

Abstract
The present study was undertaken to determine whether Ca2+-calmodulin-dependent protein kinase II (CaMKII) participates in the regulation of vascular smooth muscle contraction, and if so, to investigate the nature of the downstream effectors.
The contractility of isolated ferret aorta was measured while inhibiting CaMKII either with antisense oligodeoxynucleotides against CaMKII or with the CaMKII inhibitor KN93.
Treatment with antisense oligodeoxynucleotides against CaMKII resulted in, on average, a decrease in protein levels of CaMKII to 56 % of control levels and significantly decreased the magnitude of the contraction in response to 51 mm potassium physiological saline solution (KCl). Contraction in response to the phorbol ester DPBA was not significantly affected.
The CaMKII blocker KN93 also resulted in a significant decrease in the force induced by 51 mm KCl but caused no significant change in the contraction in response to DPBA or the α-adrenoceptor agonist phenylephrine.
During contraction with 51 mm KCl, both CaMKII and mitogen-activated protein kinase (MAPK) activity increased, as determined by phospho-specific antibodies. The MAPK phosphorylation level was inhibited by KN93, PD098059 (a MAPK kinase (MEK) inhibitor) and calcium depletion.
Myosin light chain (LC20) phosphorylation also increased during contraction with KCl and the increase was significantly blocked by PD098059 as well as by both KN93 and antisense oligodeoxynucleotides to CaMKII.
The data indicate that CaMKII plays a significant role in the regulation of smooth muscle contraction and suggest that CaMKII activates a pathway by which MAPK activation leads to phosphorylation of LC20 via activation of myosin light chain kinase.
Ca2+-calmodulin-dependent protein kinase II (CaMKII) is a multifunctional serine/threonine kinase with diverse biological targets including synaptic transmission, gene expression, cell cycle control and hormone production (see Schulman, 1993, for review). CaMKII was first isolated from brain and was subsequently found to consist of a family of isoforms (Tobimatsu & Fujisawa, 1989; Schworer et al. 1993). CaMKII is present in vascular smooth muscle, with the δ and γ isoforms of CaMKII predominating (Zhou & Ikebe, 1994; Singer et al. 1997). Several smooth muscle proteins have been identified as in vitro substrates for CaMKII including myosin light chain kinase (MLCK) (Stull et al. 1993), myosin light chains (LC20) (Edelman et al. 1990), caldesmon (Ngai & Walsh, 1984) and calponin (Winder & Walsh, 1993). As a result, several laboratories have suggested that CaMKII might play a role in the regulation of vascular smooth muscle contractility.
Several reports have suggested that CaMKII might modulate the Ca2+ sensitivity of the contractile apparatus by either the phosphorylation (and consequent desensitization to Ca2+-calmodulin) of MLCK (Tansey et al. 1992) or the direct phosphorylation (and consequent sensitization to Ca2+-calmodulin) of LC20 (Edelman et al. 1990). These reports have remained controversial, in part because key experiments in which CaMKII activity was blocked were performed only in cultured smooth muscle cells which are non-contractile (Tansey et al. 1992). Mitogen-activated protein kinase (MAPK) has been shown to be activated by CaMKII in both cultured rabbit aortic smooth muscle tissue (Muthalif et al. 1996; Katoch et al. 1999) and cultured rat aortic vascular smooth muscle cells (Abraham et al. 1997). In fresh, contractile smooth muscle from the ferret aorta, MAPK has been shown to phosphorylate caldesmon and increase contractility (Khalil & Morgan, 1993; Dessy et al. 1998). Similarly, activated extracellular-regulated kinase (ERK)2 MAPK added to Triton-permeabilized airway smooth muscle increases contractility (Gerthoffer et al. 1997). MAPK also causes phosphorylation of caldesmon in colonic smooth muscle and disinhibited caldesmon in an in vitro motility assay (Gerthoffer et al. 1996). The role of MAPK in regulating contractility remains controversial, however, since Nixon et al. (1995) showed that caldesmon could be phosphorylated by MAPK in Triton-permeabilized rabbit vascular strips in the absence of a change in contractility.
Thus, there are many possible sites of action of CaMKII in the smooth muscle cell. However, the physiological relevance of this kinase has been called into question because of its relatively low affinity for calmodulin (CaM). The affinity of CaMKII for CaM was reported to be in the range 20–100 nM at saturating calcium concentrations, about a factor of 10 lower than that of other CaM-activated kinases such as MLCK (Kamm & Stull, 1989; Schulman & Hanson, 1993). However, once CaMKII is activated by interacting with the Ca2+-CaM complex and undergoing autophosphorylation in a co-operative manner, the phosphorylated enzyme has an autonomous activity that persists even in the absence of Ca2+-CaM (Hanson et al. 1994). This property may allow CaMKII to exert a physiologically important, prolonged action long after a transient calcium signal (see Singer et al. 1996, for review).
In the present study, we provide evidence for a CaMKII-dependent component of contractile force in ferret aorta smooth muscle. Furthermore, we show that MAPK activation as well as phosphorylation of LC20 are linked to CaMKII as downstream events in a signalling cascade.
