

Abstract
Store-mediated Ca2+ entry is the main pathway for Ca2+ influx in platelets and many other cells. Several hypotheses have considered both direct and indirect coupling mechanisms between the endoplasmic reticulum and the plasma membrane. Here we pay particular attention to new insights into the regulation of store-mediated Ca2+ entry: the role of the cytoskeleton in a secretion-like coupling model. In this model, Ca2+ entry may be mediated by a reversible trafficking and coupling of the endoplasmic reticulum with the plasma membrane, that shows close parallels to the events mediating secretion. As with secretion, the actin cytoskeleton plays an inhibitory role in the activation of Ca2+ entry by preventing the approach and coupling of the endoplasmic reticulum with the plasma membrane, making cytoskeletal remodelling a key event in the activation of Ca2+ entry. We also review recent advances investigating the regulation of store-mediated Ca2+ entry by small GTPases and phosphoinositides, which might be involved in the store-mediated Ca2+ entry pathway through roles in the remodelling of the cytoskeleton.
Cytosolic Ca2+ is a key regulatory factor and perhaps the most widely used means of controlling cellular function. Increases in intracellular free Ca2+ concentration ([Ca2+]i) can initiate and modulate many different events, including short-term responses, such as muscle contraction (Reembold, 1992) and secretion (Brown et al. 1985) and long-term processes like cell growth (Means, 1994). Cells can increase [Ca2+]i by releasing compartmentalised Ca2+ from intracellular stores or by evoking the entry of extracellular Ca2+ across the plasma membrane. Ca2+ release from the internal stores is usually transient; however, many cellular processes, as well as the refilling of the intracellular stores, require a sustained increase in cytosolic Ca2+, and therefore Ca2+ entry into the cell plays an important role.
In excitable cells, such as neurons, muscle and some endocrine cells, Ca2+ entry generally occurs through voltage-operated Ca2+ channels, Ca2+-selective channels that become briefly activated during action potentials (Tsien et al. 1995). On the other hand, in non-excitable cells like blood and epithelial cells, voltage-operated Ca2+ channels are not expressed. Several different mechanisms for Ca2+ entry in non-excitable cells have been proposed; however, store-mediated Ca2+ entry (SMCE), where Ca2+ entry is controlled by the filling state of the intracellular Ca2+ stores, appears to be the predominant pathway.
Activation of SMCE
The mechanism by which information is transmitted from the intracellular Ca2+ stores to the plasma membrane, and therefore, how SMCE is activated, is not well understood. Several hypotheses have been proposed that can be divided into those that consider a direct interaction (conformational coupling) between proteins in the intracellular Ca2+ compartments and plasma membrane and those that propose the existence of a diffusible messenger (Berridge, 1995; Sage, 1997; Pareck & Penner, 1997). These hypotheses include: (1) release of a non-protein calcium influx factor (CIF) from depleted stores (Radriamampita & Tsien, 1993); (2) changes in cGMP (Pandol & Schoeffield-Payne, 1990); (3) generation of a product of cytochrome P450 that activates calcium influx (Álvarez et al. 1992); (4) a tyrosine phosphorylation-dependent step (Sargeant et al. 1993a,b; Jenner et al. 1994); (5) activation of a pathway involving a small GTP-binding protein (Fasolato et al. 1993; Bird & Putney, 1993; Xu et al. 1996; Rosado & Sage, 2000a); (6) a Ca2+- calmodulin-dependent step (Gailly et al. 1996; Vaca, 1996); (7) insertion of channels by intracellular trafficking and vesicle fusion (Somasundaram et al. 1995); (8) physical coupling between the Ins(1,4,5)P3 receptor on the endoplasmic reticulum and a Ca2+ channel (perhaps the Ins(1,3,4,5)P4 receptor) in the plasma membrane (Irvine, 1990; Berridge, 1995).
Some of these observations have not gained wide acceptance. The effect of inhibitors of cytochrome P450, such as the imidazole antimycotics econazole and miconazole, could also be mediated by inhibition of tyrosine kinase activity (Sargeant et al. 1994) and these drugs have also been shown to inhibit voltage-operated Ca2+ channels (Álvarez et al. 1992). The effect of primaquine as an inhibitor of vesicular trafficking has been reinterpreted as direct inhibition of the Ca2+ influx channels (Gregory & Barrit, 1996).
A new model has recently been proposed in smooth muscle cell lines and platelets (Patterson et al. 1999; Yao et al. 1999; Rosado et al. 2000), where some of the hypotheses mentioned above may be integrated. This new secretion-like coupling model is based on a physical but reversible interaction between the intracellular Ca2+ compartments and the plasma membrane, which involves a physical trafficking of the Ca2+ stores (endoplasmic reticulum or dense tubular system) towards the plasma membrane.
Role of the actin cytoskeleton in the activation and maintenance of SMCE in platelets
Platelets are small discs of approximately 0.5 μm × 3.0 μm. In response to different stimuli, the shape of the platelet changes from a smooth disc to an irregular form showing multiple projections. These modifications are mediated by reorganisation of the actin cytoskeleton. The actin cytoskeleton in platelets is a highly dynamic structure, organised into two major structures: a cytoplasmic actin network and a membrane-associated cytoskeleton (Ma & Abrams, 1999).
