxygen tension. We next placed the probe, lacking enzymes in both barrels, on the surface of the slice in area CA1 and made the slice hypoxic. In these cases (Fig. 3A), in the absence of enzymes, the probe recorded a signal comparable to that seen in the absence of the hippocampal slice (40 ± 43 pA; mean ±s.e.m.; n = 7 from 5 differential and 2 single-ended measurements). The slice does not therefore release non-specific electroactive interferents that could confound adenosine measurements. Nevertheless, the depression of the simultaneously recorded fEPSP (Fig. 3A) indicates that the slices were indeed exposed to a depth of hypoxia sufficient to evoke an adenosine-mediated response. In contrast, in the same series of experiments, with the appropriate enzyme combination in both barrels (Fig. 3B), the probe records a polarisation that develops shortly after the onset of the hypoxic episode (attributable largely to the presence of a dead space in the perfusion tubing leading to the recording chamber) and continues during the course of the hypoxic episode. Under these conditions the signal recorded at the end of the 5 min hypoxic episode measured 1226 ± 423 pA (n = 7 from 5 double- and 2 single-ended measurements). On return to normoxic conditions, polarisation of the sensor reversed (Fig. 3B).
The differential polarisation of the probe during hypoxia was greatly reduced (67 ± 6.5 %; range = 28-86 %; n = 9) by the application of coformycin (500 nM to 2 μM; Fig. 3C). When optimally balanced, the probe primarily responds to the production of adenosine since in 4 of 9 cases coformycin blocked probe polarisation by >80 %. Furthermore, in single-ended mode, where the probe measures both adenosine and inosine, coformycin reduced the signal by 53 ± 5.5 % (range = 38-73 %; n = 6) indicating that even under these conditions more than half of the signal is due to adenosine. The dependence of the signal on the presence of the complete enzyme combination (criterion 1) and its attenuation by coformycin (criterion 2), a selective inhibitor of AD, indicates that the major part of the signal recorded by the probe during hypoxia does indeed result from the accumulation of extracellular adenosine.
To test the validity of the subtraction process (criterion 3) in reducing the contribution of inosine to the recorded signal we performed experiments in which both barrels were loaded with identical enzyme complements. Under these conditions, and when both barrels were optimally balanced with respect to each other (see Methods), the inclusion of identical enzyme complements in both barrels should give rise to no net differential signal during hypoxia. However, simultaneously, a signal should be recorded from either barrel.
Figure 4 shows an experiment in which both barrels were loaded with NP and XO. A differential signal was recorded between the barrels while a single-ended output was simultaneously recorded from one barrel. The induction of hypoxia clearly gave rise to a strong signal from the single-ended recording (Fig. 4A; upper trace), probably reflecting inosine production by endogenous adenosine deaminase in the slice. However, only a small net signal was seen in the differential recording (Fig. 4A; lower trace) indicating that the signals from each barrel had effectively cancelled the other out. Similar observations were made in three experiments where the net difference signal, in the absence of AD, measured -7 ± 105 pA whilst the simultaneously recorded signal from one barrel measured 853 ± 205 pA. On loading with the complete enzyme cascade, the net difference signal increased to 347 ± 37 pA (Fig. 4B and C). Coformycin reduced the latter signal to 100 ± 30 pA (71 % block; Fig. 4D).
Figure 4. Dependence of hypoxic signal on adenosine deaminase.
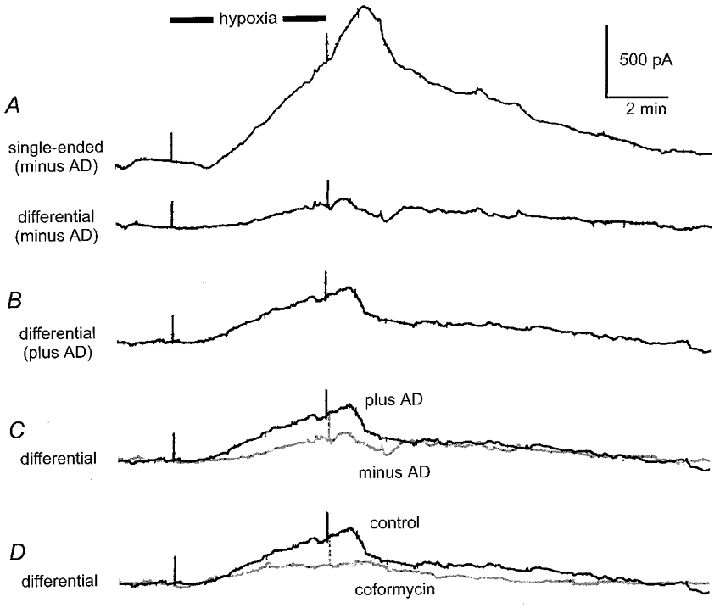
A, simultaneously recorded hypoxic signals recorded with symmetrical loading of sensor (nucleoside phosphorylase and xanthine oxidase in both barrels). Upper trace (single-ended) shows the output from one barrel whilst the lower trace (differential) shows the differential output between the two barrels. B, differential recording with adenosine deaminase loaded into one barrel. C, superimposition of differential traces with (plus AD) and without (minus AD, in grey) adenosine deaminase. D, coformycin (in grey) greatly reduced the differential signal. All traces taken from the same experiment.
Measurement of adenosine production during hypoxia
Having established that on average around 70 % of the current generated by the sensor is due to detection of adenosine, and that the remainder is probably contamination of this signal by detection of inosine, we have used the sensor to study adenosine production from the hippocampal slice during hypoxia. In the following experiments we report results as units of concentration of adenosine (see Methods for details of calibration). All measurements were made from the very first hypoxic episode delivered to a naïve slice and all experimental results reported in this section are from a different data set from those reported above.
