

Abstract
The present work was carried out in order to determine whether a decrease in cardiac Na+-Ca2+ exchanger (NCX) activity observed in diabetes is caused by a reduction in NCX protein and mRNA levels and to elucidate the significance of this decrease in alterations in [Ca2+]i homeostasis in diabetic cardiomyocytes.
The NCX current was significantly reduced in ventricular myocytes freshly isolated from streptozotocin-induced diabetic rat hearts, and its current density was about 55 % of age-matched controls.
Diabetes resulted in a 30 % decrease in cardiac protein and mRNA levels of NCX1, a NCX isoform which is expressed at high levels in the heart.
The reduced NCX current and the decreased protein and mRNA levels of NCX1 in diabetes were prevented by insulin therapy.
Although both diastolic and peak systolic [Ca2+]i were not different between the two groups of myocytes, increasing external Ca2+ concentration to high levels greatly elevated diastolic [Ca2+]i in diabetic myocytes. Inhibition of NCX by reduction in extracellular Na+ by 50 % could produce a marked rise in diastolic [Ca2+]i in control myocytes in response to high Ca2+, as seen in diabetic myocytes. However, cyclopiazonic acid, an inhibitor of sarcoplasmic reticulum Ca2+ pump ATPase, did not modify the high Ca2+-induced changes in diastolic [Ca2+]i in either control or diabetic myocytes.
Only in papillary muscles from diabetic rats did the addition of high Ca2+ cause a marked rise in resting tension signifying a partial contracture that was possibly due to an increase in diastolic [Ca2+]i.
In conclusion, the diminished NCX function in diabetic myocytes shown in this study results in part from the decreased levels of cardiac NCX protein and mRNA. We suggest that this impaired NCX function may play an important role in alterations in Ca2+ handling when [Ca2+]i rises to pathological levels.
In cardiac myocytes, a rise in [Ca2+]i plays a key role in excitation-contraction coupling. During membrane depolarization, Ca2+ enters from the extracellular fluid via L-type Ca2+ channels; this triggers Ca2+ release from the sarcoplasmic reticulum (SR), leading to contraction (Callewaert, 1992; Barry & Bridge, 1993). Subsequently, two major processes can contriute to lowering [Ca2+]i and promoting relaxation: Ca2+ uptake into the SR and extrusion to the extracellular space (Puglisi et al. 1996). The NCX and sarcolemmal Ca2+ pump are responsible for extruding Ca2+ from heart cells (Puglisi et al. 1996). The capacity of the sarcolemmal Ca2+ pump to transport Ca2+ from the cell is extremely limited and its contribution to the regulation of [Ca2+]i appears to be functionally negligible (Hammes et al. 1998), although the sarcolemmal Ca2+ pump has been recently proposed to have a significant role in [Ca2+]i homeostasis under Na+-free conditions (Choi & Eisner, 1999). Therefore, NCX is considered to be the dominant Ca2+ efflux mechanism. Although three mammalian isoforms of NCX have been cloned (Nicoll et al. 1990, 1996; Li et al. 1994), NCX1 is expressed at high levels in the heart (Kofuji et al. 1992; Quednau et al. 1997).
Diabetes mellitus has been shown to be associated with heart failure of unknown origin, which is termed diabetic cardiomyopathy (Fein & Sonnenblick, 1985). It has been suggested that the development of diabetic cardiomyopathy may result partly from altered intracellular Ca2+ homeostasis (Dhalla et al. 1985). Indeed, activities of the sarcolemmal Ca2+ pump (Heyliger et al. 1987; Makino et al. 1987), the Na+,K+-ATPase (Pierce & Dhalla, 1983), the Na+-H+ exchanger (Lagadic-Grossman et al. 1988), and the SR Ca2+ pump (Penpargkul et al. 1981; Ganguly et al. 1983; Lopaschuk et al. 1983) have been demonstrated to be depressed in the hearts of experimental diabetic animals. In addition, depressed NCX activity without change in affinity to Ca2+ has been previously reported in diabetic rat hearts (Makino et al. 1987). However, no study has been done to determine whether the lower activity of NCX in diabetic myocardium is due to a decrease in NCX protein or compositional modifications in the sarcolemmal membranes. Furthermore, until now, it has been unclear whether depressed NCX activity is significantly involved in alterations in intracellular Ca2+ handling in diabetic cardiomyocytes.
In the present work, we assessed whether NCX function is indeed depressed in ventricular myocytes isolated from streptozotocin-induced diabetic rat hearts. For this purpose, we compared the NCX current in diabetic myocytes with that in control myocytes using whole-cell patch-clamp techniques. In addition, we determined, for the first time, whether cardiac NCX protein and mRNA are altered in diabetes. Finally, our objective was to determine whether dysfunction of NCX, if any, results in alterations in Ca2+ handling and contraction in diabetic myocytes and papillary muscles, especially when [Ca2+]i rises to pathological levels.
METHODS
Induction of diabetes
All procedures were in accordance with the regulations laid down by the Hokkaido University School of Medicine Animal Care and Use Committee.
Male Wistar rats, 8 weeks old and 180–200 g in body weight, were anaesthetized with diethyl ether and received a single tail-vein injection of streptozotocin (45 mg kg−1, Sigma Chemical Co.). Streptozotocin was dissolved in a citrate buffer solution (0.1 M citric acid and 0.2 M sodium phosphate, pH 4.5). Age-matched control rats received an equivalent volume of the citrate buffer solution alone. Both groups of animals were maintained on the same diet and water ad libitum until they were used 4–6 weeks later. This period of diabetes was chosen because our previous studies have consistently characterized cardiac alterations during this period (Gando et al. 1997; Tamada et al. 1998). All animals injected with streptozotocin developed severe diabetes, as indicated by increased serum glucose levels. Mean serum glucose levels were 177 ± 12 and 605 ± 9 mg dl−1 for control rats (n = 26) and diabetic rats (n = 26), respectively. Some diabetic rats were treated with subcutaneous injection of insulin (NPH Iszilin, 3–4 U day−1, Shimizu Pharmaceutical Co. Ltd). Insulin therapy was begun 1 day after streptozotocin injection and was continued up to the day before the animals were killed. Serum glucose levels were significantly improved in insulin-treated diabetic rats (148 ± 15 mg dl−1, n = 15,P < 0.001).
On the day of the experiments, rats were usually anaesthetized with diethyl ether and killed by exsanguination. When used for isolation of ventricular myocytes, rats were anaesthetized with sodium pentobarbitone (50–60 mg kg−1i.p.), ventilated with an artificial respirator, and then the heart was removed quickly following opening of the chest (Tamada et al. 1998).
Isolation of ventricular myocytes
Briefly, single ventricular myocytes were obtained from the rat heart by an enzymatic dispersion method (collagenase, 0.03–0.05 % w/v, Wako Pure Chemical). The composition of the basic solution and further details of the procedure have been described previously (Tamada et al. 1998). Ca2+-tolerant rod-shaped ventricular myocytes were used on the day of isolation. As we have previously reported in detail (Tamada et al. 1998), no difference in the morphological parameters was observed between control and diabetic cells.
