

Abstract
The modulation of voltage-dependent Ca2+ currents (ICa) by adenosine was investigated in magnocellular neurones acutely dissociated from the rat hypothalamic supraoptic nucleus (SON) by using the whole-cell patch-clamp technique.
Adenosine dose dependently and reversibly inhibited ICa elicited by depolarizing voltage steps from a holding potential of -80 mV to potentials ranging from -30 to +20 mV. The mean (± s.e.m.) maximum inhibition rate was 36.1 ± 4.1 % (n= 6) at -20 mV and the EC50 was 9.8 × 10−7 M (n= 6).
The inhibition of ICa by adenosine was completely reversed by the selective A1 receptor antagonist 8-cyclopentyl theophylline (CPT), and was mimicked by the selective A1 receptor agonist N6-cyclohexyladenosine (CHA).
The inhibition by CHA was strongly reduced when ICa was inhibited by ω-conotoxin GVIA, a blocker of N-type Ca2+ channels.
The adenosine-induced inhibition of ICa was largely reversed by a depolarizing prepulse to +150 mV for 100 ms, which is known to reverse the inhibition of Ca2+ channels mediated by G-protein βγ subunits.
The adenosine receptor-mediated inhibition of ICa was not abolished by intracellularly applied preactivated pertussis toxin (PTX).
Using immunohistochemistry, Gzα-like immunoreactivity (a PTX-resistant inhibitory G-protein) was observed throughout the SON.
These results suggest that adenosine modulates the neuronal activity of SON neurones by inhibiting N-type voltage-dependent Ca2+ channels via A1 receptors which are coupled to PTX-resistant G-proteins.
Magnocellular neurones in the hypothalamic supraoptic nucleus (SON) synthesize arginine vasopressin and oxytocin. These peptides are released into the systemic circulation from axon terminals located in the neurohypophysis, and the released amount is closely related to the electrical activity of SON neurones (Bourque, 1991; Leng & Brown, 1997). This electrical activity is regulated by humoral factors and synaptic neurotransmitters/neuromodulators, such as glutamate and γ-aminobutyric acid, various peptides and other physiological active substances including purinergic agents (Hatton, 1990; Brann, 1995; Shibuya et al. 1999).
In various regions of the mammalian central nervous system (CNS), adenosine is well known to act as a neurotransmitter/neuromodulator (Fredholm & Dunwiddie, 1988). Adenosine modulates neural excitability and acts as a neural sleep factor in basal forebrain and mesopontine cholinergic neurones (Rainnie et al. 1994), as an endogenous anticonvulsant in the hippocampus and amygdala (Dragunow, 1988), as a modulator of LTP in the hippocampus (de Mendonća & Ribeiro, 1994) and as a neuroprotective agent in the hippocampus (Dragunow & Faull, 1988). At present, classification of adenosine receptors is based on both their protein sequences and their affinity for a variety of ligands (Fredholm et al. 1994). Four subtypes of adenosine receptor have been cloned and pharmacologically characterized: A1, A2a, A2b and A3 receptors. A1 and A3 receptors are thought to be coupled to Gi and/or Go proteins, while A2a and A2b are coupled to Gs protein (Fredholm et al. 1994; Palmer & Stiles, 1995). The various effects of adenosine on neuronal activity are thought to occur through the modulation of Ca2+ channels and/or K+ channels (Fredholm & Dunwiddie, 1988; Fredholm et al. 1994; Palmer & Stiles, 1995). It has been reported that adenosine modulates voltage-dependent calcium channels (VDCCs) in the postsynaptic membrane of many different neuronal cell types, such as hippocampus, brainstem and superior cervical ganglion (Mogul et al. 1993; Zhu & Ikeda, 1993; Umemiya & Berger, 1994; Fleming & Mogul, 1997).
Previous binding, immunohistochemistal and in situ hybridization studies have reported that the level of the various adenosine receptors in rat brain hypothalamus is very low. Only the A1 receptor subtype has been detected in the SON using both in situ hybridization and immunohistochemistry (Reppert et al. 1991; Rivkees et al. 1995). In addition, no reports on the effects of adenosine in SON neurones have been published as yet.
In SON neurones, Ca2+ entry through VDCCs plays a crucial role in the generation and/or maintenance of their action potential firing patterns, which are closely linked to arginine vasopressin and oxytocin secretion. For example modulation of VDCCs alters the bursting properties these cells, the action potential plateau and the width of the action potential (Bourque & Renaud, 1985; Andrew, 1987).
In the present study, we investigated the expression of adenosine receptors in the SON using reverse transcription-polymerase chain reaction (RT-PCR) techniques. In addition, we studied the effects of adenosine on the VDCCs in acutely dissociated SON neurones using the whole-cell patch-clamp technique.
METHODS
Preparation of SON slices
Young adult male Wistar rats at 5–8 weeks of age (125-250 g) were used in all experiments. All animal procedures in these studies were carried out in accordance with the guidelines on the use and care of laboraory animals as set out by the Physiological Society of Japan and approved by the animal care committee at this institution. For preparation of SON slices, rats were first stunned by a blow to the neck and then immediately decapitated. The brain was quickly removed and cooled in a bathing solution at 4°C for approximately 1 min. The bathing solution contained (mM): NaCl, 124; KCl, 5; MgSO4, 1.3; KH2PO4, 1.24; CaCl2, 2; NaHCO3, 25.9; and glucose, 10. The osmolality, as determined with a One-Ten osmometer (Fiske Associates, Norwood, USA), was 300–310 mosmol kg−1. This bathing solution was continuously oxygenated and equilibrated with a mixture of 95 % O2-5 % CO2. A block containing the hypothalamus was cut from the brain and glued to the stage of a vibratome-type slicer (DSK-2000; DOSAKA EM Co., Ltd, Kyoto, Japan), and then immersed in 4°C bathing solution. Coronal slices of 300 μm thickness containing the SON were cut from this brain block and then carefully trimmed around the SON using a circular micropunch (i.d. 1.0 mm) and fine scissors so as not to include any extra-SON tissue. For RT-PCR, these trimmed slices were immediately frozen by transferring them into liquid nitrogen. For electrical recordings, these trimmed slices were pre-incubated in the bathing solution at room temperature (23-25°C) for at least 1 h and then transferred to a digestion solution for dissociation of individual SON neurones.
RT-PCR
Total RNA was extracted from the SON slices, using the RNeasy mini kit (Qiagen, Hilden, Germany). RT-PCR was then performed with a thermal cycler (Perkin-Elmer, CA, USA) using a RT-PCR kit (Toyobo, Osaka, Japan). The specificities of the four independent sense and antisense primer pairs for A1, A2a, A2b and A3 receptors used in the present study have previously been described (Dixon et al. 1996). Reverse transcription of the total RNA (0.5 μg) from each slice was performed in a final volume of 20 μl using random primers and Moloney murine leukaemia virus reverse transcriptase with the RT-PCR kit. PCR was performed with a RT-PCR buffer containing 1 μm primers, 1 mm each deoxynucleotide triphosphate, 2.5 U recombinant Taq DNA polymerase and each transcribed cDNA, in a final volume of 50 μl. Single-stranded cDNA products were denatured and subjected to PCR amplification (30 cycles). Each PCR cycle consisted of denaturation at 94°C for 45 s, annealing at 64°C for 50 s and finally extension at 72°C for 65 s. Primers for a house-keeping gene, glyceraldehyde 3-phosphate dehydrogenase (G3PDH), were used as an internal standard. The PCR products were separated by electrophoresis on a 2 % agarose gel and visualized by ethidium bromide staining.
Acute dissociation of SON magnocellular neurones
After incubation at room temperature for at least 1 h, the trimmed slices were transferred into a digestion solution for 90 min at 28°C. This digestion solution was the same as the bathing solution except that it also contained trypsin (0.1 mg ml−1, Sigma type XI). The slices were then transferred into a bathing solution which contained trypsin inhibitor (1 mg ml−1; Nacalai Tesque, Kyoto, Japan) for 10 min at 28°C. They were then placed back into the bathing solution for up to 8 h until use. Individual trimmed slices were transferred to a culture dish and then mechanically dissociated by trituration with fire-polished glass pipettes (tip i.d. ranging from 250 to 650 μm) in a standard recording solution (described below), continuously oxygenated with humidified 100 % O2. The dissociated neurones were left for at least 5 min before recordings were commenced to enable them to adhere to the bottom of the culture dish.
Recording solutions
Standard recording solution (Hepes-buffered solution; HBS) contained (mM): NaCl, 140; KCl, 5; MgCl2, 1; CaCl2, 2; Hepes, 10; and glucose, 11.1. The osmolality was 300–310 mosmol kg−1 and pH was adjusted to 7.4 with NaOH. HBS was continuously oxygenated with 100 % O2 throughout the experiments.