Temporal relationship between adenosine production and the depression of the fEPSP during hypoxia
During episodes of hypoxia, levels of adenosine rose steadily in a fashion that closely paralleled the inhibition of excitatory synaptic transmission and did not appear to reach an asymptote or diminish in magnitude. This was true for both 5 and 10 min episodes of hypoxia (Fig. 5). For 5 min episodes adenosine reached a peak level of 5.6 ± 1.2 μM (n = 16, measured at the end of the episode, Fig. 5A). This compared to a peak level of 9.1 ± 1.6 μM (n = 10, Fig. 5B) achieved by the end of a 10 min episode. The extent of adenosine production seems therefore to scale with the duration of the hypoxic stimulus.
Figure 5. Adenosine release during hypoxia.
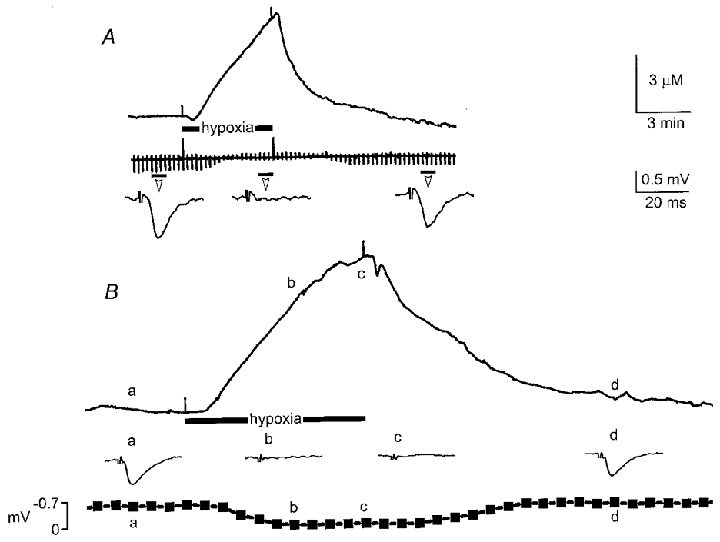
A, representative increase in extracellular adenosine during a 5 min hypoxic episode and reversible depression of fEPSP as indicated by the continuous chart trace and averages of four consecutive fEPSPs taken at times indicated by the short black bar under the chart trace. B, rise in extracellular adenosine during a 10 min exposure to hypoxia. Inset traces are averages of four consecutive fEPSPs (at 15 s intervals) taken at the times indicated by the letters a, b, c and d. The bottom graph shows the time course of the hypoxic depression of the averaged fEPSP synchronised with the start of the hypoxic episode. Note the close correlation between the increase in extracellular adenosine and the depression of the fEPSP. Scale bars refer to both A and B.
By measuring both the amplitude of the fEPSP and the level of extracellular adenosine at 1 min intervals throughout 5 min hypoxic episodes, we quantified the temporal relationship between depression of the fEPSP and production of adenosine. As might be expected, given the dependence of synaptic depression during hypoxia on adenosine A1 receptors (Fowler, 1989; Katchman & Hershkowitz, 1993a), we found a sigmoidal relationship between the depression of the fEPSP and production of extracellular adenosine (Fig. 6). Analysis of eight hypoxic episodes from separate slices gave an apparent IC50 of 3.0 ± 0.6 μM (range 0.4 to 5.1 μM; n = 8) for the action of endogenous adenosine on synaptic transmission. Both diffusional loss and uptake of adenosine will distort this apparent IC50 and cause it to be higher than the real value (the binding affinity of adenosine for the receptor). Nevertheless this apparent IC50 compares favourably with the estimate of 3 μM obtained in the presence of adenosine uptake inhibitors (Dunwiddie & Diao, 1994). This suggests that the sensor, although on the surface of the slice, is giving a reasonable measurement of the adenosine levels at their site of action.
Figure 6. Relationship between the adenosine signal recorded by sensor and hypoxic depression of the simultaneously recorded fEPSP.
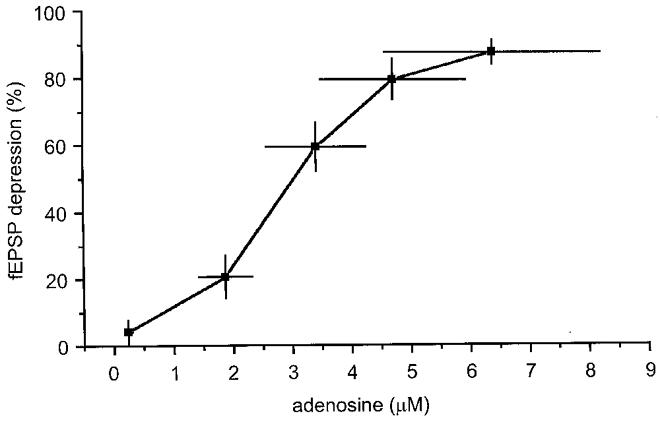
Pooled data from eight experiments in which adenosine concentration (in micromolar) is plotted, at 1 min intervals, against the depression of the simultaneously recorded fEPSP during a 5 min episode. Note the sigmoidal relationship with an IC50 of approximately 3 μM. Horizontal and vertical bars represent 1 s.e.m.