METHODS
Tissue preparation and contraction measurements
Adult ferrets were killed with chloroform in a ventilation hood in agreement with procedures approved by the Institutional Animal Care and Use Committee. The thoracic aorta was quickly removed and immersed in ice-cold and oxygenated (95 % O2-5 % CO2) physiological saline solution (PSS) composed of (mM): NaCl 120, KCl 5.9, NaHCO3 25, dextrose 11.5, CaCl2 2.5, MgCl2 1.2 and NaH2PO4 1.2 (pH 7.4). The aorta was cleaned of all adherent connective tissue, and the endothelium was removed by gentle abrasion with a rubber policeman. Circular strips (3 mm wide) of aorta were prepared as previously described (Jiang & Morgan, 1989) and attached to a force transducer. They were allowed to equilibrate at 37°C for at least 1 h and then challenged with a depolarizing solution containing 51 mm KCl (PSS in which 51 mm NaCl was stoichiometrically replaced by KCl; KCl PSS).
Oligodeoxynucleotide loading procedure and organ culture
The antisense 21-mer oligodeoxynucleotide designed against CaMKII (5′-GC GGG TGC AGG TGG CGG TGG T-3′) was derived from a sequence reported for a rat CaMKII γ isoform (Zhou & Ikebe, 1994). The sequence includes residues neighbouring the start codon, which has been shown to improve the antisense efficacy. Fluorescein isothiocyanate (FITC)-tagged, phosphorothioate antisense oligodeoxynucleotide derivatives were synthesized as well as the sense (5′-AC CAC CGC CAC CTG CAC CCG C-3′) and random sequence (5′-GT GTG GCG GTG GAC GTG GGC G-3′) counterparts using a Millipore Expedite nucleic acid synthesis system, and were cleaved from the reaction column by 30 % ammonium hydroxide and purified by a Poly-Pak cartridge (Millipore).
A method originally developed to load aequorin into smooth muscle and referred to as a ‘chemical loading procedure’ (Morgan & Morgan, 1982), has been used in many laboratories to load diverse substances such as aequorin, heparin or DNA into smooth muscle tissue (Morgan et al. 1984; Kobayashi et al. 1989; Lesh et al. 1995; Earley et al. 1998) and is sometimes referred to as a ‘reversible permeabilization method’ even though evidence is lacking that the cell membrane is permeabilized and the mechanism of the loading procedure is unknown. Briefly, muscles are soaked for 30–120 min in each of a series of four solutions at 2°C. The compositions of the solutions are (mM): Solution I: EGTA 10, Na2ATP 5, KCl 120, MgCl2 2 and Tes 20; Solution II: EGTA 0.1, Na2ATP 5, KCl 120, MgCl2 2 and Tes 20, with 25 μm oligodeoxynucleotide; Solution III: EGTA 0.1, Na2ATP 5, KCl 120, MgCl2 10 and Tes 20; Solution IV: NaCl 120, KCl 5.8, dextrose 11, NaHCO3 25, MgCl2 10 and NaH2PO4 1.4. The [Ca2+] is then raised in solution IV from a background concentration of approximately 10−6 M to the normal value of 2.5 mm in gradual steps in order to avoid damage to the preparation from the ‘Ca2+-paradox’ (Zimmerman & Hülsmann, 1966). Tissues are kept overnight at room temperature in a mixture of PSS and Dulbecco's Modified Eagle's Medium (1:1) in the presence of penicillin (25 U ml−1), streptomycin (25 mg ml−1) and nystatin (50 U ml−1). This procedure is repeated daily for 3 days in order to obtain a sufficient loading of oligodeoxynucleotides during several half-lives of the target protein. In this study the viability of the preparation and contractile function were tested daily by measuring the response to KCl PSS. The effect of the relative amount of CaMKII on the contraction was tested on day 4.
Measurement of LC20 phosphorylation
Muscle strips were quick frozen by immersion for 60 min in acetone containing 10 % trichloroacetic acid (TCA) and 10 mm dithiothreitol (DTT) precooled with dry ice. Muscles were stored at -80°C until use. Tissues were brought to room temperature in acetone- TCA-DTT, then ground with glass pestles and washed 3 times with ether to remove TCA. Tissues were extracted in 100 μl of sample buffer containing 20 mm Tris base-23 mm glycine (pH 8.6), 8.0 M urea, 10 mm DTT, 10 % glycerol and 0.04 % bromophenol blue. Urea-extracted samples (20 μl) were electrophoresed at 400 V for 2.5 h after a 30 min pre-run on mini polyacrylamide gels (1.0 mm thickness) containing 10 % acrylamide-0.27 % bisacrylamide, 40 % glycerol, 20 mm Tris base and 23 mm glycine, pH 8.6. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes and immunoblotted with a specific LC20 antibody (1:1500, Sigma). Anti-mouse IgG (goat) conjugated with horseradish peroxidase was used as a secondary antibody (1:2000, Calbiochem). The bands containing myosin light chain were detected with enhanced chemiluminescence (ECL; Super Signal, Pierce) visualized on films, acquired by Adobe Photoshop and then analysed by NIH Image. Care was taken to ensure that saturation of the signal did not occur at any step in the processing.
Western blot protocol
Samples were homogenized in a buffer containing 20 mm Mops, 4 % SDS, 10 % glycerol, 10 mm DTT, 20 mmβ-glycerophosphate, 5.5 μm leupeptin, 5.5 μm pepstatin, 20 KIU aprotinin, 2 mm Na3VO4, 1 mm NaF, 100 μm ZnCl2, 20 μm phenylmethylsulphonyl fluoride (PMSF) and 5 mm EGTA. Protein-matched samples (modified Lowry protein assay, DC Protein Assay Kit, Bio-Rad) were subjected to SDS-PAGE (Protogel, National Diagnostics), transferred to PVDF membranes and immunoblotted as described above. For the measurement of phospho-MAPK and phospho-CaMKII levels, muscles were quick frozen as above for LC20 phosphorylation. Similarly, densitometry of protein-matched (modified Lowry protein assay, DC Protein Assay Kit) immunostained samples was performed and analysed by NIH Image, taking care to ensure that saturation of the signal did not occur at any step in the processing. The success of protein matching was confirmed by Napthol Blue Black staining of the membrane and densitometry of the actin band.
