

Abstract
Acidosis alters the transient outward current, ito, in the heart. We have studied the mechanism underlying the effect of acidosis on one of the K+ channels, Kv1.4 (heterologously expressed in Xenopus laevis oocytes), known to underlie ito.
At pH 6.5, wild-type Kv1.4 current was inhibited during repetitive pulsing, in part as a result of a slowing of recovery from N-type inactivation.
Acidosis still caused slowing of recovery after deletion of just one (either the first or second) of the N-terminal inactivation ball domains. However, deletion of both the N-terminal inactivation ball domains greatly reduced the inhibition.
As well as the N-terminus, other parts of the channel are also required for the effect of acidosis, because, whereas the transfer of the N-terminus of Kv1.4 to Kv1.2 conferred N-type inactivation, it did not confer acidosis sensitivity.
Replacement of an extracellular histidine with a glutamine residue (H508Q) abolished the slowing of recovery by acidosis. Reduction of C-type inactivation by raising the bathing K+ concentration or by the mutation K532Y also abolished the slowing.
It is concluded that binding of protons to H508 enhances C-type inactivation and this causes a slowing of recovery from N-type inactivation and, thus, an inhibition of current during repetitive pulsing.
In many species, the transient outward K+ current (ito) is found throughout the heart – in the atrial and ventricular muscle, the sinoatrial and atrioventricular nodes and Purkinje fibres – although its density varies in different regions (Campbell et al. 1995). This current is an important determinant of the early repolarization phase of the cardiac action potential (Campbell et al. 1995). Kv1.4, Kv4.2 and Kv4.3 have been suggested to be responsible for ito (Comer et al. 1994; Fiset et al. 1997; Johns et al. 1997; Barry et al. 1998; Brahmajothi et al. 1999; Wickenden et al. 1999; Guo et al. 1999; Wang et al. 1999; Xu et al. 1999). There are functional differences in ito between species and between regions of the heart and it is possible that Kv4.2 and Kv4.3 underlie a phenotype of ito which shows fast recovery from inactivation (in human and ferret ventricular sub-epicardium and rat right ventricular free wall) and Kv1.4 underlies a phenotype which shows slow recovery from inactivation (in human and ferret ventricular sub-endocardium and rat ventricular septum) (Brahmajothi et al. 1999; Wickenden et al. 1999; Kaab et al. 1999). In addition, Kv1.4 expression is known to be high in the neonate and during cardiac hypertrophy (Xu et al. 1996; Guo et al. 1998). Given its important role in determining the configuration of the cardiac action potential, and the heterogeneity of ito in different regions of the heart, interventions that affect the function of the channels underlying ito may have important effects on action potential dispersion and, hence, arrhythmogenesis. One such intervention is acidosis, which has previously been shown to alter ito (Stengl et al. 1998; Hulme & Orchard, 2000). The site of action of acidosis on ito is unknown. In the present study, we have investigated the possible molecular mode and site of action of acidosis on Kv1.4, one of the channels thought to underlie ito in heart. Abstracts of this work have previously been presented to The Physiological Society (Owen et al. 1997; Claydon et al. 1999).
METHODS
Molecular biology
Experiments were carried out on Kv1.4 from rat and ferret, rKv1.4 and fKv1.4, respectively. The mutants fKv1.4 Δ2-146, rKv1.4 Δ2-39 and rKv1.4 Δ39-162 have been described previously (Rasmusson et al. 1995; Kondoh et al. 1997). To construct the chimera Kv1.4 (1-185):Kv1.2 (41-C-terminus), part of the 5′ non-coding region and the coding region for the N-terminal 185 amino acids of rat Kv1.4 was amplified by PCR. The sense primer (5′-GGGGGCCCACAACTGGAACCAGCCATTT-3′) contained an Apa I site at the 5′ end and the antisense primer (5′-AAGGCCTGACACATTTATTACCACGCG-3′) contained a Stu I site. The amplified PCR fragment was digested with Apa I and Stu I and ligated to rat Kv1.2 in pBluescript between Apa I and Stu I sites (the Apa I site is located in the multiple cloning site of the vector and the Stu I site is at nucleotide 120 of Kv1.2). The mutant rKv1.4 H508Q was constructed by overlap extension PCR, using a pair of complementary primers (5′-CTACCACCCAGTTCCAAAGCA-3′ and 5′-TGCTTTGGAACTGGGTGGTAG-3′) containing the desired base changes as described before (Mikaelian & Sergeant, 1996). The mutants rKv1.4 K532Q and rKv1.4 K532Y were constructed using the Stratagene QuikChange site-directed PCR mutagenesis technique (Stratagene, CA, USA) according to the instructions of the manufacturer. Sense primers for rKv1.4 K532Q and rKv1.4 K532Y were 5′-GCTACGGGGACATGCAGCCCATCACAG-3′ and 5′-GCTACGGGGACATGTATCCCATCACAGTGGG-3′, respectively. In all cases, vectors (pBluescript SK− for fKv1.4 and pBluescript SKII+ for rKv1.4) containing the cDNA sequences were linearized with a restriction endonuclease (Eco RI for rKv1.4 and Asp 718 for fKv1.4) and cRNA was prepared from these templates with either T3 (fKv1.4) or T7 (rKv1.4) RNA polymerase (Stratagene). Transcribed RNA was diluted in DEPC-treated water at a final concentration of 50 ng μl−1 (rKv1.4) or 100 ng μl−1 (fKv1.4).
Electrophysiology
Xenopus laevis toads were terminally anaesthetized by immersion in tricaine methanesulphonate (2 mg ml−1, Sigma) in accordance with the Home Office Animals (Scientific Procedures) Act of 1986. Stage V-VI oocytes were isolated and then defolliculated using a combination of collagenase treatment (1 h in 1 mg ml−1 collagenase type 1a; Sigma) and manual defolliculation. Defolliculated oocytes were incubated in Barth's medium for 2–24 h at 19°C before injection. Barth's medium contained (mM): 88 NaCl, 1 KCl, 2.4 NaHCO3, 0.82 MgSO4, 0.33 Ca(NO3)2, 0.41 CaCl2, 20 Hepes and 1.25 sodium pyruvate, with 0.1 mg ml−1 neomycin (Sigma) and 100 units/0.1 mg ml−1 penicillin/streptomycin mix (Sigma), titrated to pH 7.4 using NaOH. Oocytes were injected with 50 nl of cRNA encoding rKv1.4 (2.5 ng) or fKv1.4 (5 ng) using a Drummond digital microdispenser (Broomall, USA). In control experiments, oocytes were injected with 50 nl of diethyl pyrocarbonate (DEPC)-treated water; in these cases, currents were negligible compared to currents in cells injected with cRNA. Currents were recorded 16–72 h later using the two-electrode voltage clamp technique. Microelectrodes with a resistance of 0.6-3 MΩ (tip diameter, 1–5 μm) filled with 3 M KCl were used. Membrane currents were recorded using a GeneClamp 500 amplifier (Axon Instruments) with computer-driven voltage protocols (Clampex software and Digidata 1200 interface, Axon Instruments). Currents were recorded during 200 ms pulses to potentials between -80 and +60 or +90 mV from a holding potential of -80 mV to construct current-voltage relationships. The pulse frequency was 0.5, 0.067 or 0.004 Hz (corresponding to pulse intervals of 2, 15 and 240 s). A conventional conditioning-test pulse protocol was used to measure the recovery from inactivation. Currents were recorded during a 200 ms test pulse to +40 mV at different intervals following a 200 ms conditioning pulse to +40 mV (holding potential, -80 mV). The conditioning pulse frequency was 0.067 Hz (pulse interval, 15 s), except in the case of rKv1.4 Δ2-39 and rKv1.4 K532Q when it was 0.004 Hz (pulse interval, 240 s) and rKv1.4 Δ39-162 (at pH 6.5 only) when it was 0.008 Hz (pulse interval, 120 s). Experiments were performed at room temperature (20-22°C). During experiments, oocytes were perfused with ND96 solution (mM: 96 NaCl, 3 KCl, 1 MgCl2, 2 CaCl2 and 5 Hepes, titrated to pH 6.0-8.9 using NaOH) at a flow of 0.5 ml min−1. Oocytes were normally perfused with ND96 solution at pH 7.4 to obtain control recordings, and recordings were subsequently made in ND96 solution at another pH. After a change in bath solution, 3 min was allowed for equilibration before recordings were made (bath volume, 400 μl).