Ca2+ current (ICa) recording solution was HBS with 0.001 mm tetrodotoxin (TTX), and Mg2+ was omitted. The pH and osmolality were the same as for standard HBS. ICa recording solution was also continuously oxygenated with 100 % O2. The pipette solution used for ICa measurements was designed to block K+ currents and contained (mM): tetraethylammonium hydrochloride (TEA-Cl), 100; 4-aminopyridine (4-AP), 5; MgCl2, 1; CaCl2, 1; EGTA, 10; Hepes, 10; Mg-ATP, 2; creatine phosphate di-Tris salt, 20; and Na2-GTP, 0.3. The osmolality was 280–290 mosmol kg−1 and pH was adjusted to 7.2 with Tris base. ICa was pharmacologically isolated by inclusion of TTX in the ICa recording solution to block Na+ currents. K+ currents were also blocked by inclusion of TEA-Cl and 4-AP in the pipette solution. Leak and capacitative currents were cancelled by off-line subtraction of Ni+ (100 μm)- and Cd2+ (200 μm)-insensitive currents. ‘Rundown’ of ICa throughout the 80 min recording period was effectively reduced by using an ATP-regenerating system. Pertussis toxin (PTX) was intracellularly applied using a modified method of Kakehata et al. (1993). PTX was preactivated by adding it to pipette solution containing dithiothreitol (5 mm) and then left for 30 min at 37°C. It was then diluted in additional pipette solution containing β-nicotinamide adenine dinucleotide (NAD, 5 mm) before being added to the pipette. The final concentration of PTX was 1 μg ml−1.
Whole-cell patch-clamp recordings
Dissociated neurones were perfused with the standard recording solution at a flow of 1.5 ml min−1. The solution in the dish was kept at a constant volume by a low pressure aspiration system. Magnocellular neurones were identified using an upright microscope (Nikon, Tokyo, Japan). Only whole-cell tight-seal (1-5 GΩ) patch-clamp recordings were made from neurones which had the typical morphological appearance of magnocellular SON neurones: a large soma of > 15 μm diameter. Over 96 % of the neurones selected by the above criteria were thought to be neurosecretory cells (Oliet & Bourque, 1992). Patch-clamp electrodes were made from glass capillaries (GD-1.5, Narishige, Tokyo, Japan) using a horizontal pipette puller (P-87, Sutter Instrument Co., Novoto, USA). They had a final resistance of between 6 and 8 MΩ when filled with the pipette solution. All electrophysiological recordings were carried out at room temperature (23-25°C). Recording of membrane currents was generally delayed for 5–10 min after membrane rupture until the ICa reached a steady level; however, the effects of PTX on the adenosine-induced inhibition of VDCCs were examined shortly after membrane rupture because PTX is known to show an effect within a few minutes (Kakehata et al. 1993). Currents were filtered at 5 kHz and recorded using a patch-clamp amplifier (Axopatch 200A, Axon Instruments Inc.), and were digitized at 10 kHz using pCLAMP software (version 6.0.3; Axon Instruments Inc.) for subsequent off-line analysis. The standard recording solution was switched to the ICa recording solution immediately after successful membrane rupture using a peristaltic pump. The time required for the complete change of ICa recording solution and addition of drugs was estimated to be a few seconds.
Pharmacological agents
Adenosine, N6-cyclohexyladenosine (CHA), N6-[2-(3,5-dimethoxyphenyl)-9-ethyl]adenosine (DPMA), N6-[2-(4-aminophenyl)ethyl]-adenosine (APNEA), 8-cyclopentyl-3,7-dihydro-1,3-dimethyl-1H-purine-2,6-dione (CPT) and 3,7-dimethyl-1-propargylxanthine (DMPX) were purchased from Research Biochemicals International (MA, USA). Trypsin, NAD and creatine phosphate di-Tris salt were from Sigma. TTX was from Sankyo Co., Ltd (Tokyo, Japan). All of the peptide toxin Ca2+ channel blockers were from Peptide Institute (Osaka, Japan). PTX was from Seikagaku Co., Ltd (Tokyo, Japan). All other chemicals were from Nacalai Tesque.
Analysis
Data were analysed using AxoGraph software (version 3.6; Axon Instruments Inc.). ICa was measured between 3 and 8 ms after the depolarizing voltage steps and averaged for further analysis. The magnitude of ICa inhibition was expressed as percentage inhibition of the control current, which was the averaged current measured just before application of drugs and after complete recovery from the drug effects. Dose-response curves were fitted with the empirical Hill equation using the least-squares method (AxoGraph, version 3.6).
All data shown represent the mean ±s.e.m. Statistical comparisons were performed using non-parametric tests (Mann-Whitney U test). P < 0.05 was regarded as a statistically significant difference.
Immunohistochemistry
Rats were deeply anaesthetized by intraperitoneal injection of sodium pentobarbital (50 mg kg−1), and then perfused transcardially with 100 ml of 0.1 M phosphate buffer (pH 7.4) containing heparin (500 U (500 ml)−1) and 150 ml of fixative containing 4 % paraformaldehyde and 0.2 % picric acid in 0.1 M phosphate buffer.
After postfixation and cryoprotection in 20 % sucrose, 40 μm thick serial sections were cut using a microtome and processed as free-floating sections. The primary Gzα subunit antibody was a rabbit anti-Gzα subunit, N-terminal (amino acids 3–18) antiserum (Calbiochem-Novabiochem Corporation, San Diego, CA, USA), diluted 1:1000 in 0.1 M phosphate-buffered saline (PBS) containing 0.3 % Triton X-100. After treatment with 1 % hydrogen peroxidase for 1 h at room temperature, the sections were incubated with the primary antibody solution at 4°C for 5 days. The avidin-biotin peroxidase complex (Vectastain ABC kit, Vector Laboratories Inc., Burlingame, USA) in the sections was visualized with 0.02 % 3,3′-diaminobenzidine (DAB) for 20–30 min. This experiment was then repeated twice, independently.
Control sections were incubated with the same diluted Gzα subunit antiserum after it had been preincubated with synthetic peptide corresponding to the N-terminal sequence (amino acids 3–18) of the Gzα subunit (10−4 M) (Yanaihara Institute Inc., Shizuoka, Japan).
RESULTS
RT-PCR analysis of adenosine receptors in the SON
To identify subtypes of adenosine receptor in the SON, RT-PCR using four pairs of primers was performed (Table 1). In contrast to previous in situ hybridization studies, we observed that mRNAs for A1, A2a, A2b and A3 receptors were expressed in the SON (Fig. 1), although the expression level of the A3 subtype was very low.
Table 1.
Individual adenosine receptor primers for RT-PCR
| Primer | Source | Position(bp) | Sequence(5′ to 3′) | Predicted length(bp) |
|---|---|---|---|---|
| A1 | M64299 | 630–649(S) | CTC CAT TCT GGC TCT GCT CG | 207 |
| 836–818(AS) | ACA CTG CCG TTG GCT CTC C | |||
| A2a | L08102 | 407–423(S) | CCA TGC TGG GCT GGA ACA | 150 |
| 556–536(AS) | GAA GGG GCA GTA ACA CGA ACG | |||
| A2b | M91466 | 139–156(S) | TGG CGC TGG AGC TGG TTA | 160 |
| 298–281(AS) | GCA AAG GGG ATG GCG AAG | |||
| A3 | M94152 | 238–256(S) | AGA AGC TAG GTC CAC TGG C | 665 |
| 902–880(AS) | GCA CAT GAT AAC CAG GGG GAT GA |
Sources are described by the GenBank accession number. S, sense; AS, antisense.
Figure 1. RT-PCR analysis of adenosine receptor mRNAs expressed in the rat SON.
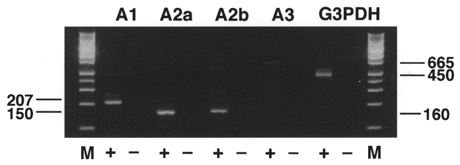
Total RNA from rat SON was reverse transcribed (+) or not (−), then amplified by PCR with each of the primer pairs described in Table 1. Primers for G3PDH were used as an internal control and generated a 450 bp fragment. Amplification products were electrophoresed on a 2 % agarose gel and visualized by ethidium bromide staining. Lane M, Hi-Lo DNA ladder marker (Avetech, Tokyo, Japan). When PCR from each sample was performed without prior reverse transcription, there was no amplification product, indicating that the bands appearing on the gel were not derived from genomic DNA. The sizes of the bands are given in base pairs.
