

Abstract
Kir2.1 channels are blocked by Rb+ and Cs+ in a voltage-dependent manner, characteristic of many inward rectifier K+ channels. Mutation of Ser165 in the transmembrane domain M2 to Leu (S165L) abolished Rb+ blockage and lowered Cs+ blocking affinity. At negative voltages Rb+ carried large inward currents.
A model of the Kir2.1 channel, built by homology with the structure of the Streptomyces lividans K+ channel KcsA, suggested the existence of an intersubunit hydrogen bond between Ser165 and Thr141 in the channel pore-forming P-region that helps stabilise the structure of this region. However, mutations of Thr141 and Ser165 did not produce effects consistent with a hydrogen bond between these residues being essential for blockage.
An alternative alignment between the M2 regions of Kir2.1 and KcsA suggested that Ser165 is itself a pore-lining residue, more directly affecting blockage. We were able to replace Ser165 with a variety of polar and non-polar residues, consistent with this residue being pore lining. Some of these changes affected channel blockage.
We tested the hypothesis that Asp172 – a residue implicated in channel gating by polyamines – formed an additional selectivity filter by using the triple mutant T141A/S165L/D172N. Large Rb+ and Cs+ currents were measured in this mutant.
We propose that both Thr141 and Ser165 are likely to provide binding sites for monovalent blocking cations in wild-type channels. These residues lie beyond the carbonyl oxygen tunnel thought to form the channel selectivity filter, which the blocking cations must therefore traverse.
Inward rectifier potassium (Kir) channels are crucial for setting resting membrane potentials and controlling excitability in many mammalian cell types (Nichols & Lopatin, 1997; Reimann & Ashcroft, 1999). Kir channels do this by allowing potassium to flow at membrane potentials around the equilibrium potential for K+ (EK). However, these channels are gated by intracellular Mg2+ (Matsuda et al. 1987) and polyamines (Ficker et al. 1994; Lopatin et al. 1994), and close under sufficient depolarisation. Gating by Mg2+ and polyamines also results in the channels being opened by an increase in extracellular [K+] (Hagiwara & Yoshii, 1979; Leech & Stanfield, 1981; Lopatin & Nichols, 1996).
Functional Kir channels are believed to be formed as homo- or heterotetramers (Yang et al. 1995), each channel subunit possessing two membrane-spanning regions (M1 and M2) with a pore-forming loop between them (the P- or H5 region; e.g. Kubo et al. 1993).
Inward rectifier K+ channels show characteristically low permeance to Rb+ (Adrian, 1964; Standen & Stanfield, 1980). They are blocked in a voltage-dependent way by both Rb+ (Standen & Stanfield, 1980) and Cs+ (Hagiwara et al. 1976; Gay & Stanfield, 1977). This blockage is sufficiently characteristic to have been used to help identify newly cloned inward rectifier K+ channels after expression (e.g. Kubo et al. 1993). However, while blockage is found in most, it is not found in all members of the family. Kir4.1, for example, has a lower affinity for blockage by Cs+ than does Kir2.0 (Bond et al. 1994). One difference between the two subfamilies is that a serine residue in M2 of Kir2.0 is replaced by leucine in Kir4.0. We have here examined in Kir2.1 the effects on blockage by Rb+ and Cs+ of replacing this serine (Ser165) by leucine (S165L). We show that this mutation reduces the affinity for blockage by Cs+ and abolishes Rb+ blockage.
The crystal structure of KcsA, the K+ channel of Streptomyces lividans (Doyle et al. 1998), suggests that the selectivity filter is composed solely of carbonyl oxygens in the P-region. Since this structure became available, we have modified an earlier model of Kir2.1 (Dart et al. 1998) using the structure of KcsA as a template (Leyland et al. 1999). Such modelling led us to consider whether the role of Ser165 is to help stabilise the channel selectivity filter, formed by the P-region, in which blockage might be expected to occur. Our experiments lead us to conclude that, while a residue in the P-region (Thr141) is also involved in channel blockage, the effect of Ser165 does not result from an interaction with this residue. Our results also confirm that the residue Asp172, closely involved in channel gating (Stanfield et al. 1994) by polyamines (Fakler et al. 1994), also forms part of an internal selectivity filter (Reuveny et al. 1996; Abrams et al. 1996). However, the impact of Asp172 on the permeability of extracellular cations is substantial only when Ser165 has also been replaced by Leu. Nonetheless, all these residues affecting Rb+ and Cs+ blockage lie beyond the primary selectivity filter, formed by the backbone carbonyl oxygens of the peptide bonds between residues in the signature sequence TxGYG (Doyle et al. 1998). The blocking cations must traverse this selectivity filter to sites regulating their blockage or permeance.
Some of this work has been published in abstract form (Thompson et al. 1999).
