

Abstract
In the present study, we investigated the mechanisms involved in the induction of apoptosis by the Ca2+-ATPase inhibitor thapsigargin (TG), in androgen-sensitive human prostate cancer LNCaP cells.
Exposure of fura-2-loaded LNCaP cells to TG in the presence of extracellular calcium produced an increase in intracellular Ca2+, the first phase of which was associated with depletion of intracellular stores and the second one with consecutive extracellular Ca2+ entry through plasma membrane, store-operated Ca2+ channels (SOCs).
For the first time we have identified and characterized the SOC-mediated membrane current (Istore) in prostate cells using whole-cell, cell-attached, and perforated patch-clamp techniques, combined with fura-2 microspectrofluorimetric and Ca2+-imaging measurements.
Istore in LNCaP cells lacked voltage-dependent gating and displayed an inwardly rectifying current-voltage relationship. The unitary conductance of SOCs with 80 mM Ca2+ as a charge carrier was estimated at 3.2 ± 0.4 pS. The channel has a high selectivity for Ca2+ over monovalent cations and is inhibited by Ni2+ (0.5–3 mM) and La3+ (1 μM).
Treatment of LNCaP cells with TG (0.1 μM) induced apoptosis as judged from morphological changes. Decreasing extracellular free Ca2+ to 200 nM or adding 0.5 mM Ni2+ enhanced TG-induced apoptosis.
The ability of TG to induce apoptosis was not reduced by loading the cells with intracellular Ca2+ chelator (BAPTA-AM).
These results indicate that in androgen-sensitive prostate cancer cells the depletion of intracellular Ca2+ stores may trigger apoptosis but that there is no requirement for the activation of store-activated Ca2+ current and sustained Ca2+ entry in induction and development of programmed cell death.
Prostate cancer is the second highest cause of cancer death in men (Woolf et al. 1995; Parker et al. 1997). Androgen withdrawal therapy is commonly used to delay the progression of the disease (Montironi et al. 1994). However, prostate cancer under hormonal ablation therapy will in most cases exhibit androgen-independent characteristics and the tumours will continue to progress. The androgen-insensitive prostate cancer cells are characterized by a very low proliferation rate that renders the typical chemotherapy agents ineffective. For this reason, targeting programmed cell death, or apoptosis, may be particularly relevant for prostate cancer therapy.
It has now been established that Ca2+ ions are major players in an intracellular signalling system that translates extracellular stimuli into the regulation and control of cellular events leading to programmed cell death (for a review see McConkey & Orrenius, 1997; see also Dowd, 1995; Berridge et al. 1998). Increases in intracellular Ca2+ concentration ([Ca2+]i) have been shown to trigger apoptosis (Martikainen et al. 1991; Juin et al. 1998) and numerous apoptosis inducers increase [Ca2+]i (McConkey et al. 1989; Spielberg et al. 1991). However, the precise mechanism(s) by which Ca2+ ions trigger apoptosis remain poorly understood. Ca2+ stores are intracellular compartments characterized by their high intraluminal Ca2+ content and their participation in the regulation of [Ca2+]i through rapid Ca2+ accumulation and release (Gill et al. 1996; Berridge, 1997). The depletion of Ca2+ stores induces a ‘Ca2+-refilling mechanism’, a plasma membrane Ca2+ entry initially called capacitative Ca2+ influx by Putney (1986, 1990) or store-operated Ca2+ current (Istore). This mechanism has been demonstrated in a variety of non-excitable cells (for review see Parekh & Penner, 1997). Store-operated Ca2+ channels (SOCs) have been shown to be involved in controlling many important physiological and physiopathological functions: secretion, gene transcription, cell cycle, proliferation and also apoptosis (Parekh & Penner, 1995; Fanger et al. 1995; Berridge, 1995a; McConkey & Orrenius, 1997; Santella, 1998). While the implication of Ca2+ ions in the induction of apoptosis is now generally accepted, the data concerning the role of store-operated current in this process are rather contradictory and confusing. Two hypotheses have been proposed. The first assumes that apoptosis may be triggered by endoplasmic reticulum (ER) calcium pool depletion without any requirement for the cytosolic Ca2+ elevation due to store-operated Ca2+ entry (He et al. 1997; Bian et al. 1997). Moreover, according to this hypothesis the capacitative Ca2+ current may be important for optimal ER pool filling and apoptosis inhibition. The second hypothesis, on the contrary, assumes that a sustained elevation in cytosolic Ca2+ to a critical level is the initiator of apoptosis (Dowd et al. 1992; Furuya et al. 1994; Wang et al. 1999).
