

Abstract
We compared sarcomere length (SL) dependence of the Ca2+-force relation of detergent-extracted bundles of fibres dissected from the left ventricle of wild-type (WT) and transgenic mouse hearts expressing slow skeletal troponin I (ssTnI-TG). Fibre bundles from the hearts of the ssTnI-TG demonstrated a complete replacement of the cardiac troponin I (cTnI) by ssTnI.
Compared to WT controls, ssTnI-TG fibre bundles were more sensitive to Ca2+ at both short SL (1.9 ± 0.1 μm) and long SL (2.3 ± 0.1 μm). However, compared to WT controls, the increase in Ca2+ sensitivity (change in half-maximally activating free Ca2+; ΔEC50) associated with the increase in SL was significantly blunted in the ssTnI-TG myofilaments.
Agents that sensitize the myofilaments to Ca2+ by promoting the actin-myosin reaction (EMD 57033 and CGP-48506) significantly reduced the length-dependent ΔEC50 for Ca2+ activation, when SL in WT myofilaments was increased from 1.9 to 2.3 μm.
Exposure of myofilaments to calmidazolium (CDZ), which binds to cTnC and increases its affinity for Ca2+, sensitized force developed by WT myofilaments to Ca2+ at SL 1.9 μm and desensitized the WT myofilaments at SL 2.3 μm. There were no significant effects of CDZ on ssTnI-TG myofilaments at either SL.
Our results indicate that length-dependent Ca2+ activation is modified by specific changes in thin filament proteins and by agents that promote the actin-myosin interaction. Thus, these in vitro results provide a basis for using these models to test the relative significance of the length dependence of activation in situ.
Although the experiments of Frank (1885) and Starling (Patterson et al. 1914) were done around the turn of the 20th century, as we enter the 21st century, the molecular mechanism by which the mammalian heart is able to increase its ability to generate pressure as a function of filling volume remains unclear. Even though there are complex relations between ventricular pressure and myocyte tension, as well as between volume and sarcomere length, it is clear that ultimately the volume-end-systolic pressure (ESP) relation depends on the number of strong, force-generating crossbridges reacting with actin as sarcomere length changes. Sarcomere length may affect the number of strong crossbridges by changes in filament overlap (Allen & Kentish, 1985; Fuchs & Wang, 1997), changes in inter-filament spacing (Harrison et al. 1988; McDonald & Moss, 1995; McDonald et al. 1997), and changes in amounts of Ca2+ released to the myofilaments (Allen & Kurihara, 1982). The relative role of these mechanisms is not certain, yet it is generally held that a length-dependent alteration in myofilament response to Ca2+ is a main determinant of the shape of the ascending limb of the sarcomere length-tension relation in heart muscle. The molecular mechanism for the length dependence of Ca2+ activation is also controversial (Solaro & Van Eyk, 1996). However, there is substantial evidence that changes in inter-filament spacing, which are likely to change the probability of a myosin head reacting with the thin filament in a force-generating complex, are primarily responsible for the dependence of myofilament Ca2+ activation on sarcomere length (Harrison et al. 1988; McDonald & Moss, 1995; McDonald et al. 1997). Results from investigations of this idea indicate that it is the rate of crossbridge binding that is varied with lattice spacing, and that crossbridge binding may itself promote activation by enhancing the Ca2+ affinity of cardiac troponin C (cTnC) and by cooperatively activating the binding of near-neighbour crossbridges (McDonald & Moss, 1995; Fukuda et al. 2000).
Investigation of the relative significance of length-dependent activation in the Frank-Starling relation would benefit from an ability to modify length-dependent activation in situ in a specific manner. Transgenesis involving over-expression of myofilament proteins offers one approach. In the present experiments, we compared length-dependent Ca2+ activation in heart myofilaments from wild-type (WT) ventricles, and from transgenic mice (ssTnI-TG) demonstrating specific isoform switching of the thin filament protein, cTnI, with the embryonic isoform, ssTnI. Length-dependent Ca2+ activation was significantly blunted in the preparations from the ssTnI-TG hearts. In a second line of experiments, we used a pharmacological approach to alter length-dependent activation in the myofilaments. Our results suggest that it is possible to substantially reduce or eliminate length-dependent activation as a variable in the in situ beating heart.
METHODS
Force measurements
We measured the Ca2+-force relations of fibre bundles dissected from papillary muscles from the left ventricle of hearts of wild-type (WT) and transgenic (ssTnI-TG) mice in which ssTnI was over-expressed and replaced cTnI. Both WT and TG mice were CD-1 strain. The generation of these transgenic mice, using the α-myosin heavy chain promoter that restricts expression to the heart, and the analysis of their hearts and myofilaments have been previously reported (Fentzke et al. 1999). The ssTnI-TG mice are fertile and viable; their hearts show no signs of hypertrophy or any detectable histo-pathology. We (Fentzke et al. 1999) have generated mice that over-express ssTnI with and without a FLAG epitope tag. Our previous studies with myofilaments from each of these mice demonstrated no detectable effects of the tag. Nevertheless, in the present experiments we have studied TG mouse heart preparations without the FLAG epitope. Experiments were conducted in compliance with animal care policies of the Animal Care Committee of the University of Illinois at Chicago.