pH measurement
Intracellular pH was measured using the pH-sensitive fluorescent dye BCECF (Molecular Probes). Oocytes were incubated in ND96 solution (pH 7.4) with 70 μm BCECF AM for 20 min. After dye loading, oocytes were washed for 10 min with ND96 solution (pH 7.4). Oocytes were placed in an experimental chamber and held in place using a blunt electrode. ND96 solution at 21–22°C was perfused at a rate of 2 ml min−1. Oocytes were viewed using a Diaphot TMD epifluorescence microscope (Nikon, Tokyo, Japan). The focus was set at the centre of the bottom of the cell. Fluorescence at 540 nm was measured in response to excitation at 440 and 490 nm (Cairn Research, Kent, UK). The fluorescence ratio (490 nm/440 nm), a measure of intracellular pH, was recorded from oocytes during perfusion of ND96 solution titrated to pH 8.5, 7.4 and 6.5, and ND96 solution (pH 7.4) with 10 mm NH4Cl (NaCl concentration was reduced by the same amount). After a change in bath solution, 15 min was allowed for equilibration before recordings were made.
Analysis and statistics
Data were analysed using pCLAMP software (Axon Instruments). In each case (except fKv1.4 Δ2-146) current was measured as the difference between peak outward current at the start of the pulse and current at the end of the pulse. fKv1.4 Δ2-146 current was measured as peak outward current at the start of the pulse. For current-voltage relationships, currents were normalized to current at +90 mV (+60 mV for fKv1.4 Δ2-146) at pH 7.4 (Inorm). For recovery from inactivation, test pulse current (as a fraction of current during the preceding conditioning pulse) was plotted against the preceding test interval. Data are shown as means ±s.e.m. (n= number of oocytes). Statistical analysis was carried out using SigmaStat (Jandel Scientific Software, CA, USA). In the figures, the pulse interval and n are shown when appropriate, current- voltage relationships are fitted with lines to guide the eye, and recovery from inactivation is fitted with a single exponential function. Curve fitting was carried out using SigmaPlot (Jandel Scientific) or pCLAMP software.
RESULTS
Acidosis-induced inhibition of Kv1.4 caused by a slowing of recovery from inactivation
rKv1.4 current was recorded during 200 ms pulses from -80 mV to potentials up to +90 mV at a pulse frequency of 0.5 Hz. Figure 1A and B shows rKv1.4 current at +90 mV and mean current-voltage relationships at pH 8.5, 7.4 and 6.5. rKv1.4 current was inhibited during acidosis at all potentials in a voltage-independent manner. Inactivation of rKv1.4 current was also accelerated by acidosis: at +90 mV, the time constant of inactivation was 56 ± 6, 59 ± 8 and 48 ± 6 ms at pH 8.5, 7.4 and 6.5, respectively (n= 5; ANOVA, P < 0.05 for pH 6.5 vs. pH 7.4). The effect of acidosis was frequency dependent: Fig. 1C shows mean current-voltage relationships when the pulse frequency was decreased to 0.067 Hz. Under these conditions, the effect of acidosis was almost eliminated. The frequency dependence suggests that the inhibition of Kv1.4 is the result of a slowing of recovery from inactivation.
Figure 1. Effect of pH on wild-type rKv1.4.
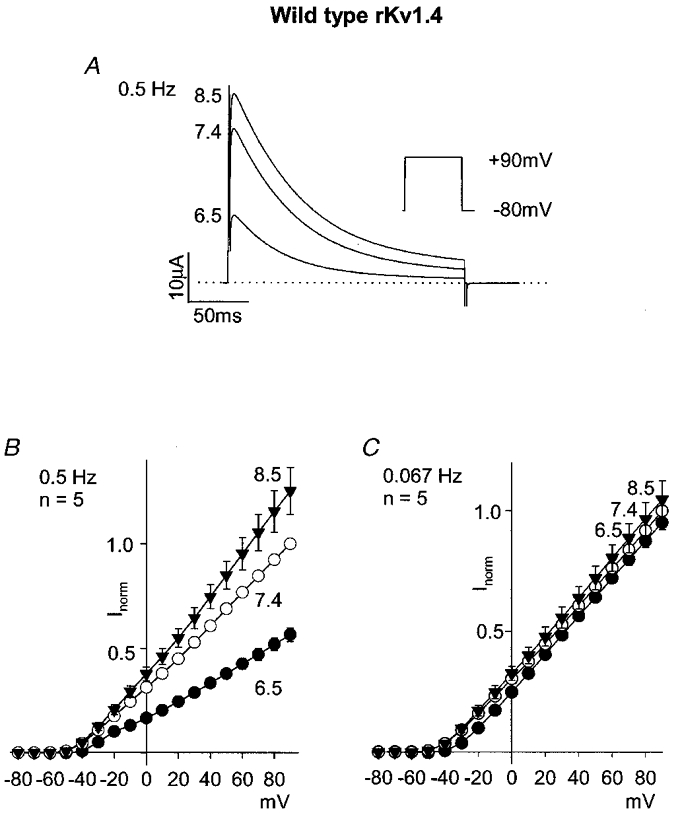
A, currents at pH 8.5, 7.4 and 6.5 during a pulse to +90 mV (protocol shown in inset; pulse frequency, 0.5 Hz). B and C, mean current-voltage relationships at pH 8.5 (▾), 7.4 (○) and 6.5 (•) at pulse frequencies of 0.5 (B) and 0.067 Hz (C).