This last hypothesis is based on experiments where apoptosis was induced by the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA pump) inhibitor thapsigargin (TG) in androgen-insensitive human prostate cancer cells from the TSU-Pr1, DU-145 and PC-3 cell lines (Furuya et al. 1994; Wang et al. 1999). However, nothing is known about apoptosis-inducing Ca2+ signalling in androgen-sensitive prostate cancer cells, where the androgen receptor plays a critical role in regulating growth and differentiation. The study of Ca2+-regulating mechanisms involved in apoptosis in androgen-dependent human prostate cancer cells could be of great importance as it was shown by Gong et al. (1995) that, in such cells, intracellular calcium is a potent regulator of androgen receptor gene expression. It has been found in this work that the calcium ionophore A23187 and thapsigargin down-regulate steady-state androgen receptor mRNA levels. On the other hand, androgen depletion is known to induce apoptosis in androgen-sensitive cancer cells and this mechanism involves Ca2+ signals (Isaacs et al. 1992). The transition of prostate cancer cells from androgen sensitivity to androgen insensitivity may also involve modifications in Ca2+ homeostasis and, probably, in the functioning of store-operated channels. Membrane current initiated by these channels, assumed to play an essential role in cancer cell apoptosis, has never been characterized using patch-clamp techniques in both androgen-sensitive and -insensitive prostate cells. In view of the fact that abnormalities in this current may give rise to human disorders, it is important to understand how this current is regulated and how it affects prostate cell behaviour.
In this work we identify the mechanism by which thapsigargin induces apoptosis in androgen-sensitive human prostate cancer LNCaP cells. We characterize for the first time the store-operated Ca2+ current in prostate cancer cells, using patch-clamp and fluorimetric (fura-2) single-cell techniques. We also show that the depletion of intracellular Ca2+ stores in androgen-sensitive prostate cancer cells may trigger apoptosis without the activation of a store-activated Ca2+ current or sustained Ca2+ entry. Our results provide new information on the link between Ca2+ pools and apoptosis of cancer cells, suggesting evidence for a potentially important signalling pathway involved in the transition from hormone-sensitive to hormone-insensitive prostate cancer.
METHODS
Cell lines
LNCaP cells from the American Type Culture Collection were grown in RPMI 1640 (Biowhittaker, Fontenay sous Bois, France) supplemented with 5 mM L-glutamine (Sigma, L'Isle d'Abeau, France) and 10 % fetal bovine serum (Seromed, Poly-Labo, Strasbourg, France). The culture medium also contained 50 000 IU l−1 penicillin and 50 mg l−1 streptomycin. Cells were routinely grown in 50 ml flasks (Nunc) and kept at 37°C in a humidified incubator in an air-CO2 (95 %-5 %) atmosphere. For electrophysiological experiments, the cells were subcultured in Petri dishes (Nunc) coated with polyornithine (5 mg l−1, Sigma) and used after 4–6 days.
Recording solutions
Bath Ringer solution contained (mM): 140 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 0.3 Na2HPO4, 0.4 KH2PO4, 4 NaHCO3, 5 glucose and 10 Hepes (pH 7.3 ± 0.01 with NaOH). In perforated-patch experiments, the recording pipette was filled with an artificial intracellular saline containing (mM): 55 CsCl, 70 Cs2SO4, 7 MgCl2, 1 CaCl2, 5 D-glucose and 10 Hepes (pH 7.2 with CsOH) with nystatin (200 μg ml−1). In whole-cell experiments, the recording pipette was filled with an artificial intracellular saline containing (mM): 140 CsCl, 2 MgCl2, 1 CaCl2, 10 EGTA and 5 Hepes (pH 7.3 ± 0.01 with CsOH); osmolarity 290 mosmol l−1. In cell-attached experiments, the pipette solution contained 80 mM CaCl2 as a charge carrier plus (mM): TEA-Cl 30, glucose 5, Hepes 10 and 4,4′-diisothiocyanostilbene-2,2′-disulphonic acid (DIDS) 0.1 (pH 7.3 with TEA-OH). The presence in the pipette of the K+ channel blocker TEA and the Cl− channel blocker DIDS ensured maximal suppression of potentially contaminating K+ and Cl− single-channel activities. All experiments were performed at room temperature (20–22°C).
Electrophysiological recording
The electrodes were pulled on a PIP 5 (HEKA, Germany) puller in two stages from borosilicate glass capillaries (1.5 mm in diameter; BBL, WPI, USA) to a tip diameter of 1.5–2.0 μm. The cultures were viewed under phase contrast with an Axiovert 135 (Zeiss, Germany) inverted microscope. Electrodes were positioned with List-Medical (Germany) micromanipulators. Grounding was achieved through a silver chloride-coated silver wire inserted into an agar bridge.
Perforated-patch recordings were performed with 200 μg ml−1 nystatin in the pipette, which was first back-filled with normal Ringer solution to allow reliable seal formation. Series resistance had a steady value of 20–100 MΩ. Perforated-patch and whole-cell recordings were carried out using an Axopatch-200B amplifier (Axon Instruments). Stimulus control, as well as data acquisition and processing were carried out with a PC computer (IBM), fitted with a Digidata 1200 series interface, using pCLAMP 6 software (Axon Instruments, interface and software). The activity of single, store-dependent, plasma membrane Ca2+ channels was recorded in the cell-attached configuration using a HEKA PC-9 amplifier. The currents in response to voltage-clamp pulses were low-pass filtered at 1.5 kHz and digitized at 10 kHz. Under such filtering conditions the root mean square noise, σ, was 0.09 pA. The single-channel data were analysed using PulseFit and Origin 5 software. The techniques have previously been described in detail (Skryma et al. 1994; Prevarskaya et al. 1995).
Data and statistical analysis
Results were expressed as means ± standard deviation where appropriate. Each experiment was repeated several times. Student's t test was used for statistical comparison among means and differences, with P < 0.05 considered significant.
