

Abstract
Many physiological and behavioural functions have circadian rhythms – endogenous oscillations with a period of approximately 24 h that can occur even in the absence of sleep. We determined whether there is an endogenous circadian rhythm in breathing, metabolism and ventilatory chemosensitivity in humans.
Ten healthy, adult males were studied throughout 4 days in a stable laboratory environment. After two initial baseline days (16 h wakefulness plus 8 h sleep) that served to achieve a steady state, subjects were studied under constant behavioural and environmental conditions throughout 41 h of wakefulness. Ventilation, metabolism and the magnitude of the hypercapnic ventilatory response (HCVR) were measured every 2 h. Individuals’ data were aligned according to circadian phase (core body temperature minimum; CBTmin) and averaged.
In the group average data, there was a significant and large amplitude circadian variation in HCVR slope (average of ±0.4 l min−1 mmHg−1; corresponding to ±12.1 % of 24 h mean), and a smaller amplitude rhythm in the HCVR x-axis intercept (average of ±1.1 mmHg; ±2.1 % of 24 h mean).
Despite a significant circadian variation in metabolism (±3.2 % of 24 h mean), there were no detectable rhythms in tidal volume, respiratory frequency or ventilation. This small discrepancy between metabolism and ventilation led to a small but significant circadian variation in end-tidal PCO2(PET,CO2; ±0.6 mmHg; ±1.5 % of 24 h mean).
The circadian minima of the group-averaged respiratory variables occurred 6-8 h earlier than CBTmin, suggesting that endogenous changes in CBT across the circadian cycle have less of an effect on respiration than equivalent experimentally induced changes in CBT.
Throughout these circadian changes, there were no correlations between HCVR parameters (slope or x-axis intercept) and either resting ventilation or resting PET,CO2. This suggests that ventilation and PET,CO2 are little influenced by central chemosensory respiratory control in awake humans even when at rest under constant environmental and behavioural conditions.
The characteristic change in PET,CO2 during non-rapid eye movement sleep was shown to be independent of circadian variations in PET,CO2, and probably reflects a change from predominantly behavioural to predominantly chemosensory respiratory control.
This study has documented the existence and magnitude of circadian variations in respiration and respiratory control in awake humans for the first time under constant behavioural and environmental conditions. These results provide unique insights into respiratory control in awake humans, and highlight the importance of considering the phase of the circadian cycle in studies of respiratory control.
In many animals, including humans, there is a robust diurnal rhythm in respiratory control which is thought to be induced by the behavioural rhythm of sleep and wakefulness (reviewed by Phillipson & Bowes, 1986). For instance, during sleep there is a systematic increase in the arterial PCO2(Pa,CO2) and a systematic decrease in ventilation, metabolism and ventilatory chemosensitivity (e.g. Douglas et al. 1982a; Berger & Phillips, 1988; Schafer, 1998). Although many physiological and behavioural functions have circadian rhythms – endogenous oscillations with a period of approximately 24 h that can occur in the absence of sleep – no studies have determined whether there exists a circadian rhythm in respiration and respiratory control in the absence of sleep and without simultaneous changes in behaviour or the environment. A circadian rhythm in respiratory control could be caused by direct neural influences on the brainstem respiratory complex from the circadian pacemaker located in the suprachiasmatic nuclei (e.g. via the paraventricular nuclei of the hypothalamus and/or the reticular formation) or indirect influences on metabolism and/or respiratory control via circadian rhythms in other variables. For example, circulating hormones and body temperature have prominent endogenous circadian rhythms (e.g. Czeisler et al. 1989) and these variables also affect respiratory control when they are manipulated experimentally (e.g. Koepchen, 1953; Vejby-Christensen & Strange Petersen, 1973; Petersen & Vejby-Christensen, 1977; Baker et al. 1996). Nonetheless, it has yet to be determined whether the respiratory responses to endogenous changes in these other variables that naturally occur over the circadian cycle would be different from the respiratory responses to equivalent experimentally induced changes in these variables (in the former case the control system ‘set-point’ may change over the circadian cycle inducing a change in the measured variable; in the latter case, the variable departs from the control system ‘set-point’).
We sought to answer the following specific questions in humans. (i) What are the magnitudes of any circadian rhythms of resting metabolism, ventilation, Pa,CO2 and hypercapnic ventilatory response (HCVR)? A large circadian variation in respiratory variables would have widespread implications for basic studies of respiratory control and assessment of patients with respiratory problems. (ii) What is the phase relationship between known circadian rhythms (e.g. core body temperature and plasma cortisol) to any circadian changes in breathing, metabolism or respiratory control? A close temporal relationship of circadian changes among variables could suggest a mechanistic link. (iii) Do circadian changes in respiratory control contribute to the increase in Pa,CO2 and the decrease in HCVR slope that have been documented to occur during sleep? If the magnitude of any circadian rhythm in respiratory variables is out of phase with or is less than the changes that occur during sleep, this would indicate independent sleep and circadian influences upon respiration. Part of this study has previously been published in abstract form (Spengler et al. 1997).
METHODS
The study was approved by the internal review board at Brigham and Women's Hospital, and was carried out in accordance with the Declaration of Helsinki. Subjects were informed of the procedures and possible risks, and each subject provided written consent prior to participation. The study was conducted on 10 healthy, adult male subjects: their age was 23.7 ± 3.9 years (mean ±s.d.), their height was 176.3 ± 6.4 cm, their weight was 73.9 ± 11.3 kg (body mass index (BMI) = 23.8 ± 3.3 kg m−2) and they had normal lung function (vital capacity 5.5 ± 0.9 l, forced expiratory volume in 1 s 4.5 ± 0.8 l and peak flow 10.2 ± 1.9 l s−1). Individuals with evidence of significant psychopathology were excluded.
Ambulatory monitoring
To ensure a stable circadian baseline before entry into the laboratory, subjects were asked to establish a regular sleep-wake schedule with their habitual bedtimes and waketimes varying by no more than 1 h each day for at least 2 weeks prior to the laboratory study. This schedule was verified by time-logged telephone calls of bedtimes and waketimes for 2 weeks as well as activity monitoring for 3-7 days (wrist-worn Actigraph; Ambulatory Monitoring, Ardsley, NY, USA). For 1 week prior to the study, subjects abstained from caffeinated drinks, smoking and any medication.
Laboratory baseline monitoring
Subjects were then studied in an individual laboratory suite isolated from sunlight and external time cues, including clocks, radios, television, visitors, mail and sunlight, but maintained contact with staff members who were trained to avoid communicating the time of day. The laboratory temperature was maintained at approximately 23°C. The laboratory protocol included two initial baseline days (16 h wakefulness plus 8 h sleep opportunity) that enabled the subjects to acclimatise to the environment and become familiarised with the equipment and respiratory tests (see below), and to assist in achieving a relatively steady state.
Laboratory constant routine protocol
Subjects woke up at their usual time on the morning of the third day in the laboratory and remained in bed and awake in a semi-recumbent position for 41 h in an established constant routine protocol (Mills et al. 1978; Czeisler et al. 1986). This constant routine protocol was performed under the same time-isolation conditions as the ‘laboratory baseline monitoring’ and was designed to ‘unmask’ underlying circadian rhythms as it reduces or eliminates influences on breathing from the environment, varied behaviours and sleep. During the constant routine, small identical snacks were given every 2 h composed of approximately 25 % fat, 50 % carbohydrate and 25 % protein. The overall calories were calculated using the Harris-Benedict formula with an activity factor of 1.4 (Harris & Benedict, 1919). The subjects also received 3.5 l of fluids per 24 h period. Experimenters were present in the laboratory throughout the constant routine to ensure that the subjects remained awake, and this was verified from continuous recordings of two electroencephalograms (EEG), two electro-oculograms and a submental electromyogram (Nicolet Biomedical Inc., Madison, WI, USA).