Materials
Rabbit polyclonal phospho-MEK1/2 antibody (1:1000), rabbit polyclonal p44/p42 MAPK antibody (1:1000) and mouse monoclonal phospho-p44/p42 (Thr202/Tyr204) MAPK antibody (1:4000) were obtained from New England Biolabs (Beverly, MA, USA). KN93 was obtained from RBI (Natick, MA, USA) and PD098059 was obtained from Sigma. The CaMKIIγ antibody was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA) and used at 1:500. The affinity-purified pan-CaMKII antibody was made to a peptide sequence in the catalytic domain shared by all isoforms of CaMKII as previously described (CK2-CAT; Singer et al. 1997). The affinity-purified phospho-CaMKII antibody was produced using similar methods to a Keyhole limpet haemocyanin (KLH)-coupled peptide corresponding to the conserved amino acid sequence surrounding the phosphorylated Thr287 residue in the CaMKII δ- and γ-subunits (HRQET(p)VDCLKKF). The antibody was specific for the phosphorylated form of CaMKII and did not cross-react with unphosphorylated kinase or kinase phosphorylated on other residues, such as Thr306 and Thr307 (D. Van Riper & H. A. Singer, unpublished data). The phospho-CaMKII antibody was used at 1:500 and the pan-CaMKII antibody was used at 1:500.
Statistics
Each set of data was expressed as a mean ±s.e.m. Student's t test was used to determine the statistical difference of the means between groups of two.
RESULTS
Antisense oligonucleotides to CaMKII γ isoform decrease contractility
As described in Methods, aortic strips were loaded with phosphorothioate antisense oligonucleotides to a γ isoform of CaMKII (Zhou & Ikebe, 1994) or the appropriate controls. To confirm adequate loading, all oligos were FITC labelled and the fluorescence of the preparation quantified at the end of the experiment as shown in Fig. 1A. A highly statistically significant increase in fluorescence was obtained after loading with FITC-tagged antisense or random sequence oligos in comparison to the sham-loaded preparations. Similar amounts of the antisense and random sequence oligos were taken up by the tissue. It was also noted that uniform fluorescence was observed throughout the thickness of the muscle in all cases (data not shown).
Figure 1. Effect of CaMKII antisense loading on fluorescence, contractility and CaMKII protein level.
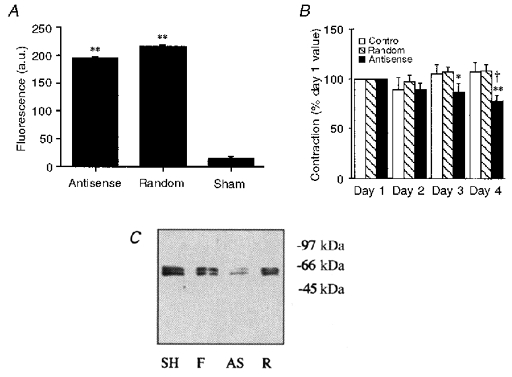
A, quantification of the fluorescence (in arbitrary units, a.u.) of FITC-oligonucleotide-loaded muscles versus sham-loaded muscles. ** P < 0.01. B, magnitude of the steady-state (10-15 min) contractile response to 51 mm KCl on the indicated number of days after loading with oligonucleotides. Forces are normalized to the amplitude of contraction of each muscle in response to 51 mm KCl on day 1 (n= 8–13 animals). * P < 0.05, ** P < 0.01 compared to same muscle on day 1. † P < 0.05 compared to random sequence-loaded muscles on day 4. C, Western blot for CaMKII γ isoforms in paired samples: sham-loaded time controls, SH; fresh tissue, F; antisense-loaded muscles, AS; and random sequence-loaded muscles, R (markers on the right are in kDa).
Contractility was assessed daily by a standard challenge with 51 mm KCl. The muscles loaded with the antisense oligos showed statistically significant decreases in the amplitude of the KCl-induced contraction on days 3 and 4 (Fig. 1B). In contrast, there was no statistically significant change in contractility in preparations loaded with the random oligonucleotide sequence or sham loaded (control). Western blots of day 4 muscles were obtained to monitor the protein levels of CaMKII. As shown in Fig. 1C, two bands of 51 and 58 kDa were consistently seen with an antibody to γ isoforms of CaMKII. The same bands were also detected by a pan-CaMKII antibody (data not shown). Densitometry was performed to quantify the sum of the two CaMKII γ isoform bands. On average, CaMKII protein levels were decreased by antisense loading to 51 ± 6.5 % (n= 13) of those in paired, random sequence-loaded muscles.
The day 4 oligonucleotide-treated muscles were challenged with a range of concentrations of KCl as shown in Fig. 2. In all cases the contractility of the antisense-treated muscles was significantly decreased as compared to that of random sequence-treated muscles. In contrast, when muscles were challenged with 1 μm DPBA, a phorbol ester, there was no statistically significant decrease in the amplitude of the contractile response of muscles loaded with antisense oligos compared to random sequence-loaded muscles: day 4 antisense-treated muscles displayed a DPBA-induced contraction that was 83 ± 10 % of the contraction on day 1 in response to 51 mm KCl; random sequence-treated muscles displayed a DPBA-induced contraction that was 91 ± 12 % of the day 1 contraction in response to 51 mm KCl.