We rapidly excised the hearts of adult mice weighing 28-38 g that had been anaesthetized with methoxyflurane. The heart was immediately placed in an ice cold relaxing solution (HR) containing (mM): 53 KCl, 10 EGTA, 20 Mops, 1 free Mg2+, 5 MgATP2−, creatine phosphate 12, and 10 IU ml−1 creatine phosphokinase at pH 7.0. The ionic strength of all solutions was 0.15 M and all solutions contained the protease inhibitors pepstatin A (2.5 μg ml−1), leupeptin (1 μg ml−1) and phenylmethylsulfonyl fluoride (50 μm). The heart was pinned to the base of a plastic chamber filled with HR. To avoid damage, the papillary muscles of the left ventricle were exposed by approaching the chamber through the right ventricle and septum. The isolated papillary muscles were placed in a separate plastic chamber, and free running longitudinal strands were visualized under the dissecting microscope. Fibre bundles with lengths of 4-5 mm and diameters of 150-200 μm were then prepared from these strands. The bundles were mounted between a micro-manipulator and a force transducer using a cellulose-acetate glue as previously described (Wolska et al. 1999). Measurements of isometric tension were carried out at room temperature and displayed on a chart recorder. Membranes in the fibre bundles were extracted with detergent for 30 min at room temperature by immersion in the HR solution containing 1 % Triton X-100. Following extraction, the fibre bundles were washed in HR and the sarcomere length was initially adjusted to 1.85 ± 0.1 μm using laser diffraction patterns as previously described (Wolska et al. 1999). Once resting isometric tension was stable, the fibre bundles were bathed in ‘low EGTA’ relaxing solution (LR), which was prepared by replacing the 10 mM EGTA in HR with 0.1 mM EGTA. An initial activation of force was induced by switching the bath to one containing the maximum activating solution, which was HR containing CaCl2 to bring the Ca2+ concentration to 3.2 × 10−5 m. The fibre bundles were then relaxed in HR and then in LR. Although there is some internal shortening of the preparation during activation, this test contraction was shown in pilot experiments, as measured by microscopy (Reiser et al. 1994; Wattanapermpool et al. 1995), to aid in maintaining the SL that had been established in HR during the contraction. Force development by the fibre bundles was then activated by sequential immersion in contracting solutions of varying Ca2+ concentrations between 1.0 × 10−8 and 3.2 × 10−5 M. After this series of contractions, the fibre bundles were relaxed in HR, adjusted to a SL of 2.3 ± 0.1 μm, and the series of contractions was repeated over a range of Ca2+ concentrations. After this second series of contractions, the fibre bundles were immersed in maximally activating Ca2+ concentration. Fibre bundles demonstrating greater than 10 % rundown of maximum force were not included in the analysis. In the experiments in which Ca2+-sensitizing agents were tested at SL 2.3 ± 0.1 μm, we performed a third series of contractions in solutions of varying Ca2+ concentrations containing the drug under study, followed by a test maximum contraction. When the effect of these agents was studied at short SL, the Ca2+ sensitizer was added after determination of the Ca2+-force relation at SL 1.85 ± 0.1 μm. Myofilament proteins were analysed by SDS-PAGE, performed according to the method of Laemmli (1970).
Solutions and reagents
A computer program (Godt & Lindley, 1982) was used to calculate the amount of CaCl2 required to be added to HR to achieve free Ca2+ concentrations ranging from 1 × 10−8 to 3.2 × 10−5 M. CGP-48506 (CGP) was synthesized at the Department of Cardiovascular Chemistry Research, Novartis. The active enantiomer EMD 57033 (EMD) was synthesized at the Chemical Department of the Pharmaceutical Research Division of E. Merck, Darmstadt, Germany. Stock solutions of 3 mM CGP and 10 mM EMD were diluted in DMSO. All control experiments contained an equivalent amount of DMSO. Calmidazolium (CDZ; 1-[bis-(4-chlorophenyl)methyl]-3-[2-(2,4-dichlorophenyl)-2-[(2,4-dichlorophenyl)methoxy]ethyl]-1H-imidazolinium chloride) was obtained from Sigma.
Statistical analysis
Isometric tensions (T) were plotted as a function of the log [Ca2+] and fitted to the Hill equation using non-linear least squares regression analysis using Prism software (GraphPad version 2.0). Isometric tensions measured at sub-maximally activating Ca2+ concentrations were expressed as a percentage of the maximum tension (Tmax)such that, percentage maximum tension (%T) =T/Tmax× 100. Data in which measurements were made in the same fibre bundle at different sarcomere lengths or with and without drugs were analysed individually using Student's paired t test. Values are expressed as means ±s.e.m. Statistical significance was set at P < 0.05. When three or more groups were analysed, a one-way repeated measure ANOVA was used with a Tukey's multiple comparison test. Statistical significance was set at P < 0.05.
RESULTS
In our first series of experiments, we compared Ca2+-force relations of myofilaments in fibre bundles prepared from WT and ssTnI-TG hearts. In confirmation of our earlier report (Fentzke et al. 1999), Fig. 1 illustrates that there is essentially complete replacement of cTnI with ssTnI in the TG hearts. Figure 2 shows results of experiments in which we determined the length dependence of the Ca2+-force relations in fibre bundles isolated from WT and ssTnI-TG ventricles. Figure 2 depicts data from the WT preparations, which demonstrated that as SL was decreased from 2.3 to 1.9 μm there was a rightward shift of the Ca2+-force relation (ΔEC50= 0.80 μm Ca2+). This shift is similar to that determined in other species (Allen & Kentish, 1985; Akella et al. 1995). Also illustrated in Fig. 2, the ΔEC50 (0.28 μm Ca2+) for the length dependence of the Ca2+-force relations determined on preparations from ssTnI-TG heart was significantly less than that of the WT controls. At each SL, the Ca2+-force relations of the myofilaments from ssTnI-TG hearts demonstrated more sensitivity to Ca2+ than the WT myofilaments. These data confirm our earlier report (Fentzke et al. 1999) that specific isoform switching of cTnI with ssTnI sensitized force developed by myofilaments of detergent-extracted single myocytes to Ca2+. However, the data shown in Fig. 2 extend these earlier results by showing that the degree of sensitization to Ca2+ is length dependent. For example at SL 1.9 μm, compared to controls, myofilaments containing ssTnI were more sensitive to Ca2+ and demonstrated a leftward shift in the pCa-force relation with a ΔEC50 of 1.47 μm Ca2+. However, at SL 2.3 μm the ΔEC50 was reduced to 1.15 μm Ca2+.
Figure 1. SDS-PAGE of myofibrillar protein expression in wild-type (WT) and transgenic (Tg) hearts.
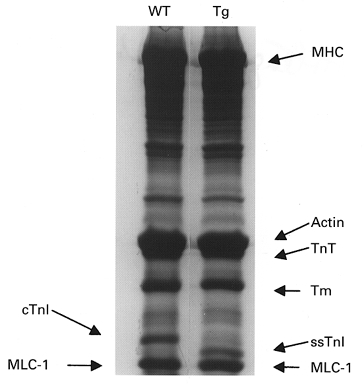
Left and right lanes show the protein content of myofibrils obtained from hearts of WT (left) and Tg (right) mice. MHC, myosin heavy chain; TnT, troponin T; Tm, tropomyosin; cTnI, cardiac troponin I; ssTnI, slow skeletal muscle troponin I; MLC-1, myosin light chain 1.