In a model based on vesicular transport, the cytoskeleton might be involved in supporting the trafficking and, as with secretion, the membrane skeleton might play an important role. Our recent studies performed in human platelets have shown that inhibition of actin filament polymerisation using the cell-permeant toxins cytochalasin D or latrunculin A had a biphasic effect with time on SMCE induced by the sarco/endoplasmic reticulum Ca2+-ATPase thapsigargin. A short preincubation with these inhibitors (1 min) increased SMCE (Fig. 1A), whilst slightly inhibiting thapsigargin-evoked actin polymerisation. In contrast, when actin polymerisation was abolished after longer exposure to the inhibitors (40 min) calcium entry was reduced by 50 % (Fig. 1B) (Rosado & Sage, 2000a). The effect of 40 min preincubation of human platelets with cytochalasin D was also demonstrated by fluorescein isothiocyanate (FITC)-labelled phalloidin staining of F-actin (Fig. 2). Cytochalasin D did not reduce the actin filament content in resting platelets but, as shown in Fig. 2, induced reorganisation of the membrane skeleton into dense foci. The lack of effect of cytochalasin D on the filament content of resting platelets is in agreement with previous results (Fox & Phillips, 1981) and confirms that if ‘treadmilling’ (the net polymerisation at one end of the filament and depolymerisation at the other) occurs in resting platelets, it must occur very slowly. In the secretory model, the actin cytoskeleton localised under the plasma membrane prevents secretory granules from reaching their exocytotic destination (Muallem et al. 1995). A similar phenomenon could explain the potentiation of SMCE after a short preincubation of platelets with cytochalasin D or latrunculin A. Since these compounds inhibit actin polymerisation in a time-dependent manner (Rosado & Sage, 2000a; Rosado et al. 2000), it is likely that as these agents diffuse into the cell they initially affect the actin filaments near the plasma membrane, thus facilitating the coupling between the dense tubular system (the intracellular Ca2+ store in platelets) and the plasma membrane. A role for the actin cytoskeleton in SMCE has also been proposed in vascular endothelial cells (Holda & Blatter, 1997) and type I astrocytes (Grimaldi et al. 1999). In contrast to observations in platelets, astrocytes and endothelial cells, in NIH 3T3 cells and DDT1MF-2 and A7r5 muscle cell lines, disruption of the actin filament network with these agents did not modify Ca2+ entry (Pedrosa-Ribeiro et al. 1997; Patterson et al. 1999). The reason for these differences is not clear. A possible explanation may lie in the different distribution of the actin cytoskeleton in these cells. In platelets, vascular endothelial cells and astrocytes, actin filaments are located mainly in the cell periphery as a submembrane skeleton and a dynamic actin filament polymerisation is needed upon activation (Jones & Cowan, 1983; Simonescu & Simonescu, 1983; Hartwig, 1992). In contrast, smooth muscle and NIH 3T3 cells have a more evenly distributed and organised actin cytoskeletal architecture (Pedrosa-Ribeiro et al. 1997; Patterson et al. 1999). Thus, reorganisation of a dense subplasmalemmal actin filament network described in platelets, endothelial cells and astrocytes might be essential in allowing transport and coupling between the intracellular Ca2+ compartments and the plasma membrane in these cells but not others, explaining the differences in the effects observed after treatment with cytochalasin D or latrunculin A on SMCE in different cells.
Figure 1. Effects of cytoskeletal modifiers on SMCE in human platelets.
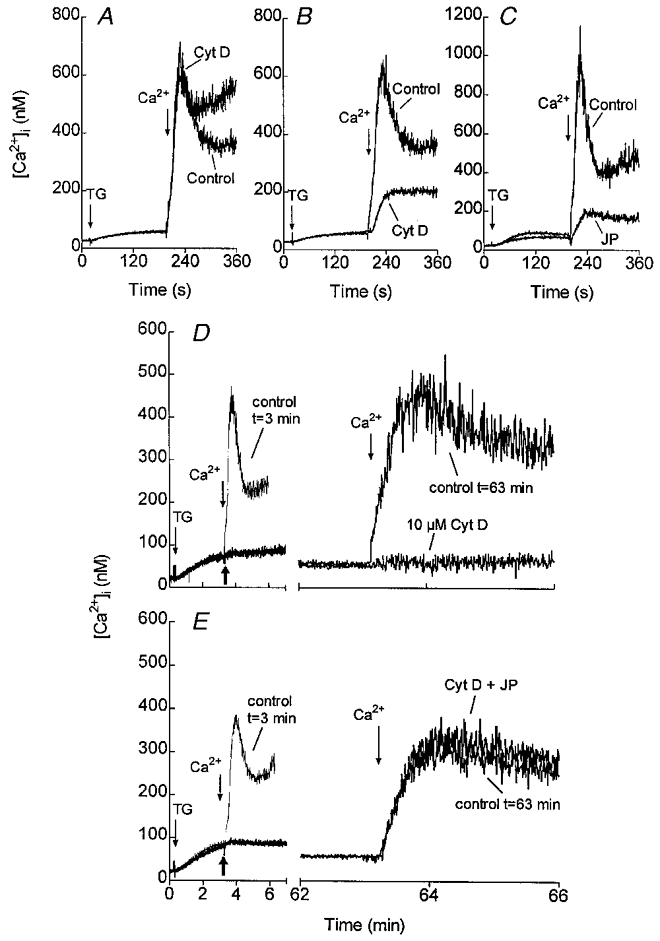
Fura-2-loaded human platelets were incubated at 37 °C either in the presence of 10 μm cytochalasin D (Cyt D) for 1 min (A) or 40 min (B), or in the presence of 10 μm jasplakinolide (JP) for 30 min (C) or the vehicles as controls. At the time of experiment 100 μm EGTA was added. Cells were then stimulated with 200 nM thapsigargin (TG) and 3 min later CaCl2 (final concentration 300 μm) was added to the medium to initiate Ca2+ entry. D and E, human platelets suspended in a Ca2+-free medium were stimulated with TG (200 nM) and 3 min later 10 μm Cyt D (D), 10 μm Cyt D plus 10 μm JP (E), or the vehicle (control t= 63 min) were added as indicated by the thick arrows. CaCl2 (300 μm) was added to the medium at the same time, as a control (control t= 3 min) or 1 h after Cyt D, Cyt D plus JP or the vehicle to initiate Ca2+ entry. Adapted from Rosado et al. (2000).