METHODS
Chinese hamster ovary cells (CHO-K1/SF, European Collection of Animal Cell Cultures) were cultured in minimum essential medium alpha (Gibco) supplemented with 10 % (w/v) fetal calf serum. Site-directed mutagenesis of murine Kir2.1 was performed using the QuikChange mutagenesis kit (Stratagene). Mutations were verified by sequencing the entire Kir2.1 coding region. Wild-type or mutant Kir2.1 cDNA was subcloned into the vector pCDNA3 and co-transfected into CHO cells with a plasmid containing the cDNA for enhanced green fluorescent protein (EGFP) as a transfection marker, using FuGENE 6 transfection reagent (Roche). Successfully transfected cells were identified 24–48 h later under fluorescence microscopy.
Whole-cell currents were recorded using an Axopatch 200A amplifier (Axon Instruments). Currents were filtered at 5 kHz (-3 dB, 4-pole Bessel), digitised at 10 kHz using a Digidata 1200 interface (Axon Instruments), and analysed on a Pentium PC using software written by Dr N. W. Davies (Department of Cell Physiology and Pharmacology, University of Leicester). The pipette (intracellular) solution contained (mM): Hepes, 10; EDTA, 10; and K+ (as KCl and KOH), 140; pH adjusted to 7.2 with KOH. External solutions contained (mM): Hepes, 10; KCl (or RbCl), 70; N-methyl-D-glucamine (NMDG), 70; CaCl2, 2; and MgCl2, 2; pH adjusted to 7.2 with HCl. Where Cs+ concentrations greater than 300 μm were required, NMDG chloride was replaced with CsCl to preserve osmolarity. RbCl was also substituted for some of the NMDG chloride when solutions were required to contain 70 mm K+ as well as Rb+. The calculated junction potential between the pipette (140 mm K+) and bath (70 mm K+) solutions used with all cells during sealing was 4.1 mV (pipette negative; using the program JPCalc, P. H. Barry, University of New South Wales, Australia). No correction was applied for this junction potential.
Experiments were carried out at room temperature (18-22°C). A multiline gravity-driven perfusion system was used to apply different solutions to individual cells under whole-cell clamp. To compare current-voltage relationships from different cells, currents for each cell were normalised to that obtained at -132 mV in 70 mm[K+]o. Results are given as means ±s.e.m. Where appropriate, results were compared using Student's t test. Significance was assumed at P < 0.05.
Structural models of Kir2.1 were constructed by comparative modelling (MODELLER; Sali & Blundell, 1993) using the crystal structure of KcsA (Doyle et al. 1998; Protein Data Bank accession code 1BL8) as a template. Two sequence alignments were used for M2 (Fig. 3C). One was based on that given by Doyle et al. (1998) for the alignment of ROMK1 and KcsA. The alternative alignment was based on mutational analysis of Kir2.1 by Minor et al. (1999) and on our own alignment of KcsA and Kir2.1 using ClustalW (Thompson et al. 1994), which gives the same result as that proposed by Minor et al. (1999). This alignment places a given residue in Kir2.1 three positions (i.e. almost one complete turn of the helix) nearer to the C-terminus of M2 when compared with the alignment based on that of Doyle et al. (1998). Restraints were applied within MODELLER to represent: 4-fold symmetry; a salt bridge between Glu138 and Arg148 in adjacent subunits (Yang et al. 1997); and, for the model based on the alignment of Minor et al. (1999), but not for that based on the alignment of Doyle et al. (1998), interactions between residues within Kir2.1 identified by mutagenesis (Minor et al. 1999).
Figure 3. Schematic representation of the molecular models of Kir2.1 illustrating the positions of Thr141, Ser165 and Asp172.

A, model based on the sequence alignment of Doyle et al. (1998), with the putative intersubunit hydrogen bond between the side chains of Thr141 and Ser165 shown in orange. The position of Asp172 is also shown for one subunit. The backbone carbonyl oxygen atoms, believed to confer K+ selectivity (Doyle et al. 1998), are shown in pink. The peptide bonds between the residues GYG (Gly, Tyr, Gly) in the K+ channel signature sequence contribute the uppermost two rings of carbonyl oxygens. B, model based on the sequence alignment of Minor et al. (1999), showing how Ser165 has moved away from Thr141 by almost one turn (3 residues) of the M2 helix and cannot form the intersubunit hydrogen bond suggested by A. In both A and B, the front subunit has been cut away for clarity. The peptide backbones of the remaining three subunits are shown in red, yellow and cyan. Also for clarity, note that B has been rotated about the vertical axis with respect to A. C, alignment of the M2 region of KcsA with that of Kir2.1, following Doyle et al. (1998; above, residues 155–181) and Minor et al. (1999; below, residues 152–178). The single letter amino acid code is used; S165 is shown in bold.
RESULTS
Mutation S165L alters block by Cs+ and Rb+
As we have previously demonstrated (Abrams et al. 1996), Rb+ and Cs+ block Kir2.1 channels in a steeply voltage-dependent manner similar to that reported in native tissues (Hagiwara et al. 1976; Gay & Stanfield, 1977; Standen & Stanfield, 1980). Blockage by Rb+ increases with hyperpolarisation, but at sufficiently negative voltages, Rb+ passes through the channel to the intracellular solution (Fig. 1A and C). The permeability ratio (PRb/PK) was calculated from the shift in reversal potential (ΔVrev) when Rb+ is substituted for external K+ (Hille, 1992); that is, from
| (1) |
where R, T and F have their usual thermodynamic meanings. The reversal potential shifted by -14.7 ± 1.1 mV (n= 10); thus PRb/PK= 0.57 ± 0.03.