Measurement of standard markers of the circadian pacemaker
Two robust markers of the endogenous circadian pacemaker were also measured: core body temperature (CBT) was recorded continuously using a rectal temperature sensor (Yellow Springs Instrument Company, Yellow Springs, OH, USA) and plasma cortisol concentration was determined from 1.75 ml blood sampled from an indwelling venous catheter every 30 min. Cortisol analysis was performed with a paramagnetic, chemoluminescent immunoassay (Beckman Coulter, Miami, FL, USA).
Respiratory measurements
In addition, measurements of resting ventilation, resting metabolism and the magnitude of the HCVR were performed every 2 h. For these respiratory measurements, subjects breathed through a mouthpiece with a nose-clip in place. To ensure a steady state of relaxed wakefulness for resting measurements, subjects had their eyes open and focused upon a picture directly in front of them for 10 min, and only data from the fifth to ninth minute were analysed. Since drowsiness can affect breathing (Bulow, 1963), drowsiness was quantified by documenting the average number of EEG theta waves (3-7 Hz) in each 30 s epoch during periods of resting respiratory measurements (greater than 45 theta waves within 30 s usually indicates significant drowsiness; Rechtschaffen & Kales, 1968). During this time, subjects breathed via a calibrated turbine for measuring volume and tidal gases were sampled using calibrated fast-responding O2 (paramagnetic) and CO2 (infrared) analysers that compensated for alterations in barometric pressure (OxyconAlpha system; Erich Jaeger GmbH, Würzburg, Germany). Breath-by-breath values for minute ventilation (VE), tidal volume (VT), respiratory frequency (fR), end-tidal partial pressure of CO2 (PET,CO2, a non-invasive estimate of Pa,CO2), oxygen consumption (VO2) and carbon dioxide production (VCO2) were then calculated. VO2 and VCO2 were determined from the volume of these gases expired over each breath (the program accounted for the phase delay between air volume and gas concentrations – due to instrument response time and transport between the mouthpiece and the gas sensors).
The magnitude of HCVR (change in ventilation relative to the change in PET,CO2) was measured by means of the Read rebreathing method (Read, 1967). Subjects breathed via a mouthpiece with a nose-clip in place for 2 min before being switched into a low-resistance spirometer circuit (Warren E. Collins, Braintree, MA, USA) that was initially filled with a mixture of 7 % CO2 and 93 % O2 to a volume equal to the subject's vital capacity plus 1 l. These standard gas concentrations were used so that the test was started at a PCO2 which approximated mixed-venous PCO2 and hyperoxia diminished peripheral chemoreceptor stimulation, thereby testing central chemoreceptor sensitivity (Read, 1967). Subjects re-breathed from the spirometer until PET,CO2 reached 65 mmHg. Tidal gases were analysed by an instrument that compensated for alterations in barometric pressure (a calibrated Cardiocap II; Datex Medical Instruments, Tewksbury, MA, USA). Spirometer volume and signals were recorded on a Macintosh computer (Superscope II, GW Instruments, Cambridge, MA, USA) and analysed using an objective technique for determining the threshold and slope of the HCVR (Lorinc et al. 1991). Extrapolation of the linear part of the HCVR relationship enabled determination of the theoretical intercept of the central ventilatory chemoreflex with the x-axis.
To determine the magnitude of the usual sleep-induced increase in PET,CO2 (for comparison with any circadian changes in PET,CO2), PET,CO2 was measured from tidal gases sampled at a nostril immediately before sleep and during sleep throughout a night in the laboratory in a subgroup of six of the same subjects.
Analysis of circadian rhythmicity
To further ensure circadian measurements were made in basal conditions, the first 5 h of constant routine data were excluded from all analyses to eliminate any residual effects of sleep on respiration. For comparison among variables sampled at different frequencies, 2 h averages of all variables were calculated, centred around the minimum CBT for every subject. The circadian phase and period of the CBT rhythm were estimated for each subject by a two-harmonic regression analysis of the temperature data using a non-linear least-squares method (Czeisler et al. 1989; Brown, 1992). This established technique constrains the circadian period to be within the normal physiological range of 24.0-24.3 h (Czeisler et al. 1999). Allowing this small degree of flexibility of circadian period results in a more accurate estimate of circadian phase. The period was used as a constant in the subsequent analysis of the circadian rhythmicity of the other variables (see below), which was performed on both individuals’ data and group mean data. The group mean period of subjects’ CBT rhythms was 24.08 h. For group analyses, individuals’ data were aligned with respect to their subject-specific minimum CBT and averaged, resulting in 16 sequential group average data points equally spaced across approximately 1.33 circadian cycles. The circadian phases and amplitudes of the respiratory, hormonal and EEG variables were estimated by cosinor analysis (Nelson et al. 1979), using the following equation: y =M+Acos(ω t+ψ) +st (where y is the selected variable, M is mesor, A is the amplitude of circadian rhythm (peak minus mean), ω is 2π/period of circadian rhythm, ψ is the phase of the circadian rhythm, t is time (h) and s is the underlying linear trend (h−1)). As the period of the circadian rhythm was known (from analysis of CBT), it was possible to estimate the parameters of the equation using multiple linear regression.
The extent to which the multiple linear regression for each variable could be used as a predictive model was estimated by calculating the fraction of the variability in y that was explained by the cosinor model (i.e. R2) The statistical significance of the model was determined from analyses of variance, with probability derived from the F ratio (mean square error from the multiple linear regression model (3 degrees of freedom) divided by the residual mean square error (12 degrees of freedom); Statview, Abacus Concepts, Berkeley, USA). To detect the existence of circadian rhythms without interference from an underlying linear trend in the data (e.g. due to effects of sleep deprivation), cosinor analysis was also applied on the detrended data (i.e. y–st). For this analysis, there were also 16 data points for group data (i.e. 15 total degrees of freedom). Statistically significant circadian oscillations were acknowledged to have occurred if P < 0.05 for F2/13. The phase shift between the circadian rhythms of CBT and the other variables was estimated as the time lag between minima of the fitted rhythms.
RESULTS
Confirmation of relaxed wakefulness
Relaxed wakefulness was confirmed during all measurement periods by EEG analysis. The threshold for significant drowsiness (45 theta waves within 30 s) was not exceeded in any subject. Indeed, theta rhythm rarely occurred, occupying less than 2 % of the EEG records at all circadian phases.
Magnitude of circadian rhythm of the respiratory variables
Group mean data of core body temperature, cortisol and selected respiratory variables are shown in Table 1 and Fig. 1. As shown in Table 1, the multiple linear regression analyses explained between 42 and 72 % of the total variation in the respiratory data. The analyses of variance of the full model as well as the analyses of detrended data confirmed significant group mean circadian variations in HCVR slope, HCVR x-axis intercept, VO2 and VCO2, but there were no statistically significant circadian rhythms in VE, VT, fR or the respiratory exchange ratio. The amplitudes of the significant circadian rhythms of the respiratory variables ranged from ±1.5 to ±12.1 % of the 24 h means. The magnitudes of the circadian changes (peak to trough) in those respiratory variables that exhibited significant circadian rhythms were always greater (range 2-46 times) than any underlying linear trend within these variables over 24 h. The small linear trends are probably attributable to the effects of prolonged wakefulness.