Recovery from inactivation at -80 mV was studied using a conventional conditioning-test pulse protocol (Fig. 2A); conditioning pulses were applied at a frequency of 0.067 Hz. Figure 2B shows the time course of recovery at pH 8.5, 7.4 and 6.5; mean rKv1.4 current during the test pulse, expressed as a fraction of the current during the conditioning pulse (I/Imax) at the same pH, is plotted against the test interval between the conditioning and test pulses. Recovery from inactivation was slowed during acidosis: the time constant of recovery was 1.1 ± 0.1, 1.4 ± 0.1 and 2.6 ± 0.2 s at pH 8.5, 7.4 and 6.5 (n= 5; ANOVA, P < 0.05 for pH 6.5 vs. pH 7.4; see also Table 1). The acidosis-dependent inhibition of rKv1.4 current during repetitive pulsing at a pulse frequency of 0.5 Hz (Fig. 1A and B) must in part be the result of the slowing of recovery (this is considered in full in the Discussion). The inset in Fig. 2B shows the mean current amplitude (in μA) during the test pulse after the test interval of 7.5 s (the longest test interval used) at pH 8.5, 7.4 and 6.5. Current amplitude was reduced in acidosis; the significance of this is considered in the Discussion.
Figure 2. Effect of pH on the recovery of wild-type rKv1.4 from inactivation.
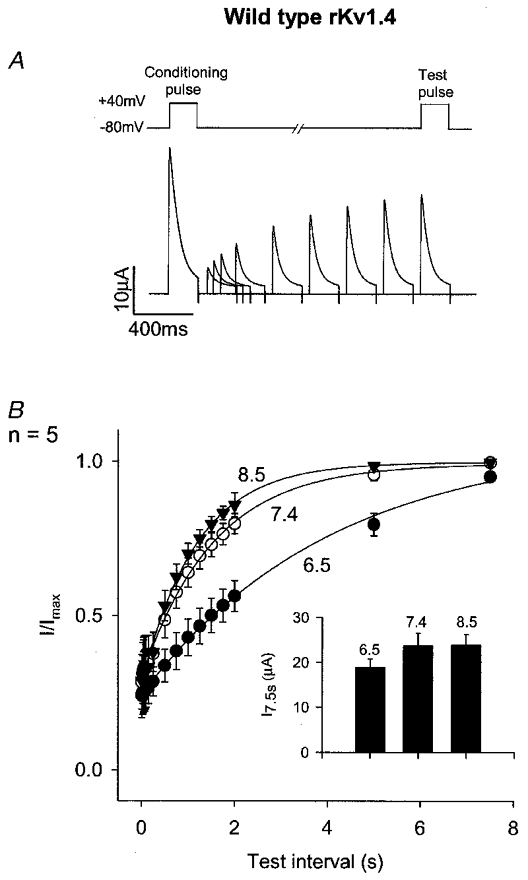
A, protocol. Top, schematic trace of membrane potential. Test pulses were applied at different test intervals after conditioning pulses (conditioning pulse frequency, 0.067 Hz). Bottom, superimposed examples of conditioning and test pulse currents at different test intervals at pH 7.4. B, mean time course of recovery from inactivation at -80 mV at pH 8.5 (▾), 7.4 (○) and 6.5 (•). Inset, mean current amplitude (in μA) during the test pulse after a test interval of 7.5 s at the pH shown.
Table 1.
Time constants of recovery from inactivation at −80 mV
| Channel/conditions | pH 8.5(% change) | pH 8.5(s) | pH 7.4(s) | pH 6.5(s) | pH 6.5(% change) | n |
|---|---|---|---|---|---|---|
| Wild-type rKv1.4 | −23 | 1.1 ± 0.1 | 1.4 ± 0.1 | 2.6 ± 0.2 | 84 | 5 |
| Wild-type fKv1.4 | −9 | 1.6 ± 0.1 | 1.8 ± 0.1 | 5.3 ± 0.6 | 201 | 5 |
| rKv1.4 Δ2–39 (Δball 1) | — | — | 53.0 ± 4.9 | 69.7 ± 9.3 | 32 | 5 |
| rKv1.4 Δ39–162 (Δball 2) | −39 | 2.9 ± 0.3 | 4.8 ± 0.9 | 14.9 ± 0.6 | 310 | 5 |
| rKv1.4:Kv1.2 | −7 | 1.2 ± 0.3 | 1.3 ± 0.2 | 1.2 ± 0.2 | −9 | 5 |
| rKv1.4 H508Q | −20 | 1.7 ± 0.2 | 2.1 ± 0.3 | 1.8 ± 0.2 | −13 | 5 |
| Wild-type rKv1.4, 99 mm K+ | −15 | 0.2 ± 0.01 | 0.2 ± 0.02 | 0.2 ± 0.01 | 8 | 4 |
| rKv1.4 K532Q | — | — | 12 ± 4 | — | — | 3 |
| rKv1.4 K532Y | −14 | 0.7 ± 0.1 | 0.8 ± 0.2 | 0.7 ± 0.2 | −12 | 4 |
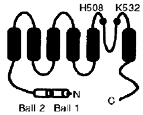
Percentage changes are given with respect to values at pH 7·4. n, number of oocytes studied. Some time constants for rKv1.4 Δ2–39 and rKv1.4 K532Q are missing, because the slow recovery of rKv1.4 Δ2–39 precluded measurement of recovery under all conditions and rKv1.4 K532Q current was too small in acidosis to be measured. The inset shows the membrane topology of Kv1.4 and the locations of mutations made.
Involvement of the N-terminus
Since inactivation of Kv1.4 is largely N-type (Rasmusson et al. 1995), the effect of acidosis could be the result of a modification of N-type inactivation. This was explored using an N-terminal deletion mutant of fKv1.4 (ferret clone), fKv1.4 Δ2-146, that has previously been shown to lack N-type inactivation (Rasmusson et al. 1995). Control experiments showed that the response of wild-type fKv1.4 to acidosis was similar to the response of rKv1.4 (rat clone): a decrease in current amplitude during repetitive pulsing and a slowing of recovery from inactivation (Table 1; ANOVA, P < 0.05 for pH 6.5 vs. pH 7.4). Figure 3 shows wild-type fKv1.4 (Fig. 3A) and fKv1.4 Δ2-146 (Fig. 3C) current at +60 mV (the loss of N-type inactivation in the case of the mutant channel is evident) and mean current- voltage relationships at pH 8.5, 7.4 and 6.5 (Fig. 3B and D). The effect of acidosis on the mutant channel current amplitude was less than the effect on the wild-type channel current amplitude. This is also shown by the dose-response curves in Fig. 3E in which the current at +60 mV (normalized to the theoretical maximum current) is plotted against the H+ concentration. Although this shows that the response of the mutant channel to acidosis was reduced, it also shows that it was not abolished – the dose-response curve was shifted to the right by one log unit (wild-type fKv1.4, pKa (-log dissociation constant) 7.4; fKv1.4 Δ2-146, pKa 6.1). Nevertheless, these data support the hypothesis that acidosis inhibits Kv1.4 current in part by slowing recovery from N-type inactivation.
Figure 3. Effect of pH on wild-type fKv1.4 and mutant fKv1.4 lacking the N-terminus (fKv1.4 Δ2-146).