Fluorescence measurements of [Ca2+]i with fura-2
For fura-2 measurements, cells were excited alternately at 340 and 380 nm. Emitted fluorescence was long-pass filtered at 510 nm, captured and analysed by a photomultiplier-based system (Photon Technologies International Ltd, Princeton, NJ, USA). [Ca2+]i was calculated from the ratio of the emitted fluorescence, excited by 340 and 380 nm light, using the Grynkiewicz, Poenie & Tsien equation (Grynkiewicz et al. 1985). For microfluorimetric measurements, cells were grown on glass coverslips for at least 3 days before the experiment, loaded for 30 min with the acetoxymethyl ester derivative of the dye (5 μM fura-2 AM), and subsequently washed three times with a dye-free solution.
Determination of apoptosis
Cells were seeded in 8-chamber culture slides (Lab-Tek) in RPMI medium containing 10 % fetal calf serum. After 24 h, cells were treated with Ni2+ or thapsigargin for varying periods of time. For morphological analysis, at the end of the treatment, cells were fixed with ice-cold methanol for 10 min and washed twice with phosphate-buffered saline (PBS). Cells were then stained with 5 μg ml−1 Hoescht 33258 for 10 min at room temperature and mounted in glycergel (DAKO). Nuclear morphology was displayed on an Olympus BH-2 fluorescence microscope (405–435 nm). The percentage of apoptotic cells was determined by counting at least 500 cells in random fields. Apoptosis was also detected by the TUNEL technique (terminal deoxynucleotide transferase-mediated dUTP-biotin nick-end labelling) using an apoptosis detection kit (Boehringer Mannheim). Following the TUNEL reaction, which detects strand breaks, cells were counterstained with 0.1 % Methyl Green for 10 min. The free calcium concentration was assayed in solutions used for apoptosis experiments using the fura-2 fluorescence equipment described above. The fluorescence of culture medium containing no added calcium was measured, as was that of chelated serum with EGTA at various concentrations (0.1 and 1 mM) with 5 μM fura-2 pentapotassium salt. Free calcium concentration was found to be 200 and 20 nM in the presence of 0.1 and 1 mM EGTA, respectively. Using serial dilutions, we assumed that the free calcium contamination in the culture medium containing no added calcium and chelated serum was around 5–10 μM. The free calcium concentration of the Ringer solution used for calcium measurements was 1 μM with no added CaCl2 and no EGTA.
It should be noted that the incubation with the high doses (more than 1 mM) of Ni2+ was toxic for LNCaP cells (the percentage of cells incorporating Trypan Blue was enhanced by 20 ± 5 % after incubation with 1 mM Ni2+ for 24 h). As 0.5 mM Ni2+ inhibited Istore but was not toxic in long-term experiments this concentration was selected as a standard dose for all studies of apoptosis.
Chemicals
All chemicals were bought from Sigma except for fura-2 AM, fura-2 pentapotassium salt, SK&F 96365 and thapsigargin, which were purchased from Calbiochem.
RESULTS
SERCA pump inhibitors induce a biphasic Ca2+ rise in LNCaP cells
The [Ca2+]i resting level of LNCaP cells in a solution containing 2 mM CaCl2 was about 81 ± 7 nM (n = 103) and remained stable during the recording for up to 60 min.
A common means of discharging the Ca2+ stores is to inhibit SERCA pump activity (Premack et al. 1994; Parekh & Penner, 1996). Potent, selective Ca2+ pump inhibitors such as thapsigargin (Thastrup et al. 1990) or cyclopiazonic acid (CPA; Mason et al. 1991) deplete intracellular Ca2+ pools and concomitantly promote a sustained capacitative Ca2+ entry (Huang & Putney, 1998). This makes Ca2+ pump inhibitors useful tools for studying controlled intracellular calcium changes and their consequences in cell physiology.
Exposure of fura-2-loaded LNCaP cells to 0.1 μM thapsigargin in the presence of 2 mM extracellular calcium produced a large initial increase in intracellular Ca2+ as a result of the depletion of intracellular stores (Fig. 1A). This was followed by a sustained plateau, corresponding to a Ca2+ influx. This depletion-activated Ca2+ entry was confirmed by the fact that 3 mM Ni2+ (Fig. 1B) and 1 μM La3+ (data not shown) blocked the sustained Ca2+ rise. Decreasing extracellular free Ca2+ concentration to 1 μM by removing CaCl2 from the bath also suppressed Ca2+ entry while reintroducing extracellular Ca2+ restored Ca2+ (Fig. 1C).
Figure 1. Stimulation of Ca2+ influx by thapsigargin in LNCaP cells.

A, thapsigargin (0.1 μM) induces an increase in Ca2+ in LNCaP cells and stimulates Ca2+ release with consecutive Ca2+ entry. B, the action of thapsigargin (0.1 μM) in Ca2+-containing medium in the presence of Ni2+ (3 mM). C, dependence of thapsigargin-induced Ca2+ influx on extracellular Ca2+. Removing Ca2+ from the external medium (measured free Ca2+ in these conditions was 1 μM) blocks thapsigargin-stimulated Ca2+ entry. Thapsigargin (0.1 μM) was added at the time indicated by arrows. Testing solutions were applied from a puffing pipette during the periods indicated by bars.