Influence of extracellular Ca2+ in adenosine release
To test the dependence upon extracellular Ca2+ of the release of adenosine during hypoxia, we preincubated slices in ACSF where extracellular Ca2+ had been replaced (by 2 mM Mg2+) for 3 to 6 h prior to delivery of a hypoxic episode. Under these conditions, hypoxia elicited much greater release of adenosine (48.9 ± 17.7 μM, n = 6; P = 0.001; unpaired t test, Fig. 7) compared to control, 2 mM extracellular Ca2+ (5.6 ± 1.2 μM; n = 16). Robust synaptic transmission could be recorded following the reintroduction of 2 mM extracellular Ca2+ indicating that this treatment had not irreversibly damaged the slice.
Figure 7. Extracellular Ca2+ is not required for hypoxic adenosine release.
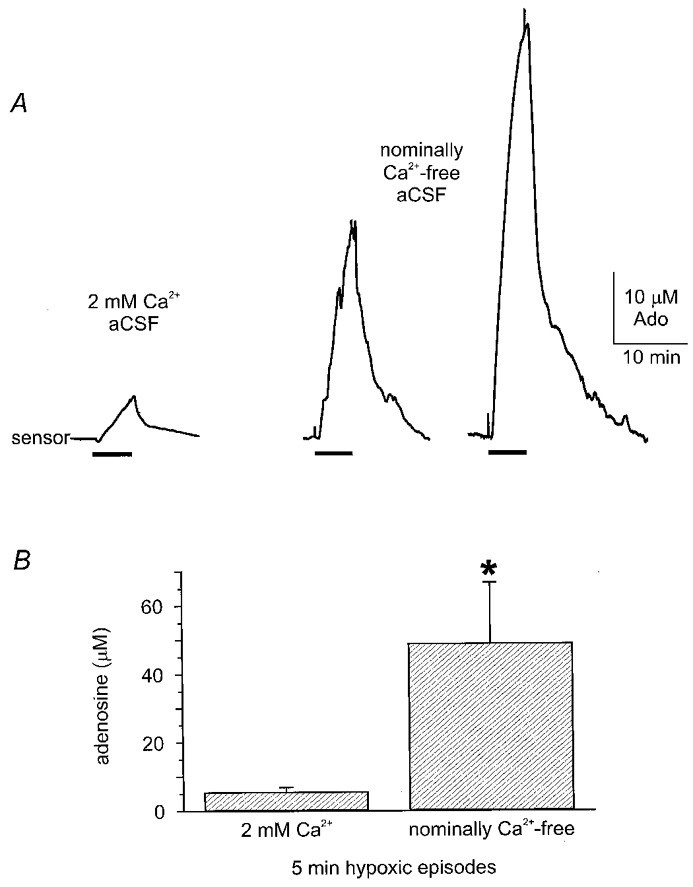
A, three examples of hypoxic adenosine release from three separate experiments (5 min exposure). Left-most in 2 mM Ca2+-containing ACSF. Two traces on the right are from two separate experiments recorded from slices pre-incubated in nominally Ca2+-free ACSF (for 3-6 h). Note the massive increase in release of adenosine during hypoxia in nominally Ca2+-free ACSF. B, pooled data showing hypoxic adenosine release is significantly greater (P = 0.001, unpaired t test) in nominally Ca2+-free ACSF (n = 6) compared to 2 mM extracellular Ca2+ (n = 16).
As our data suggest that removal of extracellular Ca2+ potentiates adenosine release, we next examined whether raising Ca2+ might exert an inhibitory influence. We therefore preincubated slices in ACSF containing 4 mM extracellular Ca2+ before delivering a hypoxic stimulus. Under these conditions adenosine release induced by hypoxia measured 3.5 ± 0.6 μM (n = 4, Fig. 8A), an ∼40 % reduction from control, consistent with the notion that extracellular Ca2+ inhibits the production of adenosine during hypoxia.
Figure 8. Elevated extracellular Ca2+ inhibits adenosine release during hypoxia and slows the rate of depression of the fEPSP.
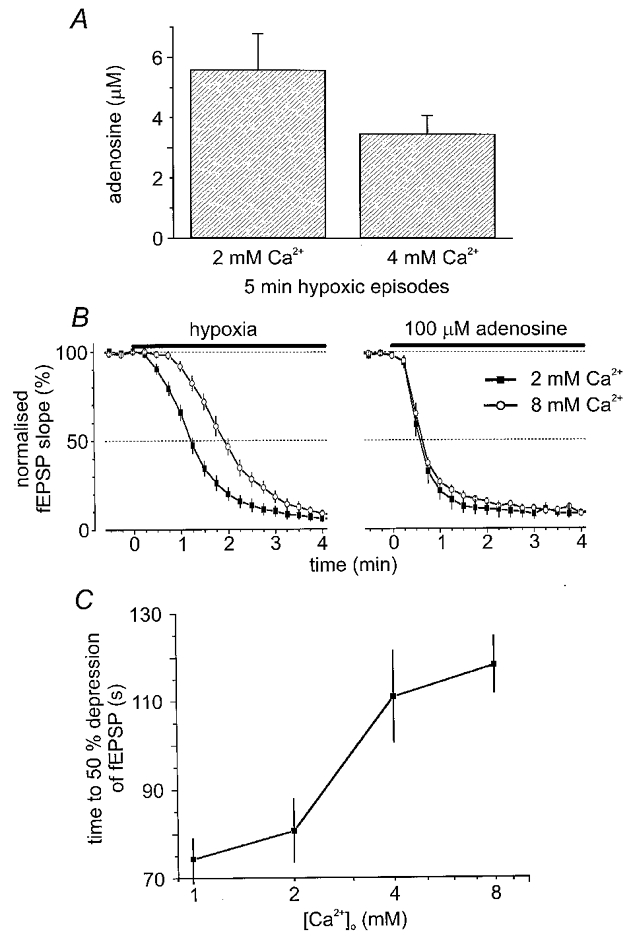
A, sensor measurements of hypoxic adenosine release in 2 mM (n = 16) and 4 mM (n = 4) extracellular Ca2+ showing a trend for reduced adenosine release in high Ca2+. B, in a parallel series of experiments the effect of hypoxia and exogenous adenosine on synaptic transmission in different extracellular Ca2+ concentrations was examined. B left, at time zero hypoxia (black bar) was induced in 2 mM Ca2+ ACSF (filled symbols; n = 18) and 8 mM Ca2+ ACSF (open symbols; n = 7). Note the pronounced retardation in the rate of depression of the fEPSP. B right, in contrast, synaptic transmission was equally sensitive to exogenous adenosine (100 μM; black bar) in both 2 mM Ca2+ ACSF (filled symbols; n = 6) and 8 mM Ca2+ ACSF (open symbols; n = 5). C, concentration dependence of the effects of extracellular Ca2+ on the time to 50 % depression of the fEPSP (1 mM, n = 13; 2 mM, n = 18; 4 mM, n = 14; 8 mM, n = 7). In B and C fEPSPs measured approximately 1 mV.