Figure 2. Effect of cytoskeletal modifiers on the reorganisation of the actin cytoskeleton in human platelets.
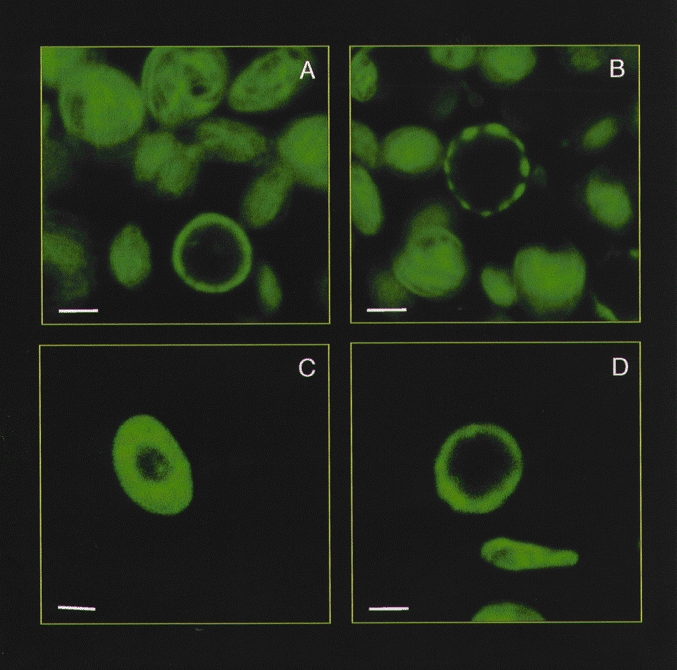
Confocal microscopy of human platelets stained with FITC-labelled phalloidin, an actin-binding fungal toxin. A, control conditions. B, platelets treated with 10 μm cytochalasin D for 40 min. C, platelets treated with 10 μm jasplakinolide for 30 min. D, platelets treated with 100 μm calyculin A for 2 min. The bars represent 1 μm.
Based on the concept that the actin cytoskeleton close to the plasma membrane might exert a regulatory effect on Ca2+ entry, experiments were performed to investigate the effect of stabilisation of the actin filament network which lies close to the plasma membrane. The stabilisation of cortical filaments was achieved by two independent means: induction of actin polymerisation using jasplakinolide and by phosphatase inhibition, which facilitates phosphorylation-dependent association of the actin-binding ezrin, radixin and moesin (ERM) proteins with the plasma membrane (Patterson et al. 1999). Jasplakinolide, a cell-permeant peptide that induces actin polymerisation and stabilisation of cortical filaments, increased actin filament content by 100 % (Rosado et al. 2000). Given that it has been established that almost half of the actin is unpolymerised in resting platelets (Hartwig, 1992), our findings suggest that jasplakinolide induced full actin polymerisation (Rosado et al. 2000), in agreement with the results obtained in smooth muscle A7r5 and DDT1MF-2 cell lines (Patterson et al. 1999). The structural changes induced by jasplakinolide are shown in Fig. 2. After treatment for 30 min with jasplakinolide the actin filaments became organised exclusively in a tight cortical layer. Calyculin A, an inhibitor of protein phosphatases 1 and 2 also induced reorganisation of the actin filaments by facilitating phosphorylation of the ERM proteins (Patterson et al. 1999). Phosphorylation of the ERM proteins activates their association, and thus the association of actin filaments, with the plasma membrane (Matsui et al. 1998). After treatment with calyculin A, actin filaments appear tightly condensed at the plasma membrane (Patterson et al. 1999; Fig. 2) without any increase in actin polymerisation (Rosado et al. 2000). Both experimental protocols resulted in a significant reduction in SMCE in platelets as well as in smooth muscle A7r5 and DDT1MF-2 cell lines (Fig. 1C; Patterson et al. 1999; Rosado et al. 2000). The findings described above suggest that redistribution of the actin filaments into a cortical layer prevents the coupling between the Ca2+ compartments (dense tubular system or endoplasmic reticulum) and the plasma membrane.
In contrast with some previous models, our results suggest that SMCE is unlikely to be mediated by a diffusible messenger in these cells. Such a factor should easily be able to reach the plasma membrane after redistribution of the actin filaments to the plasma membrane, since InsP3, generated by agonist activation, can still diffuse from the plasma membrane to the intracellular stores to stimulate the release of Ca2+ in the presence of jasplakinolide in platelets and smooth muscle A7r5 and DDT1MF-2 cell lines (Patterson et al. 1999; Rosado et al. 2000).
These findings are still compatible with two possible models for the activation of SMCE: physical coupling between the endoplasmic reticulum and the plasma membrane and the exocytotic insertion of vesicle-carried channels into the plasma membrane. To ascertain the more likely mechanism involved in the activation of Ca2+ entry, we investigated the effect of cytoskeletal modifications after activation of SMCE. Once calcium entry is activated, one would postulate that contact between the intracellular Ca2+ compartments and the plasma membrane would be easily interrupted by disruption of an actin-provided scaffold, which might be required to support the dense tubular system in a position close to the plasma membrane. In contrast, a model based on the insertion of channels in the plasma membrane is not expected to be affected by disruption of the actin cytoskeleton once the channels have been inserted. Treatment of human platelets with cytochalasin D or latrunculin A abolished Ca2+ entry previously stimulated by depletion of the intracellular Ca2+ stores (Fig. 1D; Rosado et al. 2000). Consistent with the above, jasplakinolide did not modify SMCE and reversed the inhibitory effect of cytochalasin D when added together (Fig. 1E; Rosado et al. 2000). These findings suggest that a model based on a direct and reversible coupling between the endoplasmic reticulum and the plasma membrane which requires a mechanical support is the most likely model for SMCE in these cells (Fig. 1E; Rosado et al. 2000).