Figure 2. Effect of CaMKII antisense- or random sequence loading on steady-state (10-15 min) contraction versus KCl concentration.
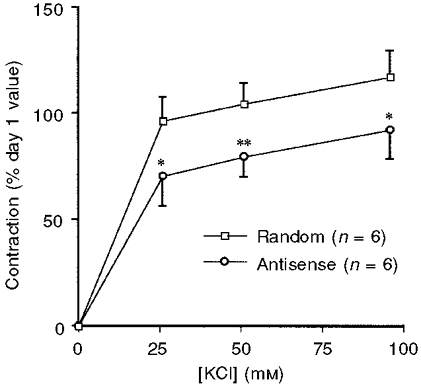
Forces are normalized to the day 1 response of the same muscle to 51 mm KCl (n= 6). * P < 0.05, ** P < 0.01.
Antisense oligonucleotides to CaMKII decrease LC20 phosphorylation
Since CaMKII has been reported to phosphorylate LC20 at the same sites as MLCK (Edelman et al. 1990), we measured LC20 phosphorylation levels in antisense-treated muscles quick frozen after 5 min exposure to 51 mm KCl. LC20 phosphorylation was 30 ± 1.9 % in sham-loaded muscles, 30 ± 2.1 % in random sequence-loaded muscles and 21 ± 2.1 % (n= 14) in antisense-loaded muscles. LC20 phosphorylation was significantly less in the antisense-treated muscles than that in both the random sequence-treated muscles (P < 0.01) and the control sham-loaded muscles (P < 0.005).
Antisense oligonucleotides to CaMKII decrease MAPK phosphorylation
Since MAPK has been reported to be downstream of CaMKII in signalling pathways in cultured smooth muscle (Abraham et al. 1997), we also measured MAPK phosphorylation levels in quick-frozen muscles after 5 min exposure to 51 mm KCl on day 4 in sham-loaded, random sequence-loaded and antisense-loaded muscles. On average, normalizing to the densitometry values from random sequence-loaded muscles, the antisense-treated muscles had phospho-MAPK levels that were 54 % (4890 ± 853 a.u. (n= 9) versus 9020 ± 942 a.u. (n= 11), P= 0.005) of those in random sequence-loaded muscles. In comparison, the level in sham-loaded muscles was 89 % (8040 ± 1065 (n= 9), P > 0.05) of that in random sequence-loaded muscles.
KN93 inhibits contractility of ferret aorta preparations
KN93 is a pharmacological inhibitor of CaMKII (Sumi et al. 1991). KN92 is an inactive analogue of KN93. For comparison with the results from antisense-treated muscles, we also investigated the effects of KN93 and KN92 in muscles studied on the day of removal from the animal. As shown in Fig. 3, 30 μm KN93 caused a statistically significant decrease in the amplitude of the contraction in response to 51 mm KCl at all time points, between 1 min and 30 min after the addition of 51 mm KCl, compared to parallel vehicle- or 30 μm KN92-treated muscles. In contrast, when the α-adrenoceptor agonist phenylephrine was used to initiate contraction, there was no significant difference between KN93- and vehicle-treated muscles (vehicle, 96 ± 10 % of the initial 51 mm KCl response; KN93, 90 ± 4.6 % of control KCl response; n= 4). Similarly, when 5 μm of the phorbol ester DPBA was used to initiate contraction, there was no significant difference between the KN93- and vehicle-treated muscles (vehicle, 134 ± 7.7 % of the initial KCl response; KN93, 127 ± 9.2 % of the initial KCl response; n= 4).
Figure 3. Effect of 30 μm KN93, KN92 or vehicle treatment on the contractile response to 51 mm KCl.
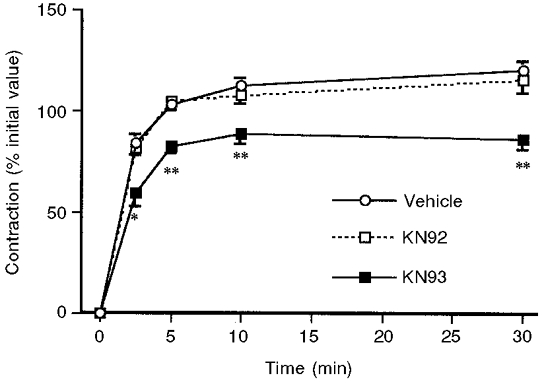
Forces are normalized to the steady-state amplitude of the contraction in response to 51 mm KCl of each muscle before treatment (n= 5–9). * P < 0.05, ** P < 0.01.
KN93 decreases LC20 phosphorylation
It is generally accepted that Ca2+-CaM-dependent activation of MLCK and the subsequent phosphorylation of the 20 kDa myosin light chains is a major regulatory mechanism of smooth muscle contractility. For this reason, we measured light chain phosphorylation levels in the presence and absence of KN93, as shown in Fig. 4. When 51 mm KCl was added at time 0, there was a rapid increase in LC20 phosphorylation, which was observed as early as 1 min after stimulation, and which persisted for at least 30 min. In parallel KN93-treated muscles, there was a statistically significant decrease in light chain phosphorylation levels at 1 and 5 min compared with control. This decrease was not statistically significant at 30 min.
Figure 4. Effect of KN93 on LC20 phosphorylation.