To see whether these reductions in adenosine release by high external Ca2+ could have physiological consequences, we examined the hypoxic depression of fEPSPs at varying levels of extracellular Ca2+. As the competitive and selective A1 adenosine receptor antagonist DPCPX (200 nM) prevents all but 5.3 ± 1.2 % (n = 5) of the depression of the fEPSP after 2 min of hypoxia (data not shown, but see Fowler, 1989; Katchman & Hershkowitz, 1993a) the depression of the fEPSP during the first few moments of hypoxia is essentially solely due to the activation of presynaptic adenosine A1 receptors. Thus the rate of onset of synaptic depression will reflect the rate of adenosine accumulation in the synaptic cleft. We therefore tested whether different concentrations of extracellular Ca2+ could influence the rate at which hypoxia depressed synaptic transmission.
Increasing extracellular Ca2+ delayed the hypoxic depression of the fEPSP (Fig. 8B left, 8C). The time to 50 % depression of synaptic transmission by hypoxia was significantly greater (P = 0.05; one-way ANOVA) in high Ca2+ (4 mM: 110.9 ± 10.5 s, n = 14; or 8 mM: 118.1 ± 6.4 s, n = 7) than in normal (2 mM: 80.7 ± 7.2 s, n = 18) or low (1 mM: 74.3 ± 4.6 s, n = 13) extracellular Ca2+ (Fig. 8B left, 8C). This is consistent with the inhibition of adenosine release by elevated Ca2+ reported with the probe measurements.
However an alternative hypothesis is that the raised extracellular Ca2+ could reduce the sensitivity of synaptic transmission to inhibition by adenosine (cf. Dunwiddie, 1984). We therefore tested the efficacy of exogenous adenosine (100 μM) at depressing synaptic transmission in either 2 mM or 8 mM extracellular Ca2+. In these experiments the fEPSP had a mean amplitude of around 1 mV and we measured both the time to 50 % depression of the fEPSP and the maximal depression of the fEPSP (Fig. 8B, right). Changing extracellular Ca2+ had no significant effect on these measures. Time to 50 % depression of transmission occurred after 37.5 ± 4.1 s (n = 6) in 2 mM Ca2+ and 38.4 ± 1.5 s (n = 5) in 8 mM Ca2+. Three minutes after application of 100 μM adenosine, the depression of synaptic transmission had asymptoted to close to 90 % of control in both 2 mM (92 ± 3 %; n = 6) and 8 mM (89 ± 1 %; n = 5) extracellular Ca2+. This shows that, in our experiments, the sensitivity of synaptic transmission to adenosine is unchanged in high Ca2+ conditions and that the reduced release of adenosine makes a major contribution to the slower onset of synaptic depression during hypoxia.
DISCUSSION
The actions of adenosine within the CNS are ubiquitous and varied. Its production and release by tissues of the central nervous system is therefore of fundamental importance. Given this vital role it is not surprising that adenosine receptors have attracted considerable interest. However, there are conflicting results regarding the sources of adenosine (ATP, cAMP), the signals for release (depolarisation, glutamate receptor activation, metabolic stress), the mechanism for release (Ca2+- and Na+-dependent and -independent mechanisms, reverse transport), the nature of the released molecule (ATP, cAMP, adenosine) and the cellular source of adenosine in the hippocampus (glia, interneurones, pyramidal neurones). It is likely that different challenges under different experimental conditions recruit different mechanisms thus complicating the study of adenosine production and release. Furthermore, more complex challenges (e.g. hypoxia/ischaemia) may recruit multiple interconnected pathways for adenosine production and release. Thus, information on the kinetics, localisation and cellular sources of adenosine release are required. HPLC techniques have been used until now to detect adenosine within collected superfusate. However, given the limited time and no spatial resolution this technique offers, this information has remained elusive. We have described a novel means by which to determine the release of adenosine into the extracellular space. This method permits direct, online and continuous measurement of adenosine from intact brain slices whilst simultaneously monitoring, for example, synaptic transmission.
Specificity of adenosine sensor
Our controls (no enzymes in probe, identical complement of enzymes in both barrels, coformycin) indicate that the probe signal is not an artefact generated by an electroactive interferent released by the slice during hypoxia. Instead the probe measured a slowly developing signal during hypoxia which was: dependent on the presence of the complete enzyme combination, greatly attenuated by coformycin, scaled in a sigmoidal fashion with the depression of the simultaneously recorded fEPSP and reversed with the recovery of synaptic transmission. These data indicate that the signal recorded by the probe during hypoxia reflects adenosine production and release.