Measurement of the NCX current
In order to record the NCX current under whole-cell voltage-clamp, Axopatch 200B and pCLAMP version 6 (Axon Instruments) were used. The NCX current was recorded by ramp pulses from the holding potential of −60 mV to +50 mV, then to −100 mV and back to −60 mV at a steady speed of 0.67 V s−1. The descending portion of the ramp (from +50 mV to −100 mV) was used to plot the current-voltage (I-V) relation curve. The NCX current was generated by loading a high concentration of Na+ into the cell via the pipette solution and rapidly increasing the external Ca2+ concentration from 0 to 5 mM. The pipette solution contained (mM): 90 CsCl, 50 NaCl, 5.0 MgATP, 3.0 MgCl2, 20 BAPTA, 13 CaCl2 (calculated free Ca2+ concentration of 433 nM), and 20 Hepes (pH 7.2 with CsOH). The external solution contained (mM): 140 NaCl, 0 or 5.0 CaCl2, 1.0 MgCl2, 5.0 CsCl, 5.0 Hepes (pH 7.2 with CsOH), 0.01 nifedipine, 1.0 ouabain, and 0.005 ryanodine. The current magnitude was measured at +40 mV. The capacitance of the cell was calculated from the capacitive current at 0 mV recorded during the ramp pulse. The current density was obtained by dividing the current magnitude by the capacitance of each cell. The net NCX current was measured as a difference current between those at 0 and 5 mM Ca2+, which was confirmed as a 5 mM NiCl2-sensitive current. The temperature of perfusate was kept constant at 36 ± 1°C.
Measurement of indo-1 fluorescence
Single ventricular myocytes bathed in Kraft-Brühe (KB) solution were loaded with the fluorescent Ca2+ probe indo-1 by incubation with 5 μM indo-1 AM (Dojin) and 0.02 % Pluronic F-127 (Molecular Probes) for 10 min at room temperature, followed by wash-out with KB solution for 60 min. KB solution contained (mM): 70 KOH, 50 L-glutamic acid, 40 KCl, 20 taurine, 20 KH2PO4, 3.0 MgCl2, 10 Hepes (pH 7.4), and 1 % bovine serum albumin. Small aliquots of loaded myocytes were placed in the experimental chamber filled with a modified Tyrode solution, allowed to settle for 5 min, and superfused with Tyrode solution for at least 15 min. The composition of Tyrode solution was (mM): 143 NaCl, 5.4 KCl, 1.3 CaCl2, 0.5 MgCl2, 0.33 NaH2PO4, 5.5 glucose, and 5.0 Hepes, with pH adjusted to 7.4 with NaOH. Myocytes were then field stimulated at a rate of 0.5 Hz by a pair of platinum electrodes connected to an electronic stimulator (SEN-7203, Nihon Kohden) through an isolation unit (SS-104J, Nihon Kohden).
The microfluometry system (OSP100-CA, Olympus) was used to provide and control ultraviolet light of 360 nm with a monochromator for excitation of indo 1 from a 75 W xenon arc lamp. The excitation light beam was directed into an inverted microscope (IX-70, Olympus) equipped for epifluorescence measurements. Emitted fluorescence signals from single indo-1 AM (the acetoxymethyl ester form of indo 1)-loaded myocytes were digitalized at 200 Hz, and the ratio of indo-1 emission at 410 nm to that at 485 nm was recorded. The ratio of indo-1 emission at the two wavelengths was calculated after subtracting the background autofluorescence. Intracellular binding and compartmentalization of this indicator prevent accurate in vivo calibrations being obtained (Spurgeon et al. 1990). Additionally, the Ca2+-binding affinities for certain proteins may vary with disease, and it is thus questionable to assume that the dissociation constant for Ca2+ is the same among different populations of cells. Therefore, our results with indo-1 AM-loaded myocytes are expressed as the fluorescence ratio rather than as absolute Ca2+ concentration.
The experiments were implemented at a temperature of 23°C to minimize loss of the Ca2+ indicator from myocytes. The fluorescence ratio data were processed and stored on an IBM AT-type microcomputer using OSP-100 CA software with A/D conversion (Olympus).
Western blot analysis
In this study we used a mouse monoclonal antibody (6H3) against NCX1, which was kindly provided by Drs M. Shigekawa and T. Iwamoto, Department of Molecular Physiology, National Cardiovascular Centre Research Institute, Osaka, Japan (Iwamoto et al. 1998).
After the rat hearts had been removed and rinsed in sterilized water on ice, ventricles were dissected free of connective tissue, major vessels and atria. Ventricles were minced with scissors, homogenized, and then centrifuged at 6000 g for 15 min to pellet any insoluble material. The protein concentration of the supernatant was determined by the method of Lowry et al. (1951) with bovine serum albumin as standard. Samples (20 μg) were run on SDS-PAGE, using 8 % polyacrylamide gel, and electrotransferred to polyvinylidine difluoride filter (PVDF) membrane. To reduce non-specific binding, the PVDF was blocked for 120 min at room temperature in Tris-buffered saline (TBS: 20 mM Tris-HCl, 500 mM NaCl, pH 7.5) containing 3 % gelatin. Thereafter, the PVDF was washed for 5 min three times in TBS-Tween buffer (TTBS: 20 mM Tris-HCl, 500 mM NaCl, 0.05 % Tween 20, pH 7.5) and incubated overnight with mouse monoclonal anti-NCX1 antibody at 1:1000 dilution in TTBS containing 1 % gelatin. After extensive washing with TTBS, the PVDF was incubated with horseradish peroxidase-conjugated anti-mouse antibody (Bio-Rad) diluted at 1:3000 in TTBS containing 1 % gelatin at room temperature for 120 min. Then, the PVDF was washed for 5 min twice in TTBS and washed for 5 min in TBS. The blots were visualized using the enhanced chemiluminescence method according to the manufacturer's (Bio-Rad) instructions.
Total RNA extraction and Northern blot analysis
Total RNA was extracted from ventricular myocardium using a guanidinium thiocyanate-phenol-chloroform method according to the protocol of Chomczinski & Sacchi (1987). Briefly, the frozen ventricles were placed in 1 ml of ISOGEN (Nippon Gene) and homogenized with a polytron. Subsequently, 200 μl chloroform was added; the mixture was shaken vigorously for 15 s and was kept at room temperature for a few minutes. The mixture was centrifuged at 12 000 g for 15 min at 4°C, the aqueous phase was transferred into a fresh tube, 500 μl isopropanol was added, and the sample was centrifuged at 12 000 g for 15 min. The resulting pellet was washed twice with 75 % ethanol by vortexing and subsequent centrifugation at 7500 g for 5 min at 4°C. Total RNA was resuspended in nanopore water. The amount of RNA present was determined by UV absorption. The ratio of optical density at 260 nm to that at 280 nm was ∼1.98–2.14 in all specimens. The amounts of RNA obtained were 538 ± 7 and 353 ± 9 μg per heart in control and diabetic groups, respectively (n = 5). However, when quantified by dividing the total RNA amount by each heart weight, the values were similar in control and diabetic groups (0.72 ± 0.01 vs. 0.70 ± 0.01 μg mg−1).