Figure 2. Ca2+-force relations of wild-type (WT) and transgenic (TG) skinned fibre bundles over-expressing ssTnI at sarcomere lengths (SL) of 1.9 and 2.3 μm.
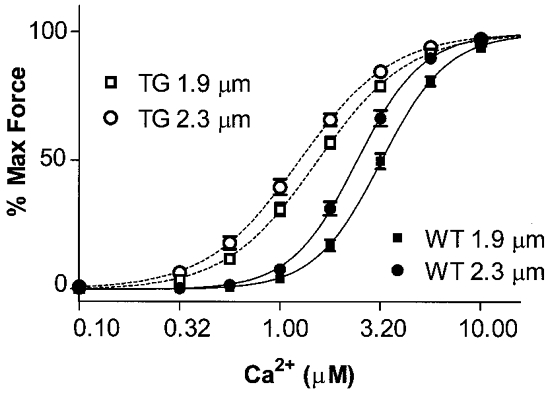
The half-maximal activating Ca2+ concentration (EC50) for WT skinned myofilaments was 3.20 ± 0.24 μm at SL 1.9 μm (n = 28, ▪) and 2.40 ± 0.16 μm at SL 2.3 μm (n = 25, •); for TG skinned myofilaments, the EC50 was 1.53 ± 0.08 μm at SL 1.9 μm (n = 28, □) and 1.25 ± 0.09 μm at SL 2.3 μm (n = 25, ○) (P < 0.05).
Data in Fig. 2 indicate that specific substitution of a thin filament protein in the myofilament lattice is able to alter length-dependent activation. One mechanism by which this may occur is by a relative increase in the binding rate of crossbridges when cTnI is replaced by ssTnI. We employed a pharmacological approach as an alternative mechanism to promoting crossbridge binding to the thin filament. In one set of experiments, we compared length-dependent activation of the myofilaments in the presence of the thiadiazinone EMD 57033, a pharmacological agent that increase rates of crossbridge binding to actin (Solaro et al. 1993; Tobias et al. 1996; Kraft & Brenner, 1997). Data shown in Fig. 3A compare the effect of EMD on the Ca2+-force relation of WT myofilaments at SL 1.9 μm and SL 2.3 μm. In WT myofilaments (Fig. 3A), sensitization to Ca2+ by EMD was length dependent. At a SL of 1.9 μm, the mean EC50 in the presence of EMD was 1.69 μm Ca2+ compared to 3.20 μm in the absence of the drug. At a SL of 2.3 μm the EC50 in the presence of EMD was 1.58 μm Ca2+ compared to 2.40 μm Ca2+ in the absence of EMD. Therefore in the WT myofilaments at the shorter SL, EMD induced a ΔEC50= 1.51 μm Ca2+, whereas at the longer SL it induced a ΔEC50= 0.82 μm Ca2+. This differential effect of EMD at the two lengths resulted in a significant blunting of length-dependent Ca2+ activation of force in WT myofilaments similar to that associated with isoform switching of TnI. The ΔEC50 for Ca2+ activation induced by a change in SL from 1.9 μm to 2.3 μm was 0.11 μm Ca2+, when measured in the presence of EMD, compared to 0.80 μm Ca2+ for the control WT preparations and 0.28 μm Ca2+ for ssTnI-TG preparations in the absence of drug. In addition, the sensitizing effect of EMD was less in the ssTnI-TG myofilaments than in the WT myofilaments. In ssTnI-TG myofilaments, the ΔEC50 induced by EMD was 0.45 μm Ca2+ at a SL of 1.9 μm and 0.25 μm Ca2+ at SL 2.3 μm. Moreover, as shown in Fig. 3B, treatment of ssTnI-TG myofilaments with EMD essentially ablated length-dependent activation (ΔEC50= 0.08 μm Ca2+).
Figure 3. The effect of 3 μm EMD 57033 on Ca2+-force relations of WT and TG skinned myofilaments at SL 1.9 μm and SL 2.3 μm.
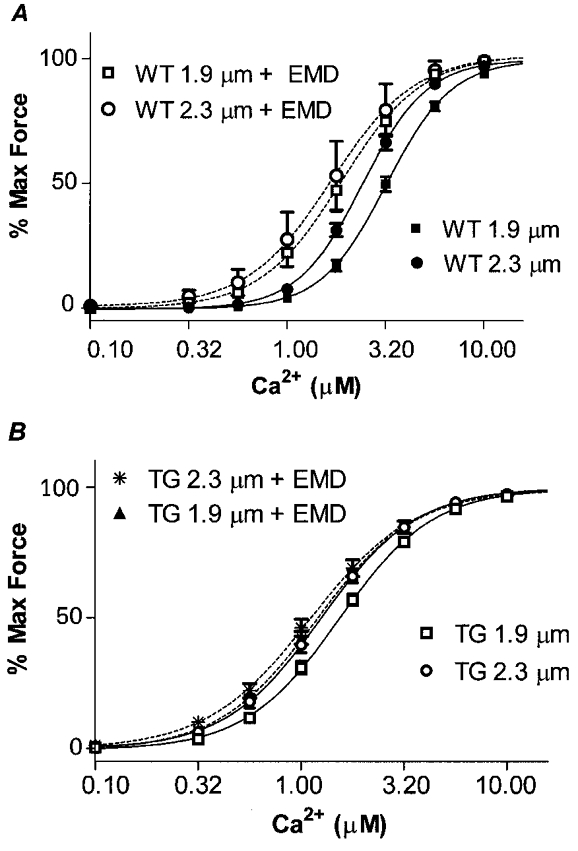
A, WT controls, EC50 3.20 ± 0.24 μm at SL 1.9 μm (n = 28, ▪) and 2.40 ± 0.16 μm at SL 2.3 μm (n = 25, •). WT + EMD, EC50 1.69 ± 0.33 μm at SL 1.9 μm (n = 5, □) and 1.58 ± 0.24 μm at SL 2.3 μm (n = 6, ○). B, TG controls, EC50 1.53 ± 0.08 μm at SL 1.9 μm (n = 28, □) and 1.25 ± 0.09 μm at SL 2.3 μm (n = 25, ○). TG + EMD, EC50 1.08 ± 0.05 μm at SL 1.9 μm (n = 6, ▴) and 1.00 ± 0.14 μm at SL 2.3 μm (n = 7, *).