In contrast to Xenopus spinal cord (Dale, 1998) where coformycin virtually abolished the adenosine signal produced by fictive locomotion, in hippocampus higher concentrations of coformycin (up to 2 μM) caused a reduction in the hypoxic adenosine signal of around 70 %. The incomplete block may be due to inosine production by residual endogenous adenosine deaminase or to the concentrations of coformycin used in this study being less than that required for full inhibition of the endogenous deaminase. This may reflect differences in tissue volume between the Xenopus spinal cord and hippocampal slices and/or the presence of an uptake system for coformycin which sequesters and effectively reduces its concentration. Furthermore, inosine can be produced independently from the breakdown of adenosine from the action of AMP deaminase (EC 3.5.4.6) to yield IMP from AMP with subsequent dephosphorylation to inosine by 5′-nucleotidase (EC 3.1.3.5) (Lloyd et al. 1993). In addition, although we recorded in differential mode for the most part, a truly differential signal requires that the two barrels be perfectly balanced. In practice this may not always occur since one barrel may be (1) intrinsically more sensitive than the other, (2) may have a slower response onset than the other or (3) may be in preferential contact with the hippocampal slice. Thus, there may be some small error associated with estimates of absolute amounts of adenosine recorded by the sensor and hence the proportion of block of the signal by adenosine deaminase inhibition. However, what is clear is that the sensor does not record a signal in the absence of enzymes thereby eliminating the possibility that non-specific electroactive interferents are being released from the hippocampus during hypoxia or under basal conditions.
Advantages and limitations of sensor
The sensor represents a significant technical advance in the study of purine release. Its compact size allows placement on defined regions within the exposed CNS. While this study concentrates on area CA1, release of adenosine has been recorded from other areas such as the dentate gyrus (B. Frenguelli & N. Dale, unpublished observations) and exposed brain stem nuclei in vivo (Thomas et al. 2000). The sensor therefore allows greater spatial resolution of adenosine release than any other currently available method. The direct, on-line measurement of adenosine release obviates the protracted procedures associated with HPLC and dialysis measurements and the continuous nature of the read-out gives much improved temporal resolution of adenosine release.
Nevertheless, the sensor suffers some limitations mainly connected to its size. The width of the dual barrelled sensor (500 μm) precludes its insertion into tissue without causing considerable damage and possible consequent alteration of physiological function. To avoid such damage we have only used the sensor to measure adenosine release from the surface of the hippocampal slice (see Methods). The diffusion path from the centre of the slice could therefore be as long as 200 μm. This would represent the upper limit of the diffusion path length on the assumption that only cells within the centre of the slice release adenosine. However there is no reason to suppose that only the centre of the slice releases adenosine. Indeed patch-clamp recordings (B. G. Frenguelli, unpublished observations) suggest that even superficial neurons undergo profound adenosine-mediated synaptic depression during hypoxia. Thus, the whole thickness of the slice apart from any dead cells on its surface would be expected to respond to hypoxia by releasing adenosine. The effective diffusion path from the nearest cells to the probe is therefore likely to be much shorter than the maximal path length (200 μm).
There will be several kinds of loss of adenosine along this diffusion path: diffusion away from the sensor; diffusional obstacles such as tortuosity (Rusakov & Kullmann, 1998), which may change during hypoxia/ischaemia (Nicholson & Sykova, 1998); metabolism by endogenous adenosine deaminase and adenosine kinase (Pak et al. 1994; Lloyd & Fredholm, 1995); and reuptake by nucleoside transporters (Thorn & Jarvis, 1996). Clearly the concentration of adenosine within the synaptic structures will be higher than that recorded by the probe. Furthermore the exact time course of changes in adenosine concentration at the synapse will differ from those recorded at the surface. Nevertheless four aspects of our evidence suggest that measurements at the slice surface may give a reasonable, if filtered, picture of the changes in adenosine levels within the neuropil. Firstly there is a close temporal correlation between the changes in adenosine concentration and the depression of synaptic transmission, even when this is recorded by electrodes up to 200 μm away from the sensor. Secondly, our calculation of the apparent IC50 for adenosine of 3 μM is comparable to the values reported by others when adenosine uptake is blocked (Dunwiddie & Diao, 1994). This suggests that adenosine uptake during hypoxia does not appreciably distort our signal. Thirdly, the reduction of adenosine release in the presence of 4 mM extracellular Ca2+ reported by the sensor is reflected in a slowed rate of depression of synaptic transmission. Fourthly, our measurements are in broad agreement with estimates for the concentration of extracellular adenosine during hypoxia/ischaemia in vivo, which vary between 5 and 40 μM (Schubert et al. 1994), and in vitro, where levels may reach 30 μM (Latini et al. 1999).
Adenosine release
Our work with the adenosine sensor indicates that the release of adenosine is directly related to the depression of excitatory synaptic transmission confirming the important role of adenosine and A1 receptor activation in the hypoxic depression of excitatory transmission in area CA1. Our data greatly strengthen the conclusions obtained from indirect pharmacological experiments which repeatedly show that in area CA1 hypoxia depresses excitatory synaptic transmission by an A1 receptor-dependent mechanism (Fowler, 1989; Katchman & Hershkowitz, 1993a). This argues strongly against the interpretation from early HPLC analysis that the release of adenosine occurs after the depression of synaptic transmission (Pedata et al. 1993; Fowler, 1993; Lloyd et al. 1993). It is likely that this latter interpretation stems from the lower temporal resolution of HPLC methods and indeed a recent HPLC study, with improved time resolution, confirms that adenosine release is coincident with the depression of transmission (Latini et al. 1998).