To ensure that the effects of thapsigargin resulted from its action on intracellular Ca2+-ATPase, we also examined the effects of CPA, a structurally unrelated agent that similarly inhibits the SERCA pump. CPA (10 μM) produced similar effects on [Ca2+]i to those of thapsigargin, also inducing an initial Ca2+ mobilization followed by Ca2+ entry (not shown, n = 11).
Depletion of intracellular Ca2+ stores activates a Ca2+ current through store-operated channels
We used the perforated-patch recording technique combined with [Ca2+]i measurement to compare the kinetics of the [Ca2+]i increase induced by TG, and the development of the Istore Ca2+ current. As illustrated in Fig. 2, within 15 ± 6 s, TG stimulated an initial [Ca2+]i increase due to the mobilization of Ca2+ from intracellular stores. An inward current appeared within 25 ± 5 s after TG application (n = 11). Decreasing extracellular free Ca2+ concentration to 1 μM by removing CaCl2 from the bath inhibited the second phase of the [Ca2+]i increase induced by TG and the corresponding inward current (Fig. 2A, n = 7). When Ca2+ was added again the sustained plateau was restored and the Ca2+ current was induced. Istore was also blocked by Ni2+ in the range of 0.5–3 mM. However, the rate of blockade was concentration dependent; 3 mM Ni2+ completely inhibited the current within 1–2 min whereas 10–15 min were required to completely inhibit the current with 0.5 mM Ni2+. Figure 2B demonstrates the effect of TG on [Ca2+]i and Istore in cells incubated in 0.5 mM Ni2+ during 15 min where only Ca2+ mobilization from intracellular stores was observed without any stimulation of store-operated current. Moreover, when Ni2+ was present in the extracellular solution, TG only induced an initial Ca2+ rise due to Ca2+ mobilization, but Istore was not stimulated (n = 9,Fig. 2B). Likewise, TG induced a monophasic increase in [Ca2+]i under conditions when extracellular Ca2+ was removed from the bath solution (not shown, n = 7).
Figure 2. Patch-clamp recordings of store-operated Ca2+ current in LNCaP cells.
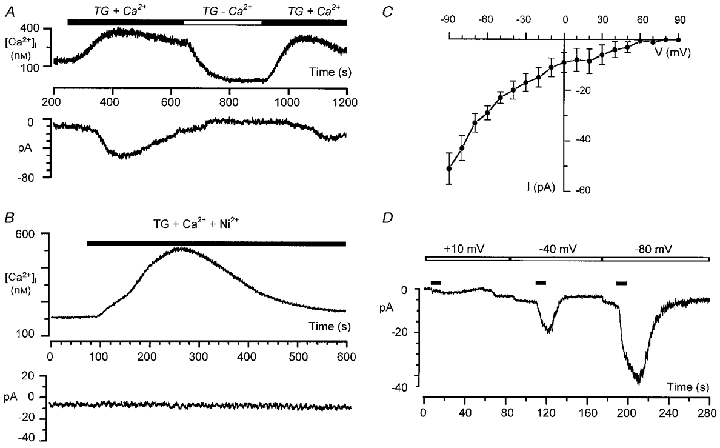
A and B, combined perforated-patch and microspectrofluorimetric single-cell recordings of the thapsigargin-activated store-operated Ca2+ current at a holding potential of −80 mV. A, reversible blockade of Ca2+ current by removing Ca2+ from external medium (measured free Ca2+, 1 μM). B, thapsigargin induces Ca2+ mobilization but does not induce store-operated Ca2+ current in the presence of 0.5 mM Ni2+. C, current- voltage relationship of the store-operated Ca2+ current (current is inwardly rectifying and not activated by depolarization). D, whole-cell patch-clamp recordings of store-operated Ca2+ current at different holding membrane potentials. The horizontal filled bars indicate the episodes of 22 mM CaCl2 application.
We used the whole-cell patch-clamp technique to study the voltage dependence of Istore in LNCaP cells. Ca2+ stores were emptied by incubating cells with 0.1 μM TG for 15 min in the low free Ca2+ (1 μM) bath solution while dialysing the interior of the cell with a strongly buffered Ca2+ solution (see Methods). Istore was elicited by adding 22 mM CaCl2 to the bath at different holding membrane potentials. An example of such an experiment and its current-voltage (I–V) relationship are shown in Fig. 2C and D. The store-operated calcium channels in LNCaP cells were non-conducting at very depolarized potentials. In contrast, the current amplitude increased following membrane hyperpolarization and the I–V relationship displayed an inward rectification (Fig. 2C).
Changing external Na+ (replaced by choline) had no significant effect on Istore in both whole-cell (n = 5) and perforated-patch experiments (n = 3). In all cells studied (n = 7, perforated-patch combined with fura-2 experiments) Istore was also insensitive to one of the suggested capacitative Ca2+ current inhibitors, SK&F 96365 (100 μM).