Figure 1. Rb+ does not block Kir2.1 S165L channels.
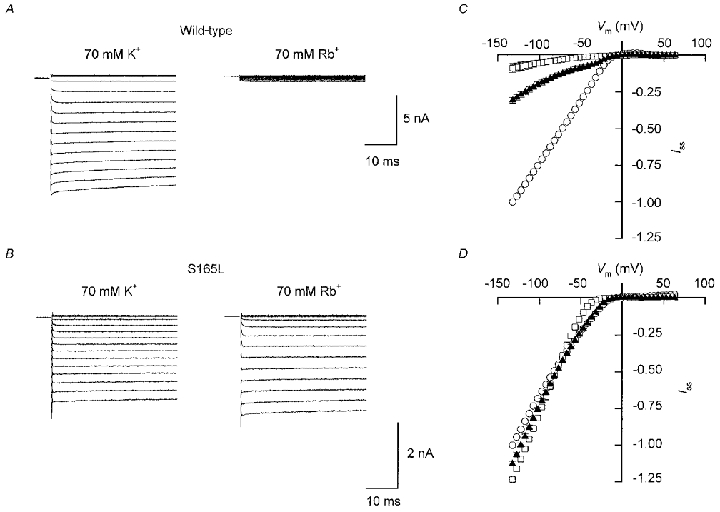
A and B, K+ and Rb+ currents activated by stepping from -17 mV (EK) to a range of potentials between +58 and -132 mV in 70 mm K+- or 70 mm Rb+-containing solution for Kir2.1 wild-type (A) and S165L (B). C and D, normalised steady-state current (Iss)-voltage relationships for A and B, respectively (○, 70 mm K+; ▴, 70 mm K+ and 10 mm Rb+; □, 70 mm Rb+).
Blockage by Cs+ develops exponentially over a few milliseconds and simply increases as the membrane potential is more hyperpolarised (Fig. 2A and C).
Figure 2. Cs+ blockage of Kir2.1 wild-type and S165L.

A and B, K+ currents activated by stepping from -17 mV to a range of potentials between +58 and -132 mV for Kir2.1 wild-type (A) and S165L (B). C and D, normalised steady-state current-voltage relationships for wild-type (C) and S165L (D) (○, control; •, 30 μm; □, 100 μm; ▪, 300 μm; ▵, 1 mm; and ▴, 10 mm Cs+). E, steady-state current, obtained in the presence of Cs+, plotted as a fraction of that obtained under control conditions against Cs+ concentration for Kir2.1 wild-type (filled symbols) and S165L (open symbols). The results were fitted to a Hill-Langmuir equation with a Hill coefficient of 1 at each voltage. The voltages are -97 mV (circles), -107 mV (squares), -117 mV (triangles) and -127 mV (inverted triangles). F, voltage dependence of KD(V) for wild-type (•) and S165L (○). Lines are fits to eqn (2). The values obtained for KD(0) and δ′ are summarised in Table 1.
The mutation S165L radically altered channel blockage by both Rb+ (Fig. 1B and D) and Cs+ (Fig. 2B and D, Table 1). First, the ability of Rb+ to block the channel was removed. When Rb+ was added at 10 mm to the 70 mm K+ extracellular solution, there was an increase, rather than a reduction in current amplitude, suggesting that Rb+ had become a permeant ion. To confirm this suggestion, experiments were performed in which extracellular K+ was replaced with 70 mm Rb+. Here, the current carried by Rb+ (IRb) was larger than that carried by K+ (IK) over a range of hyperpolarising potentials. While for the wild-type channel IRb/IK= 0.082 ± 0.03 (n= 11) at -132 mV, for the mutant S165L IRb/IK was 1.23 ± 0.01 (n= 7), a 15-fold increase. Though Rb+ now moved more rapidly through the mutant channel at negative voltages than did K+, the change in reversal potential found when external K+ was replaced by Rb+ was unaltered. ΔVrev was -15.5 ± 0.8 mV (n= 17), giving PRb/PK= 0.55 ± 0.02 (eqn (1)).
Table 1.