Table 1.
Magnitude of circadian rhythms derived from cosinor analyses
| Amplitude | Trend | Phase difference vs. CBT | ||||
|---|---|---|---|---|---|---|
| Variable | 24 h mean | (%24 h mean) | (% 24 h mean) | R2 | F ratio | |
| Pet, CO2 | 40·8 mm Hg | 0·6 mm Hg | − 0·008 mm Hg h−1 | 0·66 | 8·4** | −7·9 h |
| (1·5%) | (0·02% h−1) | |||||
| V̇e | 8·31 min−1 | 0·21 min−1 | 0·0131 min−1h−1 | 0·42 | 2·9 | −7·3 h |
| (2·4%) | (0·16% h−1) | |||||
| V̇O2 | 278 ml | 9 ml | 0·349 ml h−1 | 0·70 | 9·3** | −7·8 h |
| (3·2%) | (0·13%h−1) | |||||
| V̇CO2 | 246 ml | 8 ml | 0·380 ml h−1 | 0·67 | 8·1** | −8·0 h |
| (3·2%) | (0·15% h−1) | |||||
| HCVR slope | 3·31 min−1mmHg−1 | 0·41 min−1mmHg−1 | 0·021 l min−1mmHg-1h−1 | 0·72 | 11·3*** | −5·7 h |
| (12·1%) | (0·64% h−1) | |||||
| HCVR x-intercept | 50·9 mmHg | 1·1 mmHg | 0·002 mmHg h−1 | 0·46 | 3·7* | −8·4 h |
| (2·2%) | (0% h−1) | |||||
| Cortisol | 10·6 μg dl−1 | 5·5 μg dl−1 | 0·064 μg dl−1h−1 | 0·91 | 41·3*** | −7·9 h |
| (51·9%) | (0·60 h−1) | |||||
| CBT | 37·1 °C | 0·3 °C | −0·001 °C h−1 | 0·90 | 35·9*** | 0 h |
Mean daa of end-tidal PCO2 (Pet, CO2), minute ventilation (V̇E), O2 uptake (V̇O2), CO2 production (V̇CO2), hypercapnic ventilatory response (HCVR; slope and x-axis intercept), cortisol concentration and core body temperature (CBT) were calculated from 24 h data centred around the time of hte minimum CBT (CBTmin). Circadian amplutude and linear trend are derived from ultiple linear regression analysis of 32 h group mean data (aligned to CBTmin and averaged). R2 represents the ratio of the variation explanined by the non-linear regression analysis to the total variation in the data. The F ratio establishes whether addition of parameters that describe circadian rhythmicity reduces the sum of sqauared errors in comparison with a simple linear regression model that assumes on circandian rhythmicity (significance levels of circadian rhythmicity are shown: P < 0·05)
Mean daa of end-tidal PCO2 (Pet, CO2), minute ventilation (V̇E), O2 uptake (V̇O2), CO2 production (V̇CO2), hypercapnic ventilatory response (HCVR; slope and x-axis intercept), cortisol concentration and core body temperature (CBT) were calculated from 24 h data centred around the time of hte minimum CBT (CBTmin). Circadian amplutude and linear trend are derived from ultiple linear regression analysis of 32 h group mean data (aligned to CBTmin and averaged). R2 represents the ratio of the variation explanined by the non-linear regression analysis to the total variation in the data. The F ratio establishes whether addition of parameters that describe circadian rhythmicity reduces the sum of sqauared errors in comparison with a simple linear regression model that assumes on circandian rhythmicity (significance levels of circadian rhythmicity are shown: P < 0·01).
Mean daa of end-tidal PCO2 (Pet, CO2), minute ventilation (V̇E), O2 uptake (V̇O2), CO2 production (V̇CO2), hypercapnic ventilatory response (HCVR; slope and x-axis intercept), cortisol concentration and core body temperature (CBT) were calculated from 24 h data centred around the time of hte minimum CBT (CBTmin). Circadian amplutude and linear trend are derived from ultiple linear regression analysis of 32 h group mean data (aligned to CBTmin and averaged). R2 represents the ratio of the variation explanined by the non-linear regression analysis to the total variation in the data. The F ratio establishes whether addition of parameters that describe circadian rhythmicity reduces the sum of sqauared errors in comparison with a simple linear regression model that assumes on circandian rhythmicity (significance levels of circadian rhythmicity are shown: ***P < 0·001).
The phase difference represents the time between CBTmin and the minimum of each respiratory variable as derived from the linear regression analysis.
Figure 1.
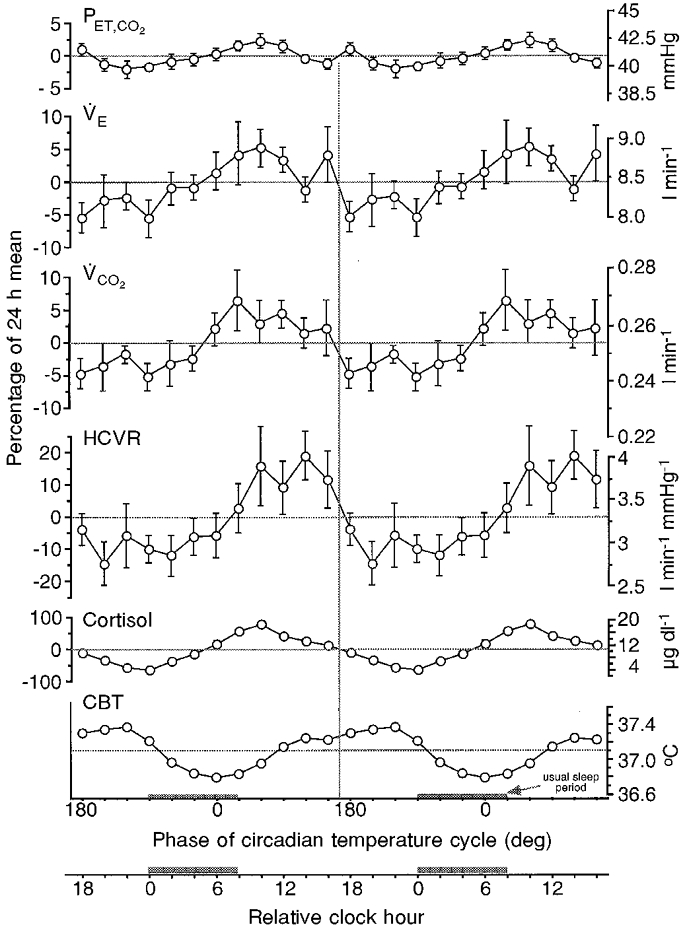
Circadian rhythms of respiration detected with a constant routine protocol
Shown are the group mean levels (±s.e.m.) of end-tidal PCO2 (PET,CO2), ventilation (VE), CO2 production (VCO2), the slope of the line that describes the hypercapnic ventilatory response (HCVR), plasma cortisol concentration and core body temperature (CBT). Individuals’ data (n = 10) were aligned with respect to the reference circadian rhythm – core body temperature minimum (CBTmin) – and averaged. The s.e.m. of all data are shown, but are small and indistinguishable from the mean at most time points for PET,CO2, cortisol and CBT. Wider variation occurred in the other variables. The ordinate is expressed as the percentage deviation from the 24 h mean (left) and in absolute units (right). Data from 24 h collected during the constant routine and centred around the CBTmin are ‘double plotted’ for ease of visualising circadian rhythms (i.e. the 24 h data on the left are reproduced on the right). The abscissa is expressed in degrees (with CBTmin assigned a phase of 0 deg) and in relative clock hour. The shaded bars on the abscissa represent the time of the subjects’ usual sleep episodes (though sleep did not occur during this constant routine). Small differences occur between the values in Table 1 and the impression from this figure because the values in the table were derived from the regression analysis, whereas the actual mean data are plotted in this figure.