The activity of single store-operated Ca2+ channels
The cell-attached patch-clamp configuration was used to study the unitary SOC activity in LNCaP cells. In the presence of 0.1 μM TG in the Ca2+-free bath solution and 80 mM CaCl2 in the pipette, under conditions that ensured inhibition of all membrane currents except those of Ca2+, we recorded a unitary activity in an inward direction that had a resolvable amplitude only at potentials more negative than the resting potential of the cell. In order to examine this type of activity at a broad range of membrane potentials and at the same time measure its I–V relationship, we employed a complex pulse protocol consisting of two steady levels of hyper- and depolarizing potentials connected to each other with a voltage ramp (Vr). The pulse protocol together with the single-channel traces are shown in Fig. 3A. In order to construct the I–V relationship, we thoroughly inspected each trace and used the cursor procedure to select those portions of the traces in response to voltage ramps that contained openings of only one channel (overlapping openings and closed states were discarded). Single-channel amplitudes corresponding to each ramp voltage were averaged, then the resulting curve was smoothed and placed in the I–V coordinates (Fig. 3B). The resultant I–V plot showed strong rectification in the inward direction. The currents at potentials close to and more positive than the resting potential were beyond the resolution limit, therefore the slope conductance was determined by the linear fit of the I–V plot at potentials of −40 to −120 mV (relative to Vr) where unitary amplitudes could be adequately resolved. This linear fit produced a slope conductance value of 3.2 ± 0.4 pS (n = 5).
Figure 3. The activity of single store-dependent Ca2+ channels.

A, single-channel recordings obtained in response to the consecutive application of voltage-clamp pulses (shown in the upper panel, 10 s interpulse interval) to the cell-attached patch in TG-treated LNCaP cells; superimposed continuous lines indicate zero current levels; downward deflection corresponds to the inward current. The recordings presented probably reflect the activity of two store-dependent Ca2+ channels. Dotted lines indicate two levels of single-channel amplitudes of −0.35 and −0.7 pA. B, the I–V relationship of the single store-dependent Ca2+ channel; the I–V plot was constructed from the ramp portions of single-channel recordings by selecting the parts corresponding to the opening of one channel (overlapping openings and closed states were discarded) with subsequent averaging and digital smoothing of the resulting curve to reduce noise; superimposed linear fit provides the value of the slope conductance (g0) of 3.2 pS.
The following lines of evidence support our notion that the unitary activity described thus far is associated with the activation of single store-dependent, plasma membrane Ca2+ channels. (i) This activity was only observed in TG-treated cells; no similar activity was found in the cells under normal conditions. (ii) The activity shows strong inward rectification, typical of Ca2+-selective channels. (iii) Neither K+ (Skryma et al. 1997, 1999) nor Cl− (Y. M. Shuba, unpublished observation) channels known to be present in LNCaP cells would be capable of producing a similar type of activity under these experimental conditions used. (iv) SOC has no similarity to the TG-independent Ca2+-permeable 23 pS cation channel recently reported by Gutiérrez et al. (1999) in LNCaP cells.
Thapsigargin induces apoptosis in LNCaP cells
Hoescht staining was used to determine apoptosis induced by TG treatment. Under control conditions, in the absence of TG, the percentage of apoptotic cells was less than 1 %. Typical apoptotic features induced by treatment with 0.1 μM TG for 24 h are shown in Fig. 6C. TG-induced apoptosis was also detected by the TUNEL technique (Fig. 7C). TG induced apoptosis in a time-dependent manner (Fig. 4A), with 7, 24 and 77 % of cells reaching apoptosis at 24, 30 and 48 h, respectively.
Figure 6. LNCaP prostate cells (staining with 5 μg ml−1 Hoescht) undergoing apoptosis due to treatment with thapsigargin.

A, control cells; B, cells treated with 0.5 mM Ni2+ (24 h); C, cells treated with 0.1 μM TG (24 h); D, cells treated with 0.1 μM TG + 0.5 mM Ni2+ (24 h).
Figure 7. Thapsigargin-induced apoptosis detection in LNCaP prostate cells using the TUNEL technique.

A, control cells; B, cells treated with 0.5 mM Ni2+ (24 h); C, cells treated with 0.1 μM TG (24 h); D, cells treated with 0.1 μM TG + 0.5 mM Ni2+ (24 h).
Figure 4. Temporal changes in apoptosis and in the cytosolic Ca2+ concentration of the LNCaP cells under physiological conditions of 2 mM extracellular free Ca2+.

A, temporal changes in apoptosis of the LNCaP cells (estimated by the number of apoptotic cells) treated with 0.1 μM TG, 0.5 mM Ni2+ or 0.1 μM TG combined with 0.5 mM Ni2+. B, temporal changes in cytosolic Ca2+ concentration of the LNCaP cells treated with 0.1 μM TG, 0.5 mM Ni2+ or 0.1 μM TG combined with 0.5 mM Ni2+.
Sustained elevation of cytosolic Ca2+ due to SOC activation is not required for induction of apoptosis by thapsigargin
As it has previously been reported that Istore activation, leading to a sustained increase in cytosolic Ca2+, is required for TG-induced apoptosis in androgen-insensitive prostate cancer cells (Furuya et al. 1994), we tested this hypothesis in androgen-sensitive prostate cancer LNCaP cells.