Cs+ and Rb+ blockage in wild-type and in mutant channels
| Channel mutant | IRb/IK(−132 mV) | KD(−97 mV)Cs+ | KD(0 mV)Cs+ | δ′ |
|---|---|---|---|---|
| Kir2.1 wild-type | 0.082 ± 0.03(11) | 100.6 ± 7 μm(4) | 52 mm | 1.57 |
| T141A | 0.36 ± 0.03(6)*** | 4.82 ± 0.7 mm(5)** | 213 mm | 0.97 |
| T141V | 0.23 ± 0.02(6)** | 2.91 ± 0.6 mm(10)* | 176 mm | 1.02 |
| T141S | 0.11 ± 0.02(3) | 72·8 ± 13 μm(3) | 51 mm | 1.70 |
| S165A | 0.061 ± 0.004(5) | 26.1 ± 4 μm(4)*** | 16 mm | 1.63 |
| S165V | 0.12 ± 0.006(4) | 125 ± 37 μm(4) | 64 mm | 1.64 |
| S165F | 0.081 ± 0.005(4) | 15.3 ± 0.4 μm(5)*** | 7 mm | 1.73 |
| S165L | 1.23 ± 0.01(7)*** | 18.3 ± 4 mm(4)* | 2.5 M | 1.19 |
| S165I | 0.11 ± 0.01(5) | 102 ± 9 μm(5) | 162 mm | 1.95 |
| S165T | 0.098 ± 0.002(6) | 107 ± 25 μm(4) | 36 mm | 1.54 |
| S165N | 0.12 ± 0.006(3) | 49.6 ± 4 μm(3)* | 20 mm | 1.54 |
| T141A/S165L | 7.84 ± 0.7(10)*** | Cs+ permeable | n.d. | n.d. |
| T141S/S165T | 0.148 ± 0.01(5) | 144 ± 19 μm(4) | 28 mm | 1.36 |
| D172N | 0.10 ± 0.04(5) | 93 ± 8 μm(3) | 19 mm | 1.32 |
| S165L/D172N | 2.10 ± 0.08(4)*** | 25.1 ± 4 mm(4)** | 763 mm | 0.90 |
| T141A/D172N | 0.43 ± 0.03(5)*** | 4.8 ± 1 mm(5)* | 285 mm | 1.05 |
| T141A/S165L/D172N | 19.4 ± 3(4)*** | Cs+ permeable | n.d. | n.d. |
KD(0 mV) and δ′ calculated by fitting KD(V) to membrane potential using eqn (2). n.d., not determined in channels where Cs+ fails to block. Numbers in parentheses are number of cells
P < 0·0001
P < 0·001
P < 0·01: other values not significantly different from wild-type
In addition, channels formed from S165L mutant subunits had a much reduced affinity for Cs+ as a blocking cation. At -97 mV, this reduction in affinity was some 180-fold: KD(-97 mV) was 18.3 ± 3.5 mm (n= 4) in mutant channels and 101 ± 7.1 μm (n= 4) in wild-type (Fig. 2E, Table 1). The voltage dependence of the KD values obtained for each mutant was calculated using:
| (2) |
where KD(V) is the voltage-dependent equilibrium dissociation constant, whose value is KD(0) at 0 mV. δ′ gives the fraction of the electrical field through which Cs+ travels to its blocking site. The high value for δ′ (1.57 in wild-type; Table 1) results from the K+ channel being a multi-ion pore, with voltage moving permeant ions as well as the blocking ion to bring Cs+ to its site of action (Hille & Schwarz, 1978; Abrams et al. 1996; see also Stampe et al. 1998). S165L gave δ′= 1.19 (Fig. 2F, Table 1), indicating that Cs+ may now block less deeply within the channel. In addition, the altered kinetics results in blockage by Cs+ appearing to be instantaneous, whereas it took some time to reach completion in wild-type Kir2.1.
Does S165 hydrogen bond with T141?
Figure 3 shows models of Kir2.1 topology built using the KcsA structure as a template (see Methods). One model (Fig. 3A) suggests that Ser165 might influence channel blockage indirectly by helping to stabilise the lower part of the selectivity filter, this stabilising effect arising through a hydrogen bond between Ser165 and Thr141 of adjacent subunits. The mutation S165L would remove this putative hydrogen bond.
To test this hypothesis, a number of mutants were made. First, the residues at positions 141 and 165 were exchanged (T141S/S165T) to conserve any hydrogen bond within an altered structure. Then hydrophobic residues were substituted for Thr141 and Ser165 to disrupt this putative hydrogen bond. The double mutant T141S/S165T showed blockage by Rb+ and Cs+ that was entirely comparable to that seen in wild-type (Table 1). Similarly, replacement of Thr141 by Ser (T141S) or Ser165 by Thr (S165T) had little or no effect on blockage by either cation (Table 1).
However, replacement of Thr141 with alanine (T141A) gave channels with reduced Rb+ and Cs+ blockage (Fig. 4, Table 1), though the results did not fully emulate those obtained with the mutant S165L. Thus, while Rb+ still blocked when added at 10 mm to the 70 mm K+ extracellular solution, blocking affinity was reduced. To illustrate this reduction in blockage, we have plotted the current obtained in the presence of Rb+ as a fraction of that in the absence of the blocking cation (Fig. 4B). In addition, in the mutant T141A, Rb+ currents were increased in size when Rb+ replaced K+ as the permeant cation. However, these currents were still smaller than those carried by K+ (at -132 mV, IRb/IK= 0.36 ± 0.03; n= 6).
Figure 4. Cs+ and Rb+ blockage in mutants of Thr141 and Ser165.