RNA (30 μg per lane) was subjected to electrophoresis (75 mA for 120 min) on 1.2 % agarose-6.5 % formaldehyde gels and then transferred to a Hybond-N+ nylon membrane (Amersham). The membrane was prehybridized in pre-warmed rate-enhanced hybridization buffer (Rapid-hyb buffer, Amersham) at 42°C for 1 h. The NCX1 cDNA probe (bases 240–737 of the coding region) was also kindly provided by Drs M. Shigekawa and T. Iwamoto (Iwamoto et al. 1996). The probe was labelled with [32P]dCTP (3000 Ci mmol−1, New England Nuclear) using a random primer labelling system (Rediprime, Amersham). After being hybridized in the buffer containing 32P-labelled probe (109 c.p.m. ml−1) at 42°C for 2 h, the membrane was washed with 5 SSC-0.1 % SDS at room temperature and with 0.1 SSC-0.1 % SDS twice at 42°C. The NCX1 mRNA was quantified by counting the radioactivity using Fujix BAS 2000 (Fuji Photo Film) and was expressed as photo-stimulated luminescence (PSL) per square millimetre (Motoji et al. 1995). In order to control for differences in RNA conditions, the membranes were sequentially probed for β-actin using an oligonucleotide probe (40mer, New England Nuclear). The membranes were stripped of the NCX1 probe by soaking the membranes twice for 20 min in 0.5 % SDS which had been brought to the boil. The oligonucleotide probe for β-actin was labelled with [α-32P]dATP (6000 Ci mmol−1, Amersham) using an oligonucleotide 3′-end labelling system (New England Nuclear), and subsequent hybridization of the β-actin probe was performed as described above. Thus, each membrane was sequentially probed for the mRNA of NCX1 and then normalized using the mRNA of the constitutively expressed protein β-actin.
Organ bath experiments
Experiments were performed as described previously (Gando et al. 1997). Briefly, left ventricular papillary muscles were isolated from the hearts. The bathing solution contained (mM): 119 NaCl, 4.8 KCl, 1.3 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 24.9 NaHCO3, 10.0 glucose, continuously gassed with 95 % O2-5 % CO2, and was maintained at a temperature of 35°C. Isometric force of contraction was measured after the muscle was preloaded to 0.5 g. We have confirmed that this resting tension produced > 90 % maximal force development in papillary muscles from both control and diabetic animals, based on resting tension/developed tension curves (Gando et al. 1997). The muscle was electrically stimulated at 1 Hz with rectangular pulses of 5 ms duration (3F46, Sanei-Sokki), the voltage being 1.5 times greater than threshold. The preparations were allowed to equilibrate for at least 60 min before any experimental procedure was applied.
Statistics
All data are quoted as means ±s.e.m. along with the number of observations (n). Statistical significance was estimated by Student's t test or ANOVA followed by Scheffé's multiple comparisons test to locate differences between groups. Differences were considered significant at a level of P < 0.05.
RESULTS
NCX current
The NCX current was recorded in single ventricular myocytes from control and diabetic rats under the whole-cell voltage clamp. As shown in Fig. 1A–C, the NCX current was induced by increasing the external Ca2+ concentration from 0 to 5 mM in the presence of 50 mM Na+ in the pipette solution. The I-V curve obtained at 5 mM Ca2+ shows a typical outward current which increased at more positive voltages, indicating that the NCX current was generated (Kimura et al. 1986). That this current was the NCX current was confirmed by applying 5 mM NiCl2 in the presence of 5 mM Ca2+, which suppressed the current to the same level as that at 0 mM Ca2+. The anticipated reversal potential is −170 mV with stoichiometry of 3Na+:1Ca2+ under our ionic conditions. The measured reversal potential for the NCX current was more positive to the anticipated value: it was −96 ± 10 mV (n = 6) in control and −89 ± 10 mV (n = 6) in diabetic myocytes. However, this is not particularly surprising because the measured reversal potential typically would fall towards the holding potential possibly due to Na+ and Ca2+ movement by NCX. The membrane potential at which the NCX current values are given was +50 mV. The current was measured at +40 mV, and the value was divided by the capacitance of the cell. This result is summarized in Fig. 1D. The mean capacitances of control and diabetic myocytes were 134.4 ± 14.8 pF (n = 6) and 146.8 ± 18.0 pF (n = 6), respectively, and the values were not significantly different. The mean NCX current density was 3.50 ± 0.45 pA pF−1 (n = 6) in control myocytes and 1.94 ± 0.37 pA pF−1 (n = 6) in diabetic myocytes. Thus, the density of the NCX current in diabetic myocytes was about 55 % of that in controls. The diminished NCX current density was significantly improved by insulin therapy (4.82 ± 0.85 pA pF−1, n = 4).
Figure 1. Assessment of the NCX current in control and diabetic rat ventricular myocytes.
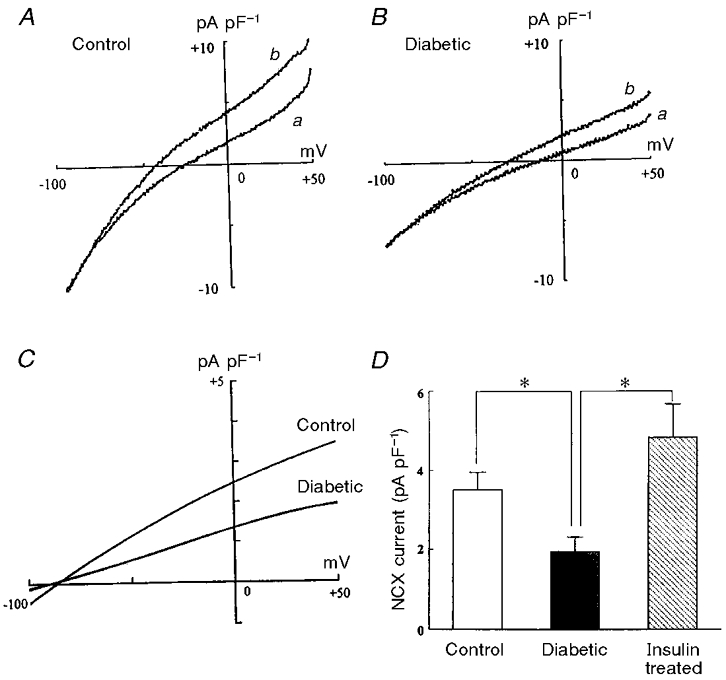
A and B represent the I–V curves obtained at an external Ca2+ concentration of 0 mM (a) and 5 mM (b) in control and diabetic myocytes, respectively. The ordinate shows the current density. The difference current between the I–V curves at 0 and 5 mM Ca2+ indicates a net NCX current. In C, the difference currents in control and diabetic myocytes are shown. Note that the net NCX current is apparently smaller in diabetic cells than in control. D, bar graph comparing the NCX current density in control, diabetic and insulin-treated diabetic myocytes. The net NCX current was estimated as a difference current between the currents at +40 mV under external Ca2+ concentrations of 0 and 5 mM. Values are the result of dividing the net NCX current by the capacitance of each cell, and represent means ±s.e.m. of 4–6 cells from at least 3 rats. *P < 0.05vs. the value with diabetes. There was no significant difference between control and insulin-treated diabetic groups.