To determine whether the Istore in LNCaP cells is important in apoptosis induction by TG, we examined the ability of TG to induce apoptosis under different experimental conditions. In patch-clamp experiments, Istore was completely abolished by 0.5 mM Ni2+ (Fig. 2B). According to the hypothesis stated above, TG-induced apoptosis should decrease in cells treated with 0.5 mM Ni2+. Unexpectedly, TG-induced apoptosis was significantly potentiated by treatment with 0.5 mM Ni2+ (Fig. 4A) whereas cell treatment with 0.5 mM Ni2+ alone did not induce apoptosis in LNCaP cells (Figs 4A, 6B and 7B). This enhancement of cell death was mostly visible after 24 and 30 h (25 vs. 7 % and 41 vs. 24 % of apoptotic cells for TG + Ni2+ and TG at 24 and 30 h, respectively). It therefore seems that Ni2+ does not enhance cell death any further after 48 h treatment (84 vs. 77 % of apoptotic cells for TG + Ni2+ and TG, respectively). Apparently, blocking the store-operated Ca2+ current facilitates and accelerates TG-induced apoptosis. Figure 6D shows the apoptotic features of LNCaP cells after combined treatment with TG and Ni2+ for 24 h. These results were also confirmed by the TUNEL technique. Figure 7 shows the apoptosis detection in LNCaP cells treated with TG (Fig. 7C) and with TG combined with Ni2+ (Fig. 7D) for 24 h. To assess how [Ca2+]i is affected by these apoptosis-inducing conditions, we compared cytoplasmic Ca2+ levels in LNCaP cells after 24, 30 and 48 h treatment with TG, Ni2+ and TG combined with Ni2+. [Ca2+]i remained at high levels throughout the 48 h, and even longer when cells were treated with TG alone, while the [Ca2+]i level in cells treated with Ni2+ or TG combined with Ni2+ was almost the same as that of the control (Fig. 4B). This indicates that a sustained high level of [Ca2+]i is not required for induction of apoptosis by TG and its subsequent development.
To confirm that blocking Ca2+ influx potentiates rather than inhibits TG-induced apoptosis, we then assessed the influence of decreasing the extracellular Ca2+ concentration. The Ca2+-deprived medium contained no added Ca2+ and dialysed serum was used to remove Ca2+ contaminants (as well as other low molecular weight species) in addition to 0.1 mM EGTA to buffer the free calcium concentration in the extracellular solution (see Methods). This gave a Ca2+ concentration of 200 nM. As shown in Fig. 5, the Ca2+-deprived medium potentiated the apoptosis induced by TG in a time-dependent manner without having any action on its own. The potentiation of TG-induced apoptosis due to decreasing the extracellular calcium was similar to that caused by nickel. Indeed, we observed that TG-induced apoptosis was enhanced by 340 % (34 h) and 170 % (30 h) by a Ca2+-deprived solution compared with 310 % (24 h) and 160 % (30 h) by nickel. Reduction to even lower Ca2+ levels with 1 mM EGTA (giving 20 nM free Ca2+) in the external medium also led to an enhancement of TG-induced apoptosis. However, in this condition, the Ca2+-deprived medium had an apoptotic action on its own (20, 28 and 88 % of apoptotic cells at 24, 30 and 48 h, respectively; data not shown). The effects of Ni2+ and low extracellular Ca2+ were really due to enhancement of TG-induced cell apoptosis and not due to necrosis. This was confirmed by an increase in the morphological indicators of apoptosis, i.e. the extent of internucleosomal degradation and DNA ladder formation (data not shown). Furthermore, the percentage of cells excluding Trypan Blue was not affected by incubation with 0.5 mM Ni2+ or 200 nM free Ca2+ medium for more than 72 h.
Figure 5. Temporal changes in the apoptosis of LNCaP cells under conditions of low extracellular free Ca2+.

Cells were treated with 0.1 μM TG n 2 mM free Ca2+, with 0.1 μM TG in 200 nM free Ca2+ or with 200 nM free Ca2+ medium
Transient [Ca2+]i increase due to depletion of Ca2+ stores is not responsible for apoptosis induction by thapsigargin
As thapsigargin is still capable of inducing a rise in [Ca2+]i in the absence of extracellular Ca2+, due to a passive leak of Ca2+ through the ER membrane, we checked whether this initial transient increase in [Ca2+]i was involved in apoptosis induction. To eliminate the transient rise in [Ca2+]i, LNCaP cells were incubated for 20 min in the presence of 50 μM BAPTA-AM, an intracellular Ca2+ chelator, and in the 200 nM free Ca2+ medium (with 0.1 mM EGTA). This procedure resulted in sufficient intracellular Ca2+ buffering to completely eliminate the transient rise in [Ca2+]i that occurred when thapsigargin was added to a Ca2+-free medium (not shown). In BAPTA-loaded cells, [Ca2+]i was 69 ± 9 nM. [Ca2+]i was 65 ± 7 nM 150 s after TG addition, a concentration not different from that before TG addition. BAPTA-AM treatment for 20 min without TG did not induce apoptosis in LNCaP cells (< 0.1 % of apoptotic cells in control after 48 h and < 0.1 % of apoptotic cells after 48 h following 20 min incubation with BAPTA-AM). The cells were then treated with 0.1 μM TG for 48 h and the percentage of apoptotic cells was compared with that of cells not loaded with BAPTA. The ability of TG to induce apoptosis in LNCaP cells was not reduced by BAPTA loading in these conditions (70 ± 5 % apoptotic cells in BAPTA-loaded, TG-treated cells, as compared with 71 ± 8 % in TG-treated cells in the 200 nM free Ca2+ medium).