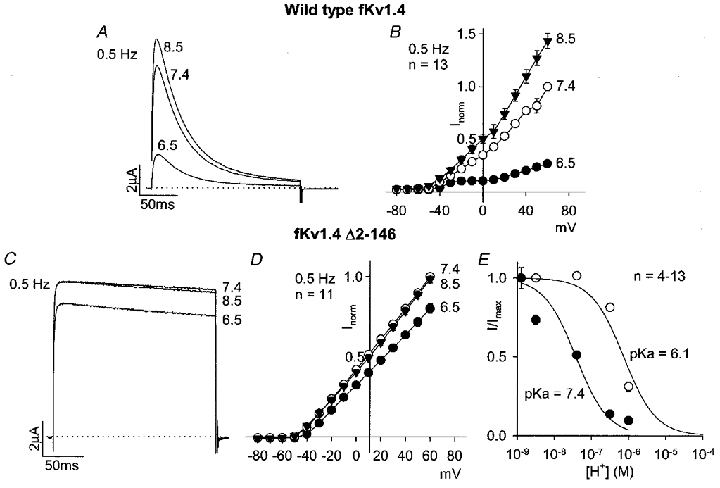
A and C, wild-type fKv1.4 (A) and fKv1.4 Δ2-146 (C) currents at pH 8.5, 7.4 and 6.5 during a pulse to +60 mV (pulse frequency, 0.5 Hz). B and D, mean current-voltage relationships for wild-type fKv1.4 (B) and fKv1.4 Δ2-146 (D) at pH 8.5 (▾), 7.4 (○) and 6.5 (•) at a pulse frequency of 0.5 Hz. E, mean dose-response curves for the effect of pH on wild-type fKv1.4 (•) and fKv1.4 Δ2-146 (○). Current at +60 mV at 0.5 Hz (normalized to the theoretical maximum current under alkaline conditions) is plotted against H+ concentration (on a logarithmic scale). Data were fitted with typical dose-response curves (assuming a Hill coefficient of 1); pKa values are shown.
There are known to be two inactivation ball domains on the N-terminus of Kv1.4 (Kondoh et al. 1997). Figure 4 shows the effect of acidosis on two deletion mutants of rKv1.4 lacking either the first or the second inactivation ball domain, rKv1.4 Δ2-39 and rKv1.4 Δ39-162 (Kondoh et al. 1997), respectively. Acidosis inhibited both mutant channel currents (Fig. 4A, B, D and E). Recovery from inactivation of both mutant channels was slower than that of the wild-type channel (Fig. 4C and F, Table 1) – in the case of the mutant channel lacking the first inactivation ball domain, rKv1.4 Δ2-39, the time constant of recovery from inactivation at pH 7.4 was 53.0 ± 4.9 s rather than 1.4 ± 0.1 s in the case of the wild-type channel. On going from pH 7.4 to pH 6.5, the time constant of recovery from inactivation was slowed by 84 % in the case of the wild-type channel, 32 % in the case of the mutant channel lacking the first inactivation ball domain, rKv1.4 Δ2-39, and 310 % in the case of the mutant channel lacking the second inactivation ball domain, rKv1.4 Δ39-162 (Fig. 4C and F, Table 1). This suggests that the effect of acidosis on recovery from inactivation requires only one inactivation ball domain, either the first or the second. The insets in Fig. 4C and F show that the current amplitude (in μA) during the test pulse after the longest test interval (240 or 7.5 s) was still reduced in acidosis with rKv1.4 Δ2-39 and rKv1.4 Δ39-162.
Figure 4. Effect of pH on mutants of rKv1.4 lacking the first (rKv1.4 Δ2-39) or second (rKv1.4 Δ39-162) N-terminal inactivation ball domain.
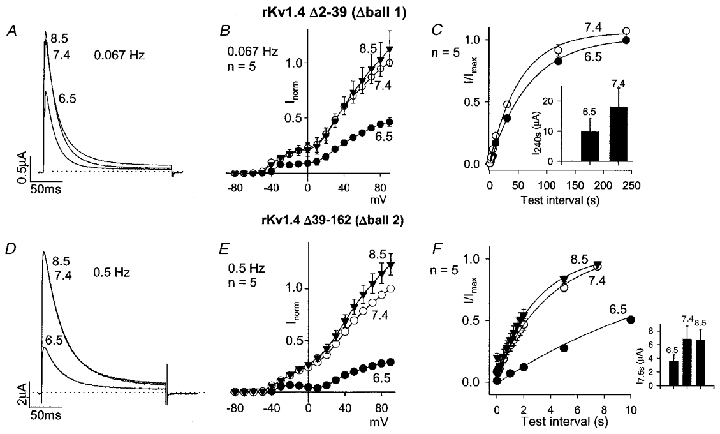
A and D, rKv1.4 Δ2-39 (A) and rKv1.4 Δ39-162 (D) currents at pH 8.5, 7.4 and 6.5 during a pulse to +90 mV (pulse frequency, 0.067 or 0.5 Hz as shown; low pulse frequency used in the case of rKv1.4 Δ2-39, because otherwise current amplitude was too small as result of slow recovery from inactivation). B and E, mean current-voltage relationships for rKv1.4 Δ2-39 (B) and rKv1.4 Δ39-162 (E) at pH 8.5 (▾), 7.4 (○) and 6.5 (•) at a pulse frequency of 0.067 or 0.5 Hz as shown. C and F, mean time course of recovery of rKv1.4 Δ2-39 (C) and rKv1.4 Δ39-162 (F) from inactivation at -80 mV at pH 8.5 (▾; F only), 7.4 (○) and 6.5 (•). Note that test intervals up to 240 s in duration were used for rKv1.4 Δ2-39 (C), because recovery from inactivation was slow. Conditioning pulse frequency, 0.004 (C), 0.067 (F; pH 7.4 and 8.5) or 0.008 Hz (F; pH 6.5). Inset, mean current amplitude (in μA) during the test pulse after a test interval of 240 (C) or 7.5 s (F) at the pH shown.
Involvement of H508
If the N-terminus is in part or in total responsible for the acidosis sensitivity of Kv1.4 (as the experiments above suggest), then transfer of the N-terminus to another channel should confer acidosis sensitivity. Kv1.2 shows minimal inactivation (C-type; no N-type) and is not sensitive to acidosis (Steidl & Yool, 1999). The N-terminus (amino acids 1–40) of Kv1.2 was replaced by the N-terminus (amino acids 1–185) of rKv1.4. The resulting chimera showed fast (presumably N-type) inactivation (Fig. 5A). Despite the apparently successful transfer of N-type inactivation to Kv1.2, the chimeric channel was little affected by acidosis: there was little effect on current amplitude during repetitive pulsing (Fig. 5A and B) and little or no effect on recovery from inactivation (Fig. 5C, Table 1). The inset in Fig. 5C shows that the current amplitude (in μA) during the test pulse after the longest test interval (7.5 s) was also little affected by acidosis. This suggests that other parts of Kv1.4 in addition to the N-terminus are required for the effect of acidosis.
Figure 5. Effect of pH on rKv1.4:Kv1.2 chimera – rKv1.4 (1-185):Kv1.2 (41-C-terminus).