A, families of currents, recorded in the presence of 70 mm Rb+, activated by stepping from -17 mV to a range of voltages between +58 and -132 mV. The dashed line indicates the current level obtained at -132 mV in 70 mm K+. B, the current in the presence of 10 mm Rb+, expressed as a fraction of that found in control, 70 mm K+ solution, for wild-type (▪), S165A (▿) and T141A (•). C, currents activated by voltage steps from -17 mV to -102, -112, -122 and -132 mV in the presence of external, 70 mm K+ solution containing 300 μm Cs+. The dashed line indicates the current recorded for each mutation at -132 mV in control solution. D, steady-state current at -97 mV, obtained in the presence of Cs+, plotted as a fraction of that obtained under control conditions against Cs+ concentration for Kir2.1 wild-type (▪) and S165A (▿) and T141A (•). The results were fitted to a Hill-Langmuir equation with a Hill coefficient of 1. The KD values obtained are summarised in Table 1.
The affinity for Cs+ was substantially reduced, with KD(-97 mV) = 4.82 ± 0.7 mm, δ′= 0.97 (n= 5; see Fig. 4D and Table 1). A similar result was obtained with a second non-polar substitution for Thr141; in the mutant T141V, Rb+ currents were increased in amplitude (at -132 mV, IRb/IK= 0.23 ± 0.02; n= 6) and Cs+ blocking affinity was reduced (KD(-97 mV) = 2.91 ± 0.6 mm; n= 10).
While non-polar substitutions for Thr141 reduced channel blockage, replacement of Ser165 with Ala (S165A) was not consistent with the hydrogen bond hypothesis (Fig. 4, Table 1). Channels formed from S165A subunits were blocked by Rb+ much as wild-type (at -132 mV, IRb/IK= 0.061 ± 0.004; n= 5). The affinity for Cs+ as a blocking cation was somewhat higher than that of wild-type channels (KD(-97 mV) = 26.1 ± 4.3 μm, δ′= 1.63; n= 4). Thus a hydrogen bond between Ser165 and Thr141 cannot be essential for channel blockage.
Other substitutions for Ser165 and channel blockage
If specific side chain-side chain interactions are involved in permitting Ser165 to produce channel blockage by Rb+ and Cs+, such blockage would be affected by most substitutions. In addition, in KATP channels, formed by Kir6.0 subunits, where both Rb+ and Cs+ block (Spruce et al. 1987; Quayle et al. 1988), the residue equivalent to Ser165 is Asn. Accordingly, we investigated the effects of replacement of Ser165 with each of a number of other polar and non-polar residues. The substitutions tested were Asn (S165N), Thr (S165T), Val (S165V), Ile (S165I) and Phe (S165F), the last three substitutions being by hydrophobic residues given in order of increasing size of side chain. Rb+ and Cs+ blocked all of the channels formed from these mutant subunits. In each case, Rb+ currents were a small fraction of the K+ currents at negative voltages, and all showed high-affinity blockage by Cs+ over the voltages examined (KD(-97 mV) ranging from 15.3 ± 0.4 μm for S165F to 125 ± 37 μm for S165V; Table 1). Cs+ affinity was significantly increased for S165F and S165N. Thus all substitutions for Ser165 that we have attempted result in channels that retain function, consistent with the residue being pore lining (Minor et al. 1999). Some of these substitutions raised the affinity for Cs+ while the mutation S165L reduced the affinity from that found in wild-type. Only in S165L did Rb+ carry a substantial current.
Mutation of both T141 and S165 permits Cs+ to permeate
The failure of the hypothesis that Ser165 exerts its effects through a hydrogen bond with Thr141 suggests that these two residues each contribute a site at which blockage by Rb+ and Cs+ can occur. We therefore constructed a double mutant in which Thr141 was replaced by Ala, while Ser165 was replaced by Leu. This double mutant (T141A/S165L; Fig. 5) had substantially increased Rb+ currents compared with those in S165L. These currents were 7.84 (± 0.69)-fold (n= 10) larger than K+ currents at -132 mV. Further, Cs+ was now able to permeate the channel at voltages negative to -90 mV (Fig. 5B). Cs+ currents increased in size over a few milliseconds at the start of the hyperpolarising pulse, consistent with channel open probability (Popen) increasing, perhaps because of the release of gating polyamines. At -132 mV, Cs+ currents were nearly half as large as K+ currents (ICs/IK= 0.47 ± 0.04; n= 5).
Figure 5. Rb+ and Cs+ permeance of Kir2.1 T141A/S165L and T141A/S165L/D172N.

Does Asp172 act as an internal selectivity filter?