HCVR slope had the largest respiratory circadian rhythm with a group average amplitude of ±12.1 % of the 24 h mean. Slight circadian phase or period differences among subjects meant that circadian rhythm amplitudes within individual subjects were generally larger than the group average. Individuals’ amplitudes ranged from ±0.05 to ±1.33 l min−1 mmHg−1, with an average of ±0.56 l min−1 mmHg−1 (±15.2 % of 24 h mean). For the group, there was a small but significant circadian rhythm in HCVR x-axis intercept (average of ±1.1 mmHg; ±2.2 % of 24 h mean) and resting PET,CO2 (average of ±0.6 mmHg; ±1.5 % of 24 h mean). Circadian amplitudes of individuals’PET,CO2 rhythms were still small, ranging from ±0.3 to ±2.4 mmHg (average ±1.0 mmHg; ±2.4 % of 24 h mean). As there was no significant circadian rhythm in VE, the circadian rhythm of PET,CO2 appeared to be caused by a circadian rhythm in metabolism (amplitudes for VO2 and VCO2 were ±3.2 % of 24 h mean).
Relationships among variables
The times between the CBTmin and the minimum of each respiratory variable as derived from the regression analysis are shown in Table 1 (Phase difference). The circadian minima of the respiratory variables occurred close to the minimum of the plasma cortisol rhythm, but 6-8 h earlier than the minimum of the CBT rhythm. The relationship among variables across the circadian cycle was further explored within each subject by calculating correlations between variables (at zero lag). Graphical results from a typical subject are presented in Fig. 2 and a group correlation matrix is presented in Table 2. A number of striking observations emerge. There was a significant positive correlation between HCVR x-axis intercept and HCVR slope in all 10 subjects (median Pearson's product-moment correlation coefficient of +0.81). There was a significant positive correlation between ventilation and metabolism in all 10 subjects (median of +0.70 for VEvs. VO2 and +0.91 for VEvs. VCO2). There was a significant negative correlation between VE/VCO2 and PET,CO2 (median of -0.68, significant in 7 out of 10 subjects), indicating that as metabolism rose, ventilation rose less, inducing an increase in PET,CO2. These three relationships were also the only ones that emerged as significant in the example subject in Fig. 2 wherein they are shown as regression lines.
Figure 2.
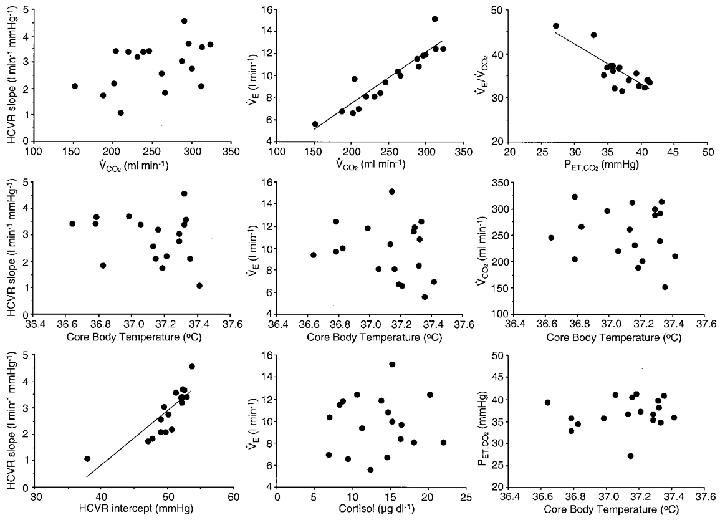
Relationship between variables throughout a constant routine protocol
Relationship among pairs of variables across 36 h of a constant routine protocol in a single representative subject (VE, PET,CO2, HCVR slope and x-axis intercept, VCO2, plasma cortisol concentration and CBT). Whenever significant correlations occurred (P < 0.05) these are indicated with the regression line of the relationship. These graphs show that there were significant positive correlations between HCVR slope and HCVR x-axis intercept, and between VE and VCO2, and there was a significant negative correlation between VE/VCO2 and VCO2, indicating that metabolism increases more than VE, inducing an increase in PET,CO2. These data also show that across the circadian cycle, ventilation and metabolic rate were not correlated with CBT, cortisol concentration or HCVR. In addition, there were no significant correlations between HCVR and either PET,CO2 or VE (not shown).
Table 2.
Correlations between pertinent variables throughout the constant routine protocol
| Variables | HCVR slope | HCVR x-intercept | V̇E | V̇E/V̇CO2 | V̇O2 | V̇CO2 | PET,CO2 | Cortisol | CBT |
|---|---|---|---|---|---|---|---|---|---|
| HCVR slope | — | 10 | 3 | 0 | 2 | 2 | 0 | 1 | 0 |
| HCVR x-intercept | 0·8075 | — | 1 | 0 | 1 | 1 | 3 | 2 | 1 |
| V̇E | −0·4030 | −0·3005 | — | 3 | 10 | 10 | 5 | 1 | 2 |
| V̇E/V̇CO2 | 0·0200 | −0·0190 | 0·1165 | — | 2 | 4 | 7 | 2 | 1 |
| V̇O2 | −0·2610 | −0·2275 | 0·6980 | −0·4385 | — | 10 | 1 | 1 | 1 |
| V̇CO2 | −0·3420 | −0·2335 | 0·9075 | −0·3730 | 0·8470 | — | 2 | 0 | 1 |
| PET,CO2 | 0·0855 | 0·1325 | −0·4480 | −0·6815 | 0·0030 | −0·1290 | — | 4 | 1 |
| Cortisol | 0·2525 | 0·1465 | 0·1290 | −0·2970 | 0·l3780 | 0·2790 | 0·1930 | — | 2 |
| CBT | 0·0200 | −0·1120 | −0·1005 | 0·1435 | −0·0825 | −0·0355 | −0·0935 | −0·3520 | — |
For each pair of variables (see Table 1 for abbreviations) within each subject, Pearson's corrleation coefficient (r, at zero lag between variables) was calculated from 18 measurements across 1·5 circadian cycles. Numbers in the top right half of the table represent the number of subjects (out of the 10) who had significant correlations (P < 0·05 when r larger than 0·47 for n = 18). Numbers in the lower left half of the table represents the median correlation coefficient of the 10 subjects. Data for specific pairs of variables are shown in bold when more than half of the subjects had a significant correlation.
Important pairings of variables that failed to yield significant correlations included metabolic rate vs. HCVR slope (median correlation coefficient of -0.26 for VO2vs. HCVR and -0.34 for VCO2vs. HCVR; significant in only 2 out of 10 subjects), CBT vs. metabolism (median of -0.08 for CBT vs. VO2 and -0.04 for CBT vs. VCO2; significant in only 1 out of 10 subjects) and resting PET,CO2vs. HCVR (median of 0.09 for HCVR slope vs. PET,CO2 and 0.13 for HCVR x-axis intercept vs. PET,CO2; significant in 0 and 3 subjects, respectively). Similarly, throughout these circadian changes, there were no significant correlations between resting ventilation and either HCVR slope or HCVR x-axis intercept. In all subjects, the concurrent changes in HCVR slope and x-axis intercept across the circadian cycle gave the impression that as the HCVR changed, it ‘pivoted’ around a specific PCO2 well above the resting PET,CO2. This is depicted for one typical subject in Fig. 3 (along with an estimate of the PCO2 at the central chemoreceptors; see Discussion).