NCX1 protein and mRNA
Immunoblot analysis using the mouse monoclonal antibody against NCX1 showed a single band with a molecular mass of 120–140 kDa, which is referred to as the NCX1 protein (Iwamoto et al. 1998), in crude membranes from both control and diabetic rat ventricular myocardium (Fig. 2A). However, immunodetectable NCX1 was found at significantly lower levels in diabetic myocardium compared with controls (Fig. 2B). Densitometric quantification of the signal revealed a decrease to 69 ± 1 % (n = 5,P < 0.001) of control in diabetic myocardium. This reduced level of the NCX1 protein observed in diabetes was reversed by insulin therapy (90 ± 1 %, n = 5).
Figure 2. Immunoblot analysis of NCX1 in ventricular myocardium from control and diabetic rats.
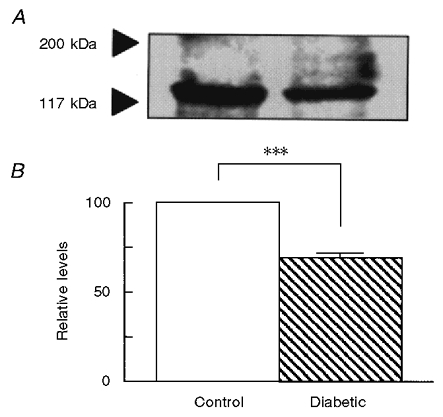
A, representative Western blot indicating clearly a decrease in the single protein band with an apparent molecular mass of 120–140 kDa in diabetes. B, bar graph comparing the immunostained band for NCX1 in the two groups of ventricular myocardium. Densitometric results are expressed as a percentage of the band obtained with control in each of the experiments. Bars are the means ±s.e.m. of 5 experiments. ***P < 0.001.
Representative Northern blots, which had been sequentially hybridized using probes for NCX1 and β-actin, of total RNA isolated from control and diabetic rat ventricular myocardium are shown in Fig. 3A. The densitometric analysis on the Northern blot autoradiograms revealed no significant difference in the absolute integrated optical density values obtained for the β-actin probe between control and diabetic myocardium (55.2 ± 1.1 vs. 52.6 ± 0.9 PSL mm−2, n = 5). Thus, the steady-state mRNA level for the constitutively expressed protein β-actin remained unchanged in diabetes. When the data were presented as the ratio of the absolute integrated optical density of the NCX1 probe to that of the β-actin probe, the relative content of NCX1 mRNA in diabetes was significantly diminished from control level to 70 ± 3 % (n = 5) (Fig. 3B). Insulin therapy significantly prevented the diminished level of cardiac NCX1 mRNA observed in diabetes (Fig. 5).
Figure 3. Northern blot analysis of the expression of NCX1 mRNA in ventricular myocardium from control, diabetic and insulin-treated diabetic rats.
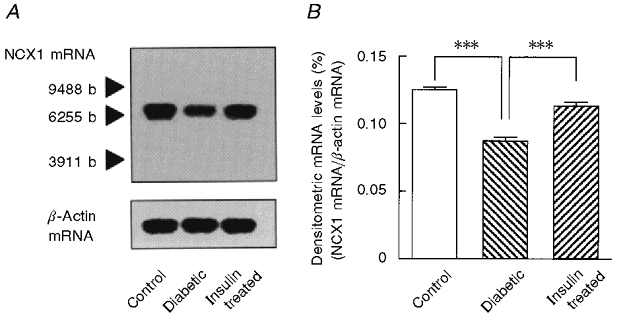
A, representative autograph of Northern blot analysis of NCX1 mRNA and β-actin mRNA expression. The blots had been hybridized sequentially using probes for NCX1 and β-actin. The positive signal localized at the molecular size appropriate for the specific mRNA with minimal background. The standard size is indicated as number of bases (b). While there appeared to be no relative change in the abundance of mRNA for β-actin, the relative content of NCX1 mRNA appeared to be decreased in diabetes. B, bar graph showing quantification of the steady-state levels of NCX1 mRNA in total myocardial RNA from control, diabetic and insulin-treated diabetic rats. The quantitative analysis was made using a Fujix BAS 2000. The steady-state levels of NCX1 mRNA are normalized to those of β-actin mRNA. Bars are means ±s.e.m. of 5 experiments. ***P < 0.001vs. the value with diabetes.
Figure 5. Changes in diastolic and peak systolic [Ca2+]i in response to different external Ca2+ concentrations in control and diabetic rat ventricular myocytes.
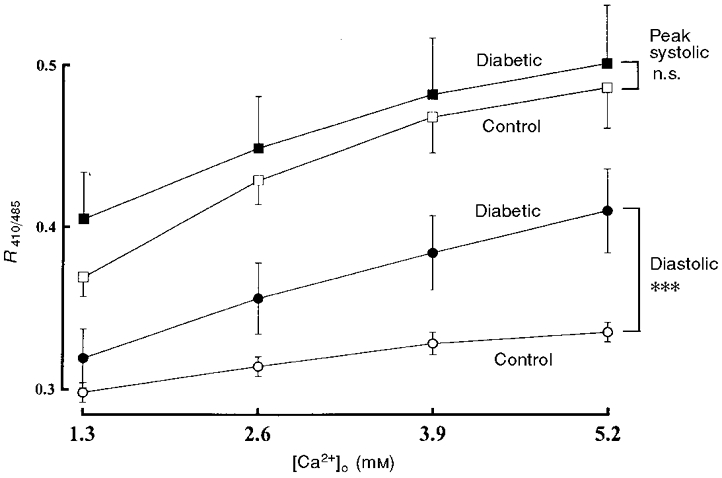
The [Ca2+]i transients were elicited by a field stimulation of 0.5 Hz. The values of diastolic and peak systolic [Ca2+]i when external Ca2+ concentration is increased stepwise from 1.3 mM to 2.6, 3.9 and 5.2 mM are shown. Data represent means ±s.e.m. of 5 cells from at least 3 rats. n.s., not significant. ***P < 0.001.
Changes in [Ca2+]i in response to external Ca2+ concentrations
As reported in our previous work (Tamada et al. 1998), both control and diabetic rat ventricular myocytes showed qualitatively similar changes in the [Ca2+]i transient when stimulated electrically at 0.5 Hz. Thus, diastolic [Ca2+]i and peak systolic [Ca2+]i, as monitored by the ratio of the fluorescence at 410 nm to that at 485 nm, did not differ between control (0.306 ± 0.005 and 0.377 ± 0.008, n = 28) and diabetic (0.327 ± 0.009 and 0.399 ± 0.010, n = 28) myocytes.
Figure 4 shows typical changes in the [Ca2+]i transient elicited by a stepwise increase in external Ca2+ concentration from 1.3 to 5.2 mM in control and diabetic myocytes. With increasing external Ca2+ concentration, peak systolic [Ca2+]i was progressively elevated in both groups of myocytes. The relationship between the magnitude of peak systolic [Ca2+]i and external Ca2+ concentration for diabetic myocytes was identical to that for controls, at least in the Ca2+ concentration range tested (Fig. 5). The change in diastolic [Ca2+]i in response to increasing external Ca2+ concentration was modest in control myocytes. On the other hand, an increase in external Ca2+ concentration resulted in a marked rise in diastolic [Ca2+]i in diabetic myocytes (Fig. 5).