Ca2+ dependence of release
The dependence of adenosine production during hypoxia on external Ca2+ is a key issue. A dependence on Ca2+ would occur if, for example, adenosine in the synaptic cleft arose from the exocytotic release of ATP and subsequent metabolism by ecto-enzymes to adenosine. This suggestion is made more plausible by the description of increased vesicular release of glutamate and GABA during the early stages of hypoxia (Hershkowitz et al. 1993; Katchman et al. 1994). However, the issue of synaptic release of ATP in area CA1 is extremely contentious. A recent paper demonstrates that ATP release in the CA1 region is Ca2+ independent and largely reflects electroporation of axons (Hamann & Attwell, 1996) whereas another demonstrates synaptically activated P2X currents (Pankratov et al. 1998).
Our observations with prolonged (3-6 h) Ca2+removal show that the production of adenosine during hypoxia does not require the presence of extracellular Ca2+. This observation is very important because it indicates that a major component of adenosine release during hypoxia must be via non-exocytotic mechanisms and further implies that the trigger for adenosine release cannot depend upon exocytosis or external Ca2+ either. Glutamate, which is released during hypoxia, has been proposed to act as a trigger for adenosine release. Our results strongly suggest that if this is the case, the release of glutamate itself during hypoxia must be independent of extracellular Ca2+ (Kauppinen et al. 1988; Ikeda et al. 1989) perhaps requiring release of Ca2+ from internal stores (Katchman & Hershkowitz, 1993b; Belousov et al. 1995); or reversed glutamate uptake (Rossi et al. 2000).
Although adenosine production during hypoxia does not require Ca2+, the amount of adenosine release is regulated by external Ca2+. In nominally Ca2+-free ACSF, adenosine release was greatly enhanced relative to that occurring in ACSF containing 2 mM Ca2+. The converse manipulation of elevating extracellular Ca2+ to 4 mM reduced the production of adenosine during hypoxia relative to the control. By varying the level of extracellular Ca2+ we found that the rate of depression of the fEPSP during hypoxia slowed as the concentration of Ca2+ increased, an observation also made in spinal cord (Czeh & Somjen, 1990). This indirectly supports our evidence that Ca2+ inhibits adenosine production, as the slower accumulation of adenosine would result in a slower rate of synaptic depression. However an alternative and plausible explanation for this observation is that the higher levels of Ca2+ reduce the ability of adenosine receptors to inhibit synaptic transmission (Dunwiddie, 1984). This possibility is rendered unlikely by our data demonstrating that synaptic transmission in both high (8 mM) and normal (2 mM) Ca2+ was equally sensitive to exogenous adenosine. We therefore conclude that Ca2+ inhibits the production of adenosine during hypoxia. It is not clear why a discrepancy exists between our findings and those of Dunwiddie (1984) but differences between the methods of stimulation or age of the animals employed in the respective studies may contribute.
Our findings extend those of a previous HPLC study in which both the basal and ischaemia-induced outflow of adenosine was increased in Ca2+-free conditions (Pedata et al. 1993). The underlying mechanism for the facilitatory effect of Ca2+ removal remains unknown but is unlikely to involve cell lysis or gross pathological change as reperfusion of the slice with control ACSF restored robust synaptic transmission. Some component in the production or release of adenosine must be Ca2+ sensitive, e.g. an inhibitory tone provided by ambient Ca2+ on enzymes involved in the production of adenosine; or Ca2+-dependent inhibition of transporters for adenosine or nucleotide precursors, which is exaggerated in elevated extracellular Ca2+.
The Ca2+-sensitive inhibition of adenosine production may well be physiologically important. For example, the local concentration of extracellular Ca2+ decreases profoundly during hypoxia (Martin et al. 1994) and this may serve to promote adenosine release for the duration of hypoxic/ischaemic episodes (Arlinghaus & Lee, 1996). Furthermore, activity-dependent decreases in extracellular Ca2+ have been described (Somjen, 1980; Rusakov et al. 1999) which may contribute to the enhanced adenosine tone observed during repetitive activity (Mitchell et al. 1993) and the termination of epileptiform activity (Dunwiddie, 1999).
Acknowledgments
We thank The Royal Society, The Wellcome Trust, Tenovus (Tayside) and The Anonymous Trust (Dundee) for financial support. We are grateful to Mr John Bell (Sycopel International Ltd) for help with the biosensor probes. B.G.F. is a Caledonian Research Foundation Fellow.