Effects of adenosine on voltage-dependent Ca2+ currents
To investigate the effects of adenosine on VDCCs in SON neurones, Ca2+ currents were elicited from a holding potential (Vh) of -80 mV to depolarized test potentials (ranging from -60 to +50 mV) for 50 ms. Figure 2A shows a representative ICa and also the time course of ICa inhibition by adenosine. Mean values between 3 and 8 ms (•) and between 24 and 34 ms (○) at a test potential of -20 mV were used to evaluate ICa amplitude. Application of 10−5 M adenosine reversibly inhibited evoked ICa in all cells tested (n= 6) with a rapid onset and a relatively slow recovery. Figure 2B shows superimposed current tracings before, during and after application of 10−5 M adenosine. In addition to inhibiting the steady-state current, adenosine also slowed the activation of ICa (‘kinetic slowing’). Such characteristics of ICa inhibition by adenosine were observed in all six SON neurones from which we recorded. Figure 2C 1 shows the averaged 3–8 ms current-voltage relationships obtained from these six SON neurones.
Figure 2. Inhibition of ICa by adenosine in SON magnocellular neurones.
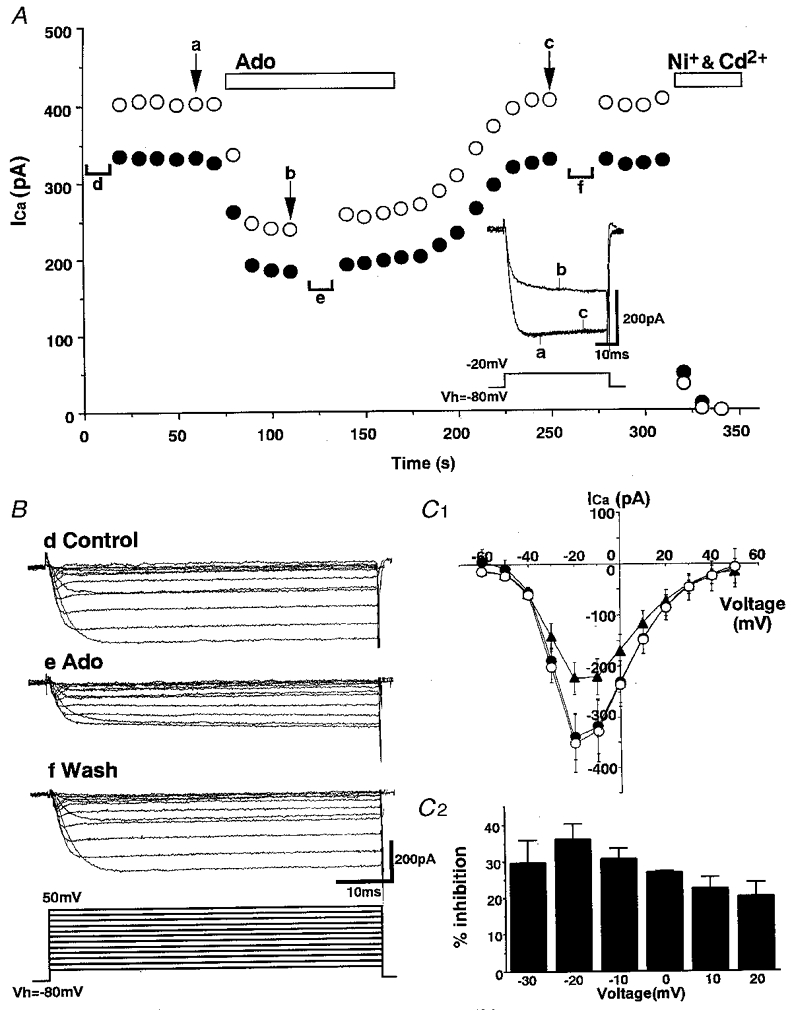
A, a representative experiment illustrating the time course of adenosine (Ado, 10−5 M)-induced inhibition of ICa. Leak currents were cancelled by subtraction of the current remaining after application of Ni+ (10−4 M) and Cd2+ (2 × 10−4 M). ICa values between 3 and 8 ms (•), and between 24 and 34 ms (○), after the beginning of the test pulse were averaged. Inset, superimposed ICa traces obtained at times a, b and c. B, representative ICa traces in response to voltage steps (to between -60 mV and +50 mV) from Vh= -80 mV before (Control), during and after (Wash) application of 10−5 M adenosine. The traces were obtained at times d, e and f in A. C 1, summary of the current-voltage relationship of ICa before (•), during (▴) and after (○) application of 10−5 M adenosine. ICa amplitude was calculated as the averaged current between 3 and 8 ms after the beginning of the test pulse (n= 6). Error bars indicate s.e.m.C 2, voltage dependence of the adenosine (10−5 M)-induced inhibition of ICa. Data are shown as the mean ±s.e.m. of values obtained from six neurones.
Figure 2C 2 shows summary data for the voltage dependence of the inhibition of ICa by adenosine. Adenosine significantly inhibited ICa at voltages between -30 and +20 mV with a peak inhibition at -20 mV. The mean maximum inhibition at -20 mV was 36.1 ± 4.1 % (n= 6).
The dose-response relationship of the adenosine-induced inhibition of ICa was studied using voltage steps to -20 mV from a holding potential of -80 mV. Figure 3A demonstrates that the inhibition of ICa by adenosine was dose dependent. This concentration dependence of the adenosine block was fitted with the empirical Hill equation by the least-squares method (Fig. 3B). Inhibition of ICa by the A1 receptor agonist CHA showed a similar dose dependence but CHA was more potent by about one order of magnitude. The half-maximal concentration (EC50) for the inhibition of ICa by adenosine was 1.0 × 10−6 M (n= 6) and by CHA was 5.8 × 10−8 M (n= 5) (Fig. 3B). Maximum inhibition was observed at 10−5 M (32.2 ± 0.9 %) in the case of adenosine, and at 10−6 M (35.0 ± 2.0 %) in the case of CHA. Recently, it was reported that in the SON, adenosine inhibits presynaptic neurotransmitter release via an A1 receptor (Oliet & Poulain, 1999). IC50 values were reported as 12.7 × 10−6 M for inhibition of IPSCs and 12.8 × 10−6 M for inhibition of EPSCs. Our present results indicate that the postsynaptic response to adenosine in the SON appeared at a concentration about one order of magnitude lower than that for the presynaptic response. In both cases, the adenosine-induced response was reversed by CPT, the A1-selective antagonist. These results suggest that adenosine affects both pre- and postsynaptic A1 receptors in the SON.
Figure 3. Dose-response relationship of adenosine-induced inhibition of ICa.
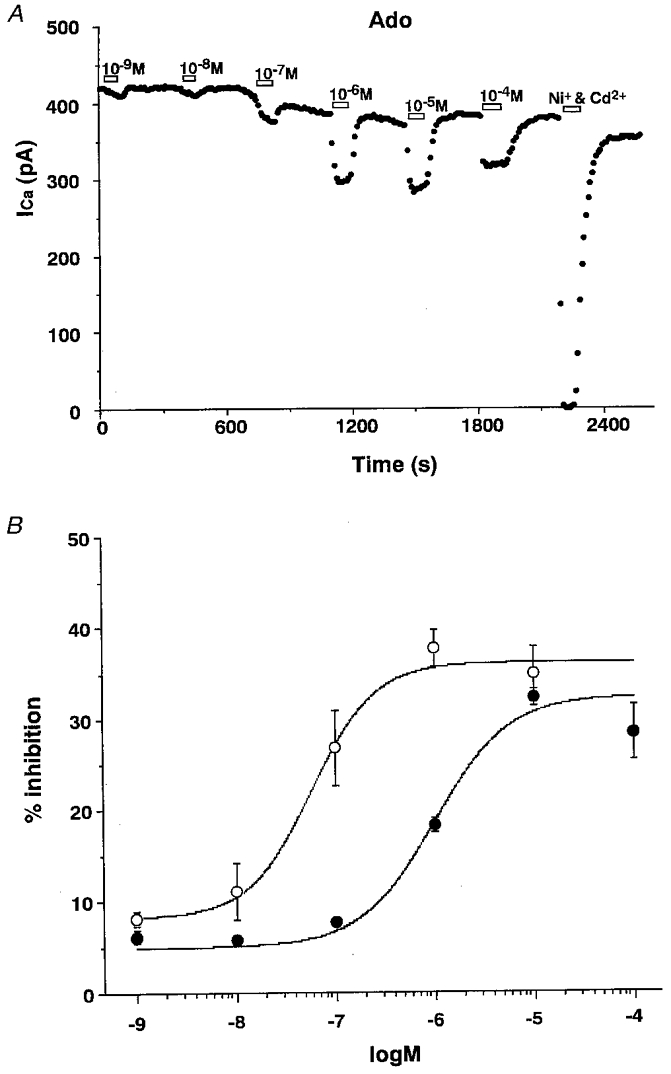
A, a representative experiment illustrating the time course of the dose dependence of inhibition of ICa by adenosine (Ado, 10−9 to 10−4 M). B, dose-response curves of ICa inhibition induced by adenosine (•) and CHA (○). Curves represent the Hill fits to the mean data using the least-squares method. The Hill coefficients obtained from the curves were 1.17 (adenosine) and 1.36 (CHA). Data are shown as the mean ±s.e.m. of values obtained from seven (10−9 to 10−5 M) and six (10−4 M) experiments in adenosine, and from five (10−9 to 10−5 M) experiments in CHA.