We also tested the effect of a benzodiacine derivative, CGP-48506, on the length dependence of the Ca2+-force relation of WT and ssTnI-TG myofilaments (Fig. 4). Previous work from our laboratory (Wolska et al. 1996) demonstrated that CGP is able to increase contractility and myofilament force by a mechanism involving direct effects on the actin-myosin interaction. Unlike EMD, CGP has been shown to be devoid of any phosphodiesterase activity (Herold et al. 1995). The lack of phosphodiesterase inhibitory activity would not be expected to influence the results of our studies in detergent-extracted fibre bundles, but the difference between EMD and CGP may be of significance when the agents are applied to intact cardiac muscle preparations. Another potentially significant difference between EMD and CGP is the apparent myofibrillar receptor site. EMD has been shown to bind to the high affinity structural sites of cTnC (Pan & Johnson, 1996). On the other hand, we (Wolska et al. 1996) could find no effect of CGP on Ca2+ binding to cTnC. Figure 4 illustrates the length dependence of the increase in Ca2+ sensitivity induced by CGP. At a SL of 1.9 μm mean EC50 in the presence of CGP was 1.18 μm Ca2+ compared to 3.20 μm Ca2+ in the absence of the drug. At a SL of 2.3 μm the EC50 in the presence of CGP was 1.05 μm Ca2+ compared to 2.40 μm in the absence of CGP. Therefore at the shorter SL, CGP induced a ΔEC50= 2.02 μm Ca2+, whereas at the longer SL CGP induced a ΔEC50= 1.35 μm Ca2+. Thus, as was the case with EMD, there was a length dependence of the Ca2+ sensitization by CGP. Also, as shown in Fig. 4, the length-dependent ΔEC50 was 0.13 μm Ca2+ in the presence of CGP, compared to a ΔEC50 of 0.11 μm in the case of EMD. This differential effect occurred even though CGP induced a greater degree of Ca2+ sensitization compared to EMD.
Figure 4. The effect of 10 μm CGP-48506 on Ca2+-force relations of WT and TG skinned myofilaments at SL 1.9 μm and SL 2.3 μm.
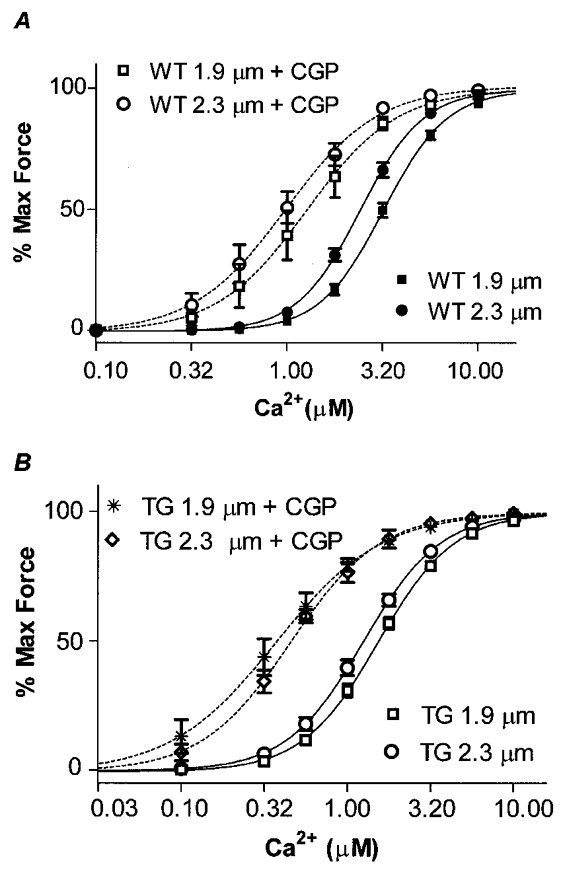
A, WT controls, EC50 3.20 ± 0.24 μm at SL 1.9 μm (n = 28, ▪) and 2.40 ± 0.16 μm at SL 2.3 μm (n = 25, •). WT + CGP, EC50 1.18 ± 0.22 μm at SL 1.9 μm (n = 7, □) and 1.05 ± 0.18 μm at SL 2.3 μm (n = 5, ○). B, TG controls, EC50 1.53 ± 0.08 μm at SL 1.9 μm (n = 28, □) and 1.25 ± 0.09 μm at SL 2.3 μm (n = 25, ○). TG + CGP, EC50 0.46 ± 0.13 μm at SL 1.9 μm (n = 4, *) and 0.39 ± 0.06 μm at SL 2.3 μm (n = 7, ⋄).
We also carried out a series of experiments on length-dependent activation employing calmidazolium (CDZ), a pharmacological agent that we (El-Saleh & Solaro, 1987) previously demonstrated binds to cTnC, increasing its affinity for Ca2+ and thereby sensitizing the myofilaments to Ca2+. Figure 5 shows results of experiments in which Ca2+-force relations were determined at long and short SL of WT fibre bundles in the presence of 20 μm CDZ. In agreement with our earlier report (El-Saleh & Solaro, 1987), 20 μm CDZ induced an increase in sub-maximal force (pCa 5.75) developed by the fibre bundles at the shorter SL of 1.9 μm. However, this sensitization by CDZ was not evident when the fibre bundles were stretched to a SL of 2.3 μm. In fact, these bundles exhibited a desensitization to Ca2+ as shown in Fig. 5 where at SL 2.3 μm the EC50 was 2.40 μm Ca2+ in controls and 3.02 μm Ca2+ in the presence of CDZ. The combination of a small sensitization at SL 1.9 μm and desensitization at the longer SL resulted in a loss of length-dependent activation in the preparations treated with CDZ. In the case of ssTnI-TG fibre bundles, there were small, but not significant effects of CDZ on the Ca2+-force relations at each SL (Fig. 5). Even so, in the case of the ssTnI-TG preparations, the length-dependent activation remained near zero in the presence of CDZ. Table 1 summarizes the values of EC50 for all the groups investigated, and compares the ΔEC50 between the two SLs studied.