References
- Anderson WW, Collingridge GL. A data acquisition program for on-line analysis of long-term potentiation and long-term depression. Society for Neuroscience Abstracts. 1997;23:665. doi: 10.1016/s0165-0270(01)00374-0. [DOI] [PubMed] [Google Scholar]
- Arlinghaus L, Lee KS. Endogenous adenosine mediates the sustained inhibition of excitatory synaptic transmission during moderate hypoxia. Brain Research. 1996;724:265–268. doi: 10.1016/0006-8993(96)00343-5. [DOI] [PubMed] [Google Scholar]
- Belousov AB, Godfraind J-M, Krnjevic K. Internal Ca2+ stores involved in anoxic responses of rat hippocampal neurons. The Journal of Physiology. 1995;486:547–556. doi: 10.1113/jphysiol.1995.sp020833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischofberger N, Jacobson KA, Von Lubitz DK. Adenosine A1 receptor agonists as clinically viable agents for treatment of ischemic brain disorders. Annals of the New York Academy of Sciences. 1997;825:23–29. doi: 10.1111/j.1749-6632.1997.tb48411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundege JM, Dunwiddie TV. Role of adenosine as a modulator of synaptic activity in the central nervous system. Advances in Pharmacology. 1997;39:353–391. doi: 10.1016/s1054-3589(08)60076-9. [DOI] [PubMed] [Google Scholar]
- Czeh G, Somjen GG. Hypoxic failure of synaptic transmission in the isolated spinal cord, and the effects of divalent cations. Brain Research. 1990;527:224–233. doi: 10.1016/0006-8993(90)91141-3. [DOI] [PubMed] [Google Scholar]
- Dale N. Delayed production of adenosine underlies temporal modulation of swimming in frog embryo. The Journal of Physiology. 1998;511:265–272. doi: 10.1111/j.1469-7793.1998.265bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N, Kuenzi FM. Ion channels and the control of swimming in the Xenopus embryo. Progress in Neurobiology. 1997;53:729–756. doi: 10.1016/s0301-0082(97)00048-8. [DOI] [PubMed] [Google Scholar]
- De Mendonça A, Ribeiro JA. Adenosine and neuronal plasticity. Life Sciences. 1997;60:245–251. doi: 10.1016/s0024-3205(96)00544-9. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV. Interactions between the effects of adenosine and calcium on synaptic responses in rat hippocampus in vitro. The Journal of Physiology. 1984;350:545–559. doi: 10.1113/jphysiol.1984.sp015217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV. Adenosine and suppression of seizures. Advances in Neurology. 1999;79:1001–1010. [PubMed] [Google Scholar]
- Dunwiddie TV, Diao L. Extracellular adenosine concentrations in hippocampal brain slices and the tonic inhibitory modulation of evoked excitatory responses. Journal of Pharmacology and Experimental Therapeutics. 1994;268:537–545. [PubMed] [Google Scholar]
- Edwards FA, Konnerth A, Sakmann B, Takahashi T. A thin slice preparation for patch clamp recording from neurones of the mammalian central nervous system. Pflügers Archiv. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- Fowler JC. Adenosine antagonists delay hypoxia-induced depressions of neuronal activity in hippocampal brain slice. Brain Research. 1989;490:378–384. doi: 10.1016/0006-8993(89)90258-8. [DOI] [PubMed] [Google Scholar]
- Fowler JC. Adenosine antagonists alter the synaptic response to in vitro ischemia in the rat hippocampus. Brain Research. 1990;509:331–334. doi: 10.1016/0006-8993(90)90560-x. [DOI] [PubMed] [Google Scholar]
- Fowler JC. Purine release and inhibition of synaptic transmission during hypoxia and hypoglycemia in rat hippocampal slices. Neuroscience Letters. 1993;157:83–86. doi: 10.1016/0304-3940(93)90648-5. [DOI] [PubMed] [Google Scholar]
- Frenguelli BG. The effects of metabolic stress on glutamate receptor-mediated depolarisations in the in vitro rat hippocampal slice. Neuropharmacology. 1997;36:981–991. doi: 10.1016/s0028-3908(97)00084-1. [DOI] [PubMed] [Google Scholar]
- Frenguelli BG, Dale N. Direct measurement of adenosine release during hypoxia in the rat hippocampal slice. The Journal of Physiology. 1999;515.P:125P. doi: 10.1111/j.1469-7793.2000.00143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene RW, Haas HL. The electrophysiology of adenosine in the mammalian central nervous system. Progress in Neurobiology. 1991;36:329–341. doi: 10.1016/0301-0082(91)90005-l. [DOI] [PubMed] [Google Scholar]
- Hamann M, Attwell D. Non-synaptic release of ATP by electrical stimulation in slices of rat hippocampus, cerebellum and habenula. European Journal of Neuroscience. 1996;8:1510–1515. doi: 10.1111/j.1460-9568.1996.tb01613.x. [DOI] [PubMed] [Google Scholar]
- Hershkowitz N, Katchman AN, Veregge S. Site of synaptic depression during hypoxia: A patch-clamp analysis. Journal of Neurophysiology. 1993;69:432–441. doi: 10.1152/jn.1993.69.2.432. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Nakazawa T, Abe K, Kaneko T, Yamatsu K. Extracellular accumulation of glutamate in the hippocampus induced by ischemia is not calcium dependent –in vitro and in vivo evidence. Neuroscience Letters. 1989;96:202–206. doi: 10.1016/0304-3940(89)90058-x. [DOI] [PubMed] [Google Scholar]
- Katchman AN, Hershkowitz N. Adenosine antagonists prevent hypoxia-induced depression of excitatory but not inhibitory synaptic currents. Neuroscience Letters. 1993a;159:123–126. doi: 10.1016/0304-3940(93)90814-2. [DOI] [PubMed] [Google Scholar]
- Katchman AN, Hershkowitz N. Early anoxia-induced vesicular glutamate release results from mobilization of calcium from intracellular stores. Journal of Neurophysiology. 1993b;70:1–7. doi: 10.1152/jn.1993.70.1.1. [DOI] [PubMed] [Google Scholar]