Role of small GTP-binding proteins in the activation and maintenance of SMCE
A role for small GTP-binding proteins in the activation of SMCE has been reported in several different cell types. Fasolato et al. (1993) have presented evidence for the involvement of small GTP-binding proteins in the activation of Ca2+ entry in RBL cells. In these cells, dialysis with the non-hydrolysable analogue of GTP, GTPγS, before the Ca2+ stores were depleted, prevented the activation of ICRAC (calcium-release activated current) when the cells were treated with ionomycin to deplete the Ca2+ stores (Fasolato et al. 1993). Similar results were reported in mouse lachrymal acinar cells (Bird & Putney, 1993), megakaryocytes (Somasundaram et al. 1995) and HeLa cells (Peppelenbosch et al. 1996).
Recent studies in human platelets (Rosado & Sage, 2000a) and HEK cells (Xu et al. 1996) have, using a different methodological approach, suggested the involvement of small GTP-binding proteins of the Ras superfamily. In these studies the activity of small GTP-binding proteins was inhibited by preventing methylation of Ras proteins using farnesylcysteine analogues. Ras proteins are post-translationally prenylated and methylated (Ando et al. 1992; Sinensky & Lutz, 1992), a modification that involves the attachment of a farnesyl or geranylgeranyl group to the protein through a thioether linkage to a cysteine residue located four residues from the C-terminus, followed by the removal of the three C-terminal amino acids and methylation of the newly exposed α-carboxyl group of the farnesyl or geranylgeranyl cysteine residue (Sinensky & Lutz, 1992; Fig. 3). Prenylation, but mainly methylation, is essential for the association of Ras proteins with the cell membrane, a process that is required for Ras protein activation (Zhang & Casey, 1996). Studies of Ras association with the cell membrane have shown that less that 20 % of the farnesylated intermediate of Ras associates with membranes, compared with an association of 80 % after farnesylation and methylation, the latter being the only reversible step in activation by prenylation/methylation (Hancock et al. 1991; Zhang & Casey, 1996). Consistent with the role of Ras proteins in SMCE, these proteins have been shown to associate with membranes after depletion of intracellular Ca2+ stores using thapsigargin in human platelets (Rosado & Sage, 2000a). This translocation is inhibited by treatment of platelets with farnesylcysteine analogues (Rosado & Sage, 2000a).
Figure 3. Overview of prenylation and methylation of Ras proteins.
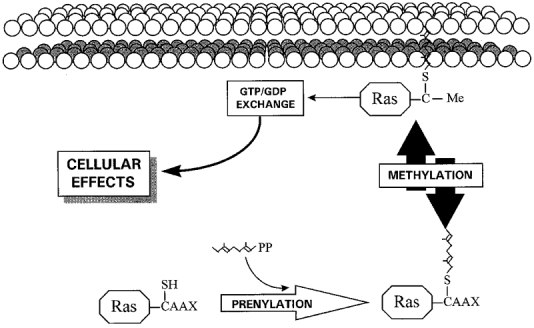
Modification of the C-terminus cysteine residue (designated ‘C’) by attachment of a farnesyl or geranylgeranyl group, followed by the removal of the three C-terminal amino acids and reversible methylation. ‘C-Me’ indicates that the carboxyl group has been methylated.
The small GTP-binding proteins play a central role in several signal transduction pathways. Several members of the Ras superfamily regulate the organisation of the actin cytoskeleton in different cell types. Rho mediates the assembly of actin filaments and focal adhesion plaques (Symons, 1996; Fox, 1999), Cdc42 leads to extension of filopodia (Ridley & Hall, 1994; Nobes & Hall, 1995; Olson et al. 1995) and Rac modulates the accumulation of actin filaments by the plasma membrane (Ridley & Hall, 1994; Nobes & Hall, 1995; Olson et al. 1995). Consistent with the above, the involvement of small GTP-binding proteins in SMCE has been shown to be partially mediated through the reorganisation of the actin cytoskeleton in human platelets (Rosado & Sage, 2000a).
Ras proteins also mediate vesicular transport through the reorganisation of the actin cytoskeleton. Rho D has recently been proposed to co-ordinate vesicular transport with the rearrangement of the actin filament network (Murphy et al. 1996) and Arf is a regulatory component in several pathways of intracellular vesicular trafficking (Moss & Vaughan, 1999). Inhibition of Arf using brefeldin A significantly reduced Ca2+ entry in human platelets, a mechanism that can be reversed by removal of the inhibitor (Rosado et al. 2000). These observations suggest a role for Arf, and thus vesicular transport, in the activation of SMCE in these cells.
The observations described above are in agreement with the existence of the secretion-like coupling model in human platelets, where Ras proteins would modulate both the rearrangement of actin filaments supporting the coupling and vesicular trafficking through the reorganisation of the actin cytoskeleton. Consistent with the inhibitory effect of actin disassembly after activation of SMCE in human platelets, inhibition of Ras proteins once Ca2+ entry was activated in these cells also impaired this mechanism, an effect that is likely to be mediated by disruption of the actin filament network supporting the coupling between the endoplasmic reticulum and the plasma membrane (Rosado et al. 2000). In contrast, inhibition of the vesicular transport once SMCE was activated has been reported to have a negligible effect, if any, on Ca2+ influx (Rosado et al. 2000). This finding agrees with the secretion-like coupling model since vesicular trafficking of the intracellular Ca2+ compartments towards the plasma membrane is only required for the activation of this mechanism but not for its maintenance.