DISCUSSION
The present study demonstrates that emptying intracellular Ca2+ stores by SERCA pump inhibitors stimulates a Ca2+ current, Istore, through specific store-operated channels (SOCs), in androgen-sensitive human prostate cancer LNCaP cells. These channels are activated by intracellular store depletion, and had not previously been identified and characterized in prostate cells.
The presence of store-operated Ca2+ entry has been documented in a large variety of cells, mainly on the basis of measurements of intracellular Ca2+ levels after store depletion by thapsigargin. The first electrophysiological demonstration of a store-operated Ca2+ current was carried out in mast cells by Hoth & Penner (1992), who called it Ca2+ release-activated current (ICRAC). ICRAC is now the best-characterized store-operated Ca2+ current. It is known to be activated by depleting intracellular Ca2+ stores and has the highest selectivity for Ca2+ over other cations (Hoth & Penner, 1992; Zweifach & Lewis, 1993; Berridge, 1995b). Recent studies using patch-clamp techniques have now clearly established the existence of a number of store-operated Ca2+ currents in several non-excitable cell types, differentiated by their unitary conductance, selectivity and pharmacology (for review see Parekh & Penner, 1997). Regardless of some differences in channel properties, store-operated channels form a family of channels characterized by several specific, common features. The first of these properties consists of current activation by emptying the intracellular calcium stores, using a variety of procedures. In our experiments, Istore was identified by emptying intracellular stores using TG. As in basophilic leukaemia (RBL) cells, or Jurkat T cells, Istore in LNCaP cells appears to be the critical Ca2+ influx pathway as LNCaP cells do not have voltage-activated Ca2+ channels (Skryma et al. 1997, 1999). The second important property of Istore is its characteristic voltage dependence. Istore could be considered as a voltage-independent Ca2+ current, as it is not gated by membrane voltage changes (Zweifach & Lewis, 1993; Hoth & Penner, 1993). However, once it has been activated by store depletion, Istore increases when the membrane potential shifts toward negative values. The current-voltage relationship also shows a pronounced inward rectification at negative voltages (Zweifach & Lewis, 1993; Hoth & Penner, 1993). The Istore reversal potential was above +50 mV, as expected for selective Ca2+ currents. Changing external Na+ had no significant effect on Istore in LNCaP cells, demonstrating that Na+ does not permeate the channel in the presence of external Ca2+. Istore was inhibited by Ni2+ and La3+, thus corresponding to the typical pharmacological profile of store-operated currents in other cells (Schlegel et al. 1993; Grudt et al. 1996). SK&F 96365, one of the proposed inhibitors of capacitative Ca2+ current, did not inhibit Istore in LNCaP cells. However, blocking the Ca2+ current with this inhibitor is not evidence for capacitative current as it can also block other channels in similar concentrations (Franzius et al. 1994).
The unitary conductance of the Istore channel was identified as approximately 3.2 pS, which is rather large in comparison with classical ICRAC single-channel conductance, which is usually under 1 pS (Zweifach & Lewis, 1993; Parekh & Penner, 1997). Large single-channel conductances of 2 and 11 pS were also observed for store-operated currents in A431 epidermal cells (Lückhoff & Clapham, 1994) and endothelial cells (Vaca & Kunze, 1995), respectively. However, in our experiments, as in other cell models (Lückhoff & Clapham, 1994), the I–V relationship for store-operated channels is non-linear. For a non-linear I–V relationship, the slope conductance is a function of potential. When comparing values obtained in different cells it is, therefore, necessary to consider not only specific ionic conditions but also the range of membrane potentials at which they were determined. This makes separating store-operated channels by their single-channel conductance quite a challenging task. Thus the properties of SOC in human cancer prostate LNCaP cells suggest that it belongs to the ‘store-operated’ channel family, but it may be not the same as classical CRAC.