Figure 3.
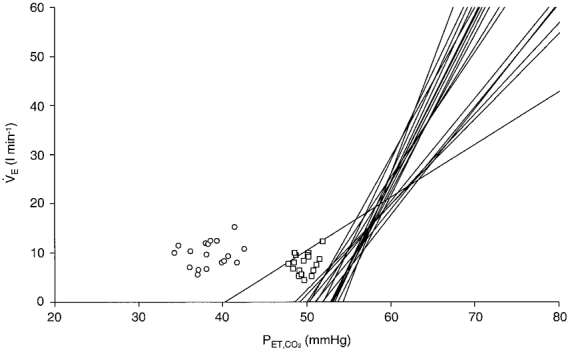
Relationship between resting and hypercapnic ventilatory responses throughout a constant routine protocol
The relationship between the HCVR (lines), spontaneous resting PET,CO2 plotted at resting ventilation (VE, ○), and estimated PCO2 at the central chemoreceptors plotted at a ventilation expected during rest when hyperoxic (□, see Discussion for details). Data are plotted across 36 h of a constant routine protocol in a single representative subject (same subject as in Fig. 2). The intersection between the HCVR and the x-axis represents the x-intercept. The graph shows that, as the HCVR slope increases, the x-axis intercept increases such that the HCVR response line appears to ‘pivot’ around a specific PET,CO2 well above both the resting PET,CO2 and the estimated PCO2 at the central chemoreceptors. We found that in all subjects, the estimated PCO2 at the central chemoreceptors was similar to the HCVR intercept but substantially lower (-5.9 mmHg) than the PCO2 required to explain the actual level of resting ventilation (minus 20 % adjustment for hyperoxia). In the typical subject, equilibrium PCO2 was less than the HCVR intercept in 11 out of 18 measurement periods, and the hyperoxic resting ventilation level at equilibrium PCO2 was substantially higher than predicted from the HCVR line in 17 out of 18 periods. These data suggest that CO2 at the central chemoreceptors at rest is not sufficient to explain the level of ventilation at rest.
Comparison of circadian and sleep-related changes in PCO2
On the baseline nights, the maximum sleep-induced change in PET,CO2 was 4.0 mmHg (range 2.9-5.0 mmHg) from relaxed wakefulness to the first stable period of slow-wave sleep and it occurred within 30 min of falling asleep in all subjects (n = 6). This sleep-induced increase in PET,CO2 was substantially larger than the circadian amplitude of PET,CO2 in any of the individuals.
DISCUSSION
This study has documented the existence and magnitude of circadian variations in various aspects of respiratory control in awake humans for the first time under constant behavioural and environmental conditions, and in the absence of sleep. We found a consistent but small amplitude circadian rhythm in mean PET,CO2, a larger amplitude rhythm in metabolism, and the largest amplitude rhythm in ventilatory chemosensitivity (HCVR slope). We also found that the characteristic change in PET,CO2 during non-rapid eye movement sleep was independent of circadian variations in PET,CO2.
Possible underlying mechanisms of circadian rhythms of respiratory control
The phase relationships among circadian rhythms may provide insight into mechanisms of respiratory control. Possible mechanisms underlying circadian rhythms in respiratory control could include changes in neural influences on the brainstem respiratory complex from the circadian pacemaker (e.g. from the suprachiasmatic nuclei via the paraventricular nuclei of the hypothalamus and/or the reticular formation), or indirect influences on metabolism and/or respiratory control via other circadian variables such as circulating hormones or body temperature.
Many endogenous and exogenously administered hormones affect both the ‘resting’Pa,CO2 and ventilatory chemosensitivity (Dempsey et al. 1986). From the group analysis, the circadian minimum of each respiratory variable occurred close to the minimum of the plasma cortisol rhythm, suggesting a possible mechanistic link between these variables, as previously suggested by Koepchen (1953). However, this issue is far from resolved because, on closer examination, we found no systematic correlation (at zero lag) within individuals between circulating cortisol and any of the respiratory variables (Table 2). One possible explanation is that endogenous changes in cortisol level across the circadian cycle have less of an effect on respiration than equivalent experimentally administered changes in cortisol.
It is also noteworthy that the circadian minimum of each respiratory variable occurred 6-8 h earlier than the minimum of the CBT rhythm (Table 1), and that individual analyses failed to find a significant correlation (at zero lag) between CBT and any of the respiratory variables as these changed throughout the circadian cycle (Table 2). Induced deviations in CBT from the regulated ‘set-point’ CBT (i.e. an increased error signal in the negative feedback control system), as occur with a hot bath or heavy exercise, affect virtually all respiratory variables (Vejby-Christensen & Strange Petersen, 1973; Petersen & Vejby-Christensen, 1977; Baker et al. 1996). However, the lack of relationship between CBT and respiratory variables in the current study implies the respiratory response to endogenous circadian changes in CBT (which may not necessitate a change in ‘error signal’ of the negative feedback control system) is different from the respiratory response to experimentally induced changes in CBT.
Additional insight into respiratory control may be provided by examining the relative amplitudes of circadian rhythms and the correlations among respiratory variables. PET,CO2 and metabolism increased at times when VE increased throughout the circadian cycle (Fig. 1). This suggests that the circadian changes in PET,CO2 were driven primarily by changes in VCO2 rather than in VE (as primary increases in VE would decrease PET,CO2). Indeed, the small discrepancy between the degree of circadian change in metabolism and ventilation (Table 1) is consistent with the small changes in PET,CO2 that we observed, without having to invoke significant changes in body stores of CO2 or alterations in metabolic substrates to explain these results (Cunningham et al. 1986). The respiratory exchange ratio did not exhibit a circadian rhythm, indicating that the substrates utilised for metabolism probably did not change substantially throughout the constant routine. This latter finding is in agreement with previous observations (Kräuchi & Wirz-Justice, 1994). In all subjects there was a very tight correlation between VCO2 and VE. We acknowledge that this correlation could be caused by random fluctuations in VE influencing VCO2 measured at the mouth (even when the tissue remains unchanged). However, this random effect is unlikely to be the reason for the tight correlations seen in our data as each datum is the mean of all breaths over a 5 min period that started after establishing a steady state, thereby reducing the effect of random fluctuations in VE on VCO2. (Indeed, if ventilation had changed to a new steady state without changes in tissue VCO2, this would affect Pa,CO2, but not necessarily VCO2 measured at the mouth.) Thus, it seems much more likely that the changes in VCO2 and VE are physiologically linked. Indeed, numerous studies involving natural changes in metabolism during exercise (e.g. Wasserman et al. 1986) or with adjusted diets (e.g. Zwillich et al. 1977), or with experimental alterations in CO2 delivered to the lungs (during extra-corporeal membrane circulation; Phillipson et al. 1981) suggest that VCO2 drives VE via a physiological reflex.
It is noteworthy that individual analyses failed to find a significant correlation between metabolic rate and HCVR as these changed throughout the circadian cycle (Table 2). Sahn et al. (1977) also found no relationship between changes in metabolic rate and HCVR, although a positive correlation is often seen between the hypoxic ventilatory chemosensitivity and metabolic rate in many situations in which metabolism changes, including exercise, hyperthermia, hyperthyroidism, myxedema and semi-starvation (reviewed by Sahn et al. 1977).