Role of phosphoinositides in the activation of SMCE
At several levels, phosphoinositides play a key role in the reorganisation of the actin filament network in different cell types, including platelets (Ma & Abrams, 1999; Sun et al. 1999). The actions of specific phosphoinositide kinases, such as phosphoinositide 3-kinase (PI3-kinase) and phosphoinositide 4-kinase (PI4-kinase) and phosphatases, leading to the regulation of D3- and D4-containing phosphoinositides, have an important effect on the dynamic reorganisation of the actin cytoskeleton. For instance, phosphatidylinositol phosphates PtdIns-3-P, PtdIns-3,4-P2 and PtdIns-3,4,5-P3 can induce actin filament uncapping, thereby facilitating actin polymerisation (Hartwig et al. 1996). In addition, PtdIns-4-P and PtdIns-4,5-P2 have been shown to regulate cytoskeletal rearrangement through their association with actin-binding proteins (Lassing & Lindberg, 1985). There is also an important interaction between the phosphoinositide kinases and the small GTP-binding proteins. PI3- and PI4-kinases have been shown to be regulated upstream by small GTP-binding proteins, such as Rac, Cdc42 and Rho in platelets and other cells (Fox, 1996; Gasman et al. 1998). On the other hand, PI3-kinase can activate Rac proteins (Hawkins et al. 1995; Ma & Abrams, 1999).
Consistent with the widely described regulatory role of phosphoinositides in actin reorganisation, we have compelling evidence that PI3-kinase and PI4-kinase activity, and thus phosphoinositides, are required for SMCE in human platelets (Rosado & Sage, 2000b). This conclusion is supported by the fact that inhibition of PI3- and PI4-kinases by two different agents, LY294002 and wortmannin, reduced store depletion-induced Ca2+ entry in a concentration-dependent manner in these cells (Rosado & Sage, 2000b). LY294002 inhibited actin polymerisation over the same concentration range in which it caused a reduction in phosphatidylinositol phosphate levels and Ca2+ entry, suggesting that these mechanisms are closely related (Rosado & Sage, 2000b). Recently, Irvine's group has demonstrated that Ins(1,3,4,5)P4 is involved in the regulation of Ca2+ entry (Cullen, 1998). PtdIns(3,4,5)P3, a product of PI3- and PI4-kinases, and Ins(1,3,4,5)P4 share a common structural feature: chemically PtdIns(3,4,5)P3 has Ins(1,3,4,5)P4 as its inositol head group, with the distinction that the 1-phosphate is esterified. This could lead us to predict that PtdIns(3,4,5)P3 is an effector molecule in the activation of SMCE by PI3- and PI4-kinases by binding to the Ins(1,3,4,5)P4 receptor. However, esterification of the 1-phosphate resulted in a decrease in binding affinity to a putative Ins(1,3,4,5)P4 receptor, whilst increasing the binding affinity to a putative PtdIns(3,4,5)P3 receptor (Irvine & Cullen, 1996). Therefore, the role of PI3- and PI4-kinases in the regulation of SMCE is more likely to be explained by a role in the reorganisation of the actin cytoskeleton, providing a mechanical support for the coupling between the endoplasmic reticulum and the plasma membrane, as previously described in this review.
It is important to note that PI4-kinase activity is essential for the synthesis of PtdIns(4,5)P2 and subsequently Ins(3,4,5)P3, which is the immediate precursor of Ins(1,3,4,5)P4. Therefore, PI4-kinase might play a central role in receptor-mediated Ca2+ entry where agonist-receptor binding activates phospholipase C, which hydrolyses PtdIns(4,5)P2, so increasing Ins(3,4,5)P3 levels. This messenger then can be phosphorylated to yield Ins(1,3,4,5)P4 (Irvine, 1990).
Conclusion
It is possible to link all of these data into a model which may explain the mechanisms involved in SMCE in human platelets and other non-excitable cells. The tentative scheme we propose is shown in Fig. 4. As illustrated in Fig. 4, depletion of intracellular Ca2+ stores induces translocation and association of small GTP-binding proteins of the Ras superfamily with the plasma membrane, a process that is required for their activation as discussed above (Zhang & Casey, 1996). Several members of the Ras family could induce polymerisation and reorganisation of the actin filament network at two different sites: the cytoplasmic actin network and the membrane-associated cytoskeleton. Reorganisation of the cytoplasmic actin network can provide a support for cellular trafficking (Murphy et al. 1996). The small GTP-binding protein Arf is also involved in intracellular transport. On the other hand, reorganisation of the membrane-associated cytoskeleton can facilitate and support the coupling between the endoplasmic reticulum and the plasma membrane. The effect of Ras proteins in the reorganisation of the actin cytoskeleton might be supported and, to some extent, mediated by activation of PI3- and PI4-kinase, and thus the synthesis of phosphoinositides. It is likely that all these components are closely related and involved in a complex signalling cascade that leads to extracellular Ca2+ entry after depletion of the intracellular Ca2+ stores in human platelets and other non-excitable cells.
Figure 4. Proposed secretion-like coupling model for SMCE in human platelets.