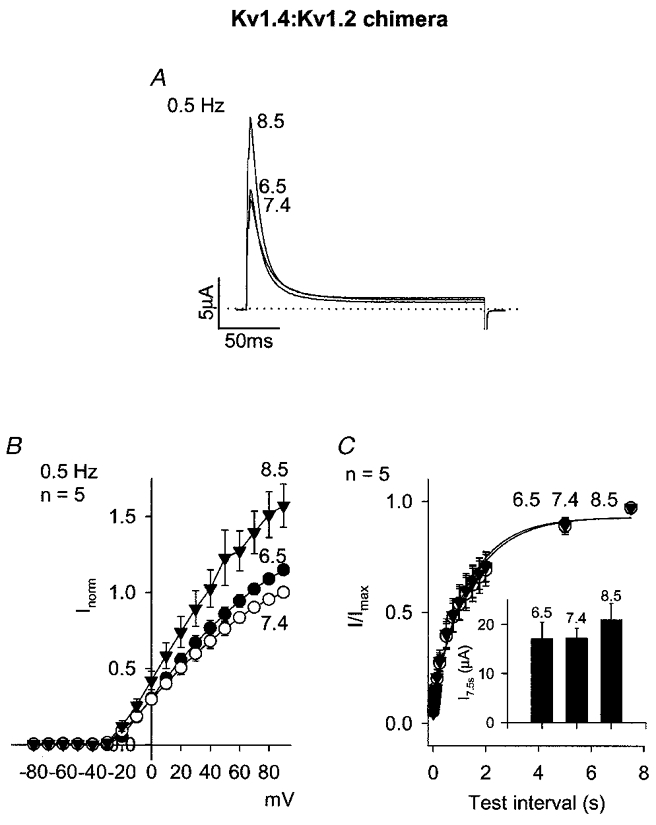
A, currents at pH 8.5, 7.4 and 6.5 during a pulse to +90 mV (pulse frequency, 0.5 Hz). B, mean current-voltage relationships at pH 8.5 (▾), 7.4 (○) and 6.5 (•) at a pulse frequency of 0.5 Hz. C, mean time course of recovery from inactivation at -80 mV at pH 8.5 (▾), 7.4 (○) and 6.5 (•). Conditioning pulse frequency, 0.067 Hz. Inset, mean current amplitude (in μA) during the test pulse after a test interval of 7.5 s at the pH shown.
Figure 6 shows the effect of acidosis on a mutant form of rKv1.4 in which an extracellular histidine (H508) was replaced by a glutamine (Q) residue. At pH 7.4, the properties of rKv1.4 H508Q were similar to those of wild-type rKv1.4. However, acidosis had no effect on either current amplitude during repetitive pulsing (Fig. 6A and B) or recovery from inactivation (Fig. 6C, Table 1). The inset in Fig. 6C shows that the current amplitude (in μA) during the test pulse after the longest test interval (7.5 s) was also unaffected by acidosis. It is possible that H508 is the proton-binding site mediating the effect of acidosis.
Figure 6. Effect of pH on a mutant of rKv1.4 lacking the extracellular histidine residue at position 508 (rKv1.4 H508Q).
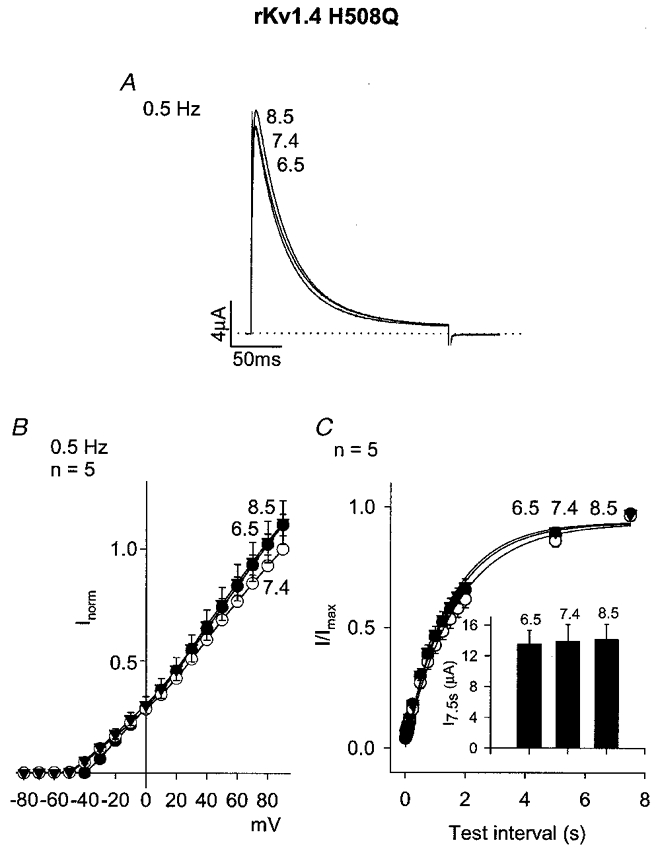
A, currents at pH 8.5, 7.4 and 6.5 during a pulse to +90 mV (pulse frequency, 0.5 Hz). B, mean current-voltage relationships at pH 8.5 (▾), 7.4 (○) and 6.5 (•) at a pulse frequency of 0.5 Hz. C, mean time course of recovery from inactivation at -80 mV at pH 8.5 (▾), 7.4 (○) and 6.5 (•). Conditioning pulse frequency, 0.067 Hz. Inset, mean current amplitude (in μA) during the test pulse after a test interval of 7.5 s at the pH shown.
Involvement of C-type inactivation
On the basis of the results presented so far, it is suggested that the binding of protons to H508 results in a slowing of recovery from N-type inactivation. Binding of protons to an extracellular residue (H508) may affect N-type inactivation by acting on C-type inactivation. This is feasible because (i) Kv1.4 is known to show C-type inactivation, (ii) C-type inactivation of Kv1.4 is known to involve extracellular residues close to H508 and (iii) recovery from C-type inactivation of Kv1.4 is known to determine recovery from N-type inactivation (Rasmusson et al. 1995). C-type inactivation of Kv1.4 is known to be reduced by raising the extracellular K+ concentration (Rasmusson et al. 1995). Figure 7 shows data obtained using wild-type rKv1.4 in the presence of a high extracellular K+ concentration (99 mm). Under these conditions, recovery from inactivation at pH 7.4 was accelerated (Fig. 7C, Table 1) consistent with a reduction of C-type inactivation (see Discussion). In the presence of 99 mm K+, acidosis had a small effect on current amplitude during repetitive pulsing (Fig. 7A and B) and no effect on recovery from inactivation (Fig. 7C, Table 1). This supports the possibility that C-type inactivation is involved. The inset in Fig. 7C shows that the current amplitude (in μA) during the test pulse after the longest test interval (7.5 s) was still affected by acidosis in the presence of 99 mm K+.
Figure 7. Effect of pH on wild-type rKv1.4 in the presence of high K+ (99 mm).