Figure 5. The effect of 20 μm calmidazolium (CDZ) on Ca2+-force relations of WT and TG skinned myofilaments at SL 1.9 μm and SL 2.3 μm.
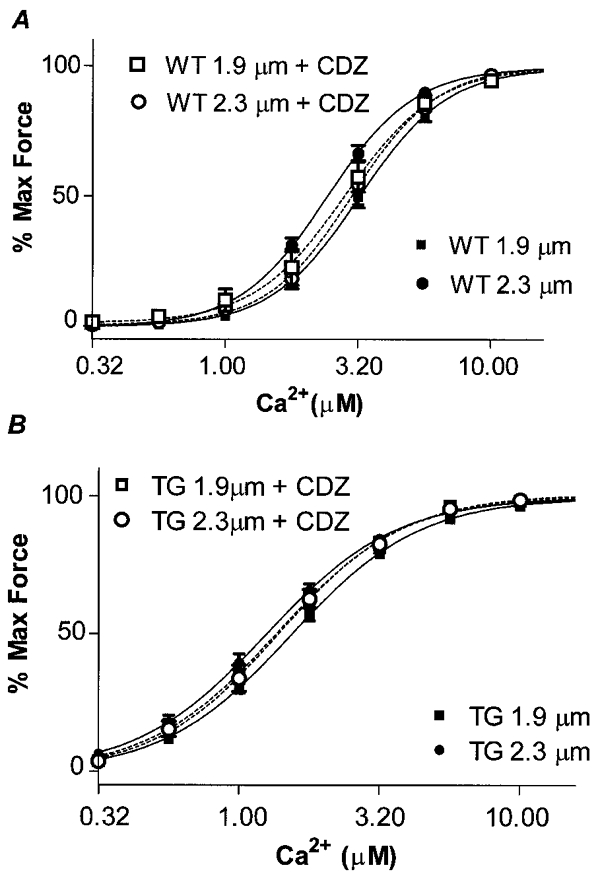
A, WT controls, EC50 3.20 ± 0.24 μm at SL 1.9 μm (n = 28, ▪) and 2.40 ± 0.16 μm at SL 2.3 μm (n = 25, •). WT + CDZ, EC50 2.89 ± 0.08 μm at SL 1.9 μm (n = 6, □) and 3.02 ± 0.07 μm at SL 2.3 μm (n = 4, ○). B, TG controls, EC50 1.53 ± 0.08 μm at SL 1.9 μm (n = 25, ▪) and 1.25 ± 0.09 μm at SL 2.3 μm (n = 20, •). TG + CDZ, EC50 1.38 ± 0.04 μm at SL 1.9 μm (n = 4, □) and 1.38 ± 0.14 μm at SL 2.3 μm (n = 5, ○).
Table 1.
Effects of inotropic agents in length dependence of Ca2+ activation of wild-type and transgenic cardiac myofilaments
| EC50 Ca2+(μM) | |||||
|---|---|---|---|---|---|
| Fibre bundle preparation | SL 1·9 μm | n | SL 2·3 μm | n | |
| Wild-type (WT) | 3·20 ±0·24 | 28 | 2·40 ± 0·162 | 25 | 0·80* |
| Transgenic (TG) | 1·53 ± 0·08 | 28 | 1·25 ± 0·092 | 25 | 0·28* |
| WT+EMD | 1·69 ± 0·33 | 5 | 1·58 ± 0·24 | 6 | 0·11* |
| TG + EMD | 1·08 ± 0·05 | 6 | 1·00 ± 0·14 | 7 | 0·08 |
| WT+CGP | 1·18 ± 0·22 | 7 | 1·05 ± 0·18 | 5 | 0·13* |
| TG+CGP | 0·46 ± 0·13 | 4 | 0·39 ± 0·06 | 7 | 0·07 |
| WT+CDZ | 2·89 ± 0·08 | 6 | 3·02 ± 0·07 | 4 | –0·13 |
| TG+CDZ | 1·38 ± 0·04 | 4 | 1·38 ± 0·14 | 5 | 0·00 |
P < 0·05; n, number of measurements.
DISCUSSION
Our results provide a new perspective on the understanding of the modulation of relations between ventricular volume and ESP and the theory that this relation is determined by the length sensitivity of myofilament Ca2+ activation. One important new finding reported here is that specific isoform switching from cTnI to ssTnI in the thin filament is able to alter length-dependent activation of cardiac myofilaments. Previous studies (Akella et al. 1995; Komukai & Kurihara, 1997) have also suggested that alterations in the thin filament structure and function may alter length-dependent activation. Yet these studies did not involve specific changes in the thin filament and may be related to mechanisms involving the thick filament and other cellular proteins, as well. Moreover, we (Wolska et al. 1999) previously reported that specific isoform switching from α-tropomyosin (α-Tm) to β-Tm in the hearts of transgenic mice induced a sensitization of the cardiac myofilaments to Ca2+ and a reduction in maximum tension, but did not alter length-dependent activation as gauged by shifts in the EC50 for activation of normalized maximum tension of skinned fibre bundles. Thus, the blunting of length-dependent activation that occurred with isoform switching of cTnI to ssTnI is not an inevitable consequence of the associated increase in Ca2+ sensitivity.
As a key thin filament component at the cross-roads of signalling between Ca2+ binding to cTnC and transmission of the signal to cTnT, Tm and actin (Solaro & Rarick, 1998), it is possible that switching of cTnI with ssTnI may affect length-dependent activation by multiple mechanisms. Possible mechanisms by which TnI isoform switching may affect length-dependent activation are couched in current theories about how variations in inter-filament spacing and strong crossbridge binding may affect myofilament activation. One theory is that strong crossbridge binding increases the affinity of cTnC for Ca2+ (Allen & Kentish, 1985; Hofmann & Fuchs, 1988). Interestingly though, slow skeletal myofilaments that naturally contain cTnC and ssTnI demonstrate length-dependent activation with no change in cTnC affinity for Ca2+, whereas cardiac myofilaments apparently do (Wang & Fuchs, 1994). Thus, the difference in length-dependent activation between myofilaments from WT and ssTnI-TG mouse hearts may be due to a difference in feedback effects of strongly bound crossbridges on cTnC Ca2+ binding. However, inasmuch as both slow and fast skeletal myofilaments demonstrate length-dependent activation with no change in TnC Ca2+ binding, we do not think this is a major mechanism.