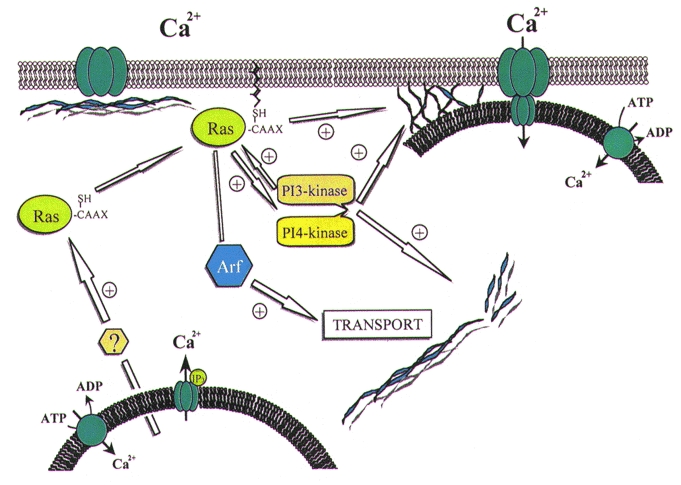
Depletion of the intracellular Ca2+ stores induces translocation and association of the small GTP-binding proteins of the Ras superfamily with the plasma membrane. Association of Ras proteins with the plasma membrane facilitates their interaction with GDP/GTP exchangers and, therefore, their activation. Ras proteins promote actin polymerisation leading the trafficking of the endoplasmic reticulum towards the plasma membrane. In addition, Arf, a member of the Ras small GTP-binding proteins, is involved in intracellular transport. Ras proteins induce reorganisation of the membrane actin filaments to support the coupling between elements in the endoplasmic reticulum (possibly the InsP3 receptors: ‘IP3’ in figure) and the Ca2+ release-activated channel (CRAC) in the plasma membrane through a protein-protein interaction. The effect of Ras proteins might be mediated to some extent by the activation of phosphoinositide 3′ and 4′ kinases, which in turn promote actin cytoskeleton rearrangement and activation of different Ras proteins.
Acknowledgments
J.A.R. is supported by a Grant of Junta de Extremadura-Consejería de Educación y Juventud and Fondo Social Europeo, Spain. We also acknowledge the support of the Wellcome Trust (051560).
References
- Álvarez J, Montero M, García-Sancho J. Cytochrome P450 may regulate plasma membrane Ca2+ permeability according to the filling state of the intracellular Ca2+ stores. FASEB Journal. 1992;6:786–792. doi: 10.1096/fasebj.6.2.1537469. [DOI] [PubMed] [Google Scholar]
- Ando S, Kaibuchi K, Sasaki T, Hiraoka K, Nishiyama T, Mizuno T, Asada M, Nunoi H, Matsuda I, Matsuura Y, Polakis P, McCormick F, Takai Y. Post-translational processing of rac p21s is important both for their interaction with the GDP/GTP exchange proteins and for their activation of NADPH oxidase. Journal of Biological Chemistry. 1992;267:25709–25713. [PubMed] [Google Scholar]
- Berridge MJ. Capacitative calcium entry. Biochemical Journal. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird GStJ, Putney JW., Jr Inhibition of thapsigargin-induced calcium entry by microinjected guanine nucleotide analogues. Evidence for the involvement of a small G-protein in capacitative calcium entry. Journal of Biological Chemistry. 1993;268:21486–21488. [PubMed] [Google Scholar]
- Brown BL, Walker SW, Tomlinson S. Calcium calmodulin and hormone secretion. Clinical Endocrinology. 1985;23:201–218. doi: 10.1111/j.1365-2265.1985.tb00216.x. [DOI] [PubMed] [Google Scholar]
- Cullen PJ. Bridging the GAP in inositol 1,3,4,5-tetrakisphosphate signalling. Biochimica et Biophysica Acta. 1998;1436:35–47. doi: 10.1016/s0005-2760(98)00149-0. [DOI] [PubMed] [Google Scholar]
- Fasolato C, Hoth M, Penner R. A GTP-dependent step in the activation mechanism of capacitative calcium influx. Journal of Biological Chemistry. 1993;268:5545–5551. [PubMed] [Google Scholar]
- Fox JEB. Platelet activation: new aspects. Haemostasis. 1996;26:102–131. doi: 10.1159/000217291. [DOI] [PubMed] [Google Scholar]
- Fox JEB. On the role of calpain and Rho proteins in regulating integrin-induced signaling. Thrombosis and Haemostasis. 1999;82:385–391. [PubMed] [Google Scholar]
- Fox JEB, Phillips DR. Inhibition of actin polymerization in blood platelets by cytochalasins. Nature. 1981;292:650–652. doi: 10.1038/292650a0. [DOI] [PubMed] [Google Scholar]
- Gailly Ph, Hermans E, Gillis JM. Role of [Ca2+]i in ‘Ca2+ stores depletion-Ca2+ entry coupling’ in fibroblasts expressing the rat neurotensin receptor. The Journal of Physiology. 1996;491:635–646. doi: 10.1113/jphysiol.1996.sp021245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasman S, Chasserot-Golaz S, Hubert P, Aunis D, Bader MF. Identification of a potential effector pathway for the trimeric Go protein associated with secretory granules. Go stimulates a granule-bound phosphatidylinositol 4-kinase by activating RhoA in chromaffin cells. Journal of Biological Chemistry. 1998;273:16913–16920. doi: 10.1074/jbc.273.27.16913. [DOI] [PubMed] [Google Scholar]
- Gregory RB, Barrit GJ. Store-activated Ca2+ inflow in Xenopus laevis oocytes: inhibition by primaquine and evaluation of the role of membrane fusion. Biochemical Journal. 1996;319:755–760. doi: 10.1042/bj3190755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi M, Favit A, Alkon DL. cAMP-induced cytoskeleton rearrangement increases calcium transient through the enhancement of capacitative calcium entry. Journal of Biological Chemistry. 1999;274:33557–33564. doi: 10.1074/jbc.274.47.33557. [DOI] [PubMed] [Google Scholar]