Relationship between the central chemoreceptive negative feedback control and resting Pa,CO2
The importance of the central chemoreceptive negative feedback control of breathing at rest (as opposed to non-chemoreceptive and/or behavioural inputs to breathing) still remains a conundrum (reviewed by Cunningham et al. 1986; Shea, 1996, 1997). It has been proposed that humans exist at an ‘equilibrium point’ at the junction of the metabolic hyperbola (effect of VE on Pa,CO2 at constant VCO2) and the HCVR response line (effect of Pa,CO2 on VE at constant VCO2). Nonetheless, we are still uncertain in awake humans whether or not the spontaneous resting PET,CO2, at which we usually exist, is on a flat portion (‘dogleg’) in the HCVR relationship (e.g. Cunningham et al. 1986), with little influence from the chemoreceptive drives to breathe. In the present study, the large circadian rhythm in HCVR slope in the face of relatively little circadian variation in spontaneous resting PET,CO2 suggests that these parameters can vary independently. Furthermore, the lack of correlation between the HCVR parameters and either resting PET,CO2 or VE in all subjects suggests that spontaneous resting ventilation and PET,CO2 in awake humans may be substantially unrelated to the central respiratory chemical control system.
To further address this important question we performed a posthoc analysis of data from the current study to estimate whether the PCO2 at the central chemoreceptors at rest is sufficient to contribute to resting ventilation via the central respiratory chemical control system (HCVR response). As the HCVR tests were performed by rebreathing from a circuit that initially contained 7 % CO2 there was rapid equilibration between PCO2 in the circuit, lungs, arterial blood, venous blood and tissues. It has been estimated that there is less than 2 mmHg difference between Pa,CO2 and brain tissue PCO2 within 20 s of initiating rebreathing (Read & Leigh, 1967). Thus, PCO2 at this equilibration point is an estimate of PCO2 at the central chemoreceptors. We compared all equilibrium PCO2 values to the PCO2 required to stimulate ventilation at rest. We assumed that if the equilibrium PCO2 was above the HCVR x-axis intercept, then this would indicate that there is some contribution to resting ventilation from the central chemoreceptors. Additionally, if the equilibrium PCO2 at resting ventilation was identical to that predicted by the HCVR relationship, this would provide evidence that the central respiratory chemical control system was entirely responsible for the level of resting ventilation. To negate any possible effect of peripheral chemoreceptor activity on resting ventilation in this latter calculation, we reduced the level of resting ventilation at the equilibrium PCO2 by 20 % (Dejours, 1962). This group analysis revealed that the estimated PCO2 at the central chemoreceptors was similar to the HCVR x-axis intercept (mean (±s.e.m.) difference of 1.8 ± 1.4 mmHg; P = 0.243, paired t test). However, the estimated PCO2 at the central chemoreceptors was substantially lower (-5.9 ± 1.1 mmHg; P = 0.002) than the PCO2 required to explain the actual level of resting ventilation (minus 20 % adjustment for hyperoxia) solely in terms of a central chemoreflex. In the typical subject shown in Fig. 3, the equilibrium PCO2 was less than the HCVR x-axis intercept in 11 out of 18 measurement periods, and the hyperoxic resting ventilation level at equilibrium PCO2 was substantially higher than predicted from the HCVR line in 17 out of 18 periods. Clearly there are numerous assumptions in our extrapolations. Indeed, there are more minor refinements that could be made. For instance, the difference between the equilibrium PCO2 and the HCVR line could be reduced by 1-2 mmHg because brain tissue PCO2 is slightly higher than Pa,CO2 during rebreathing (Read & Leigh, 1967). Also, rather than adjusting the level of resting ventilation by -20 % to account for the effect of hyperoxia during the rebreathing test, it may be more appropriate to adjust the HCVR line for the effects of hyperoxia, which would probably have shifted our measured HCVR response line to the left by 2-3 mmHg (e.g. Fig. 5 in Cunningham et al. 1986). To answer the question definitively it may be necessary in future studies to perform additional respiratory tests that differentiate between changes in the peripheral and central chemoreceptor influences (e.g. Cunningham et al. 1986; Duffin & McAvoy, 1988; Mohan & Duffin, 1997). Nonetheless, on face value the results from the current study demonstrate that CO2 at the central chemoreceptors in awake resting humans is close to the ventilatory threshold of the central chemoreflex (HCVR x-axis intercept), but insufficient to explain the level of ventilation that does occur. This finding, along with a lack of correlation between HCVR parameters and either resting PET,CO2 or VE, suggests that ventilation and PET,CO2 are little influenced by central chemosensory respiratory control in awake resting humans. Thus, other drives to breathe such as the behavioural drive to breathe – as well as possible influences from CO2 production and peripheral chemoreception – probably supervene during resting breathing. Indeed, these other drives to breathe have previously been demonstrated to result in relatively normal ventilation and in numerous behavioural conditions in awake patients who lack central ventilatory chemosensitivity (congenital central hypoventilation syndrome; reviewed by Shea, 1997).
Do circadian changes contribute to the increase in Pa,CO2 and the decrease in HCVR slope that have been documented to occur during sleep?
Our data strongly suggest that the normal sleep-induced increase in Pa,CO2 and decrease in HCVR slope (e.g. Phillipson & Bowes, 1986; Schafer, 1998) are caused by independent effects of sleep rather than an underlying circadian rhythm. That is because the sleep-induced change in PET,CO2 recorded during the baseline days in the current study (mean 4.0 mmHg; range 2.9-5.0 mmHg; maximum change occurred over 30 min period) was substantially greater and occurred much faster than any circadian change in PET,CO2 (mean ±0.6 mmHg; range 0.3-2.4 mmHg; maximum change occurred over 12 h period). HCVR was not measured during sleep in the current study, but a significant reduction in HCVR slope during sleep has been observed in many other studies (e.g. Phillipson, 1978; Gothe et al. 1981; Douglas et al. 1982b; Schafer, 1998). Since the circadian minimum of HCVR slope in our awake subjects occurred before the onset of the usual sleep period, it appears that a sleep-related reduction in HCVR slope cannot be explained by a nocturnal circadian reduction in HCVR slope. The change in PET,CO2 during non-rapid eye movement sleep probably reflects a change from predominantly behavioural respiratory control to predominantly chemosensory respiratory control.
Comparisons with other studies
A previous study also reported a pronounced circadian variation in HCVR slope (Raschke & Möller, 1989). The main differences in protocol between this earlier study and the present study are that the earlier study had a much shorter protocol (24 h), ad lib sleep was permitted, and subjects were woken immediately prior to respiratory measurements (at those circadian phases when subjects slept). One of the reasons for performing the current study was that we were concerned that respiratory variables in the earlier study would be influenced by ‘sleep inertia’ after awakening subjects for measurements during the night (for instance, a sleep-induced reduction in HCVR slope could persist into wakefulness). Such sleep inertia effects have been demonstrated to have time constants ranging from 40 min to over 1 h for many variables including reaction time, cognitive performance, vigilance and subjective alertness (Jewett et al. 1999). In addition, sleep itself has an independent effect on CBT (Gillberg & Akerstedt, 1982) which could secondarily affect metabolism, ventilation and chemosensitivity. Thus, we preferred to use a constant routine protocol to avoid these potential confounding influences. The principal difference that emerged between the present study and this previous study was that Raschke & Möller (1989) found that the minimum of the circadian rhythm in HCVR slope occurred at approximately the same time as the minimum of CBT, whereas we found the circadian minimum of HCVR slope occurred 5.8 h before the minimum of CBT. For the reasons mentioned above, we believe that the constant routine protocol is better suited to address these questions. Nonetheless, it is also worth noting that a constant routine may induce physiological changes related to the protocol itself that could affect results, such as an increase in stress because of prolonged wakefulness. We did observe a very small increase in plasma cortisol induced by the sleep deprivation, perhaps indicating an increase in stress. However, this linear trend over 24 h was equivalent to only 13 % of the peak-to-trough change in circadian rhythms in cortisol (Table 1). Furthermore, we found no correlation between cortisol and the HCVR, nor between cortisol and CBT. Thus, we do not believe that stress significantly contributed to our results or is likely to explain the different results among studies.