Pharmacological analysis of adenosine receptor subtypes
To analyse which adenosine receptor subtypes mediated the inhibition of ICa in the SON, we used the following relatively selective adenosine receptor ligands: CHA (A1 agonist), DPMA (A2 agonist), APNEA (A3 agonist), CPT (A1 antagonist) and DMPX (A2 antagonist). The effects of the agonists and antagonists were evaluated independently, and at concentrations that exhibited a maximum response (CHA, 10−6 M; DPMA, 10−6 M; APNEA, 10−6 M; CPT, 10−6 M; and DMPX, 10−5 M).
The selective A1 receptor antagonist CPT completely reversed adenosine-induced inhibition of ICa without enhancement, whereas the A2 receptor antagonist DMPX caused only a partial reversal of adenosine-induced inhibition. The effects of these antagonists on the CHA-induced inhibition were similar to the effects on adenosine-induced inhibition (Fig. 4A and B). These results suggest that adenosine induces inhibition of ICa through the A1 receptor. The relatively selective A2 agonist DPMA, however, also inhibited ICa, but the percentage inhibition by DPMA was smaller than that induced by adenosine or CHA (adenosine, 34.9 ± 4.3 %, n= 4; CHA, 37.1 ± 7.3 %, n= 6 vs. DPMA, 10.4 ± 1.4 %, n= 7; P < 0.05). Inhibition of ICa by DPMA was completely reversed by CPT but also by the A2 antagonist DMPX (Fig. 4C). These results also suggest that DPMA-induced inhibition of ICa is likely to be mediated through the A1 receptor. The relatively selective A3 agonist APNEA inhibited ICa but the inhibition was almost completely reversed by CPT and DMPX (Fig. 4C). These results suggest that adenosine-induced inhibition of VDCCs in SON neurones occurred mainly though the A1 adenosine receptor, although there was possibly a small A2 contribution, and furthermore demonstrate the lack of specificity of these ligands.
Figure 4. Pharmacological characterization of the receptor subtype involved in adenosine-induced ICa inhibition.
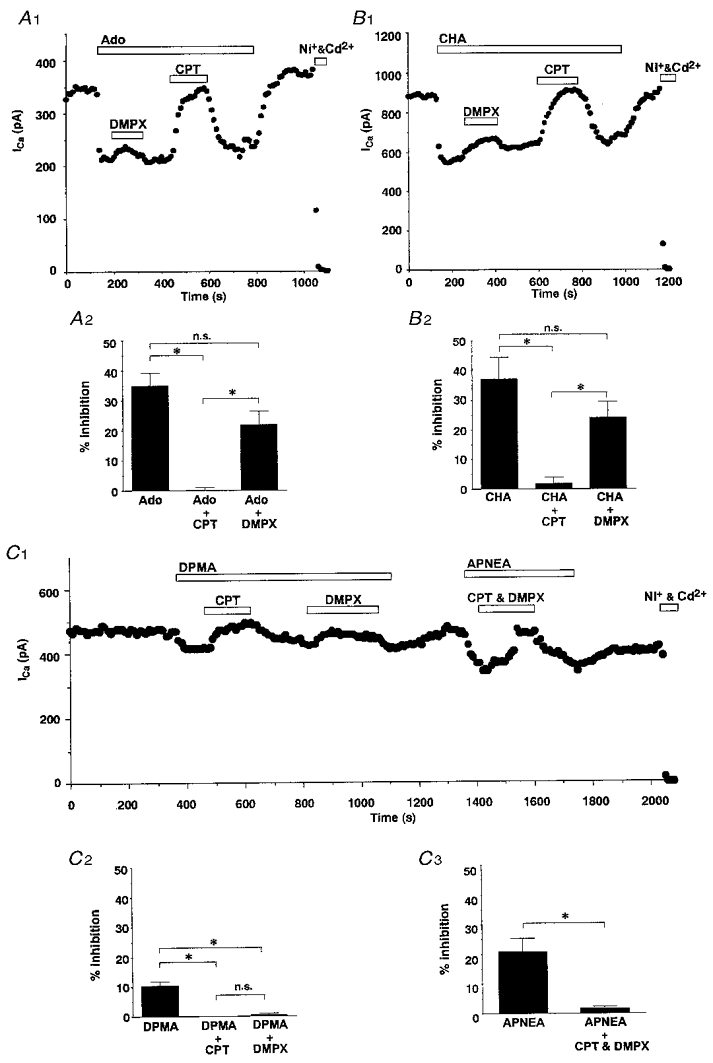
A 1 and B 1, representative experiments illustrating the effects of the A1 antagonist CPT (10−6 M) and the A2 antagonist DMPX (10−5 M) on 10−6 M adenosine (A 1)- and 10−6 M CHA (B 2)-induced ICa inhibition. A 2 and B 2, summary of the effects of CPT and DMPX on 10−6 M adenosine (A 2)- and 10−6 M CHA (B 2)-induced ICa inhibition (adenosine, n= 4; CHA, n= 5). C 1, representative experiments illustrating the effects of CPT and DMPX on DPMA (10−6 M)- and APNEA (10−6 M)-induced ICa inhibition. C 2, summary of the effects of CPT and DMPX on DPMA-induced ICa inhibition (n= 7). C 3, summary of the effects of CPT and DMPX on APNEA-induced ICa inhibition (n= 4). * P < 0.05; n.s., not significant.
Effects of blockers of Ca2+ channels on inhibition by A1 agonist
To detect which types of VDCC contributed to the component of ICa that was inhibited by adenosine, we used the selective Ca2+ channel blockers ω-conotoxin-GVIA (ω-CTx-GVIA; N-type), ω-agatoxin IVA (ω-Aga-IVA) plus ω-conotoxin MVIIC (ω-CTx-MVIIC) (P/Q-type), and nifedipine (L-type). Both the contribution of different subtypes of VDCC to the total calcium influx and the relative percentage of CHA-induced inhibition of each subtype were measured.
Figure 5A shows a representative example trace illustrating the experimental protocol and the ICa classification used in the present study. The small component of the ICa that reversed upon washout of the irreversible toxin-blockers ω-CTx-GVIA, ω-Aga-IVA and ω-CTx-MVIIC was considered to represent L-type current (α1D subunit) as described by Brinbaumer et al. (1994). The percentage of the total ICa characterized by these blockers was: N-type, 27.0 ± 3.4 %; P/Q -type, 15.0 ± 3.9 %; L-type, 16.6 ± 4.1 %; and the residual (R-type) current was 41.3 ± 4.0 % (n= 4; Fig. 5B, outer circle). The percentage inhibition (expressed as percentage of the total ICa) of each ICa subtype by CHA was: N-type, 21.1 ± 4.2 %; P/Q -type, 4.5 ± 1.8 %; L-type, 4.8 ± 0.9 %; and R-type, 6.6 ± 1.5 % (n= 4; Fig. 5B, inner circle). Thus the relative contribution of each subtype of ICa for the inhibition by CHA were calculated as: N-type, 54.7 %; P/Q -type, 12.0 %; L-type, 14.9 %; and R-type, 17.4 % (n= 4; Fig. 5C). These results indicate that N-type Ca2+ channels are the main contributors in to the inhibition of ICa by A1 receptor activation.
Figure 5. Analyses of VDCC subtypes contributing to CHA-induced ICa inhibition.