A second theory for length-dependent activation is that changes in lattice spacing alter strong crossbridge binding thereby altering the rate of binding of near-neighbour crossbridges (McDonald & Moss, 1995; McDonald et al. 1997; Fitzsimmons & Moss, 1998). A related theory is that activation of a functional unit spreads to near-neighbour functional units by strong crossbridge-induced movements of Tm that affect activity of a contiguous Tm (Tawada et al. 1975; Pan et al. 1989). One mechanism by which a change in thin filament proteins may modulate length-dependent activation is by shifting the steady-state relation between the blocked, closed and open state of the myofilaments. Lehrer (1994) suggested, for example, that isoform shifts of TnI or TnT may induce a variation in weakly bound crossbridges, thus modulating the closed state. It is possible that TnI could affect the distribution of crossbridge states by both steric and allo-steric effects. cTnI appears to bind across two actin monomers and interacts with a region of actin that is shared by a binding site for crossbridges (Solaro & Rarick, 1998). In fact, reconsideration of the steric blocking model of myofilament activation now includes the potential effect that crossbridge binding to actin may be impeded not only by Tm but also by TnI and TnT. TnT is also a long asymmetric molecule extending across two to three actin monomers (Squire & Morris, 1998; Solaro & Rarick, 1998).
A significant structural difference between cTnI and ssTnI is that the cardiac variant possesses an additional mass of 30 amino acids forming an N-terminal extension not present in either slow skeletal or fast skeletal variants. It is not known whether the cardiac variant has an extended molecular length compared to ssTnI. It is known, however, that the N-terminal extension of cTnI contains serial serine residues that are sites of protein kinase A (PKA)-dependent phosphorylation (Solaro & Rarick, 1998). Activation of PKA-dependent phosphorylation cascades in situ and in vitro have been reported to alter length-dependent activation of cardiac muscle preparations (Komukai & Kurihara, 1997; Konhilas et al. 2000). The molecular mechanism for this effect of PKA activation on length-dependent activation is not clear, but it is likely that phosphorylation of cTnI is involved. We (Dong et al. 1997) have shown that PKA-dependent phosphorylation induces global changes in cTnI that may alter the state of extension of the N-terminus. Although more work needs to be done, this structural change may prove to be functionally significant. Under the conditions of these measurements, levels of phosphorylation of cTnI and myosin binding protein C are minimal (Fentzke et al. 1999; Konhilas et al. 2000). Our results do, however, indicate that in situ length-dependent activation may be altered in the ssTnI-TG hearts not only by the TnI isoform switch itself, but also by the lack of phosphorylation sites for PKA. In any case, our results fit with the idea that shifts in the isoform of TnI increase the number of crossbridges in the closed state, thus promoting the open state and attenuating the length dependence of Ca2+ activation. Along these lines, Fitzsimmons & Moss (1998) reported that promotion of the open state by titrating the myofilaments with strongly binding myosin heads (NEM-S1), eliminated length-dependent activation.
Similar findings were recently reported by Fukuda et al. (2000), who showed that addition of MgADP to skinned fibre preparations, which promotes strong crossbridge binding to the thin filament, also reduced the length dependence of Ca2+ sensitivity.
Shifts in the steady-state relation between blocked, open and closed states also provide a straightforward explanation for the attenuation of length-dependent activation by CGP and EMD. Both CGP and EMD have been demonstrated to exert their effects by promoting the rate of crossbridge binding to the thin filaments. When this rate is enhanced pharmacologically it would be expected that the ability of a change in lattice spacing to modulate crossbridge binding rate would be reduced. Of particular interest is our finding that treatment of the fibre bundles from ssTnI-TG mouse hearts with either EMD or CGP induced a myofilament state in which there was no length-dependent activation. Alterations of length-dependent activation are predicted to affect the relation between ventricular volume and ventricular ESP. Our findings provide a condition in which it is possible to test the relative significance of length-dependent activation as a determinant of the Frank-Starling relation using various approaches. In addition, our results indicate that use of these drugs as inotropic agents may not only involve enhanced contractility but also changes in the shape of the volume-pressure relations of the ventricle. Although the manifestation of a change in pCa-force relations in the volume-pressure relations of the intact ventricle is complex, we think a simplified prediction of how a change in length-dependent activation may affect the slope of the volume-pressure relation can be made (Fig. 6). As indicated in Fig. 6, a generalized prediction is that the slope of the volume-ESP relation would decrease with reductions in the dependence of activation of the myofilaments on sarcomere length. In the extreme case, where length-dependent activation is absent, we would expect the volume-pressure loop to be dominated by changes in filament overlap and/or altered Ca2+ release.
Figure 6. Theoretical representation of the predictive association between length-dependent activation and the slope of the ventricular volume-end-systolic pressure (ESP) relation.
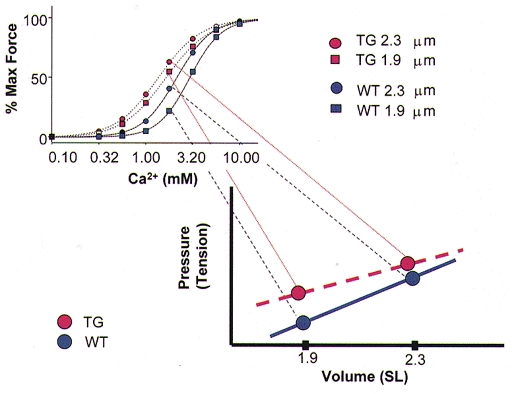
The blue data points and lines represent volume-ESP and Ca2+-force relations for the WT hearts demonstrating normal length-dependent activation. The red data points and lines represent the volume-ESP and myofilament Ca2+-force relations for ssTnI-TG hearts, and illustrate that blunting of length-dependent activation may reduce the slope of the volume-ESP relation.