Another study that used a similar, albeit shorter, constant routine protocol found, as in the current study, that the circadian minimum of metabolism occurred significantly ahead of the minimum of CBT (Kräuchi & Wirz-Justice, 1994). These authors reported a mean advance between metabolism and CBT of 2 h from cross-correlation analysis (Fig. 4; Kräuchi & Wirz-Justice, 1994) but an advance of 4-5.5 h from visual analyses of individuals’ data (Table 2; Kräuchi & Wirz-Justice, 1994). In the current study, we found a group mean advance of 7.8 h. Thus, these studies have qualitatively similar results, but the reason for the quantitative discrepancy cannot be resolved with the available data.
Implications of circadian rhythms in respiratory control
Our findings suggest that it is not crucial to consider the phase of the circadian cycle in respiratory studies when arterial blood gases are the only variables of interest, because there is little circadian variability in these measurements. On the other hand, it may be important to consider the phase of the circadian cycle, or at least the time of day, when interpreting results or designing studies in which metabolism or respiratory chemosensitivity are among the key variables. For instance, in the present data there was an average decline in HCVR slope of 30 % between 14.00 and 20.00 h (Fig. 1). This magnitude of underlying systematic circadian change could seriously affect data interpretation in virtually any long-term respiratory control study in animals or humans. It is very likely that circadian HCVR slope changes would have greatest relevance in conditions in which the chemoreceptive negative feedback system is known to predominate over other drives to breathe, such as during sleep, when at altitude, and in many respiratory disorders. For instance, a patient's own chemoreceptive drive to breathe is often a critical factor in determining when a patient is to be weaned from a ventilator (Montgomery et al. 1987). Also, the HCVR slope has been shown to be an excellent predictor of (and likely mechanism for) the development of central sleep apnoea in patients with heart failure (Javaheri, 1999). Furthermore, the circadian variation in HCVR could explain the changes in the severity of obstructive sleep apnoeas that occur across the night, because arousal from sleep during apnoea is thought to arise partly from the underlying chemoreceptive drive to breathe (Berry & Gleeson, 1997). Such considerations of circadian phase have been entirely ignored in the past.
Conclusion
This study has documented the existence and magnitude of circadian variations in various aspects of respiratory control in awake humans for the first time under constant behavioural and environmental conditions. This study has also documented that there are independent sleep and circadian influences upon respiration. The circadian rhythm of mean PET,CO2 was much smaller than the sleep-induced changes in PET,CO2 and occurred in the presence of larger circadian rhythms in metabolism and ventilatory chemosensitivity. Our data provide numerous clues as to the underlying mechanisms responsible for the circadian variations of PET,CO2, HCVR and metabolism (as discussed).
Acknowledgments
The authors would like to thank the volunteer subjects, Hance H. Oliver and David W. Rimmer for assisting with protocol development and respiratory measurements, Elita Harvey for excellent technical assistance, the technical staff of the Brigham and Women's Hospital General Clinical Research Center for aspects of data collection, Joseph Ronda for computer management, Johnette Kao for subject recruitment, and Janice Swain for design and preparation of metabolic diets. We are grateful to Jaeger/MedPoint Technologies for the loan of an OxyconAlpha metabolic system and Datex for the loan of Cardiocap II equipment. This work was supported by NIH grants HL62149 to Steven A. Shea and NCRR GCRC M01 RR02635 to the Brigham and Women's Hospital General Clinical Research Center. Christina M. Spengler was kindly supported by the Swiss Foundation for Medical-Biological Fellowships.
References
- Baker JF, Goode RC, Duffin J. The effect of a rise in body temperature on the central-chemoreflex ventilatory response to carbon dioxide. European Journal of Applied Physiology. 1996;72:537–541. doi: 10.1007/BF00242287. [DOI] [PubMed] [Google Scholar]
- Berger RJ, Phillips NH. Comparative aspects of energy metabolism, body temperature and sleep. Acta Physiologica Scandinavica. 1988;574:21–27. [PubMed] [Google Scholar]
- Berry RB, Gleeson K. Respiratory arousal from sleep: mechanisms and significance. Sleep. 1997;20:654–675. doi: 10.1093/sleep/20.8.654. [DOI] [PubMed] [Google Scholar]
- Brown EN, Czeisler CA. The statistical analysis of circadian phase and amplitude in constant-routine core-temperature data. Journal of Biological Rhythms. 1992;7:177–202. doi: 10.1177/074873049200700301. [DOI] [PubMed] [Google Scholar]
- Bulow K. Respiration and wakefulness in man. Acta Physiologica Scandinavica. 1963;59:7–99. [PubMed] [Google Scholar]
- Cunningham DJC, Robbins PA, Wolff CB. Integration of respiratory responses to changes in alveolar partial pressures of CO2 and O2 and in arterial pH. In: Cherniack NS, Widdicombe JG, editors. Handbook of Physiology, section 3, The Respiratory System, Control of Breathing. II. Baltimore: American Physiological Society; 1986. pp. 475–528. part 2. [Google Scholar]
- Czeisler CA, Allan JS, Strogatz SH, Ronda JM, Sanchez R, Rios CD, Freitag WO, Richardson GS, Kronauer RE. Bright light resets the human circadian pacemaker independent of the timing of the sleep-wake cycle. Science. 1986;233:667–671. doi: 10.1126/science.3726555. [DOI] [PubMed] [Google Scholar]
- Czeisler CA, Duffy JF, Shanahan TL, Brown EN, Mitchell JF, Rimmer DW, Ronda JM, Silva EJ, Allan JS, Emens JS, Dijk DJ, Kronauer RE. Stability, precision, and near-24-hour period of the human circadian pacemaker. Science. 1999;284:2177–2181. doi: 10.1126/science.284.5423.2177. [DOI] [PubMed] [Google Scholar]
- Czeisler CA, Kronauer RE, Allan JS, Duffy JF, Jewett ME, Brown EN, Ronda JM. Bright light induction of strong (type 0) resetting of the human circadian pacemaker. Science. 1989;244:1328–1333. doi: 10.1126/science.2734611. [DOI] [PubMed] [Google Scholar]
- Dejours P. Chemoreflexes in breathing. Physiological Reviews. 1962;42:335–358. doi: 10.1152/physrev.1962.42.3.335. [DOI] [PubMed] [Google Scholar]
- Douglas NJ, White DP, Weil JV, Pickett CK, Martin RJ, Hudgel DW, Zwillich CW. Hypoxic ventilatory response decreases during sleep in normal men. American Review of Respiratory Disease. 1982a;125:286–289. doi: 10.1164/arrd.1982.125.3.286. [DOI] [PubMed] [Google Scholar]