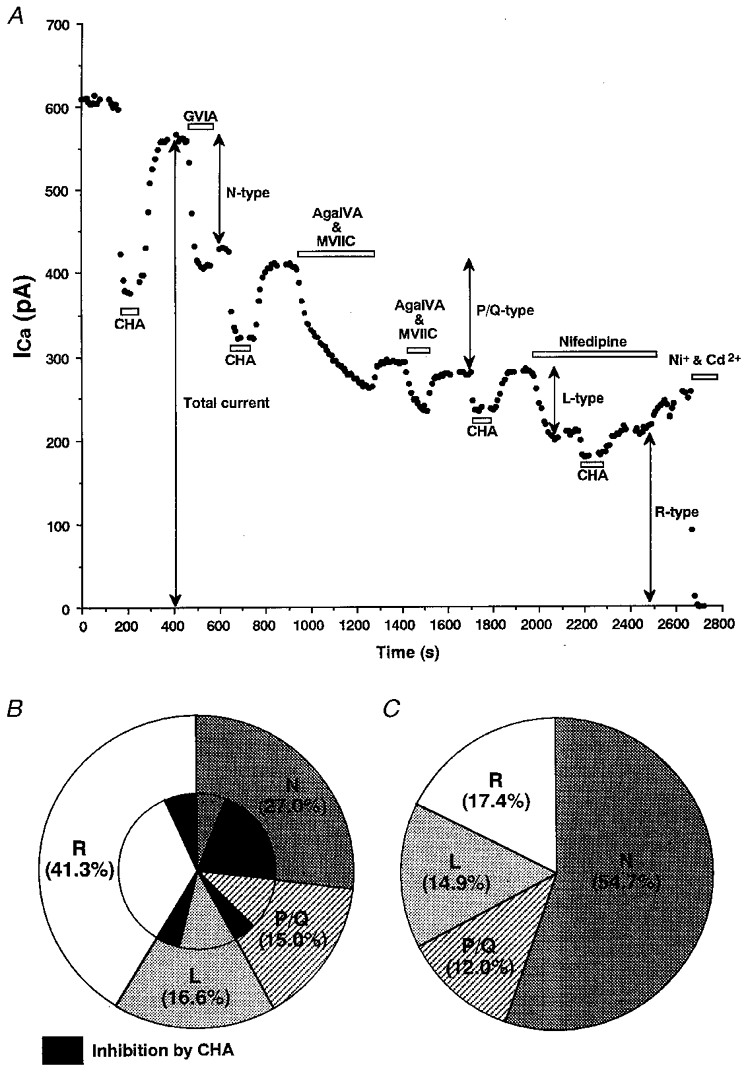
A, a representative experiment illustrating the effect of 10−6 M CHA on ICa before and after application of various selective Ca2+ channel blockers. N-type Ca2+ current was the component irreversibly blocked by ω-CTx-GVIA (10−6 M; GVIA). P/Q -type Ca2+ current was the component irreversibly blocked by a combination of ω-Aga-IVA (10−7 M; AgaIVA) and ω-CTx-MVIIC (10−6 M; MVIIC). L-type Ca2+ current was the component reversibly blocked by nifedipine (10−5 M). The residual component insensitive to these drugs was regarded as R-type. B, summary of the distribution of high-voltage-activated Ca2+ channel subtypes (outer circle) and their fractional components which were sensitive to 10−6 M CHA (inner circle, black area) from four SON neurones. C, summary data for the relative proportion of each Ca2+ channel subtype that contributed to the total CHA-induced ICa inhibition.
Effects of prepulse and dialysis with preactivated PTX on adenosine-induced inhibition of VDCCs
The rapid onset, in the order of seconds, for the inhibition of ICa by adenosine indicates that the response might be mediated by a membrane-delimited mechanism. It is well established that a large depolarizing prepulse reverses the inhibition of ICa by the membrane-delimited action of G-protein βγ subunits (Ikeda, 1996; Herlitze et al. 1996; Furukawa et al. 1998). In the present study, a prepulse to +150 mV for 100 ms reversed the majority of the adenosine-induced inhibition. In the presence of CHA, the ICa following such as a prepulse (C2) was much larger than the current observed prior to the prepulse (C1). In addition to the increased amplitude, the ICa following this prepulse displayed normal activation kinetics without the characteristic adenosine-induced ‘kinetic slowing’ (Fig. 6A). Similar results were obtained in all other SON neurones examined (n= 6).
Figure 6. Effects of a depolarizing prepulse on CHA-induced ICa inhibition.
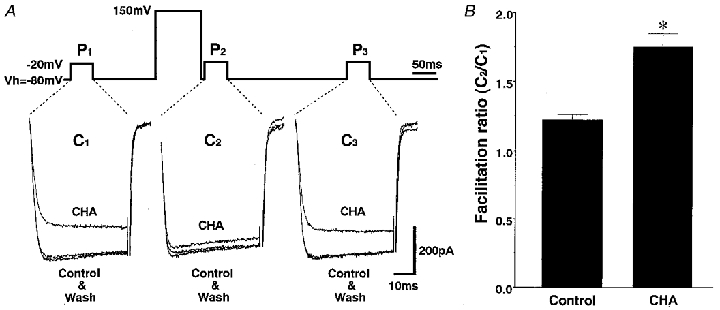
A, three consecutive current traces in response to 50 ms test pulses from Vh= -80 to -20 mV. The middle test pulse (P2) was preceded by a 5 ms prepulse to +150 mV for 100 ms. The three traces in each panel correspond to before (Control), during and after (Wash) application of 10−6 M CHA. Most of the CHA-induced ICa inhibition was reversed by a depolarizing prepulse. B, summary of the change in the facilitation ratio by CHA. ICa amplitude between 3 and 8 ms was averaged and the facilitation ratio was calculated by dividing the C2 current amplitude by the corresponding C1 current amplitude. The facilitation ratio (C2/C1) was significantly greater in the presence of CHA than without CHA (control, 1.22 ± 0.04; CHA, 1.75 ± 0.10; * P < 0.05).
The facilitation of ICa following this prepulse was investigated in both the absence and the presence of 10−6 M CHA (Fig. 6B). In the absence of CHA, the facilitation ratio was 1.22 ± 0.04 (n= 6). This facilitated current is thought to reflect tonic voltage-dependent inhibition of the VDCCs by G-protein βγ subunits (Ikeda, 1996; Herlitze et al. 1996). Application of CHA significantly increased this facilitation ratio to 1.75 ± 0.10 (n= 6, P < 0.05). These results indicate that G-proteins are involved in the inhibition of ICa by adenosine.
To investigate whether Gi/o mediates adenosine-induced inhibition of ICa in SON neurones, the effects of dialysis with preactivated PTX (1 μg ml−1) were examined. Pre-activated PTX was applied intracellularly (see Methods), because extracellular application of PTX requires too much time for the effects to appear and it was difficult to maintain good conditions for acutely dissociated neurones for such a long time. ICa was measured at times 0–20 min, 30–50 min and > 60 min after the membrane was ruptured. This time course was considered sufficient for pre-activated PTX to be dialysed into the neurone and to catalyse PTX-sensitive G-proteins. There were no significant changes in CHA-induced ICa inhibition, even more than 1 h after the cell membrane was ruptured (0-20 min, 37.1 ± 7.0 %; 30–50 min, 37.2 ± 6.4 %; 60 min, 33.2 ± 3.0 %). In addition the degree of inhibition in the 0–20 min period was similar to that observed without PTX in the pipette (Fig. 4B 2). This result indicates that the PTX resistant G-protein α subunit may mediate the CHA-induced inhibition of ICa.
Existence of Gz in SON neurones
We investigated the existence of Gzα subunit in the SON and the PVN by using immunohistochemistry. We observed Gzα-like immunoreactivity (LI) throughout the SON and PVN (Fig. 7). This result provides the first demonstration that Gzα exists in the SON and PVN. Gzα LI was also found, at similar levels, in the cortex and hippocampus (data not shown), and these observations were consistent with those of a previous study using the same antiserum for Gzα (Hinton et al. 1990).
Figure 7. Gzα-like immunoreactivity in the rat SON.
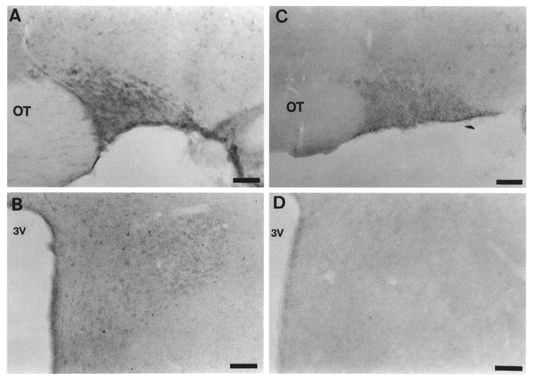
Light micrograph illustrating Gzα-like immunoreactivity in a coronal section of the SON (A) and PVN (B). The staining is found in the perikarya of SON neurones and PVN neurones. This positive staining was abolished by preincubation with Gzα (10−4 M) (C and D). OT, optic tract; 3V, third ventricle. Scale bars, 50 μm.