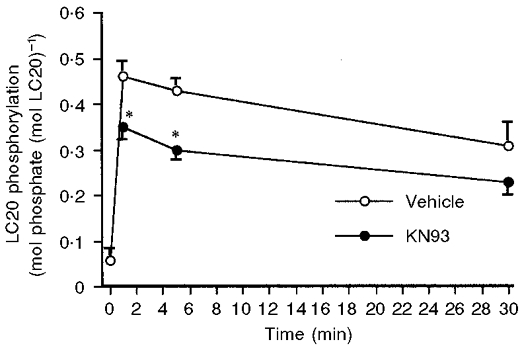
LC20 phosphorylation (moles phosphate per mole LC20) at the indicated time points after treatment with 51 mm KCl in the presence of 30 μm KN93 or vehicle (n= 4–6). * P < 0.05.
Potassium-induced depolarization activates CaMKII in ferret aortic smooth muscle
An antiphospho-CaMKII antibody was used to probe CaMKII activity levels at various time points after the addition of 51 mm KCl to ferret aortic strips (Fig. 5). The activity of this enzyme increased significantly by 1 min, continuing to increase at 5 min and remained high at 30 min after exposure to KCl. When the rate of activation of CaMKII is compared with the time course of the KCl-induced contraction (Fig. 5B), it can be seen that CaMKII activity began to increase at roughly the same time that force began to rise, and like force, CaMKII phosphorylation remained at high levels during steady-state tone.
Figure 5. Activation of CaMKII during KCl-induced contraction.
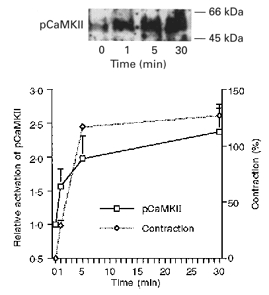
An antibody specific for autophosphorylated CaMKII (pCaMKII) was used to monitor activation of CaMKII after 0, 1, 5 and 30 min exposure to 51 mm KCl. Top panel, typical Western blot. Bottom panel, contraction and densitometry of phospho-CaMKII versus time (mean of 4 experiments).
KCl-induced MAPK activation is calcium dependent and sensitive to compound KN93
We have previously shown that phenylephrine induces MAPK activation in this tissue in a Ca2+-independent manner (Dessy et al. 1998). However, it is known that CaMK activation as well as the amplitude of the KCl-induced contraction are Ca2+ dependent. Therefore, we determined the effect of Ca2+ depletion on MAPK activation in KCl-stimulated ferret aortas using antibodies to both phospho-MEK and phospho-MAPK. As is shown in Fig. 6A, 51 mm KCl increased both phospho-MEK and phospho-MAPK levels (lane 2). Both increases were prevented by Ca2+ depletion during the KCl challenge (lane 3). Similar results were obtained in three experiments.
Figure 6. Effect of Ca2+ depletion and KN93 on MAPK activation by 51 mm KCl.
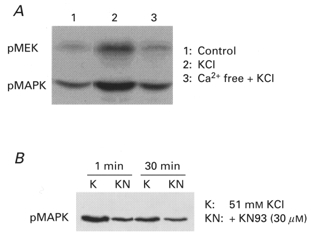
A, effect of Ca2+ depletion on MAPK activation. Resting muscles (lane 1) or muscles treated with 51 mm KCl for 5 min in normal PSS (lane 2) or Ca2+-free PSS containing 5 mm EGTA plus 51 mm KCl (lane 3). Blots were probed for phospho-MAPK and then re-probed for phospho-MEK. Similar results were seen in three experiments. B, ERK-1 activation in the presence of KCl for 1 or 30 min in the absence (K) or presence of 30 μm KN93 (KN). Similar results were seen in three experiments.
As shown in Fig. 6B, the addition of 30 μm KN93 before induction of contractions with 51 mm KCl caused a decrease in the level of phospho-MAPK both at 1 min and at 30 min after the addition of KCl. Similar results were obtained in three experiments. It was noted that KN93 brought phospho-MAPK levels to near basal values (i.e. a nearly 100 % decrease), whereas KN93 caused, at the same concentration, a 26 % decrease in contractility (Fig. 3). These findings are consistent with the CaMKII-induced activation of MAPK being only part of the mechanism by which KCl increases contractility.
If the effect of CaMKII on contractility depends on MAPK, then inhibition of MAPK should also decrease the amplitude of the contraction and reduce LC20 phosphorylation in a manner similar to that observed with KN93. Compound PD098059 is known to block the activation of MEK and MAPK (Dudley et al. 1996). As shown in Fig. 7A, PD098059 caused a significant decrease in the amplitude of the contraction in response to KCl. Similarly, when LC20 phosphorylation was measured in the tissue after 5 min exposure to 51 mm KCl, in the absence and presence of PD098059, the values were 31 ± 1.8 % (n= 8) and 24 ± 2.1 % (n= 8), respectively (Fig. 7B). Thus, these data are consistent with CaMKII modulating contraction, at least in part through activation of MAPK. It is worth noting that the effect of PD098059 on force appears somewhat larger than that of KN93 while inhibition of LC20 phosphorylation is of a similar magnitude. This raises the suggestion that MAPK does not act solely through the activation of MLCK.
Figure 7. Effect of PD098059 on contraction and LC20 phosphorylation.
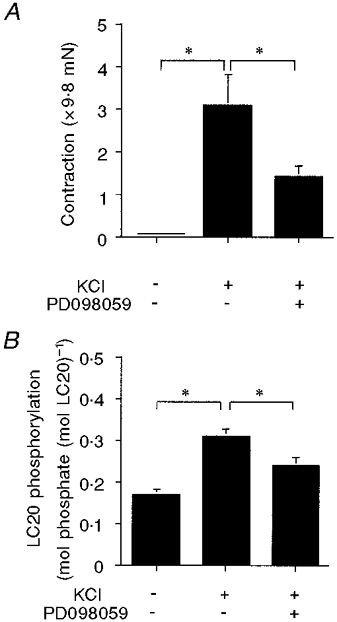
A, amplitude of the contractile response to 51 mm KCl. First bar, baseline resting tone, by definition equal to 0; second bar, control KCl response; third bar, response to KCl in the presence of 50 μm PD098059 (n= 4). B, LC20 phosphorylation. First bar, in resting muscle; second bar, during control KCl contraction; third bar, with KCl in the presence of 50 μm PD098059 (n= 7–8). * P < 0.05.