- Hancock JF, Cadwallader K, Paterson H, Marshall CJ. A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. EMBO Journal. 1991;10:641–646. doi: 10.1002/j.1460-2075.1991.tb04979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig JH. Mechanisms of actin rearrangements mediating platelet activation. Journal of Cellular Biology. 1992;118:1421–1442. doi: 10.1083/jcb.118.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig JH, Kung S, Kovacsovics T, Janmey PA, Cantley LC, Stossel TP, Toker A. D3 phosphoinositides and outside-in integrin signaling by glycoprotein IIb-IIIa mediate platelet actin assembly and filopodial extension induced by phorbol 12-myristate 13-acetate. Journal of Biological Chemistry. 1996;271:32986–32993. doi: 10.1074/jbc.271.51.32986. [DOI] [PubMed] [Google Scholar]
- Hawkins PT, Eguinoa A, Qiu RG, Stokoe D, Cooke FT, Walters R, Wennstrom S, Claesson-Welsh L, Evans T, Symons M, Stephens L. PDGF stimulates an increase in GTP-Rac via activation of phosphoinositide 3-kinase. Current Biology. 1995;5:393–403. doi: 10.1016/s0960-9822(95)00080-7. [DOI] [PubMed] [Google Scholar]
- Holda JR, Blatter LA. Capacitative calcium entry is inhibited in vascular endothelial cells by disruption of cytoskeletal microfilaments. FEBS Letters. 1997;403:191–196. doi: 10.1016/s0014-5793(97)00051-3. [DOI] [PubMed] [Google Scholar]
- Irvine RF. ‘Quantal’ Ca2+ release and the control of Ca2+ entry by inositol phosphates: a possible mechanism. FEBS Letters. 1990;263:5–9. doi: 10.1016/0014-5793(90)80692-c. [DOI] [PubMed] [Google Scholar]
- Irvine RF, Cullen PJ. Inositol phosphates – whither bound? Intracellular signalling. Current Biology. 1996;6:537–540. doi: 10.1016/s0960-9822(02)00536-5. [DOI] [PubMed] [Google Scholar]
- Jenner S, Farndale RW, Sage SO. The effect of calcium-store depletion and refilling with various bivalent cations on tyrosine phosphorylation and Mn2+ entry in fura-2-loaded human platelets. Biochemical Journal. 1994;303:337–339. doi: 10.1042/bj3030337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EG, Cowan WM. The nervous tissue. In: Weiss L, editor. The Cardiovascular System. Histology, Cell and Tissue Biology. 5. New York: Elsevier Science Publishing Co; 1983. pp. 282–370. [Google Scholar]
- Lassing I, Lindberg U. Specific interaction between phosphatidylinositol 4,5-bisphosphate and profilactin. Nature. 1985;314:472–474. doi: 10.1038/314472a0. [DOI] [PubMed] [Google Scholar]
- Ma AD, Abrams CS. Pleckstrin homology domains and phospholipid-induced cytoskeletal reorganisation. Thrombosis and Haemostasis. 1999;82:399–406. [PubMed] [Google Scholar]
- Matsui T, Maeda M, Doi Y, Yonemura S, Amano M, Kaibuchi K, Tsukita S. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin /moesin (ERM) proteins and regulates their head-to-tail association. Journal of Cellular Biology. 1998;140:647–657. doi: 10.1083/jcb.140.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means AR. Calcium, calmodulin and cell cycle regulation. FEBS Letters. 1994;347:1–4. doi: 10.1016/0014-5793(94)00492-7. [DOI] [PubMed] [Google Scholar]
- Moss J, Vaughan M. Activation of toxin ADP-ribosyltransferases by eukaryotic ADP-ribosylation factors. Molecular and Cellular Biochemistry. 1999;193:153–157. [PubMed] [Google Scholar]
- Muallem S, Kwiatkowska K, Xu X, Yin HL. Actin filament disassembly is a sufficient final trigger for exocytosis in nonexcitable cells. Journal of Cellular Biology. 1995;128:589–598. doi: 10.1083/jcb.128.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy C, Saffrich R, Grummt M, Gournier H, Rybin V, Rubino M, Auvinen P, Lutcke A, Parton RG, Zerial M. Endosome dynamics regulated by a Rho protein. Nature. 1996;384:427–432. doi: 10.1038/384427a0. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Olson MF, Ashworth A, Hall A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science. 1995;269:1270–1272. doi: 10.1126/science.7652575. [DOI] [PubMed] [Google Scholar]
- Pandol SJ, Schoeffield-Payne MS. Cyclic AMP mediates the agonist-stimulated increase in plasma membrane calcium entry in the pancreatic acinar cell. Journal of Biological Chemistry. 1990;265:12846–12853. [PubMed] [Google Scholar]
- Pareck AB, Penner R. Store depletion and calcium influx. Physiological Reviews. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Patterson RL, Van Rossum DB, Gill DL. Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell. 1999;98:487–499. doi: 10.1016/s0092-8674(00)81977-7. [DOI] [PubMed] [Google Scholar]
- Pedrosa-Ribeiro CM, Reece J, Putney JW., Jr Role of the cytoskeleton in calcium signaling in NIH 3T3 cells. An intact cytoskeleton is required for agonist-induced [Ca2+]i signaling, but not for capacitative calcium entry. Journal of Biological Chemistry. 1997;272:26555–26561. doi: 10.1074/jbc.272.42.26555. [DOI] [PubMed] [Google Scholar]