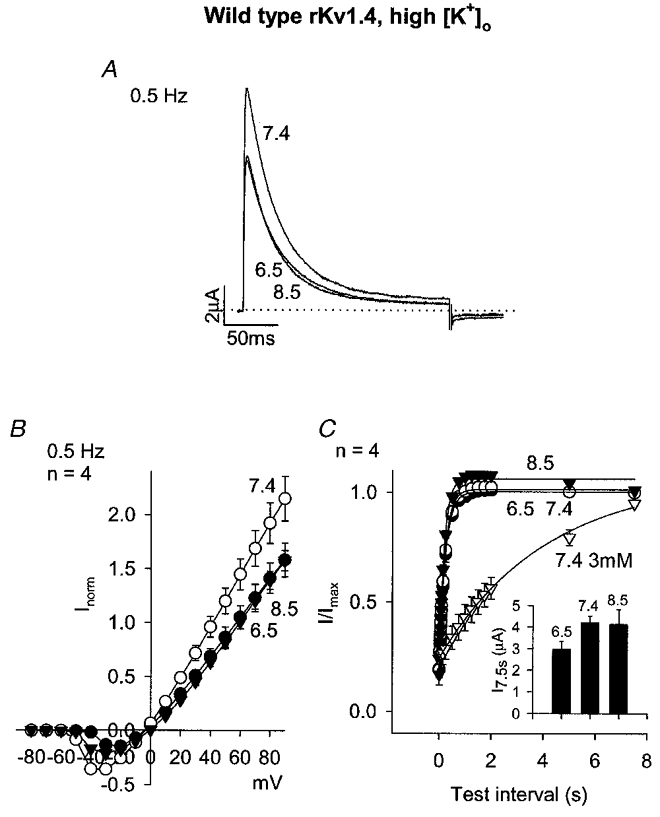
A, currents at pH 8.5, 7.4 and 6.5 during a pulse to +90 mV (pulse frequency, 0.5 Hz). B, mean current-voltage relationships at pH 8.5 (▾), 7.4 (○) and 6.5 (•) at a pulse frequency of 0.5 Hz. C, mean time course of recovery from inactivation at -80 mV at pH 8.5 (▾), 7.4 (○) and 6.5 (•) in the presence of 99 mm extracellular K+ and pH 7.4 in the presence of 3 mm (i.e. normal) extracellular K+ (▿). Conditioning pulse frequency, 0.067 Hz. Inset, mean current amplitude (in μA) during the test pulse after a test interval of 7.5 s at the pH shown (all data obtained in the presence of 99 mm K+).
K532 in Kv1.4 is known to be involved in C-type inactivation (Rasmusson et al. 1995). K532 in rKv1.4 was replaced by a glutamine or tyrosine residue. An increase of C-type inactivation is expected to slow recovery from N-type inactivation, whereas a reduction of C-type inactivation is expected to accelerate it (see Discussion). In the case of rKv1.4 K532Q, C-type inactivation was probably increased, because at pH 7.4 recovery from N-type inactivation was slowed as compared to recovery of the wild-type channel (Fig. 8C, Table 1), whereas in the case of rKv1.4 K532Y, it was probably reduced, because at pH 7.4 recovery from N-type inactivation was accelerated as compared to recovery of the wild-type channel (Fig. 8F, Table 1) (cf. López-Barneo et al. 1993; Rasmusson et al. 1995). Comparison of Fig. 8A and B (rKv1.4 K532Q) with Fig. 1A and B (wild-type rKv1.4) shows that acidosis had a greater effect on current amplitude in experiments on rKv1.4 K532Q (despite the pulse frequency being 0.004 Hz, to enhance current amplitude, in the experiments shown in Fig. 8A and B, and 0.5 Hz in the experiments shown in Fig. 1A and B). This suggests that when C-type inactivation is increased acidosis sensitivity is enhanced. With rKv1.4 K532Q, recovery from inactivation at pH 6.5 could not be studied, because the current was too small. In contrast, in the case of rKv1.4 K532Y, acidosis had little or no effect on current amplitude during repetitive pulsing (Fig. 8D and E), recovery from inactivation (Fig. 8F, Table 1) or the current amplitude (in μA) during the test pulse after the longest test interval (7.5 s; Fig. 8F, inset). This confirms the result obtained with 99 mm K+ that when C-type inactivation is reduced, acidosis sensitivity is reduced.
Figure 8. Effect of pH on mutants of rKv1.4 in which the residue known to be involved in C-type inactivation (K532) is replaced by a glutamine (rKv1.4 K532Q) or tyrosine (rKv1.4 K532Y) residue.
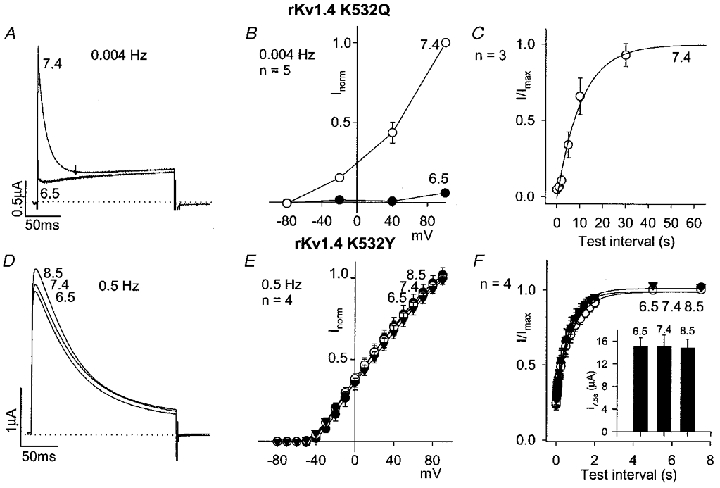
A and D, rKv1.4 K532Q (A) and rKv1.4 K532Y (D) currents at pH 8.5, 7.4 and 6.5 during a pulse to +90 mV (pulse frequency, 0.004 or 0.5 Hz as shown; low pulse frequency used in the case of rKv1.4 K532Q, because otherwise current amplitude was too small as result of slow recovery from inactivation). B and E, mean current-voltage relationships for rKv1.4 K532Q (B) and rKv1.4 K532Y (E) at pH 8.5 (▾; E only), 7.4 (○) and 6.5 (•) at a pulse frequency of 0.004 or 0.5 Hz as shown. C and F, mean time course of recovery of rKv1.4 K532Q (C) and rKv1.4 K532Y (F) from inactivation at -80 mV at pH 8.5 (▾; F only), 7.4 (○) and 6.5 (•; F only). Conditioning pulse frequency, 0.004 (C) and 0.067 Hz (F). Inset in F, mean current amplitude (in μA) during the test pulse after a test interval of 7.5 s at the pH shown.
Nature of acidosis
In the present study, pH was varied by varying the pH of the Hepes-buffered bathing solution. In four oocytes, intracellular pH was measured using BCECF AM. On switching pH from 7.4 to 6.5 for 15 min there was little or no observable change in fluorescence ratio, whereas on application of 10 mm NH4Cl for 15 min there was a substantial change. The change in fluorescence ratio on switching pH from 7.4 to 6.5 was 2.6 ± 5.8 % of that induced by NH4Cl. This suggests that intracellular pH declined by less than 0.005 pH units after switching from pH 7.4 to pH 6.5 (Sasaki et al. 1992). It is concluded that the manoeuvre used in the present study predominantly affects extracellular pH.