DISCUSSION
The present study provides the first direct evidence that adenosine A1 receptors are functionally expressed in SON neurones and, when activated by adenosine, mediate inhibition of ICa. The present study also indicates the involvement of G-proteins in the signal transduction pathway underlying this effect.
RT-PCR analysis of adenosine receptors in the SON
Classification of adenosine receptors is based on both their protein sequences and their affinity for a variety of ligands (Fredholm et al. 1994). Four subtypes of adenosine receptor have been cloned and pharmacologically characterized: A1, A2a, A2b and A3. Previous binding, immunohistochemical and in situ hybridization studies reported that the distribution of adenosine receptors was not high in rat brain hypothalamus. Only A1 receptors were detected in the SON by in situ hybridization or immunohistochemistry (Reppert et al. 1991; Rivkees et al. 1995). Recently, however, Dixon and colleagues (Dixon et al. 1996) demonstrated the existence of all these subtypes of adenosine receptor in the hypothalamus by using RT-PCR, although their distribution within individual hypothalamic nuclei was not examined. In the present study, we have identified all these subtypes of adenosine receptor in the SON by using RT-PCR, although the expression level of the A3 subtype was quite low.
Pharmacological analysis of adenosine receptor subtypes in SON neurones
Using reasonably selective agonists and antagonists, we have shown that the majority of the adenosine-induced inhibition of ICa in SON neurones was mediated by A1 receptors. The evidence for this conclusion is as follows. Inhibition of ICa by adenosine or CHA was completely reversed by the selective A1 receptor antagonist CPT. Because DMPX at 10−5 M has some affinity for the A1 receptor (Seale et al. 1988), inhibition of ICa by adenosine or CHA was partially reversed by DMPX. While DPMA induced a small inhibition, it is likely that this was mediated via A1 receptors as it was reversed by CPT. In addition, A2 receptors have been reported to enhance ICa in other preparations (Mogul et al. 1993). Although the exact affinity of APNEA for each adenosine receptor subtype is still unclear, it has been reported that APNEA also has some affinity for the A1 receptor (Armstrong & Ganote, 1994). The finding that APNEA inhibited ICa and that this was reversed by a selective A1 receptor antagonist supports this suggestion.
VDCCs in the SON
VDCCs are characterized by both their pharmacological characteristics and the molecular identity of their α1 subunits. In the SON neurones, five different high-voltage-activated (HVA) Ca2+ channels have been identified to date on the basis of pharmacology: L-, N-, P-, Q- and R-type (Fisher & Bourque, 1995; Foehring & Armstrong, 1996). Using specific Ca2+ channel blockers, we investigated their contribution to the total ICa in SON neurones.
The proportion of R-type ICa in the present study was larger (41 % of total ICa) than reported in previous studies (Fisher & Bourque, 1995; Foehring & Armstrong, 1996; Harayama et al. 1998; Soldo & Moises, 1998) although this result is in good agreement with the observation that there is a large amount of mRNA for R-type channel α subunit (α1E) in the SON (Soong et al. 1993). A developmental difference in the relative amount of VDCCs may be one of the reasons for these differences (Widmer et al. 1997). Complex factors such as different strains of rats and drug application protocol might also have contributed to the difference in proportion of HVA Ca2+ channel subtypes.
In the present study, the peak current appeared at a relatively low potential (-20 mV) when compared with these previous studies, where the peak current appeared at around -10 mV in adult rats (Fisher & Bourque, 1995; Foehring & Armstrong, 1996) and was at 0 mV in infant rats (Harayama et al. 1998; Soldo & Moises, 1998). Because R-type Ca2+ channels encoded by α1E show a relatively lower voltage threshold and peak activation than any of the other HVA Ca2+ channels (Chin et al. 1992; Soong et al. 1993; Dunlap et al. 1995), the negative shift of the current-voltage relationship in the present study may be caused by a large proportion of R-type ICa.
The Ca2+ channel subtype susceptible to inhibition by A1 agonist
In the present study, by using inhibitors of VDCCs, distinct subtypes of HVA ICa were identified in the somata of SON neurones. Although all subtypes of HVA ICa were inhibited by the A1 agonist CHA, the relative contribution of each subtype to the total CHA-induced inhibition differed. N-type Ca2+ channels made the largest contribution to the CHA-induced inhibition of ICa. Although adenosine inhibits different subtypes of VDCC in different neuronal preparations, the most common target seems to be N-type VDCCs. For example, adenosine inhibits N-type ICa in the hippocampus, brainstem and superior cervical ganglion (Mogul et al. 1993; Zhu & Ikeda, 1993; Umemiya & Berger, 1994; Fleming & Mogul, 1997). Although the specific physiological role of each subtype is not clear in the SON, it has been reported that L- or N-type channel blockers significantly reduce depolarizing afterpotentials (DAPs) (Li & Hatton, 1997). These DAPs are important in the generation and/or maintenance of the phasic firing pattern in vasopressin-secreting neurones. Thus, N-type ICa in the somata of SON neurones may make a significant contribution to the regulation of hormone release from the neurohypophysis.
Signal transduction pathway for the inhibition of VDCCs by adenosine
Adenosine-induced inhibition occurred within a few seconds and was associated with a kinetic slowing of ICa activation. In addition, the inhibition was reversed by a large depolarizing prepulse. These effects suggest that the inhibition is mediated through G-proteins via a membrane-delimited pathway without the involvement of soluble intracellular second messengers (Ikeda, 1996; Herlitze et al. 1996; Furukawa et al. 1998). In the membrane-delimited pathway, it has been suggested that there are two direct interaction pathways between G-protein subunits and VDCCs. One is a voltage-independent pathway via the G-protein α subunit and the other is a voltage-dependent pathway via the G-protein βγ subunit. In the case of SON neurones, the majority of the CHA (adenosine)-induced inhibition was reversed by a depolarizing prepulse suggesting that the inhibition of ICa by adenosine is mainly mediated by Gβγ. CHA (adenosine), however, inhibited all subtypes of HVA VDCCs in the present study (Fig. 5) and a depolarizing prepulse did not completely reverse this adenosine-induced inhibition. Recently, it has been suggested that agonist-induced inhibition of N-, P/Q – and R-type VDCCs is reversed by a large depolarizing prepulse, but inhibition of L-type VDCCs is not (Zhang et al. 1996; Qin et al. 1997; Furukawa et al. 1998). The residual component, which was not reversed by a prepulse, might therefore be L-type and/or parts of other subtypes of VDCC, which are mediated by Gα in a voltage-independent manner.
PTX resistance of SON neurones
It is widely accepted that adenosine A1 receptors couple to Gi/o proteins, which are sensitive to PTX. In the present study, however, PTX did not affect the CHA (adenosine)-induced inhibition of ICa. The technique used to intracellularly apply PTX has previously been shown to be effective in significantly diminishing 5-HT-induced inhibition of Ba2+ currents in melanotrophs (Noguchi & Yamashita, 1999). The dose used in the present study (1 μg ml−1) was considered to be sufficient to completely ribosylate Gi/o proteins in SON neurones. Similar PTX-resistant phenomena have been reported in SON and other hypothalamic neurones. In slice preparations of adult rat SON, pretreatment with PTX did not affect the baclofen-induced depression of IPSCs under the same conditions in which PTX blocked the postsynaptic GABAB response in neurones of the lateral parabrachial nucleus (Mouginot et al. 1998). In cultured hypothalamic suprachiasmatic nucleus neurones, it has been reported that baclofen-induced inhibition of postsynaptic VDCCs is not fully blocked after PTX pretreatment (Chen & van den Pol, 1998). In our previous study, baclofen-induced inhibition of ICa in dissociated SON neurones from infant rats also seemed to be relatively resistant to PTX (Harayama et al. 1998). These reports suggest that in hypothalamic neurones, including SON neurones, inhibitory G-proteins might be unusually resistant to PTX. Using in situ hybridization histochemistry, the distribution of the gene expression of G-protein subtypes in the CNS including the SON has been reported. Gs mRNA was strongly expressed in the SON, whereas Gi/o mRNA could not be detected in the SON (Noguchi & Yamashita, 1999). These reports support the possibility that the level of Gi/o protein in SON neurones is insufficient to mediate A1 receptor coupling and subsequent ICa inhibition. To address this issue, we investigated the existence of Gzα in the SON by using immunohistochemistry. The result indicates that there were Gzα subunits in SON neurones. Gzα is a member of the Gi/o protein superfamily but is resistant to PTX, and recently it has been reported that Gzα mediates agonist-induced inhibition of VDCCs (Jeong & Ikeda, 1998; Furukawa et al. 1998). At present there are no tools available to elucidate signal pathways mediated by Gzα in native cells, thus we could not provide direct evidence that Gzα mediated the adenosine-induced inhibition of ICa in SON neurones. Nevertheless, the present results are consistent with an involvement of Gzα in mediating the adenosine-induced inhibition of ICa. Further experiments are needed to more directly determine which subtypes of G-protein mediate the inhibition of ICa by adenosine.