Figure 4. Changes in the [Ca2+]i transients in response to different external Ca2+ concentrations in control and diabetic rat ventricular myocytes.
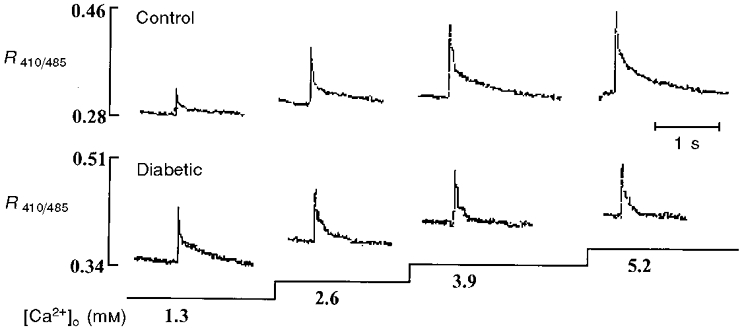
After loading with indo-1 AM, myocytes were field stimulated at 0.5 Hz. Myocytes were exposed to Tyrode solution containing different concentrations of Ca2+ during the period indicated by the horizontal bars. Note that the increases in diastolic [Ca2+]i in response to increasing external Ca2+ concentration from 1.3 mM to 2.6, 3.9 and 5.2 mM were apparently more marked in diabetic cells than in controls.
In Fig. 6 the [Ca2+]i transient change in response to an increase in external Ca2+ concentration from 1.3 to 5.2 mM is depicted in control and diabetic myocytes when the Ca2+ efflux on NCX could be greatly depressed by a reduction in external Na+ concentration by 50 %. Exposure to low Na+ medium significantly elevated the basal level of diastolic [Ca2+]i in control myocytes (0.302 ± 0.007 to 0.381 ± 0.012, 26 ± 2 %, n = 4). This effect tended to be less in diabetic myocytes (0.349 ± 0.035 to 0.400 ± 0.024, 16 ± 5 %, n = 4). Under low Na+ medium, control myocytes exhibited a pronounced increase in diastolic [Ca2+]i when changing to a high concentration of external Ca2+. Thus, exposure to high Ca2+ increased diastolic [Ca2+]i, as monitored by the ratio of fluorescence, from 0.320 ± 0.024 to 0.397 ± 0.035 (n = 5) in control myocytes in low Na+ medium, which was very similar to the change in diabetic myocytes observed in normal Na+ medium (0.319 ± 0.018 to 0.410 ± 0.026, n = 5). On the other hand, the reduction in external Na+ concentration did not enhance the high Ca2+-induced increase in diastolic [Ca2+]i in diabetic myocytes (0.357 ± 0.02 to 0.433 ± 0.027, n = 4).
Figure 6. The influence of a reduction in external Na+ concentration on the change in the [Ca2+]i transients in response to increasing external Ca2+ concentration in control and diabetic rat ventricular myocytes.
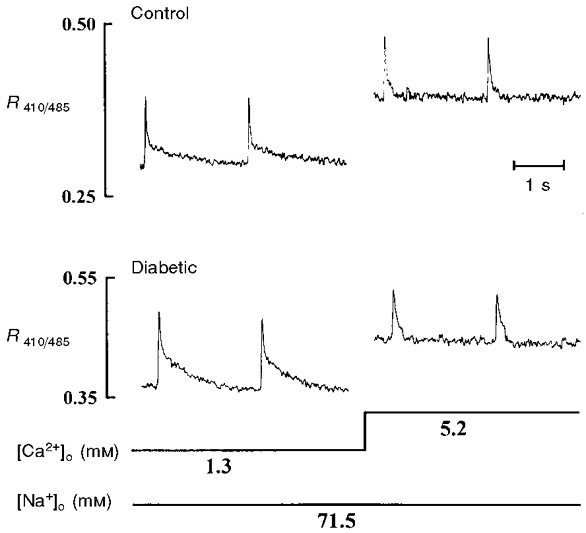
After loading with indo-1 AM, myocytes were field stimulated with 0.5 Hz in normal Tyrode solution. The external Na+ concentration was then reduced by 50 % by replacing NaCl with equimolar choline chloride. Note that the increase in diastolic [Ca2+]i was apparently the same in the two groups of myocytes when increasing the Ca2+ concentration from 1.3 to 5.2 mM.
Treatment with 1 μM cyclopiazonic acid, an inhibitor of SR Ca2+-ATPase, produced a significant increase in the basal level of diastolic [Ca2+]i in both control and diabetic myocytes (P < 0.05). In the presence of cyclopiazonic acid, elevation of external Ca2+ concentration from 1.3 to 5.2 mM increased diastolic [Ca2+]i from 0.356 ± 0.011 to 0.394 ± 0.014 (n = 5) in control myocytes and from 0.340 ± 0.021 to 0.431 ± 0.043 (n = 5) in diabetic myocytes. These changes induced by high Ca2+ were comparable to those obtained in control and diabetic myocytes under normal conditions.
Effect of high Ca2+ on force of contraction in papillary muscle
When electrically stimulated with 1 Hz, the basal force of contraction of left ventricular papillary muscles isolated from diabetic rats was not significantly different from that of muscles from control animals (Gando et al. 1997). In control muscles, the addition of 10 mM CaCl2 produced an increase in force of contraction with little change in resting tension (Fig. 7A). Thus, the effect of high Ca2+ on resting tension was never observed in any of eight control muscles. In diabetic muscles, the high Ca2+-induced positive inotropic effect was accompanied by a marked rise in resting tension (Fig. 7B). The rise in resting tension was clearly detected in three of five diabetic muscles. In the three diabetic muscles, the extent of the rise in resting tension was 44 ± 4 % when the amplitude of the basal developed tension was assigned a value of 100 %.
Figure 7. The effect of the addition of high Ca2+ concentration on force of contraction in left ventricular papillary muscles isolated from control and diabetic rats.
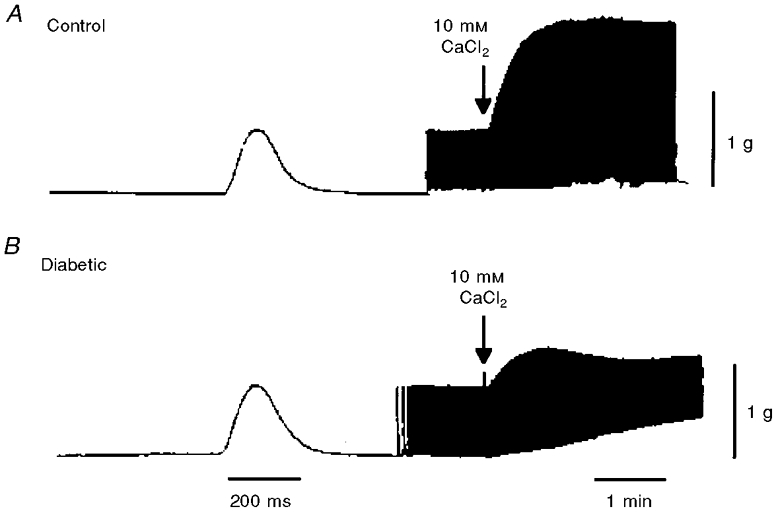
Muscles were electrically stimulated at 1 Hz. A, in control muscle, the addition of 10 mM CaCl2 caused an increase in the peak amplitude of force of contraction without any change in resting tension. B, in diabetic muscle, a marked rise in resting tension was elicited by 10 mM CaCl2.