DISCUSSION
The present study has shown that extracellular acidosis inhibits Kv1.4 current, at least in part, as a result of a slowing of recovery from N-type inactivation. The inhibition was greatly reduced after deletion of much of the N-terminus (although the slowing still occurred after deletion of only the first or the second N-terminal inactivation ball domain). Other parts of the channel must also be involved, however, because transfer of the N-terminus to Kv1.2 conferred N-type inactivation but not the acidosis sensitivity. The mutation H508Q abolished the slowing of recovery; inhibition of C-type inactivation by 99 mm K+ or by the mutation K532Y also abolished the slowing. In addition, the mutation K532Q enhanced the inhibition of current amplitude by acidosis during repetitive pulsing. On the basis of these data, it is suggested that protons bind to the extracellular residue H508, and this enhances C-type inactivation. This in turn results in a slowing of recovery from N-type inactivation (involving either the first or the second N-terminal inactivation ball domains), thus decreasing current amplitude during repetitive pulsing.
Linkage of C- and N-type inactivation
Central to our hypothesis is a linkage between C- and N-type inactivation. This was first shown by Rasmusson et al. (1995). During a 200 ms pulse, as used in the present study, inactivation is largely N-type, because inactivation is almost completely lost on removal of the N-terminus (Fig. 3A and C; Rasmusson et al. 1995). Recovery from inactivation after a 200 ms pulse (e.g. Fig. 2) must, therefore, be recovery from N-type inactivation. Rasmusson et al. (1995), however, showed that recovery from N-type inactivation is determined by the speed of recovery from C-type inactivation; in their study, recovery from N- and C-type inactivation proceeded at the same rate, and any intervention that reduced C-type inactivation (high K+, K532Y) accelerated recovery from both C- and N-type inactivation (such acceleration of recovery from N-type inactivation can also be seen in Figs 7C and 8F, and Table 1). The slowing of recovery from N-type inactivation during acidosis in the present study, therefore, is consistent with an enhancement of C-type inactivation by acidosis. There is evidence of this in other channels (see below).
Acidosis-dependent inhibition of Kv1.4
The slowing of recovery from inactivation during acidosis must in part be responsible for the inhibition of current amplitude during repetitive pulsing during acidosis. For example, during pulsing at a frequency of 0.5 Hz (pulse interval, 2 s) wild-type rKv1.4 current was decreased by 45 % at +40 mV at pH 6.5 (as compared to pH 7.4; Fig. 1A). From Fig. 2B, the slowing of recovery at pH 6.5 is expected to result in a 24 % decrease in current (as compared to that at pH 7.4) at a pulse interval of 2 s. Thus, the slowing of recovery can in part, but not in full, explain the decrease in current amplitude during repetitive pulsing. The normalization of the data in Fig. 2B (and other similar figures) conceals another effect of acidosis: the inset in Fig. 2B shows that the amplitude of rKv1.4 current (in μA) during the test pulse after a test interval of 7.5 s (longest test interval studied) was decreased by 20 % at pH 6.5. This effect is also responsible, in part, for the acidosis-dependent inhibition of current amplitude during repetitive pulsing. The decrease in wild-type rKv1.4 current after a test interval of 7.5 s could be the result of a decrease in current after full recovery from inactivation (e.g. as a result of a decrease in channel conductance) or a very slow component of recovery from inactivation during acidosis. It is possible that the decrease in current amplitude after a long test interval is related to the slowing of recovery from inactivation; both the H508Q and K532Y mutations abolished both effects (Figs 6C and 8F) suggesting that the two are related (however, raising the K+ concentration, Fig. 7C, abolished the slowing of recovery from inactivation but not the decrease in current amplitude after a long test interval; the decrease in current amplitude after a long test interval in this case was solely responsible for the inhibition of current amplitude during repetitive pulsing).
Relation to previous studies
Steidl & Yool (1999) showed that acidosis enhances (accelerates) C-type inactivation of Kv1.5 and this is the result of a histidine residue equivalent to H508 in Kv1.4 (H452 in Kv1.5). Pérez-Cornejo (1999) showed that the onset of C-type inactivation of the wild-type Shaker K+ channel is also enhanced (accelerated) by acidosis. The Shaker K+ channel, however, does not have a histidine residue at the position equivalent to H508 in Kv1.4 – instead there is a phenylalanine residue (F425). This shows that in Shaker, another residue must be responsible for the acidosis sensitivity of C-type inactivation. However, Pérez-Cornejo (1999) showed that replacement of F425 in Shaker by a histidine residue enhances the acidosis sensitivity of C-type inactivation. Padanilam et al. (1998) studied the effect of pH on human Kv1.4 and concluded that the effects of intracellular pH were mediated by a histidine residue in the N-terminus (H16). In the present study, extracellular pH was changed and this is presumably the reason why the pH sensitivity was the result of an extracellular residue (H508) rather than the intracellular residue identified by Padanilam et al. (1998). Thus, there are likely to be at least two proton-binding sites on Kv1.4. Evidence of other actions of pH has also been obtained from the present study – after deletion of the N-terminus (fKv1.4 Δ2-146), although the sensitivity of current amplitude to acidosis was reduced, current amplitude was still inhibited if the pH was reduced to 6 (Fig. 3E).
Patho-physiological significance
In contrast to the effect of acidosis on Kv1.4 current demonstrated here, acidosis does not reduce ito in rat ventricular cells (Hulme & Orchard, 2000). However, ito in rat ventricular cells is mediated by Kv4.2/Kv4.3 (Brahmajothi et al. 1999) and we have shown that acidosis does not inhibit rat Kv4.2 expressed in Xenopus oocytes (Claydon et al. 1999). Furthermore, rKv4.2 does not have a histidine residue at position 353 (the equivalent residue to H508 in Kv1.4).
In rat ventricular cells, acidosis can increase ito as a result of a depolarizing shift of the inactivation curve (Hulme & Orchard, 2000); a similar effect is observed with rat Kv4.2 expressed in Xenopus oocytes (T. W. Claydon, unpublished data). A similar shift of the inactivation curve is also seen with Kv1.4 expressed in Xenopus oocytes (T. W. Claydon, M. R. Boyett, A. Sivaprasadarao, K. Ishii & C. H. Orchard, unpublished data). In the case of Kv1.4, the shift of the inactivation curve does not result in an increase in current, because the inhibitory effect of acidosis on Kv1.4 (which this study is concerned with) dominates. The mechanism underlying the shift of the inactivation curve is unknown, but in the case of Kv1.4 we have shown that the shift of the inactivation curve and the inhibitory effect are independent (T. W. Claydon, M. R. Boyett, A. Sivaprasadarao, K. Ishii & C. H. Orchard, unpublished data). Since (i) ito is important in determining the configuration of the cardiac action potential, (ii) the effects of acidosis on Kv1.4 and Kv4.2 differ and (iii) the distribution of Kv1.4 and Kv4.2 varies in different regions of the heart, acidosis may influence the normal dispersion of action potential duration and hence be arrhythmogenic.
Acknowledgments
We would like to thank Drs D. Beech and M. Hunter for their ideas and comments and Dr M. Lancaster for his assistance with experiments. We would also like to thank the British Heart Foundation and the University of Leeds for financial support.