Physiological implications of the inhibition of VDCCs by adenosine
We observed that adenosine inhibited ICa in all the recorded cells, which were dissociated randomly from various areas of the SON. This suggests that Ca2+ currents in both vasopressin- and oxytocin-secreting neurones were modulated by adenosine.
In the hypothalamo-neurohypophysial system, the amount of hormone release from the axon terminals is highly dependent on the electrical activity of SON neurones (Bourque, 1991; Leng & Brown, 1997). Ca2+ entry through VDCCs during action potentials makes an important contribution to this electrical activity. More specifically, Ca2+ influx through VDCCs is required for action potential broadening in response to different firing frequencies, which leads to a facilitation of hormone release in both oxytocin and vasopressin neurones (Bourque & Renaud, 1985). In addition, Ca2+ influx is essential for action potential DAPs which summate potentials which then initiate phasic burst firing and vasopressin secretion (Bourque, 1986). Furthermore, the subsequent activation of Ca2+-dependent potassium conductances also contributes to the pattern and rate of firing in both oxytocin and vasopressin neurones (Kirkpatrick & Bourque, 1996). In SON neurones, protein phosphorylation, enzyme activity, gene expression and somatodendritic release of both oxytocin and vasopressin are also thought to be Ca2+-dependent processes. Ca2+ influx through VDCCs may be important for these events. Hence the inhibition of VDCCs is an important cellular mechanism whereby adenosine may contribute to the electrical activity and neurosecretion in SON neurones.
We revealed that functional adenosine receptors exist in post-synaptic membranes of SON neurones and that adenosine acts as inhibitory modulator. However, in physiological situations, where does the adenosine come from?
Recently, Oliet & Poulain (1999) revealed that in the SON, endogenous adenosine is released into the extracellular space in response to electrical field stimulation and acts as an inhibitor of neurotransmitter release from presynaptic terminals projecting to SON magnocellular neurones. While the specific origin of this endogenous adenosine is not clear, there are two general possibilities.
One is intracellular adenosine that is formed from the breakdown of ATP. In general, when cellular activity increases, intracellular ATP is metabolized to adenosine which can be directly released into the extracellular space via transport processes present in various types of cell. The possible sites that could release adenosine in the SON are neuronal terminals, glial cells and SON neurones themselves (Oliet & Poulain, 1999). If adenosine is released from the SON neurones themselves, adenosine may mediate an activity-dependent negative feedback action.
Another possible source of endogenous adenosine is from the breakdown of neuronally released ATP by ecto-nucleotidases in the extracellular space. For instance, noradrenergic fibres, which project to the SON (Pittman, 1999) and SON magnocellular neurones themselves both contain ATP in their synaptic vesicles (Troadec et al. 1998; Pittman, 1999). The released ATP can be metabolized to adenosine by ecto-ATPase in the extracellular synaptic space in the SON. In this case, adenosine may act to terminate the excitatory effect of ATP.
In conclusion, the present study has demonstrated that functional A1 adenosine receptors are present in the postsynaptic membrane of SON magnocellular neurones where they mediate inhibition of VDCCs, in particular the N-type channel, via PTX-resistant, membrane-delimited, G-proteins.
Acknowledgments
This study was supported in part by grants from the Ministry of Education, Science, Sports and Culture, Japan (to H.Y., nos 10470019 and 10218210), a research grant from the Ministry of Health and Welfare, and the Salt Science Research Foundation. We thank Dr Andrew Moorhouse (School of Physiology and Pharmacology, The University of New South Wales, Australia) and Dr Toshihisa Nagatomo (2nd Department of Internal Medicine, University of Occupational and Environmental Health) for helpful comments on this manuscript, Dr Yasuhito Uezono (Department of Pharmacology, Miyazaki Medical College, Japan) for technical advice and Ms Yuko Hara for technical assistance.
References
- Andrew RD. Endogenous bursting by rat supraoptic neuroendocrine cells is calcium dependent. The Journal of Physiology. 1987;384:451–465. doi: 10.1113/jphysiol.1987.sp016463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong S, Ganote CE. Adenosine receptor specificity in preconditioning of isolated rabbit cardiomyocytes: evidence of A3 receptor involvement. Cardiovascular Research. 1994;28:1049–1056. doi: 10.1093/cvr/28.7.1049. [DOI] [PubMed] [Google Scholar]
- Birnbaumer L, Campbell KP, Catterall WA, Harpold MM, Hofmann F, Horne WA, Mori Y, Schwartz A, Suntch TP, Tsien RW. The naming of voltage-gated calcium channels. Neuron. 1994;13:505–506. doi: 10.1016/0896-6273(94)90021-3. [DOI] [PubMed] [Google Scholar]
- Bourque CW. Calcium-dependent spike after-current induces burst firing in magnocellular neurosecretory cells. Neuroscience Letters. 1986;70:204–209. doi: 10.1016/0304-3940(86)90464-7. [DOI] [PubMed] [Google Scholar]
- Bourque CW. Activity-dependent modulation of nerve terminal excitation in a mammalian peptidergic system. Trends in Neurosciences. 1991;14:29–30. doi: 10.1016/0166-2236(91)90180-3. [DOI] [PubMed] [Google Scholar]
- Bourque CW, Renaud LP. Activity dependence of action potential duration in rat supraoptic neurosecretory neurones recorded in vitro. The Journal of Physiology. 1985;363:429–439. doi: 10.1113/jphysiol.1985.sp015720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brann DW. Glutamate: a major excitatory transmitter in neuroendocrine regulation. Neuroendocrinology. 1995;61:213–225. doi: 10.1159/000126843. [DOI] [PubMed] [Google Scholar]
- Chen G, Van Den Pol AN. Presynaptic GABAB autoreceptor modulation of P/Q -type calcium channels and GABA release in rat suprachiasmatic nucleus neurons. Journal of Neuroscience. 1998;18:1913–1922. doi: 10.1523/JNEUROSCI.18-05-01913.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin H, Smith MA, Kim HL, Kim H. Expression of dihydropyridine-sensitive brain calcium channels in the rat central nervous system. FEBS Letters. 1992;299:69–74. doi: 10.1016/0014-5793(92)80103-n. [DOI] [PubMed] [Google Scholar]
- De Mendonća A, Ribeiro JA. Endogenous adenosine modulates long-term potentiation in the hippocampus. Neuroscience. 1994;62:385–390. doi: 10.1016/0306-4522(94)90373-5. [DOI] [PubMed] [Google Scholar]
- Dixon AK, Gubitz AK, Sirinathsinghji DJ, Richardson PJ, Freeman TC. Tissue distribution of adenosine receptor mRNAs in the rat. British Journal of Pharmacology. 1996;118:1461–1468. doi: 10.1111/j.1476-5381.1996.tb15561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragunow M. Purinergic mechanisms in epilepsy. Progress in Neurobiology. 1988;31:85–108. doi: 10.1016/0301-0082(88)90028-7. [DOI] [PubMed] [Google Scholar]
- Dragunow M, Faull RL. Neuroprotective effects of adenosine. Trends in Pharmacological Sciences. 1988;9:193–194. doi: 10.1016/0165-6147(88)90079-x. [DOI] [PubMed] [Google Scholar]
- Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends in Neurosciences. 1995;18:89–98. [PubMed] [Google Scholar]
- Fisher TE, Bourque CW. Voltage-gated calcium currents in the magnocellular neurosecretory cells of the rat supraoptic nucleus. The Journal of Physiology. 1995;486:571–580. doi: 10.1113/jphysiol.1995.sp020835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming KM, Mogul DJ. Adenosine A3 receptors potentiate hippocampal calcium current by a PKA-dependent/PKC-independent pathway. Neuropharmacology. 1997;36:353–362. doi: 10.1016/s0028-3908(97)83762-8. [DOI] [PubMed] [Google Scholar]