DISCUSSION
Prior studies have shown that NCX activity is diminished in sarcolemmal fractions prepared from hearts of streptozotocin- or alloxan-induced diabetic rats (Makino et al. 1987; Kato & Kako, 1988; Pierce et al. 1990). In the present study, a significant decrease in NCX function was demonstrated in intact ventricular myocytes freshly isolated from streptozotocin-induced diabetic rats. Thus, the whole-cell voltage-clamp experiments indicated a considerable reduction in the NCX current in diabetic ventricular myocytes. This was evidenced by measuring the outward NCX current measured at +40 mV. The decreased density in the NCX current observed in diabetic myocytes was prevented by insulin therapy. This suggests that the reduction in the NCX current was a consequence of the resulting diabetic state and independent of direct cardiotoxic effect of streptozotocin. At the beginning of the high plateau of the action potential, the outward NCX current could be activated, while the inward NCX current would become active during the low late plateau (Noble et al. 1996). The action potential duration is known to be significantly prolonged in diabetic rat myocardium (Fein et al. 1983; Nobe et al. 1990). The reduced outward mode of the NCX current might contribute, at least in part, to the prolongation of action potential duration in diabetic rat ventricular myocytes. However, this is unlikely, because the outward NCX current is probably only very brief due to the large rise in local [Ca2+]i produced by the Ca2+ current and SR Ca2+ release, and because less NCX function in diabetes will also mean less inward NCX current later in the action potential throughout the [Ca2+]i transient. Thus, the prolonged action potential duration would be mainly due to a decrease in the transient outward current and the steady-state outward current (Magyar et al. 1992; Jourdon & Feuvray, 1993; Wang et al. 1995).
To our knowledge, this study is the first to demonstrate that the protein and mRNA levels of NCX1, a NCX isoform which is expressed to a great extent in myocardium (Kofuji et al. 1992; Quednau et al. 1997), were significantly diminished in diabetic rat myocardium. This suggests that the reduced NCX function observed in diabetic myocytes is partly due to the quantitative decrease in NCX machinery. In contrast, it has been reported that the NCX mRNA level is unchanged in the heart from the non-insulin-dependent diabetic rat, a condition produced by injection of streptozotocin at the neonatal stage (Schaffer et al. 1997). We found that insulin therapy restored the diminished levels of NCX1 protein and mRNA, indicating apparently that the diminished NCX mRNA level is associated with insulin deficiency. However, possible mechanisms other than the diminishment of the NCX protein may also be considered for the reduced cardiac NCX function in diabetes. Incubation with high glucose causes a decrease in NCX activity in neonatal rat cardiomyocytes (Schaffer et al. 1997). On the other hand, only acute exposure to insulin can stimulate NCX activity in sarcolemmal membrane fractions from bovine hearts (Kato & Kako, 1988) and in neonatal rat cardiomyocytes (Schaffer et al. 1997). We found that the NCX function, which was assessed by the NCX current, was nearly completely improved by insulin therapy. Therefore, the depression of cardiac NCX function observed in diabetes seems to result from both hyperglycaemia and insulin deficiency.
To date, measurements of basal [Ca2+]i in diabetic cardiomyocytes have been controversial. Noda et al. (1992) have reported that the [Ca2+]i level of quiescent myocytes of diabetic rats is lower than that of controls using fura-2. Using indo-1, Lagadic-Grossmann et al. (1996) have also shown a lower basal [Ca2+]i level in diabetic rat myocytes when stimulated at 1 Hz. However, evidence has been presented that basal [Ca2+]i levels in both quiescent and electrically stimulated diabetic rat myocytes are unchanged using fura-2 (Yu et al. 1994). Furthermore, one study using Ca2+-sensitive microelectrodes has revealed a high [Ca2+]i level in diabetic rat ventricular muscle (Lopez et al. 1988). The reasons for this controversy are not apparent, but may be related to differences in experimental models used, i.e. differences in strains, ages of animals, duration of diabetes, and experimental conditions. As previously reported in detail (Tamada et al. 1998), diastolic [Ca2+]i and peak systolic [Ca2+]i levels in diabetic rat ventricular myocytes employed in this study did not significantly differ from the corresponding values obtained in controls under physiological conditions. However, when external Ca2+ concentration was increased stepwise from 1.3 mM to 2.6, 3.9 and 5.2 mM, diabetic myocytes exhibited a marked elevation of diastolic [Ca2+]i but no significant difference in peak systolic [Ca2+]i increase compared with the changes seen in control myocytes. As a result, the amplitude of the [Ca2+]i transient in diabetic myocytes was smaller than that in controls when a high Ca2+ concentration was present in the external solution. We interpret these observations to indicate that abnormalities of intracellular Ca2+ handling in diabetic myocytes become manifest only when [Ca2+]i rises to pathological levels.
This greater susceptibility of diabetic cardiomyocytes to high Ca2+ levels could be brought about by depressed Ca2+ reuptake into the SR, mediated by the SR Ca2+-ATPase, and by diminished extrusion of Ca2+ from the cell primarily through NCX, because we have observed that the density of the L-type Ca2+ current, a major source of Ca2+ influx, is unaffected by diabetes (Tamada et al. 1998). In rat ventricular myocytes, reuptake into the SR by Ca2+-ATPase is known to contribute 87 % of the total efflux of Ca2+ from the cytosol, whereas NCX only contributes 9 % (Negretti et al. 1993). Therefore, although in diseased cardiomyocytes an imbalance may develop between the two processes of reducing [Ca2+]i, the enhanced elevation of diastolic [Ca2+]i in diabetic myocytes in response to high Ca2+ levels might be largely due to an impairment of Ca2+ reuptake by the SR. In this study, however, a marked rise in diastolic [Ca2+]i induced by exposure to high Ca2+ was observed in control myocytes where the Ca2+ efflux on NCX was expected to be strongly depressed by reduction in external Na+ concentration by 50 %. Reduction in external Na+ did not cause a further rise in diastolic [Ca2+]i in response to high Ca2+ in diabetic myocytes. Furthermore, cyclopiazonic acid, an inhibitor of SR Ca2+ pump ATPase, did not alter the high Ca2+-induced changes in diastolic [Ca2+]i in either control or diabetic myocytes, although this agent greatly increased the basal level of diastolic [Ca2+]i in both groups of myocytes as seen in the presence of low Na+. These results suggest that the striking increase in diastolic [Ca2+]i in diabetic myocytes when exposed to high Ca2+ may be principally the result of diminished Ca2+ extrusion from the cell through NCX rather than depressed Ca2+ reuptake into the SR. We observed that the addition of high Ca2+ could produce a rise in resting tension in papillary muscles isolated from diabetic rats, but not in control muscles. Fein et al. (1983) have also reported that with high levels of external Ca2+, or with ouabain, diabetic papillary muscles exhibit a much greater rise in resting tension compared with control muscles. This rise in resting tension is most likely to be caused by an increased level of diastolic [Ca2+]i. We therefore propose that the reduced capacity of diabetic cardiomyocytes to buffer abnormally high [Ca2+]i, which is possibly due to diminished NCX function, may lead to partial contracture indicated by the elevated resting tension.