References
- Barry DM, Xu H, Schuessler RB, Nerbonne JM. Functional knockout of the transient outward current, long-QT syndrome, and cardiac remodeling in mice expressing a dominant-negative Kv4 α subunit. Circulation Research. 1998;83:560–567. doi: 10.1161/01.res.83.5.560. [DOI] [PubMed] [Google Scholar]
- Brahmajothi MV, Campbell DL, Rasmusson RL, Morales MJ, Trimmer JS, Nerbonne JM, Strauss HC. Distinct transient outward potassium current (Ito) phenotypes and distribution of fast-inactivating potassium channel alpha subunits in ferret left ventricular myocytes. Journal of General Physiology. 1999;113:581–600. doi: 10.1085/jgp.113.4.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DL, Rasmusson RL, Comer MB, Strauss HC. The cardiac calcium-independent transient outward potassium current: kinetics, molecular properties, and role in ventricular repolarization. In: Zipes DP, Jalife J, editors. Cardiac Electrophysiology: From Cell to Bedside. Philadelphia, London, Toronto, Montreal, Sydney, Tokyo: WB Saunders; 1995. pp. 83–96. [Google Scholar]
- Claydon TW, Sivaprasadarao A, Lancaster MK, Boyett MR, Orchard CH. Comparison of the effects of acidosis on cloned transient outward K+ channels fKv1.4 and rKv4.2 from ferret and rat ventricle. The Journal of Physiology. 1999;521:20P. [Google Scholar]
- Comer MB, Campbell DL, Rasmusson RL, Lamson DR, Morales M J Z Y, Strauss HC. Cloning and characterization of an Ito-like potassium channel from ferret ventricle. American Journal of Physiology. 1994;267:H1383–1395. doi: 10.1152/ajpheart.1994.267.4.H1383. [DOI] [PubMed] [Google Scholar]
- Fiset C, Clark RB, Shimoni Y, Giles WR. Shal-type channels contribute to the Ca2+-independent transient outward K+ current in rat ventricle. The Journal of Physiology. 1997;500:51–64. doi: 10.1113/jphysiol.1997.sp021998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Kamiya K, Hojo M, Kodama I, Toyama J. Regulation of Kv4.2 and Kv1.4 K+ channel expression by myocardial hypertrophic factors in cultured newborn rat ventricular cells. Journal of Molecular and Cellular Cardiology. 1998;30:1449–1455. doi: 10.1006/jmcc.1998.0730. [DOI] [PubMed] [Google Scholar]
- Guo W, Xu H, London B, Nerbonne JM. Molecular basis of transient outward K+ current diversity in mouse ventricular myocytes. The Journal of Physiology. 1999;521:587–599. doi: 10.1111/j.1469-7793.1999.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme JT, Orchard CH. Effect of acidosis on transient outward potassium current in isolated rat ventricular myocytes. American Journal of Physiology. 2000;278:H50–59. doi: 10.1152/ajpheart.2000.278.1.H50. [DOI] [PubMed] [Google Scholar]
- Johns DC, Nuss HB, Marban E. Suppression of neuronal and cardiac transient outward currents by viral gene transfer of dominant-negative Kv4.2 contructs. Journal of Biological Chemistry. 1997;272:31598–31603. doi: 10.1074/jbc.272.50.31598. [DOI] [PubMed] [Google Scholar]
- Kaab S, Kartman H, Andrassy J, Hinterseer M, Barth A, Nabauer M. Presence of Kv4.3 and Kv1.4 potassium channel mRNA in human left ventricular myocardium: transmural mRNA-gradients and correlation with current properties. Circulation Research. 1999;96(suppl.):1867. [Google Scholar]
- Kondoh S, Ishii K, Nakamura Y, Taira N. A mammalian transient type K+ channel, rat Kv1.4, has two potential domains that could produce rapid inactivation. Journal of Biological Chemistry. 1997;272:19333–19338. doi: 10.1074/jbc.272.31.19333. [DOI] [PubMed] [Google Scholar]
- López-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors and Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Mikaelian I, Sergeant A. Methods in Molecular Biology: In Vitro Mutagenesis Protocols. Totowa, NJ, USA: Humana Press; 1996. pp. 193–202. [DOI] [PubMed] [Google Scholar]
- Owen JM, Hulme JT, Leach R, Findlay JBC, Hopkins P, Boyett MR, Orchard CH. Comparison of the effects of acidosis on the transient outward K+ current in ferret ventricular myocytes and a cloned transient outward K+ channel, FK1, from ferret ventricle. The Journal of Physiology. 1997;504:77P. [Google Scholar]
- Padanilam BJ, Hoshi T, Shibata EF, Lee H-C. Molecular localization of a titratable histidine residue in the intracellular pH modulation of human Kv1.4 N-type inactivation kinetics. Journal of the American College of Cardiology. 1998;31:A398. [Google Scholar]
- Pérez-Cornejo P. H+ ion modulation of C-type inactivation of Shaker K+ channels. Pflügers Archiv. 1999;437:865–870. doi: 10.1007/s004240050856. [DOI] [PubMed] [Google Scholar]
- Rasmusson RL, Morales MJ, Castellino RC, Zhang Y, Campbell DL, Strauss HC. C-type inactivation controls recovery in a fast inactivating cardiac K+ channel (Kv1.4) expressed in Xenopus oocytes. The Journal of Physiology. 1995;489:709–721. doi: 10.1113/jphysiol.1995.sp021085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Ishibashi K, Nagai T, Marumo F. Regulation mechanisms of intracellular pH of Xenopus laevis oocyte. Biochimica et Biophysica Acta. 1992;1137:45–51. doi: 10.1016/0167-4889(92)90098-v. [DOI] [PubMed] [Google Scholar]
- Steidl JV, Yool AJ. Differential sensitivity of voltage-gated potassium channels Kv1.5 and Kv1.2 to acidic pH and molecular identification of pH sensor. Molecular Pharmacology. 1999;55:812–820. [PubMed] [Google Scholar]
- Stengl M, Carmeliet E, Mubagwa K, Flameng W. Modulation of transient outward current by extracellular protons and Cd2+ in rat and human ventricular myocytes. The Journal of Physiology. 1998;511:827–836. doi: 10.1111/j.1469-7793.1998.827bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Feng J, Shi H, Pond A, Nerbonne JM, Nattel S. Potential molecular basis of different physiological properties of the transient outward K+ current in rabbit and human atrial myocytes. Circulation Research. 1999;84:551–561. doi: 10.1161/01.res.84.5.551. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Jegla TJ, Kaprielian R, Backx PH. Regional contributions of Kv1.4, Kv4.2, and Kv4.3 to transient outward K+ current in rat ventricle. American Journal of Physiology. 1999;276:H1599–1607. doi: 10.1152/ajpheart.1999.276.5.H1599. [DOI] [PubMed] [Google Scholar]
- Xu H, Dixon JE, Barry DM, Trimmer JS, Merlie JP, McKinnon D, Nerbonne JM. Developmental analysis reveals mismatches in the expression of K+ channel α subunits and voltage-gated K+ channel currents in rat ventricular myocytes. Journal of General Physiology. 1996;108:405–419. doi: 10.1085/jgp.108.5.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Guo W, Nerbonne JM. Four kinetically distinct depolarization-activated K+ currents in adult mouse ventricular myocytes. Journal of General Physiology. 1999;113:661–677. doi: 10.1085/jgp.113.5.661. [DOI] [PMC free article] [PubMed] [Google Scholar]