Both Reuveny et al. (1996) and Abrams et al. (1996) argued that Asp172, a pore-lining residue (Minor et al. 1999) lying deep in the K+ channel, contributed an internal selectivity filter. In this study, we asked whether this residue contributes significantly to channel blockage. We found that in the absence of other mutations, replacement of Asp172 by Asn had little effect on channel blockage by either Rb+ or Cs+ (Table 1). Accordingly, we constructed double and triple channel mutants, coupling the mutation D172N with T141A (T141A/D172N), S165L (S165L/D172N), or both mutations (T141A/S165L/D172N). Results from the double mutant T141A/D172N showed little change from T141A alone (Table 1). On the other hand the double mutant S165L/D172N (Table 1) showed reduced blockage by both Rb+ and Cs+ over the single mutant S165L, consistent both with Asp172 contributing to channel selectivity and with blockage at S165 occurring at a site separate from that provided by Thr141. Thus the removal of the negative charge at Asp172 increases Rb+ current and reduces Cs+ blockage, but does so substantially only in the presence of the mutant S165L (see Abrams et al. 1996).
The triple mutant T141A/S165L/D172N resulted in Rb+ currents being increased to approximately 20-fold those generated in K+ at -132 mV (Fig. 5A, Table 1). And this triple mutant allowed comparable amounts of Cs+ and K+ to pass at negative voltages, with ICs/IK= 1.09 ± 0.10 (n= 4) at -132 mV. Examination of the effect of increasing the proportion of Cs+ in the external solution showed that the reversal potential shifted as predicted assuming independence (Fig. 5C and D). These shifts in reversal potential were consistent with PCs/PK= 0.049. However, the magnitude of the current recorded at -132 mV did not follow independence, but showed an anomalous mole fraction effect, as expected for a mixture of permeant ions in a multi-ion pore (Hille & Schwarz, 1978; Stampe et al. 1998). The current was found to reach a minimum when the external solution contained approximately 52.5 mm Cs+ and 17.5 mm K+. A low concentration of K+ reduced the transfer of both K+ and Cs+. Thus the currents recorded at negative voltages in 70 mm Cs+ cannot be an artefact, for example associated with membrane breakdown at such voltages: Cs+ is a permeant cation in this mutant channel.
DISCUSSION
Our experiments identify three residues that contribute to channel blockage by Rb+ and Cs+. In the absence of other changes, Ser165 in M2 is the major residue, since its replacement by Leu (S165L) so radically alters blockage by both cations. Thr141 in the H5 or P-region is also involved. And in the presence of the mutation S165L, Asp172 – a residue of major importance in channel gating (Stanfield et al. 1994) by polyamines (Fakler et al. 1994) – also strongly affects ionic block and permeance. All three residues lie beyond the region lined by the backbone carbonyl oxygens of the peptide bonds between residues of the signature sequence TxGYG (Fig. 3) and, surprisingly, blocking cations appear to traverse this region to reach their blocking site(s).
We have considered two hypotheses for the role of Ser165. The first is that the residue exerts its effect through an interaction with Thr141 in the P-region, contributing to the stabilisation of the lowest part of this selectivity filter. This hypothesis followed our building of a homology model based on KcsA, using the alignment of M2 given by Doyle et al. (1998). The model suggested the existence of an intersubunit hydrogen bond between Ser165 and Thr141.
The apparent dependence of high-affinity Cs+ blockage on the presence of polar residues at position 141 and the lack of effect of the double mutant T141S/S165T supported the hydrogen bond hypothesis. However, several mutants of Ser165 (including S165A), designed to disrupt the putative hydrogen bond, failed to reduce Cs+ and Rb+ blockage. Thus a hydrogen bond between S165 and T141 cannot be a critical determinant of high-affinity block by Cs+ and Rb+ in Kir2.1.
The second hypothesis is that Ser165 is pore lining, as proposed by Minor et al. (1999). In this case Ser165 and Thr141 might contribute to two sites affecting blockage. This hypothesis is supported by the results obtained using the double mutant T141A/S165L. This double mutation had a greater effect than did either mutation alone, indicating that these mutations may influence ion conduction through independent processes, in turn implying that Thr141 and Ser165 either are binding sites themselves for Cs+ and Rb+or provide support for such sites.
We now consider whether Thr141 influences channel blockage through a specific side chain-side chain interaction, holding the selectivity filter at the appropriate diameter (see Doyle et al. 1998) or whether Thr141 is pore lining. The equivalent residue in KcsA (Thr74) is not pore lining (Doyle et al. 1998). However, scanning cysteine accessibility mutagenesis studies in Kir2.1 have suggested that Thr141 does have its side chain lining the pore. Thus T141C is easily accessible from the outside to Ag+, which blocks (Dart et al. 1998), or to Cd2+, which increases current (Kubo et al. 1998). Ag+ does not block Shaker channels when the equivalent residue is replaced by Cys (Lü & Miller, 1995). Thus, Kubo et al. (1998) have argued for significant structural differences between Kir2.1 channels on the one hand and voltage-gated potassium (Kv) channels and KcsA on the other. In the case of Thr141, it has been shown that the corresponding residue in ROMK2, Val121, can be replaced with Thr, producing a channel with an increased affinity for Ba2+ and Cs+ (Zhou et al. 1996). These authors propose that the increase in affinity is due to the hydroxyl of the Thr residue providing a dipole moment that helps co-ordinate cations within the pore. Our results are consistent with this possibility, since channels formed from either T141A or T141V have reduced Cs+ and Rb+ affinity, while T141S, in which the hydroxyl group is preserved, retains a high affinity for the blocking cations.