In conclusion, the present study shows that NCX is functionally impaired in ventricular myocytes isolated from streptozotocin-induced diabetic rat hearts. This impairment of NCX function results, in part, from reductions in cardiac NCX protein and mRNA. The impaired cardiac NCX function in diabetes may not disturb [Ca2+]i homeostasis and contractile performance during physiological states, but seems to play a critical role in alterations in [Ca2+]i handling when [Ca2+]i abruptly rises to pathological levels.
Acknowledgments
The authors thank Fumika Sakuraya for her excellent technical assistance, and Drs Mitsuhiro Fukao and Shigeaki Kobayashi for expert help in the performance of electrophysiological studies. This study was supported by a Grant-in-Aid for Science Research from the Ministry of Education, Science, Sports and Culture of Japan.
References
- Barry WH, Bridge JHB. Intracellular calcium homeostasis in cardiac myocytes. Circulation. 1993;87:1806–1815. doi: 10.1161/01.cir.87.6.1806. [DOI] [PubMed] [Google Scholar]
- Callewaert G. Excitation-contraction coupling in mammalian cardiac cells. Cardiovascular Research. 1992;26:923–932. doi: 10.1093/cvr/26.10.923. [DOI] [PubMed] [Google Scholar]
- Choi HS, Eisner DA. The role of sarcolemmal Ca2+-ATPase in the regulation of resting calcium concentration in rat ventricular myocytes. The Journal of Physiology. 1999;515:109–118. doi: 10.1111/j.1469-7793.1999.109ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczinski P, Sacchi N. Single step method of RNA isolation by guanidine thiocyanate-phenol-chloroform extraction. Analytical Biochemistry. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Dhalla NS, Pierce GN, Innes IR, Beamish RE. Pathogenesis of cardiac dysfunction in diabetes mellitus. Canadian Journal of Cardiology. 1985;1:263–281. [PubMed] [Google Scholar]
- Fein FS, Aronson RS, Nordin C, Miller-Green B, Sonnenblick EH. Altered myocardial response to ouabain in diabetic rats: mechanics and electrophysiology. Journal of Molecular and Cellular Cardiology. 1983;15:769–784. doi: 10.1016/0022-2828(83)90336-x. [DOI] [PubMed] [Google Scholar]
- Fein FS, Sonnenblick EH. Diabetic cardiomyopathy. Progress in Cardiovascular Diseases. 1985;27:255–270. doi: 10.1016/0033-0620(85)90009-x. [DOI] [PubMed] [Google Scholar]
- Gando S, Hattori Y, Akaishi Y, Nishihira J, Kanno M. Impaired contractile response to beta adrenoceptor stimulation in diabetic rat hearts: Alterations in beta adrenoceptors-G protein-adenylate cyclase system and phospholamban phosphorylation. Journal of Pharmacology and Experimental Therapeutics. 1997;282:475–484. [PubMed] [Google Scholar]
- Ganguly PK, Pierce GN, Dhalla KS, Dhalla NS. Defective sarcoplasmic reticular calcium transport in diabetic cardiomyopathy. American Journal of Physiology. 1983;244:E528–535. doi: 10.1152/ajpendo.1983.244.6.E528. [DOI] [PubMed] [Google Scholar]
- Hammes A, Oberdorf-Maass S, Rother T, Nething K, Gollnick F, Linz KW, Meyer R, Hu K, Han H, Gaudron P, Ertl G, Hoffmann S, Ganten U, Vetter R, Schuh K, Benkwitz C, Zimmer HG, Neyses L. Overexpression of the sarcolemmal calcium pump in the myocardium of transgenic rats. Circulation Research. 1998;83:877–888. doi: 10.1161/01.res.83.9.877. [DOI] [PubMed] [Google Scholar]
- Heyliger CE, Prakash A, McNeill JH. Alterations in cardiac sarcolemmal Ca2+ pump activity during diabetes mellitus. American Journal of Physiology. 1987;252:H540–544. doi: 10.1152/ajpheart.1987.252.3.H540. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Pan Y, Nakamura TY, Wakabayashi S, Shigekawa M. Protein kinase C-dependent regulation of Na+/Ca2+ exchanger isoforms NCX1 and NCX3 does not require their direct phosphorylation. Biochemistry. 1998;37:17230–17238. doi: 10.1021/bi981521q. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Pan Y, Wakabayashi S, Imanaga T, Yamanaka HI, Shigekawa M. Phosphorylation-dependent regulation of cardiac Na+/Ca2+ exchanger via protein kinase C. Journal of Biological Chemistry. 1996;271:13609–13615. doi: 10.1074/jbc.271.23.13609. [DOI] [PubMed] [Google Scholar]
- Jourdon P, Feuvray D. Calcium and potassium currents in ventricular myocytes isolated from diabetic rats. The Journal of Physiology. 1993;470:411–429. doi: 10.1113/jphysiol.1993.sp019866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Kako KJ. Na+/Ca2+ exhange of isolated sarcolemmal membrane: effects of insulin, oxidants and insulin deficiency. Molecular and Cellular Biochemistry. 1988;83:15–25. doi: 10.1007/BF00223194. [DOI] [PubMed] [Google Scholar]
- Kimura J, Noma A, Irisawa H. Na-Ca exchange current in mammalian heart cells. Nature. 1986;319:596–597. doi: 10.1038/319596a0. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Hadley RW, Kieval RS, Lederer WJ, Schulze DH. Expression of the Na-Ca exchanger in diverse tissues: a study using the cloned human cardiac Na-Ca exchanger. American Journal of Physiology. 1992;263:C1241–1249. doi: 10.1152/ajpcell.1992.263.6.C1241. [DOI] [PubMed] [Google Scholar]
- Lagadic-Grossmann D, Buckler KJ, Le Prigent K, Feuvray D. Altered Ca2+ handling in ventricular myocytes isolated from diabetic rats. American Journal of Physiology. 1996;270:H1529–1537. doi: 10.1152/ajpheart.1996.270.5.H1529. [DOI] [PubMed] [Google Scholar]
- Lagadic-Grossmann D, Chesnais JM, Feuvray D. Intracellular pH regulation in papillary muscle cells from streptozotocin diabetic rats: an ion-sensitive microelectrode study. Pflügers Archiv. 1988;412:613–617. doi: 10.1007/BF00583762. [DOI] [PubMed] [Google Scholar]
- Li Z, Matsuoka S, Hryshko LV, Nicoll DA, Bersohn MM, Burke EP, Lifton RP, Philipson KD. Cloning of the NCX2 isoform of the plasma membrane Na+-Ca2+ exchanger. Journal of Biological Chemistry. 1994;269:17434–17439. [PubMed] [Google Scholar]