Our data are relevant to the developmental physiology of the heart muscle in which there are natural variations in the TnI isoform population. During development, there is a progressive shift from a preponderance of ssTnI to cTnI, which is the exclusive isoform in the adult heart (Gao et al. 1995). The physiological relevance of this shift in thin filament isoforms is not clear, but one idea is that the presence of ssTnI renders the myofilaments more sensitive to Ca2+ activation and less sensitive to deactivation by acidic pH (Solaro & Van Eyk, 1996; Solaro & Rarick, 1998). This may be of some advantage to the fetal heart operating in a hypoxic environment with cells possessing scant sarcoplasmic reticulum. Data reported here also indicate that the presence of the ssTnI may modulate length-dependent activation and thus the Frank-Starling relation in a way not previously appreciated. In fact, in the developing heart, active volume-pressure relations are decreased compared to the mature heart (Friedman, 1972; Romero & Friedman, 1979). Part of the mechanism for the difference is likely to be due to the smaller size and smaller content of myofibrils in the developing myocytes. Interestingly, when volume-pressure relations are normalized for contractile/non-contractile mass, there is a similar relation between volume and pressure. Although these results indicate that the shifts in TnI isoform population may not alter the volume-pressure relation, the studies reported so far have not carefully correlated ssTnI expression with function. Our results indicate the importance of making such determinations as well as providing an approach in which length-dependent activation could be eliminated in the beating heart.
Acknowledgments
This work was supported by NIH Grants R37 HL 22231 (R.J.S.) and RO1 HL 54592 (J.M.L.). G.M.A. was supported by a Minority Individual in Postdoctoral Training Supplement to HL 22231, and NIH NHLBI Training Grant T32 HL 07692 supported K.A.P. We are grateful to Drs Anne F. Martin and Beata M. Wolska for help in the breeding of the transgenic mice. We are also grateful to Dr Sanjeev Shroff for helpful discussions.
References
- Akella AB, Ding XL, Cheng R, Gulati J. Diminished Ca2+ sensitivity of skinned cardiac muscle contractility coincident with troponin T-band shifts in the diabetic rat. Circulation Research. 1995;76:600–606. doi: 10.1161/01.res.76.4.600. [DOI] [PubMed] [Google Scholar]
- Allen DG, Kentish JC. The cellular basis of the length-tension relation in cardiac muscle. Journal of Molecular and Cellular Cardiology. 1985;17:821–840. doi: 10.1016/s0022-2828(85)80097-3. [DOI] [PubMed] [Google Scholar]
- Allen DG, Kurihara S. The effects of muscle length on intracellular calcium transients in mammalian cardiac muscle. The Journal of Physiology. 1982;327:79–94. doi: 10.1113/jphysiol.1982.sp014221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong W-J, Chandra M, Xing J, She M, Solaro RJ, Cheung HC. Phosphorylation-induced distance change in a cardiac muscle troponin I mutant. Biochemistry. 1997;36:6754–6761. doi: 10.1021/bi9622276. [DOI] [PubMed] [Google Scholar]
- El-Saleh SC, Solaro RJ. Calmidazolium, a calmodulin antagonist, stimulates calcium-troponin C and calcium-calmodulin-dependent activation of striated muscle myofilaments. Journal of Biological Chemistry. 1987;262:17240–17246. [PubMed] [Google Scholar]
- Fentzke RC, Buck SH, Patel JR, Lin H, Wolska BM, Stojanovic MO, Martin AF, Solaro RJ, Moss RL, Leiden JM. Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. The Journal of Physiology. 1999;517:143–157. doi: 10.1111/j.1469-7793.1999.0143z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimons DP, Moss RL. Strong binding of myosin modulates length-dependent Ca2+ activation of rat ventricular myocytes. Circulation Research. 1998;83:602–607. doi: 10.1161/01.res.83.6.602. [DOI] [PubMed] [Google Scholar]
- Frank O. Zur dynamik des Herzmusckels. Zietschrift fur Biologie. 1885;32:370–447. [Google Scholar]
- Friedman WF. The intrinsic physiologic properties of the developing heart. Progress in Cardiovascular Diseases. 1972;15:87–111. doi: 10.1016/0033-0620(72)90006-0. [DOI] [PubMed] [Google Scholar]
- Fuchs F, Wang YP. Length-dependence of actin-myosin interaction in skinned cardiac muscle fibres in rigor. Journal of Molecular and Cellular Cardiology. 1997;29:3267–3274. doi: 10.1006/jmcc.1997.0552. [DOI] [PubMed] [Google Scholar]
- Fukuda N, Kajiwara H, Ishiwata S, Kurihara S. Effects of MgADP on length dependence of tension generation in skinned rat cardiac muscle. Circulation Research. 2000;86:E1–6. doi: 10.1161/01.res.86.1.e1. [DOI] [PubMed] [Google Scholar]
- Gao L, Kennedy JM, Solaro RJ. Differential expression of TnI and TnT isoforms in rabbit heart during the perinatal period and during cardiovascular stress. Journal of Molecular and Cellular Cardiology. 1995;27:541–550. doi: 10.1016/s0022-2828(08)80049-1. [DOI] [PubMed] [Google Scholar]
- Godt RE, Lindley BD. Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle fibers of the frog. Journal of General Physiology. 1982;80:279–297. doi: 10.1085/jgp.80.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SM, Lamont C, Miller DJ. Hysteresis and the length dependence of calcium sensitivity in chemically skinned rat cardiac muscle. The Journal of Physiology. 1988;401:115–143. doi: 10.1113/jphysiol.1988.sp017154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold P, Herzig JW, Wenk P, Leutert T, Zbinden P, Fuhrer W, Stutz S, Schenker K, Meier M, Rihs G. 5-Methyl-6-phenyl-1,3,5,6-tetrahydro-3,6-methano-1,5-benzodiazocine-2,4-dione (BA 41899): representative of a novel class of purely calcium-sensitizing agents. Journal of Medicinal Chemistry. 1995;38:2946–2954. doi: 10.1021/jm00015a017. [DOI] [PubMed] [Google Scholar]