- Foehring RC, Armstrong WE. Pharmacological dissection of high-voltage-activated Ca2+ current types in acutely dissociated rat supraoptic magnocellular neurons. Journal of Neurophysiology. 1996;76:977–983. doi: 10.1152/jn.1996.76.2.977. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, Williams M. Nomenclature and classification of purinoceptors. Pharmacological Reviews. 1994;46:143–156. [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Dunwiddie TV. How does adenosine inhibit transmitter release? Trends in Pharmacological Sciences. 1988;9:130–134. doi: 10.1016/0165-6147(88)90194-0. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Miura R, Mori Y, Strobeck M, Suzuki K, Ogihara Y, Asano T, Morishita R, Hashii M, Higashida H, Yoshii M, Nukada T. Differential interactions of the C terminus and the cytoplasmic I–II loop of neuronal Ca2+ channels with G-protein α and βγ subunits. II. Evidence for direct binding. Journal of Biological Chemistry. 1998;273:17595–17603. doi: 10.1074/jbc.273.28.17595. [DOI] [PubMed] [Google Scholar]
- Harayama N, Shibuya I, Tanaka K, Kabashima N, Ueta Y, Yamashita H. Inhibition of N- and P/Q -type calcium channels by postsynaptic GABAB receptor activation in rat supraoptic neurones. The Journal of Physiology. 1998;509:371–383. doi: 10.1111/j.1469-7793.1998.371bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton GI. Emerging concepts of structure-function dynamics in adult brain: the hypothalamo-neurohypophysial system. Progress in Neurobiology. 1990;34:437–504. doi: 10.1016/0301-0082(90)90017-b. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein βγ subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Hinton DR, Blanks J, Fong HK, Casey PJ, Hildebrandt E, Simons MI. Novel localization of a G protein, Gz-α, in neurons of brain and retina. Journal of Neuroscience. 1990;10:2763–2770. doi: 10.1523/JNEUROSCI.10-08-02763.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Jeong SW, Ikeda SR. G protein alpha subunit Gαz couples neurotransmitter receptors to ion channels in sympathetic neurons. Neuron. 1998;21:1201–1212. doi: 10.1016/s0896-6273(00)80636-4. [DOI] [PubMed] [Google Scholar]
- Kakehata S, Nakagawa T, Takasaka T, Akaike N. Cellular mechanism of acetylcholine-induced response in dissociated outer hair cells of guinea-pig cochlea. The Journal of Physiology. 1993;463:227–244. doi: 10.1113/jphysiol.1993.sp019592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick K, Bourque CW. Activity dependence and functional role of the apamin-sensitive K+ current in rat supraoptic neurones in vitro. The Journal of Physiology. 1996;494:389–398. doi: 10.1113/jphysiol.1996.sp021500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng G, Brown D. The origins and significance of pulsatility in hormone secretion from the pituitary. Journal of Neuroendocrinolgy. 1997;9:493–513. doi: 10.1046/j.1365-2826.1997.00615.x. [DOI] [PubMed] [Google Scholar]
- Li Z, Hatton GI. Ca2+ release from internal stores: role in generating depolarizing after-potentials in rat supraoptic neurones. The Journal of Physiology. 1997;498:339–350. doi: 10.1113/jphysiol.1997.sp021862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogul DJ, Adams ME, Fox AP. Differential activation of adenosine receptors decreases N-type but potentiates P-type Ca2+ current in hippocampal CA3 neurons. Neuron. 1993;10:327–334. doi: 10.1016/0896-6273(93)90322-i. [DOI] [PubMed] [Google Scholar]
- Mouginot D, Kombian SB, Pittman QJ. Activation of presynaptic GABAB receptors inhibits evoked IPSCs in rat magnocellular neurons in vitro. Journal of Neurophysiology. 1998;79:1508–1517. doi: 10.1152/jn.1998.79.3.1508. [DOI] [PubMed] [Google Scholar]
- Noguchi J, Yamashita H. Baclofen inhibits postsynaptic voltage-dependent calcium currents of supraoptic nucleus neurons isolated from young rats. Biomedical Research. 1999;20:239–247. [Google Scholar]
- Oliet SH, Bourque CW. Properties of supraoptic magnocellular neurones isolated from the adult rat. The Journal of Physiology. 1992;455:291–306. doi: 10.1113/jphysiol.1992.sp019302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SHR, Poulain DA. Adenosine-induced presynaptic inhibition of IPSCs and EPSCs in rat hypothalamic supraoptic nucleus neurones. The Journal of Physiology. 1999;520:815–825. doi: 10.1111/j.1469-7793.1999.00815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer TM, Stiles GL. Adenosine receptors Neuropharmacology. 1995;34:683–694. doi: 10.1016/0028-3908(95)00044-7. [DOI] [PubMed] [Google Scholar]
- Pittman QJ. The action is at the terminal. The Journal of Physiology. 1999;520:629. doi: 10.1111/j.1469-7793.1999.t01-1-00629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin N, Platano D, Olcese R, Stefani E, Birnbaumer L. Direct interaction of Gβγ with a C-terminal Gβγ-binding domain of the Ca2+ channel alpha1 subunit is responsible for channel inhibition by G protein-coupled receptors. Proceedings of the National Academy of Sciences of the USA. 1997;94:8866–8871. doi: 10.1073/pnas.94.16.8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainnie DG, Grunze HC, McCarley RW, Greene RW. Adenosine inhibition of mesopontine cholinergic neurons: implications for EEG arousal. Science. 1994;263:689–692. doi: 10.1126/science.8303279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR, Stehle JH, Rivkees SA. Molecular cloning and characterization of a rat A1-adenosine receptor that is widely expressed in brain and spinal cord. Molecular Endocrinology. 1991;5:1037–1048. doi: 10.1210/mend-5-8-1037. [DOI] [PubMed] [Google Scholar]
- Rivkees SA, Price SL, Zhou FC. Immunohistochemical detection of A1 adenosine receptors in rat brain with emphasis on localization in the hippocampal formation, cerebral cortex, cerebellum, and basal ganglia. Brain Research. 1995;677:193–203. doi: 10.1016/0006-8993(95)00062-u. [DOI] [PubMed] [Google Scholar]
- Seale TW, Abla KA, Shamim MT, Carney JM, Daly JW. 3,7-Dimethyl-1-propargylxanthine: a potent and selective in vivo antagonist of adenosine analogs. Life Sciences. 1988;43:1671–1684. doi: 10.1016/0024-3205(88)90478-x. [DOI] [PubMed] [Google Scholar]
- Shibuya I, Tanaka K, Hattori Y, Uezono Y, Harayama N, Noguchi J, Ueta Y, Izumi F, Yamashita H. Evidence that multiple P2X purinoceptors are functionally expressed in rat supraoptic neurones. The Journal of Physiology. 1999;514:351–367. doi: 10.1111/j.1469-7793.1999.351ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldo BL, Moises HC. μ-Opioid receptor activation inhibits N- and P-type Ca2+ channel currents in magnocellular neurones of the rat supraoptic nucleus. The Journal of Physiology. 1998;513:787–804. doi: 10.1111/j.1469-7793.1998.787ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong TW, Stea A, Hodson CD, Dubel SJ, Vincent SR, Snutch TP. Structure and functional expression of a member of the low voltage-activated calcium channel family. Science. 1993;260:1133–1136. doi: 10.1126/science.8388125. [DOI] [PubMed] [Google Scholar]
- Troadec J-D, Thirion S, Nicaise G, Lemos JR, Dayanithi G. ATP-evoked increases in [Ca2+]i and peptide release from rat isolated neurohypophysial terminals via a P2X2 purinoceptor. The Journal of Physiology. 1998;511:89–103. doi: 10.1111/j.1469-7793.1998.089bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemiya M, Berger AJ. Activation of adenosine A1 and A2 receptors differentially modulates calcium channels and glycinergic synaptic transmission in rat brainstem. Neuron. 1994;13:1439–1446. doi: 10.1016/0896-6273(94)90429-4. [DOI] [PubMed] [Google Scholar]
- Widmer H, Amerdeil H, Fontanaud P, Desarmenien MG. Postnatal maturation of rat hypothalamoneurohypophysial neurons: evidence for a developmental decrease in calcium entry during action potentials. Journal of Neurophysiology. 1997;77:260–271. doi: 10.1152/jn.1997.77.1.260. [DOI] [PubMed] [Google Scholar]
- Zhang JF, Ellinor PT, Aldrich RW, Tsien RW. Multiple structural elements in voltage-dependent Ca2+ channels support their inhibition by G proteins. Neuron. 1996;17:991–1003. doi: 10.1016/s0896-6273(00)80229-9. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Ikeda SR. Adenosine modulates voltage-gated Ca2+ channels in adult rat sympathetic neurons. Journal of Neurophysiology. 1993;70:610–620. doi: 10.1152/jn.1993.70.2.610. [DOI] [PubMed] [Google Scholar]