- Lopaschuk GD, Tahiliani AG, Vadlamudi RVSV, Katz S, McNeill JH. Cardiac sarcoplasmic reticulum function in insulin or carnitine-treated diabetic rats. American Journal of Physiology. 1983;245:H969–976. doi: 10.1152/ajpheart.1983.245.6.H969. [DOI] [PubMed] [Google Scholar]
- Lopez JR, Banyasz T, Kavacs T, Sreter FA, Szucs G. Defective myoplasmic Ca2+ homeostasis in ventricular muscle in diabetic cardiomyopathic rats. Biophysical Journal. 1988;53:161a. (abstract) [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. Journal of Biological Chemistry. 1951;193:265–275. [PubMed] [Google Scholar]
- Magyar J, Rusznak Z, Szentesi P, Szucs G, Kovacs L. Action potentials and potassium currents in rat ventricular muscle during experimental diabetes. Journal of Molecular and Cellular Cardiology. 1992;24:841–853. doi: 10.1016/0022-2828(92)91098-p. [DOI] [PubMed] [Google Scholar]
- Makino N, Dhalla KS, Elimban V, Dhalla NS. Sarcolemmal Ca2+-transport in streptozotocin-induced diabetic cardiomyopathy in rats. American Journal of Physiology. 1987;253:E202–207. doi: 10.1152/ajpendo.1987.253.2.E202. [DOI] [PubMed] [Google Scholar]
- Motoji N, Hayama E, Shigematsu A, Tazaki S, Mori N, Miyahara J. Studies on the quantitative autoradiography. II. Radioluminography for quantitative autoradiography of 3H. Biological and Pharmaceutical Bulletin. 1995;18:94–99. doi: 10.1248/bpb.18.94. [DOI] [PubMed] [Google Scholar]
- Negretti N, O'Neill SC, Eisner DA. The relative contributions of different intracellular and sarcolemmal systems to relaxation in rat ventricular myocytes. Cardiovascular Research. 1993;27:1826–1830. doi: 10.1093/cvr/27.10.1826. [DOI] [PubMed] [Google Scholar]
- Nicoll DA, Longoni S, Philipson KD. Molecular cloning and functional expression of the cardiac sarcolemmal Na+-Ca2+ exchanger. Science. 1990;250:562–565. doi: 10.1126/science.1700476. [DOI] [PubMed] [Google Scholar]
- Nicoll DA, Quednau BD, Qui Z, Xia Y-R, Lusis AJ, Philipson KD. Cloning of a third mammalian Na+-Ca2+ exchanger, NCX3. Journal of Biological Chemistry. 1996;271:24914–24921. doi: 10.1074/jbc.271.40.24914. [DOI] [PubMed] [Google Scholar]
- Nobe S, Aomine M, Arita M, Ito S, Takaki R. Chronic diabetes mellitus prolongs action potential duration of rat ventricular muscles: circumstantial evidence for impaired Ca2+ channel. Cardiovascular Research. 1990;24:381–389. doi: 10.1093/cvr/24.5.381. [DOI] [PubMed] [Google Scholar]
- Noble D, LeGuennec J-Y, Winslow R. Functional roles of sodium-calcium exchange in normal and abnormal cardiac rhythm. Annals of the New York Academy of Sciences. 1996;779:480–488. doi: 10.1111/j.1749-6632.1996.tb44822.x. [DOI] [PubMed] [Google Scholar]
- Noda N, Hayashi H, Miyata H, Suzuki S, Kobayashi A, Yamazaki N. Cytosolic Ca2+ concentration and pH of diabetic rat myocytes during metabolic inhibition. Journal of Molecular and Cellular Cardiology. 1992;24:435–446. doi: 10.1016/0022-2828(92)93197-r. [DOI] [PubMed] [Google Scholar]
- Penpargkul S, Fein FS, Sonnenblick EH, Scheuer J. Depressed cardiac sarcoplasmic reticular function from diabetic rats. Journal of Molecular and Cellular Cardiology. 1981;13:303–309. doi: 10.1016/0022-2828(81)90318-7. [DOI] [PubMed] [Google Scholar]
- Pierce GN, Dhalla NS. Sarcolemmal Na+-K+ ATPase activity in diabetic rat heart. American Journal of Physiology. 1983;245:C241–247. doi: 10.1152/ajpcell.1983.245.3.C241. [DOI] [PubMed] [Google Scholar]
- Pierce GN, Ramjiawan B, Dhalla NS, Ferrari R. Na+-H+ exchange in cardiac sarcolemmal vesicles isolated from diabetic rats. American Journal of Physiology. 1990;258:H225–261. doi: 10.1152/ajpheart.1990.258.1.H255. [DOI] [PubMed] [Google Scholar]
- Puglisi JL, Bassani RA, Bassani JWM, Amin JN, Bers DM. Temperature and relative contributions of Ca transport systems in cardiac myocyte relaxation. American Journal of Physiology. 1996;270:H1772–1778. doi: 10.1152/ajpheart.1996.270.5.H1772. [DOI] [PubMed] [Google Scholar]
- Quednau BD, Nicoll DA, Philipson KD. Tissue specificity and alternative splicing of the Na+/Ca2+ exchanger isoforms NCX1, NCX2, and NCX3 in rat. American Journal of Physiology. 1997;272:C1250–1261. doi: 10.1152/ajpcell.1997.272.4.C1250. [DOI] [PubMed] [Google Scholar]
- Schaffer SW, Ballard-Croft C, Boerth S, Allo SN. Mechanisms underlying depressed Na+/Ca2+ exchanger activity in the diabetic rat. Cardiovascular Research. 1997;34:129–136. doi: 10.1016/s0008-6363(97)00020-5. [DOI] [PubMed] [Google Scholar]
- Spurgeon HA, Stern MD, Baartz G, Hansford RG, Talo A, Lakatta EG, Capogrossi MC. Simultaneous measurement of Ca2+, contraction, and potential in cardiomyocytes. American Journal of Physiology. 1990;258:H574–586. doi: 10.1152/ajpheart.1990.258.2.H574. [DOI] [PubMed] [Google Scholar]
- Tamada A, Hattori Y, Houzen H, Yamada Y, Sakuma I, Kitabatake A, Kanno M. Effects of β-adrenoceptor stimulation on contractility, [Ca2+]i, and Ca2+ current in diabetic rat cardiomyocytes. American Journal of Physiology. 1998;274:H1849–1857. doi: 10.1152/ajpheart.1998.274.6.H1849. [DOI] [PubMed] [Google Scholar]
- Wang DW, Kiyosue T, Shigematsu S, Arita M. Abnormalities of K+ and Ca2+ currents in ventricular myocytes from rats with chronic diabetes. American Journal of Physiology. 1995;269:H1288–1296. doi: 10.1152/ajpheart.1995.269.4.H1288. [DOI] [PubMed] [Google Scholar]
- Yu Z, Quamme GA, McNeill JH. Depressed [Ca2+]i responses to isoproterenol and cAMP in isolated cardiomyocytes from experimental diabetic rats. American Journal of Physiology. 1994;266:H2334–2342. doi: 10.1152/ajpheart.1994.266.6.H2334. [DOI] [PubMed] [Google Scholar]