- Douglas NJ, White DP, Weil JV, Pickett CK, Zwillich CW. Hypercapnic ventilatory response in sleeping adults. American Review of Respiratory Disease. 1982b;126:758–762. doi: 10.1164/arrd.1982.126.5.758. [DOI] [PubMed] [Google Scholar]
- Duffin J, McAvoy GV. The peripheral-chemoreceptor threshold to carbon dioxide in man. The Journal of Physiology. 1988;406:15–26. doi: 10.1113/jphysiol.1988.sp017365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillberg M, Akerstedt T. Body temperature and sleep at different times of day. Sleep. 1982;5:378–388. doi: 10.1093/sleep/5.4.378. [DOI] [PubMed] [Google Scholar]
- Gothe B, Altose MD, Goldman MD, Cherniack NS. Effect of quiet sleep on resting and CO2-stimulated breathing in humans. Journal of Applied Physiology. 1981;50:724–730. doi: 10.1152/jappl.1981.50.4.724. [DOI] [PubMed] [Google Scholar]
- Harris JA, Benedict FG. Standard Basal Metabolism Constants for Physiologists and Clinicians; A Biometric Study of Basal Metabolism in Man. Philadelphia: J. B. Lippincott; 1919. [Google Scholar]
- Javaheri S. A mechanism of central sleep apnea in patients with heart failure. New England Journal of Medicine. 1999;341:949–954. doi: 10.1056/NEJM199909233411304. [DOI] [PubMed] [Google Scholar]
- Jewett ME, Wyatt JK, Ritz-De Cecco A, Khalsa SB, Dijk DJ, Czeisler CA. Time course of sleep inertia dissipation in human performance and alertness. Journal of Sleep Research. 1999;8:1–8. doi: 10.1111/j.1365-2869.1999.00128.x. [DOI] [PubMed] [Google Scholar]
- Koepchen HP. Über die Wirkung von Cortison und Testosteron auf die Atmung. Pflügers Archiv. 1953;257:144–154. doi: 10.1007/BF00412468. [DOI] [PubMed] [Google Scholar]
- Kräuchi K, Wirz-Justice A. Circadian rhythm of heat production, heart rate, and skin and core temperature under unmasking conditions in men. American Journal of Physiology. 1994;267:R819–829. doi: 10.1152/ajpregu.1994.267.3.R819. [DOI] [PubMed] [Google Scholar]
- Lorinc Z, Derr J, Snider M, Lydic R. Defining origin of positive slope in hypercapnic ventilatory response curve. American Journal of Physiology. 1991;261:R747–751. doi: 10.1152/ajpregu.1991.261.3.R747. [DOI] [PubMed] [Google Scholar]
- Mills JN, Minors DS, Waterhouse JM. Adaptation to abrupt time shifts of the oscillator(s) controlling human circadian rhythms. The Journal of Physiology. 1978;285:455–470. doi: 10.1113/jphysiol.1978.sp012582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan R, Duffin J. The effect of hypoxia on the ventilatory response to carbon dioxide in man. Respiration Physiology. 1997;108:101–115. doi: 10.1016/s0034-5687(97)00024-8. [DOI] [PubMed] [Google Scholar]
- Montgomery AB, Holle RH, Neagley SR, Pierson DJ, Schoene RB. Prediction of successful ventilator weaning using airway occlusion pressure and hypercapnic challenge. Chest. 1987;91:496–499. doi: 10.1378/chest.91.4.496. [DOI] [PubMed] [Google Scholar]
- Nelson W, Liang Tong Y, Lee J-K, Halberg F. Methods for cosinor-rhythmometry. Chronobiologia. 1979;6:305–323. [PubMed] [Google Scholar]
- Petersen ES, Vejby-Christensen H. Effects of body temperature on ventilatory response to hypoxia and breathing pattern in man. Journal of Applied Physiology. 1977;42:492–500. doi: 10.1152/jappl.1977.42.4.492. [DOI] [PubMed] [Google Scholar]
- Phillipson EA. Respiratory adaptations in sleep. Annual Review of Physiology. 1978;40:133–156. doi: 10.1146/annurev.ph.40.030178.001025. [DOI] [PubMed] [Google Scholar]
- Phillipson EA, Bowes G. Control of breathing during sleep. In: Cherniack NS, Widdicombe JG, editors. Handbook of Physiology, section 3, The Respiratory System, Control of Breathing. II. Baltimore: American Physiological Society; 1986. pp. 649–689. part 2. [Google Scholar]
- Phillipson EA, Duffin J, Cooper JD. Critical dependence of respiratory rhythmicity on metabolic CO2 load. Journal of Applied Physiology. 1981;50:45–54. doi: 10.1152/jappl.1981.50.1.45. [DOI] [PubMed] [Google Scholar]
- Raschke F, Möller KH. The diurnal rhythm of chemosensitivity and its contribution to nocturnal disorders of respiratory control. Pneumologie. 1989;1:568–571. [PubMed] [Google Scholar]
- Read DJ. A clinical method for assessing the ventilatory response to carbon dioxide. Australasian Annals of Medicine. 1967;16:20–32. doi: 10.1111/imj.1967.16.1.20. [DOI] [PubMed] [Google Scholar]
- Read DJC, Leigh J. Blood-brain tissue relationships and ventilation during rebreathing. Journal of Applied Physiology. 1967;23:53–70. doi: 10.1152/jappl.1967.23.1.53. [DOI] [PubMed] [Google Scholar]
- Rechtschaffen A, Kales A. A Manual of Standardized Terminology, Techniques and Scoring System for Sleep Stages of Human Subjects. Washington DC: Public Health Service, US Government Printing Office; 1968. [Google Scholar]
- Sahn SA, Zwillich CW, Dick N, McCullough RE, Lakshminarayan S, Weil JV. Variability of ventilatory responses to hypoxia and hypercapnia. Journal of Applied Physiology. 1977;43:1019–1025. doi: 10.1152/jappl.1977.43.6.1019. [DOI] [PubMed] [Google Scholar]
- Schafer T. Variability of vigilance and ventilation: studies on the control of respiration during sleep. Respiration Physiology. 1998;114:37–48. doi: 10.1016/s0034-5687(98)00070-x. [DOI] [PubMed] [Google Scholar]
- Shea SA. Behavioural and arousal-related influences on breathing in humans. Experimental Physiology. 1996;81:1–26. doi: 10.1113/expphysiol.1996.sp003911. [DOI] [PubMed] [Google Scholar]
- Shea SA. Life without ventilatory chemosensitivity. Respiration Physiology. 1997;110:199–210. doi: 10.1016/s0034-5687(97)00084-4. [DOI] [PubMed] [Google Scholar]
- Spengler CM, Oliver H, Harvey E, Rimmer D, Czeisler CA, Shea SA. Effects of circadian rhythms and sleep deprivation upon arterial carbon dioxide regulation in humans. Sleep Research. 1997;26:755. [Google Scholar]
- Vejby-Christensen H, Strange Petersen E. Effect of body temperature and hypoxia on the ventilatory CO2 response in man. Respiration Physiology. 1973;19:322–332. doi: 10.1016/0034-5687(73)90036-4. [DOI] [PubMed] [Google Scholar]
- Wasserman K, Whipp BJ, Casaburi R. Respiratory control during exercise. In: Cherniack NS, Widdicombe JG, editors. Handbook of Physiology, section 3, The Respiratory System, Control of Breathing. II. Baltimore: American Physiological Society; 1986. pp. 595–619. part 2. [Google Scholar]
- Zwillich CW, Sahn SA, Weil JV. Effects of hypermetabolism on ventilation and chemosensitivity. Journal of Clinical Investigation. 1977;60:900–906. doi: 10.1172/JCI108844. [DOI] [PMC free article] [PubMed] [Google Scholar]