Though there are subtle effects of replacement of Asp172 by other residues (Abrams et al. 1996), mutation of this site produces substantial effects only when Ser165 has also been replaced by Leu. This suggests that Asp172 sees little of externally applied Cs+ or Rb+, unless the outer blocking sites are removed, as in S165L. This suggestion seems logical as Asp172 has already been shown to form part of an internal selectivity filter, discriminating between K+ and other cations applied to the cytoplasmic face of the membrane (Reuveny et al. 1996).
In summary, residues in the P-region (Thr141) and beyond (Ser165) are crucial for ionic blockage by Rb+ and Cs+. In two models built by homology with KcsA (Fig. 3) and in a previous model arising from scanning cysteine accessibility mutagenesis (Dart et al. 1998), both these residues (T141 and S165) lie deeper than the consensus GYG motif that forms a major part of the selectivity filter (Heginbotham et al. 1992; Doyle et al. 1998). Appropriate replacement of Ser165 (S165L) suggests this residue to be of major importance in determining blockage and permeance. None of the mutants we have examined altered the permeability ratio PRb/PK. It seems unlikely that the major part of the selectivity filter (i.e. around the GYG motif) is altered in the mutants we have studied. In the double mutant T141A/S165L, Cs+ carries measurable current through the ion channel. Rb+ and Cs+ currents are enhanced in size if Asp172 is also replaced by Asn. The ratio of permeability remains PK > PRb > PCs. However, both Rb+ and Cs+ must traverse the major part of the channel selectivity filter to reach their blocking site(s) at and just beyond the tip of the P-region. Thus, contrary to our expectations from the crystal structure of KcsA (Doyle et al. 1998), residues beyond the P-region contribute to permeation and blockage by extracellular cations. This finding may have implications for other K+ channels.
Acknowledgments
We thank the Wellcome Trust for support.
References
- Abrams CJ, Davies NW, Shelton PA, Stanfield PR. The role of a single aspartate residue in ionic selectivity and block of a murine inward rectifier K+ channel Kir2.1. The Journal of Physiology. 1996;493:643–649. doi: 10.1113/jphysiol.1996.sp021411. Erratum: The Journal of Physiology 521, 761 (1999) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrian RH. The rubidium and potassium permeability of frog muscle membrane. The Journal of Physiology. 1964;175:134–159. doi: 10.1113/jphysiol.1964.sp007508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond CT, Pessia M, Xia X-M, Lagrutta A, Kavanaugh MP, Adelman JP. Cloning and expression of a family of inward rectifier potassium channels. Receptors and Channels. 1994;2:183–191. [PubMed] [Google Scholar]
- Dart C, Leyland ML, Spencer PJ, Stanfield PR, Sutcliffe MJ. The selectivity filter of a potassium channel, murine Kir2.1, investigated using scanning cysteine mutagenesis. The Journal of Physiology. 1998;511:25–32. doi: 10.1111/j.1469-7793.1998.025bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Fakler B, Brandle U, Bond CH, Glowatzki E, Konig C, Adelman JP, Zenner H-P, Ruppersberg JP. A structural determinant of different sensitivity of cloned inward rectifier K+ channels to intracellular spermine. FEBS Letters. 1994;356:199–203. doi: 10.1016/0014-5793(94)01258-x. [DOI] [PubMed] [Google Scholar]
- Ficker E, Taglialatela M, Wible BA, Henley CM, Brown AM. Spermine and spermidine as gating molecules for inward rectifier K+ channels. Science. 1994;266:1068–1072. doi: 10.1126/science.7973666. [DOI] [PubMed] [Google Scholar]
- Gay LA, Stanfield PR. Cs+ causes a voltage-dependent block of inward K currents in resting skeletal muscle fibres. Nature. 1977;267:169–170. doi: 10.1038/267169a0. [DOI] [PubMed] [Google Scholar]
- Hagiwara S, Miyazaki S, Rosenthal NP. Potassium current and the effect of cesium on this current during anomalous rectification of the egg cell membrane of a starfish. Journal of General Physiology. 1976;67:621–638. doi: 10.1085/jgp.67.6.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara S, Yoshii M. Effects of internal potassium and sodium on the anomalous rectification of the starfish egg as examined by internal perfusion. The Journal of Physiology. 1979;292:251–265. doi: 10.1113/jphysiol.1979.sp012849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heginbotham L, Abramson T, MacKinnon R. A functional connection between the pores of distantly related ion channels as revealed by mutant K+ channels. Science. 1992;258:1152–1155. doi: 10.1126/science.1279807. [DOI] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. 2. Sunderland, MA, USA: Sinauer Associates Inc; 1992. [Google Scholar]