- Peppelenbosch MP, Tertoolen LGJ, De Vries-Smits AMM, Qiu R-G, M'Rabet L, Symons MH, De Laat SW, Bos JL. Rac-dependent and -independent pathways mediate growth factor-induced Ca2+ influx. Journal of Biological Chemistry. 1996;271:7883–7886. doi: 10.1074/jbc.271.14.7883. [DOI] [PubMed] [Google Scholar]
- Radriamampita C, Tsien RY. Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature. 1993;364:809–814. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]
- Reembold CM. Regulation of contraction and relaxation in arterial smooth muscle. Hypertension. 1992;20:129–137. doi: 10.1161/01.hyp.20.2.129. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Hall A. Signal transduction pathways regulating Rho-mediated stress fibre formation: requirement for a tyrosine kinase. EMBO Journal. 1994;13:2600–2610. doi: 10.1002/j.1460-2075.1994.tb06550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado JA, Jenner S, Sage SO. A role for the actin cytoskeleton in the initiation and maintenance of store-mediated calcium entry in human platelets. Journal of Biological Chemistry. 2000;275:7527–7533. doi: 10.1074/jbc.275.11.7527. [DOI] [PubMed] [Google Scholar]
- Rosado JA, Sage SO. Farnesylcysteine analogues inhibit store-regulated calcium entry in human platelets. Evidence for involvement of small GTP-binding proteins and actin cytoskeleton. Biochemical Journal. 2000a;347:183–192. [PMC free article] [PubMed] [Google Scholar]
- Rosado JA, Sage SO. Phosphoinositides are required for store-mediated calcium entry in human platelets. Journal of Biological Chemistry. 2000b;275:9110–9113. doi: 10.1074/jbc.275.13.9110. [DOI] [PubMed] [Google Scholar]
- Sage SO. Calcium entry mechanisms in human platelets. Experimental Physiology. 1997;82:807–823. doi: 10.1113/expphysiol.1997.sp004066. [DOI] [PubMed] [Google Scholar]
- Sargeant P, Farndale RW, Sage SO. ADP-and thapsigargin-evoked Ca2+ entry and protein tyrosine phosphorylation are inhibited by the tyrosine kinase inhibitors genistein and methyl-2,5-dihydroxycinnamate in fura-2-loaded human platelets. Journal of Biological Chemistry. 1993a;268:18151–18156. [PubMed] [Google Scholar]
- Sargeant P, Farndale RW, Sage SO. The tyrosine kinase inhibitor methyl 2,5-dihydroxycinnamate and genistein reduce thrombin-evoked tyrosine phosphorylation and Ca2+ entry in human platelets. FEBS Letters. 1993b;315:242–246. doi: 10.1016/0014-5793(93)81172-v. [DOI] [PubMed] [Google Scholar]
- Sargeant P, Farndale RW, Sage SO. The imidazole antimycotics econazole and miconazole reduce agonist-evoked protein tyrosine phosphorylation and evoke membrane depolarisation in human platelets: cautions for their use in studying Ca2+ signalling pathways. Cell Calcium. 1994;16:413–418. doi: 10.1016/0143-4160(94)90034-5. [DOI] [PubMed] [Google Scholar]
- Simonescu N, Simonescu M. Ultrastructure of the microvascular wall: functional correlations. In: Weiss L, editor. The Cardiovascular System. Histology, Cell and Tissue Biology. 5. New York: Elsevier Science Publishing Co; 1983. pp. 371–433. [Google Scholar]
- Sinensky M, Lutz RJ. The prenylation of proteins. Bioesssays. 1992;14:25–31. doi: 10.1002/bies.950140106. [DOI] [PubMed] [Google Scholar]
- Somasundaram BJ, Norman C, Mahaut-Smith MP. Primaquine, an inhibitor of vesicular transport, blocks the calcium-release-activated current in rat megakaryocytes. Biochemical Journal. 1995;309:725–729. doi: 10.1042/bj3090725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HQ, Yamamoto M, Mejillano M, Yin HL. Gelsolin, a multifunctional actin regulatory protein. Journal of Biological Chemistry. 1999;274:33179–33182. doi: 10.1074/jbc.274.47.33179. [DOI] [PubMed] [Google Scholar]
- Symons M. Rho family GTPases: the cytoskeleton and beyond. Trends in Biochemical Sciences. 1996;21:178–181. [PubMed] [Google Scholar]
- Tsien RW, Lipscombe D, Madison D, Bley K, Fox A. Reflections on Ca2+-channel diversity, 1988–1994. Trends in Neurosciences. 1995;18:52–54. [PubMed] [Google Scholar]
- Vaca L. Calmodulin inhibits calcium influx current in vascular endothelium. FEBS Letters. 1996;390:289–293. doi: 10.1016/0014-5793(96)00675-8. [DOI] [PubMed] [Google Scholar]
- Xu X, Gilbert BA, Rando RR, Chen L, Tashjian AH., Jr Inhibition of capacitative Ca2+ entry into cells by farnesylcysteine analogs. Molecular Pharmacology. 1996;50:1495–1501. [PubMed] [Google Scholar]
- Yao Y, Ferrer-Montiel AV, Montal M, Tsien RY. Activation of store-operated Ca2+ current in Xenopus oocytes requires SNAP-25 but not a diffusible messenger. Cell. 1999;98:475–485. doi: 10.1016/s0092-8674(00)81976-5. [DOI] [PubMed] [Google Scholar]
- Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annual Review of Biochemistry. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]