- Katchman AN, Vicini S, Hershkowitz N. Mechanism of early anoxia-induced suppression of the GABAA-mediated inhibitory postsynaptic current. Journal of Neurophysiology. 1994;71:1128–1138. doi: 10.1152/jn.1994.71.3.1128. [DOI] [PubMed] [Google Scholar]
- Kauppinen RA, McMahon HT, Nicholls DG. Ca2+-dependent and Ca2+-independent glutamate release, energy status and cytosolic free Ca2+ concentration in isolated nerve terminals following metabolic inhibition: possible relevance to hypoglycaemia and anoxia. Neuroscience. 1988;27:175–182. doi: 10.1016/0306-4522(88)90228-x. [DOI] [PubMed] [Google Scholar]
- Latini S, Bordoni F, Corradetti R, Pepeu G, Pedata F. Temporal correlation between adenosine outflow and synaptic potential inhibition in rat hippocampal slices during ischemia-like conditions. Brain Research. 1998;794:325–328. doi: 10.1016/s0006-8993(98)00304-7. [DOI] [PubMed] [Google Scholar]
- Latini S, Bordoni F, Pedata F, Corradetti R. Extracellular adenosine concentrations during in vitro ischaemia in rat hippocampal slices. British Journal of Pharmacology. 1999;127:729–739. doi: 10.1038/sj.bjp.0702591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd HG, Lindstrom K, Fredholm BB. Intracellular formation and release of adenosine from rat hippocampal slices evoked by electrical stimulation or energy depletion. Neurochemistry International. 1993;23:173–185. doi: 10.1016/0197-0186(93)90095-m. [DOI] [PubMed] [Google Scholar]
- Lloyd HGE, Fredholm BB. Involvement of adenosine deaminase and adenosine kinase in regulating extracellular adenosine concentration in rat hippocampal slices. Neurochemistry International. 1995;26:387–395. doi: 10.1016/0197-0186(94)00144-j. [DOI] [PubMed] [Google Scholar]
- Martin RL, Lloyd HGE, Cowan AI. The early events of oxygen and glucose deprivation: Setting the scene for neuronal death? Trends in Neurosciences. 1994;17:251–257. doi: 10.1016/0166-2236(94)90008-6. [DOI] [PubMed] [Google Scholar]
- Mitchell JB, Lupica CR, Dunwiddie TV. Activity-dependent release of endogenous adenosine modulates synaptic responses in the rat hippocampus. Journal of Neuroscience. 1993;13:3439–3447. doi: 10.1523/JNEUROSCI.13-08-03439.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, Sykova E. Extracellular space structure revealed by diffusion analysis. Trends in Neurosciences. 1998;21:207–215. doi: 10.1016/s0166-2236(98)01261-2. [DOI] [PubMed] [Google Scholar]
- Pak MA, Haas HL, Decking UKM, Schrader J. Inhibition of adenosine kinase increases endogenous adenosine and depresses neuronal activity in hippocampal slices. Neuropharmacology. 1994;33:1049–1053. doi: 10.1016/0028-3908(94)90142-2. [DOI] [PubMed] [Google Scholar]
- Pankratov Y, Castro E, Miras-Portugal MT, Krishtal O. A purinergic component of the excitatory postsynaptic current mediated by P2X receptors in the CA1 neurons of the rat hippocampus. European Journal of Neuroscience. 1998;10:3898–3902. doi: 10.1046/j.1460-9568.1998.00419.x. [DOI] [PubMed] [Google Scholar]
- Pedata F, Latini S, Pugliese AM, Pepeu G. Investigations into the adenosine outflow from hippocampal slices evoked by ischemia-like conditions. Journal of Neurochemistry. 1993;61:284–289. doi: 10.1111/j.1471-4159.1993.tb03566.x. [DOI] [PubMed] [Google Scholar]
- Porkka-Heiskanen T. Adenosine in sleep and wakefulness. Annals of Medicine. 1999;31:125–129. doi: 10.3109/07853899908998788. [DOI] [PubMed] [Google Scholar]
- Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacological Reviews. 1998;50:413–492. [PubMed] [Google Scholar]
- Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403:316–321. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- Rudolphi KA, Schubert P, Parkinson FE, Fredholm BB. Neuroprotective role of adenosine in cerebral ischaemia. Trends in Pharmacological Sciences. 1992;13:439–445. doi: 10.1016/0165-6147(92)90141-r. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM. Geometric and viscous components of the tortuosity of the extracellular space in the brain. Proceedings of the National Academy of Sciences of the USA. 1998;95:8975–8980. doi: 10.1073/pnas.95.15.8975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM, Stewart MG. Hippocampal synapses: do they talk to their neighbours? Trends in Neurosciences. 1999;22:382–388. doi: 10.1016/s0166-2236(99)01425-3. [DOI] [PubMed] [Google Scholar]
- Schubert P, Rudolphi KA, Fredholm BB, Nakamura Y. Modulation of nerve and glial function by adenosine – role in the development of ischemic damage. International Journal of Biochemistry. 1994;26:1227–1236. doi: 10.1016/0020-711x(94)90092-2. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Stimulus-evoked and seizure-related responses of extracellular calcium activity in spinal cord compared to those in cerebral cortex. Journal of Neurophysiology. 1980;44:617–632. doi: 10.1152/jn.1980.44.4.617. [DOI] [PubMed] [Google Scholar]
- Thomas T, Dale N, Spyer KM. Release of adenosine from brainstem structures during hypothalamic stimulation and hypoxia in the anaesthetised rat. The Journal of Physiology. 2000;523.P:252P. [Google Scholar]
- Thorn JA, Jarvis SM. Adenosine transporters. General Pharmacology. 1996;27:613–620. doi: 10.1016/0306-3623(95)02053-5. [DOI] [PubMed] [Google Scholar]
- Zhu PJ, Krnjevic K. Adenosine release is a major cause of failure of synaptic transmission during hypoglycaemia in rat hippocampal slices. Neuroscience Letters. 1993;155:128–131. doi: 10.1016/0304-3940(93)90689-i. [DOI] [PubMed] [Google Scholar]
- Zhu PJ, Krnjevic K. Adenosine release mediates cyanide-induced suppression of CA1 neuronal activity. Journal of Neuroscience. 1997;17:2355–2364. doi: 10.1523/JNEUROSCI.17-07-02355.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]