- Hofmann PA, Fuchs F. Bound calcium and force development in skinned cardiac muscle bundles: effect of sarcomere length. Journal of Molecular and Cellular Cardiology. 1988;20:667–677. doi: 10.1016/s0022-2828(88)80012-9. [DOI] [PubMed] [Google Scholar]
- Komukai K, Kurihara S. Length dependence of Ca(2+)-tension relationship in aequorin-injected ferret papillary muscles. American Journal of Physiology. 1997;273:H1068–1074. doi: 10.1152/ajpheart.1997.273.3.H1068. [DOI] [PubMed] [Google Scholar]
- Konhilas JP, Wolska BM, Martin AF, Solaro RJ, Detombe P. PKA modulates length-dependent activation in murine myocardium. Biophysical Journal. 2000;78:108A. [Google Scholar]
- Kraft T, Brenner B. Force enhancement without changes in cross-bridge turnover kinetics: the effect of EMD 57033. Biophysical Journal. 1997;72:272–281. doi: 10.1016/S0006-3495(97)78666-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lehrer SS. The regulatory switch of the muscle thin filament: Ca2+ or myosin heads? Journal of Muscle Research and Cell Motility. 1994;15:232–236. doi: 10.1007/BF00123476. [DOI] [PubMed] [Google Scholar]
- McDonald KS, Moss RL. Osmotic compression of single cardiac myocytes eliminates the reduction in Ca2+ sensitivity of tension at short sarcomere length. Circulation Research. 1995;77:199–205. doi: 10.1161/01.res.77.1.199. [DOI] [PubMed] [Google Scholar]
- McDonald KS, Wolff MR, Moss RL. Sarcomere length dependence of the rate of tension redevelopment and submaximal tension in rat and rabbit skinned skeletal muscle fibres. The Journal of Physiology. 1997;501:607–621. doi: 10.1111/j.1469-7793.1997.607bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan BS, Gordon AM, Luo ZX. Removal of tropomyosin overlap modifies cooperative binding of myosin S-1 to reconstituted thin filaments of rabbit striated muscle. Journal of Biological Chemistry. 1989;264:8495–8498. [PubMed] [Google Scholar]
- Pan BS, Johnson RGJ. Interaction of cardiotonic thiadiazinone derivatives with cardiac troponin C. Journal of Biological Chemistry. 1996;271:817–823. (Published erratum appears in Journal of Biological Chemistry (1996) 271, 19632) [PubMed] [Google Scholar]
- Patterson SW, Piper H, Starling EH. The regulation of the heart beat. The Journal of Physiology. 1914;48:357–379. doi: 10.1113/jphysiol.1914.sp001676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser PJ, Westfall MV, Schiaffino S, Solaro RJ. Tension production and thin-filament protein isoforms in developing rat myocardium. American Journal of Physiology. 1994;267:H1589–1596. doi: 10.1152/ajpheart.1994.267.4.H1589. [DOI] [PubMed] [Google Scholar]
- Romero TE, Friedman WF. Limited left ventricular response to volume overload in the neonatal period: a comparative study with the adult animal. Pediatric Research. 1979;13:910–915. doi: 10.1203/00006450-197908000-00008. [DOI] [PubMed] [Google Scholar]
- Solaro RJ, Gambassi G, Warshaw DM, Keller MR, Spurgeon HA, Beier N, Lakatta EG. Stereoselective actions of thiadiazinones on canine cardiac myocytes and myofilaments. Circulation Research. 1993;73:981–990. doi: 10.1161/01.res.73.6.981. [DOI] [PubMed] [Google Scholar]
- Solaro RJ, Rarick HM. Troponin and tropomyosin: proteins that switch on and tune in the activity of cardiac myofilaments. Circulation Research. 1998;83:471–480. doi: 10.1161/01.res.83.5.471. [DOI] [PubMed] [Google Scholar]
- Solaro RJ, Van Eyk J. Altered interactions among thin filament proteins modulate cardiac function. Journal of Molecular and Cellular Cardiology. 1996;28:217–230. doi: 10.1006/jmcc.1996.0021. [DOI] [PubMed] [Google Scholar]
- Squire JM, Morris EP. A new look at thin filament regulation in vertebrate skeletal muscle. FASEB Journal. 1998;12:761–771. doi: 10.1096/fasebj.12.10.761. (Published erratum appears in FASEB Journal (1998) 12, 1252.) [DOI] [PubMed] [Google Scholar]
- Tawada Y, Oara H, Ooi T, Tawada K. Non-polymerizable tropomyosin and control of the superprecipitation of actomyosin. Journal of Biochemistry. 1975;78:65–72. (Tokyo) [PubMed] [Google Scholar]
- Tobias AH, Slinker BK, Kirkpatrick RD, Campbell KB. Functional effects of EMD-57033 in isovolumically beating isolated rabbit hearts. American Journal of Physiology. 1996;271:H51–58. doi: 10.1152/ajpheart.1996.271.1.H51. [DOI] [PubMed] [Google Scholar]
- Wang YP, Fuchs F. Length, force, and Ca(2+)-troponin C affinity in cardiac and slow skeletal muscle. American Journal of Physiology. 1994;266:C1077–1082. doi: 10.1152/ajpcell.1994.266.4.C1077. [DOI] [PubMed] [Google Scholar]
- Wattanapermpool J, Reiser PJ, Solaro RJ. Troponin I isoforms and differential effects of acidic pH on soleus and cardiac myofilaments. American Journal of Physiology. 1995;268:C323–330. doi: 10.1152/ajpcell.1995.268.2.C323. [DOI] [PubMed] [Google Scholar]
- Wolska BM, Keller RS, Evans CC, Palmiter KA, Phillips RM, Muthuchamy M, Oehlenschlager J, Wieczorek DF, De Tombe PP, Solaro RJ. Correlation between myofilament response to Ca2+ and altered dynamics of contraction and relaxation in transgenic cardiac cells that express beta-tropomyosin. Circulation Research. 1999;84:745–751. doi: 10.1161/01.res.84.7.745. [DOI] [PubMed] [Google Scholar]
- Wolska BM, Kitada Y, Palmiter KA, Westfall MV, Johnson MD, Solaro RJ. CGP-48506 increases contractility of ventricular myocytes and myofilaments by effects on actin-myosin reaction. American Journal of Physiology. 1996;270:H24–32. doi: 10.1152/ajpheart.1996.270.1.H24. [DOI] [PubMed] [Google Scholar]