DISCUSSION
The main finding of the present study is that inhibition of CaMKII in differentiated smooth muscle results in inhibition of contractility. Similar results were obtained with two very different approaches, the use of antisense oligonucleotides and the pharmacological inhibitor KN93. These results were further supported by the finding that CaMKII autophosphorylation increases with a time course comparable to that of the contraction and is maintained during tone maintenance. To some degree these findings were surprising. Previous studies had suggested that CaMKII down-regulates calcium signalling via phosphorylation of MLCK (Tansey et al. 1992). This mechanism could not explain the results of the present study, since downregulation of CaMKII by pretreatment with both KN93 and antisense oligonucleotides should increase the force by this mechanism, but a decrease in force in aortic strips was seen in the present study. The results obtained in cultured cells in the study of Tansey et al. (1992) may not be directly applicable to contractile strips because in cultured smooth muscle cells smooth muscle (sm)-MLCK was almost completely replaced by non-muscle (nm)-MLCK (Stephens et al. 1999b). nm-MLCK is normally localized to the nucleus in contrast to sm-MLCK, which is localized in the cytosol, perhaps being associated with contractile filaments (Stephens et al. 1999a).
The ability of CaMKII to influence contraction appears to be mediated by MAPK. As noted above, depletion of Ca2+ significantly decreased activation of MAPK. Similarly, compound KN93, an antagonist of CaMKII, blocked activation of MAPK, contraction and phosphorylation of LC20. Consistent with this possibility, an inhibitor of MAPK activation (i.e. PD098059) also inhibited contraction and phosphorylation of LC20 in this tissue. Thus, a plausible sequence of signalling events in KCl-stimulated ferret aorta would be: Ca2+↑→ CaMKII →?→ MAPK → MLCK → LC20. We cannot rule out the possibility in the present study that KN93, PD098059 or CaMKII antisense oligonucleotides artifactually decreased Ca2+ to indirectly decrease LC20 phosphorylation levels; however, the fact that these three very different experimental manipulations produced the same results would appear to make this possibility unlikely.
We have previously reported that MAPK is activated during α-adrenoceptor agonist-induced contraction through a pathway involving protein kinase C ε (PKCε) in ferret aorta vascular smooth muscle cells (Khalil & Morgan, 1993; see Morgan & Leinweber, 1998, for review). This activation was Ca2+ independent and occurred in the absence of detectable changes in LC20 phosphorylation. The finding that depletion of calcium completely abolished the activation of MAPK during KCl-induced contractions as well as the lack of effect of KN93 and antisense to CaMKII on phorbol ester-initiated contractions in the present study suggests that two different pathways of Ca2+-independent and Ca2+-dependent MEK activation may exist during α-adrenoceptor agonist- and KCl-induced contractions, respectively, in this tissue. Similarly, in other studies, MAPK activation in cultured cells in response to ionomycin, but not phorbol esters and platelet-derived growth factor, was almost completely abolished by 30 μm of the CaMKII inhibitor KN93 (Abraham et al. 1997). Interestingly, calponin has been suggested to act as an adaptor protein assisting the translocation of MAPK to the plasmalemma in the PKC-dependent, Ca2+-independent pathway in ferret aorta (Menice et al. 1997). In contrast, during KCl-induced contractions, neither calponin (Parker et al. 1994) nor MAPK (Khalil & Morgan, 1993) appears to translocate to the plasmalemma, highlighting the differences between the two putative pathways.
Since MAPK has previously been suggested to contract smooth muscle via phosphorylation of caldesmon (in the presence of an α-adrenoceptor agonist) (Dessy et al. 1998), we were surprised to find that both KN93 and CaMKII antisense oligonucleotides significantly decreased LC20 phosphorylation. Interestingly, CaMKII has been reported to directly phosphorylate LC20 at the same sites phosphorylated by MLCK in vitro. This, however, raises the issue of the relationship of the alteration in LC20 phosphorylation levels to that of MAPK activation. A possible unifying mechanism is provided by the reports of Nguyen et al. (1999) and Morrison et al. (1996) who observed that MAPK can activate MLCK. This mechanism is consistent with the fact that PD098059, which prevents MAPK activation, also decreased LC20 phosphorylation and force production during KCl-induced contractions.
In summary, the data in the present study indicate that CaMKII plays a significant role in the regulation of smooth muscle contractility and suggest that the downstream signalling pathways include a MAPK-induced activation of MLCK.
Acknowledgments
This work was supported by grants from the PHS: NIH HL31704 and HL42293 to Kathleen G. Morgan, NIH HL56035 to John A. Badwey and NIH HL49426 to Harold A. Singer; a grant from the Pennsylvania affiliate of the AHA to Dee Van Riper; and fellowships from the Korean Science and Engineering Foundation to Inkyeom Kim and Hyun-Dong Je.