- Hille B, Schwarz W. Potassium channels as multi-ion single-file pores. Journal of General Physiology. 1978;72:409–442. doi: 10.1085/jgp.72.4.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo Y, Baldwin TJ, Jan YN, Jan LY. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 1993;362:127–133. doi: 10.1038/362127a0. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Yoshimichi M, Heinemann SH. Probing pore topology and conformational changes of Kir2.1 potassium channels by cysteine scanning mutagenesis. FEBS Letters. 1998;435:69–73. doi: 10.1016/s0014-5793(98)01038-2. [DOI] [PubMed] [Google Scholar]
- Leech CA, Stanfield PR. Inward rectification in frog skeletal muscle fibres and its dependence on membrane potential and external potassium. The Journal of Physiology. 1981;319:295–309. doi: 10.1113/jphysiol.1981.sp013909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyland ML, Dart C, Spencer PJ, Sutcliffe MJ, Stanfield PR. The possible role of a disulphide bond in forming functional Kir2.1 potassium channels. Pflügers Archiv. 1999;438:778–781. doi: 10.1007/s004249900153. [DOI] [PubMed] [Google Scholar]
- Lopatin AN, Makhina EN, Nichols CG. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 1994;372:366–369. doi: 10.1038/372366a0. [DOI] [PubMed] [Google Scholar]
- Lopatin AN, Nichols CG. Journal of General Physiology. 1996;108:105–113. doi: 10.1085/jgp.108.2.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lü Q, Miller C. Silver as a probe of pore-forming residues in a potassium channel. Science. 1995;268:304–307. doi: 10.1126/science.7716526. [DOI] [PubMed] [Google Scholar]
- Matsuda H, Saigusa A, Irisawa H. Ohmic conductance through an inward rectifier K channel and blocking by internal Mg2+ Nature. 1987;325:156–159. doi: 10.1038/325156a0. [DOI] [PubMed] [Google Scholar]
- Minor DL, Masseling SJ, Jan YN, Jan LY. Transmembrane structure of an inwardly rectifying potassium channel. Cell. 1999;96:879–891. doi: 10.1016/s0092-8674(00)80597-8. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annual Review of Physiology. 1997;59:171–191. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- Quayle JM, Standen NB, Stanfield PR. The voltage-dependent block of ATP-sensitive potassium channels of frog skeletal muscle by caesium and barium ions. The Journal of Physiology. 1988;405:677–697. doi: 10.1113/jphysiol.1988.sp017355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann F, Ashcroft FM. Inwardly rectifying potassium channels. Current Opinion in Cell Biology. 1999;11:503–508. doi: 10.1016/S0955-0674(99)80073-8. [DOI] [PubMed] [Google Scholar]
- Reuveny E, Jan YN, Jan LY. Contributions of a negatively charged residue in the hydrophobic domain of the IRK1 inwardly rectifying K+ channel to K+-selective permeation. Biophysical Journal. 1996;70:754–761. doi: 10.1016/S0006-3495(96)79615-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. Journal of Molecular Biology. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Spruce AE, Standen NB, Stanfield PR. Studies of the unitary properties of adenosine 5′-triphosphate regulated potassium channels of frog skeletal muscle. The Journal of Physiology. 1987;382:213–236. doi: 10.1113/jphysiol.1987.sp016364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampe P, Arreola J, Perez-Cornejo P, Begenisich T. Non-independent K+ movement through the pore in IRK1 potassium channels. Journal of General Physiology. 1998;112:475–484. doi: 10.1085/jgp.112.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standen NB, Stanfield PR. Rubidium block and rubidium permeability of the inward rectifier of frog skeletal muscle fibres. The Journal of Physiology. 1980;304:415–435. doi: 10.1113/jphysiol.1980.sp013333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfield PR, Davies NW, Shelton PA, Sutcliffe MJ, Khan IA, Brammar WJ, Conley EC. A single aspartate residue is involved in both intrinsic gating and blockage by Mg2+ of the inward rectifier, IRK1. The Journal of Physiology. 1994;478:1–6. doi: 10.1113/jphysiol.1994.sp020225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson GA, Leyland ML, Sutcliffe MJ, Ashmole I, Stanfield PR. Block of murine Kir2.1 by Rb+ and Cs+ is influenced by certain mutations of a serine residue, S165, in the M2 region. The Journal of Physiology. 1999;520:94P. [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. ClustalW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Jan YN, Jan LY. Determination of the subunit stoichiometry of an inwardly rectifying potassium channel. Neuron. 1995;15:1441–1447. doi: 10.1016/0896-6273(95)90021-7. [DOI] [PubMed] [Google Scholar]
- Yang J, Yu M, Jan YN, Jan LY. Stabilization of ion selectivity filter by pore loop ion pairs in an inwardly rectifying potassium channel. Proceedings of the National Academy of Sciences of the USA. 1997;94:1568–1572. doi: 10.1073/pnas.94.4.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Chepilko S, Schutt W, Choe H, Palmer LG, Sackin H. Mutations in the pore region of ROMK enhance Ba2+ block. American Journal of Physiology. 1996;40:C1949–1956. doi: 10.1152/ajpcell.1996.271.6.C1949. [DOI] [PubMed] [Google Scholar]