References
- Abraham ST, Benscoter H, Schworer CM, Singer HA. A role for Ca2+/calmodulin-dependent protein kinase II in the mitogen-activated protein kinase signaling cascade of cultured rat aortic vascular smooth muscle cells. Circulation Research. 1997;81:575–584. doi: 10.1161/01.res.81.4.575. [DOI] [PubMed] [Google Scholar]
- Dessy C, Kim I, Sougnez CL, Laporte R, Morgan KG. A role for MAP kinase in differentiated smooth muscle contraction evoked by α-adrenoceptor stimulation. American Journal of Physiology. 1998;275:C1081–1086. doi: 10.1152/ajpcell.1998.275.4.C1081. [DOI] [PubMed] [Google Scholar]
- Dudley DT, Lang P, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated kinase cascade. Proceedings of the National Academy of Sciences of the USA. 1996;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley JJ, Su X, Moreland RS. Caldesmon inhibits active crossbridges in unstimulated vascular smooth muscle: an antisense oligodeoxynucleotide approach. Circulation Research. 1998;83:661–667. doi: 10.1161/01.res.83.6.661. [DOI] [PubMed] [Google Scholar]
- Edelman AM, Lin W-H, Osterhout DJ, Bennett MK, Kennedy MB, Krebs EG. Phosphorylation of smooth muscle myosin by type II Ca2+/calmodulin-dependent protein kinase. Molecular and Cellular Biochemistry. 1990;97:87–98. doi: 10.1007/BF00231704. [DOI] [PubMed] [Google Scholar]
- Gerthoffer WT, Yamboliev IA, Pohl J, Haynes R, Dang S, McHugh J. Activation of MAP kinases in airway smooth muscle. American Journal of Physiology. 1997;272:L244–252. doi: 10.1152/ajplung.1997.272.2.L244. [DOI] [PubMed] [Google Scholar]
- Gerthoffer WT, Yamboliev IA, Shearer M, Pohl J, Haynes R, Dang S, Sato K, Sellers JR. Activation of MAP kinases and phosphorylation of caldesmon in canine colonic smooth muscle. The Journal of Physiology. 1996;495:597–609. doi: 10.1113/jphysiol.1996.sp021619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson PI, Meyer T, Stryer L, Schulman H. Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron. 1994;12:943–956. doi: 10.1016/0896-6273(94)90306-9. [DOI] [PubMed] [Google Scholar]
- Jiang MJ, Morgan KG. Agonist-specific myosin phosphorylation and intracellular calcium during isometric contractions of arterial smooth muscle. Pflügers Archiv. 1989;413:637–643. doi: 10.1007/BF00581814. [DOI] [PubMed] [Google Scholar]
- Kamm KE, Stull JT. Regulation of smooth muscle contractile elements by second messengers. Annual Review of Physiology. 1989;51:299–313. doi: 10.1146/annurev.ph.51.030189.001503. [DOI] [PubMed] [Google Scholar]
- Katoch SS, Su XL, Moreland RS. Ca2+- and protein kinase C-dependent stimulation of mitogen-activated protein kinase in detergent-skinned vascular smooth muscle. Journal of Cellular Physiology. 1999;179:208–217. doi: 10.1002/(SICI)1097-4652(199905)179:2<208::AID-JCP11>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Khalil RA, Morgan KG. PKC-mediated redistribution of mitogen-activated protein kinase during smooth muscle activation. American Journal of Physiology. 1993;265:C406–411. doi: 10.1152/ajpcell.1993.265.2.C406. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Kitazawa T, Somlyo AV, Somlyo AP. Cytosolic heparin inhibits muscarinic and alpha-adrenergic Ca2+ release in smooth muscle. Journal of Biological Chemistry. 1989;264:17997–18004. [PubMed] [Google Scholar]
- Lesh RE, Somlyo AP, Owens GK, Somlyo AV. Reversible permeabilization. A novel technique for the intracellular introduction of antisense oligonucleotides into intact smooth muscle. Circulation Research. 1995;77:220–230. doi: 10.1161/01.res.77.2.220. [DOI] [PubMed] [Google Scholar]
- Menice CB, Hulvershorn J, Adam LP, Wang C-L A, Morgan KG. Calponin and mitogen-activated protein kinase signaling in differentiated vascular smooth muscle. Journal of Biological Chemistry. 1997;272:25157–25161. doi: 10.1074/jbc.272.40.25157. [DOI] [PubMed] [Google Scholar]
- Morgan JP, DeFeo TT, Morgan KG. A chemical procedure for loading the calcium indicator aequorin into mammalian working myocardium. Pflügers Archiv. 1984;400:338–340. doi: 10.1007/BF00581571. [DOI] [PubMed] [Google Scholar]
- Morgan JP, Morgan KG. Loading of aequorin into reversibly hyperpermeable cardiac and vascular smooth muscle cells. Federation Proceedings. 1982;41:1522. [Google Scholar]
- Morgan KG, Leinweber BD. PKC-dependent signalling mechanisms in differentiated smooth muscle. Acta Physiologica Scandinavica. 1998;164:495–505. doi: 10.1046/j.1365-201X.1998.00445.x. [DOI] [PubMed] [Google Scholar]
- Morrison DL, Sanghera JS, Stewart J, Sutherland C, Walsh MP, Pelech SL. Phosphorylation and activation of smooth muscle myosin light chain kinase by MAP kinase and cyclin-dependent kinase-1. Biochemistry and Cell Biology. 1996;74:549–557. doi: 10.1139/o96-459. [DOI] [PubMed] [Google Scholar]
- Muthalif MM, Benter IF, Uddin MR, Malik KU. Calcium/calmodulin-dependent protein kinase II alpha mediates activation of mitogen-activated protein kinase and cytosolic phospholipase A2 in norepinephrine-induced arachidonic acid release in rabbit aortic smooth muscle cells. Journal of Biological Chemistry. 1996;271:30149–30157. doi: 10.1074/jbc.271.47.30149. [DOI] [PubMed] [Google Scholar]
- Ngai PK, Walsh MP. Inhibition of smooth muscle actin-activated myosin Mg2+-ATPase activity by caldesmon. Journal of Biological Chemistry. 1984;259:13656–13659. [PubMed] [Google Scholar]
- Nguyen DHD, Catling AD, Webb DJ, Sankovic M, Walker LA, Somlyo AV, Weber MJ, Gonias SL. Myosin light chain kinase functions downstream of Ras/ERK to promote migration of urokinase-type plasminogen-activator cells in an integrin-selective manner. Journal of Cell Biology. 1999;146:149–164. doi: 10.1083/jcb.146.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon GG, Iizuka K, Haystead CM, Haystead TA, Somlyo AP, Somlyo AV. Phosphorylation of caldesmon by mitogen-activated protein kinase with no effect on Ca2+ sensitivity in rabbit smooth muscle. The Journal of Physiology. 1995;487:283–289. doi: 10.1113/jphysiol.1995.sp020879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CA, Takahashi K, Tao T, Morgan KG. Agonist-induced redistribution of calponin in contractile vascular smooth muscle cells. American Journal of Physiology. 1994;267:C1262–1270. doi: 10.1152/ajpcell.1994.267.5.C1262. [DOI] [PubMed] [Google Scholar]
- Schulman H. The multifunctional Ca2+/calmodulin-dependent protein kinases. Current Opinion in Cell Biology. 1993;5:247–253. doi: 10.1016/0955-0674(93)90111-3. [DOI] [PubMed] [Google Scholar]
- Schulman H, Hanson PI. Multifunctional Ca2+/calmodulin-dependent protein kinase. Neurochemical Research. 1993;18:65–77. doi: 10.1007/BF00966924. [DOI] [PubMed] [Google Scholar]
- Schworer CM, Rothblum LI, Thekkumkara TJ, Singer HA. Identification of novel isoforms of the delta subunit of Ca2+/calmodulin-dependent protein kinase II. Differential expression in rat brain and aorta. Journal of Biological Chemistry. 1993;268:14443–14449. [PubMed] [Google Scholar]
- Singer HA, Abraham ST, Schworer CM. Calcium/calmodulin-dependent protein kinase II. In: Bárány M, editor. Biochemistry of Smooth Muscle Contraction. San Diego: Academic Press, Inc.; 1996. pp. 143–153. [Google Scholar]
- Singer HA, Benscoter HA, Schworer CM. Novel Ca2+/calmodulin-dependent protein kinase II γ-subunit variants expressed in vascular smooth muscle, brain, and cardiomyocytes. Journal of Biological Chemistry. 1997;272:9393–9400. doi: 10.1074/jbc.272.14.9393. [DOI] [PubMed] [Google Scholar]
- Stephens NL, Cui L, Cheng Z, Wang Y, Samuel SK, Davie JR. Can non-muscle light-chain kinase (NM-MLCK) act as a transcription factor? Nuclear matrix protein and DNA cross-linking studies in situ with cisplatin. Journal of Muscle Research and Cell Motility. 1999a;20:598. [Google Scholar]
- Stephens NL, Cui L, Liu G, Wang Y, Stephens D. Cytokinesis in smooth muscle cells: the roles of non-muscle myosin light-chain kinase, ‘speckles’ and ‘clustersomes’. Journal of Muscle Research and Cell Motility. 1999b;20:598. [Google Scholar]
- Stull JT, Tansey MG, Tang D-C, Word RA, Kamm KE. Phosphorylation of myosin light chain kinase: a cellular mechanism for Ca2+ desensitization. Molecular and Cellular Biochemistry. 1993;127:229–237. doi: 10.1007/BF01076774. [DOI] [PubMed] [Google Scholar]
- Sumi M, Kiuchi K, Ishikawa T, Isshii A, Hagiwara M, Nagatsu T, Hidaka H. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine content in PC12 h cells. Biochemical and Biophysical Research Communications. 1991;181:968–975. doi: 10.1016/0006-291x(91)92031-e. [DOI] [PubMed] [Google Scholar]
- Tansey MG, Word RA, Hidaka H, Singer HA, Schworer CM, Kamm KE, Stull JT. Phosphorylation of myosin light chain kinase by the multifunctional calmodulin-dependent protein kinase II in smooth muscle cells. Journal of Biological Chemistry. 1992;267:12511–12516. [PubMed] [Google Scholar]
- Tobimatsu T, Fujisawa H. Tissue-specific expression of four types of rat calmodulin-dependent protein kinase II mRNAs. Journal of Biological Chemistry. 1989;264:17907–17912. [PubMed] [Google Scholar]
- Winder SJ, Walsh MP. Calponin: thin filament-linked regulation of smooth muscle contraction. Cellular Signalling. 1993;5:677–686. doi: 10.1016/0898-6568(93)90029-l. [DOI] [PubMed] [Google Scholar]
- Zhou ZL, Ikebe M. New isoforms of Ca2+/calmodulin-dependent protein kinase II in smooth muscle. Biochemical Journal. 1994;299:489–495. doi: 10.1042/bj2990489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman AN, Hülsmann WC. Paradoxical influence of calcium ions on the permeability of the cell membranes of the isolated rat heart. Nature. 1966;211:646–647. doi: 10.1038/211646a0. [DOI] [PubMed] [Google Scholar]