Our study provides the first direct demonstration and characterization of a store-operated current in prostate cells. SOC characterization could be of great interest in prostate cancer studies as these channels were suggested to be the target of a Ca2+-influx inhibitor, which has been found, in clinical trials, to slow down the growth of certain aggressive cancer cells (Kohn et al. 1996). We studied the role of SOCs in TG-induced apoptosis in androgen-sensitive prostate cancer LNCaP cells. TG induces apoptosis in many cell types (Furuya et al. 1994; Gill et al. 1996; He et al. 1997; Bian et al. 1997). Based on the changes in Ca2+ homeostasis induced by TG, three hypotheses can be proposed to explain the apoptosis induction mechanism. Apoptosis is induced by: (i) a transient increase in [Ca2+]i due to a passive leak of Ca2+ through the ER membrane, (ii) Ca2+ pool depletion, or (iii) a sustained rise in cytosolic Ca2+ secondary to Ca2+ entry through Istore channels. In this report we show that neither the transient nor the sustained increase in [Ca2+]i is required for induction of apoptosis by TG. Our results show first that a sustained increase in [Ca2+]i via Istore activation is not required for TG to induce apoptosis in LNCaP cells because Istore inhibition by the 200 nM free Ca2+ medium or 0.5 mM Ni2+ did not abolish TG-induced apoptosis whereas the TG-increased cytosolic Ca2+ concentration was reduced to control values by the incubation in 200 nM free Ca2+ or 0.5 mM Ni2+-containing medium. Under these conditions, however, a transient rise in [Ca2+]i can still occur due to calcium mobilization from internal stores. We strongly buffered [Ca2+]i by preincubating cells with the Ca2+ chelator BAPTA-AM to eliminate the transient rise in [Ca2+]i, while the sustained rise was abolished by the absence of external calcium. This did not inhibit apoptosis induced by TG, suggesting that the transient increase in [Ca2+]i is not responsible for apoptosis induction. These results, where cytosolic Ca2+ is strongly buffered using BAPTA-AM, also exclude another possible consequence of ER depletion that might induce apoptosis: mitochondrial calcium overloading (Berridge et al. 1998; Green & Reed, 1998). These data are consistent with the findings of others on lymphoid cells (Berridge, 1995a; Bian et al. 1997) and murine hypothalamic cell lines (Wei et al. 1998). However, surprisingly, they are in contrast with those described in androgen-insensitive prostate cancer cells (Furuya et al. 1994). In these cells, as in thymocytes (Jiang et al. 1998), TG-induced apoptosis was inhibited by preincubating cells with BAPTA-AM, or by overexpressing the cytosolic Ca2+-binding protein calbindin. On the basis of these results, a sustained increase in cytosolic Ca2+, mediated by a store-operated Ca2+ current, was considered to play a role in apoptosis induction by TG (Furuya et al. 1994).
Our results therefore suggest that Ca2+ pool depletion, and not an increase in cytosolic Ca2+, induces apoptotic cell death in androgen-sensitive human prostate cancer cells. In addition, our data show that TG-induced apoptosis was enhanced (see Figs 4A and 5) by 0.5 mM Ni2+ and a low external free Ca2+ concentration (200 nM free Ca2+). Therefore, blocking capacitative Ca2+ entry potentiates apoptosis induced by TG. This reinforces the hypothesis that Ca2+ pool depletion is involved in apoptosis, since an increase in cytosolic Ca2+ due to capacitative Ca2+ entry (inhibited by Ni2+) would be required for optimal ER pool filling and apoptosis inhibition. It should be noted that decreasing external calcium to very low levels using 1 mM EGTA (20 nM free Ca2+) led to apoptosis on its own. This is certainly due to the inversion of the Ca2+ concentration gradient in comparison to physiological conditions (the intracellular Ca2+ concentration of LNCaP cells is usually about 100 nM). The intracellular Ca2+ stores are depleted in response to the decrease in cytosolic Ca2+ concentration (in turn induced by stimulation of the Ca2+ pump of the plasma membrane by low extracellular Ca2+). This calcium gradient hypothesis may also be confirmed by the fact that, under low external calcium conditions (200 nM), where values are, however, higher than the intracellular Ca2+ concentration (100 nM), apoptosis was not observed in the absence of TG (< 1 % of apoptotic cells at 48 h). Similarly, this does not occur with Ni2+ because the Ca2+ gradient is not perturbed.
Our data are in agreement with those of Distelhorst and colleagues (He et al. 1997) on WEH17.2 lymphoma cells. These authors suggested that ER calcium pool depletion by TG could trigger apoptosis and that overexpression of the Bcl-2 anti-apoptotic protein, which anchors to intracellular membranes, maintains Ca2+ homeostasis within the ER, thereby inhibiting apoptosis induction by TG.
The exact mechanism by which calcium pool depletion induces apoptosis is not known. The high levels of Ca2+ within the lumen of the ER are essential not only for Ca2+ signal transduction, but also for protein synthesis and processing, and cell division (Sambrook, 1990; Koch, 1990; Kuznetsov et al. 1992; Gill et al. 1996; Jiang et al. 1998). Three potential mechanisms by which ER depletion might contribute to apoptosis have been proposed (He et al. 1997): (i) depletion of the ER Ca2+ pool might destabilize the Ca2+-protein gel and its associated membrane, leading to vesiculation and the formation of apoptotic blebs; (ii) disruption of protein processing and transport within the ER may contribute to TG-induced apoptosis; (iii) TG-induced ER Ca2+ pool depletion releases an endonuclease into the nucleus responsible for DNA fragmentation. Thus the decline in ER calcium levels leads to the activation of stress signals that switch on the genes associated with death. Although the importance of intraluminal ER Ca2+ storage in apoptosis appears to be evident, other mechanisms depending on extracellular, cytosolic, mitochondrial or nuclear Ca2+ have been shown to contribute to apoptosis in a variety of cell models (Dowd, 1995; Marin et al. 1996; McConkey & Orrenius, 1997). Therefore, the general applicability of the store-depletion hypothesis is doubtful and the relationship between intracellular Ca2+ stores, Bcl-2 and apoptosis may be cell specific.
The reasons why the differences between androgen-dependent and androgen-independent prostate cancer cells are associated with changes in Ca2+ store-dependent mechanisms involved in apoptosis remain intriguing. It is known that such progression is also associated with expression of the intracellular membrane protein Bcl-2 (Raffo et al. 1995; Chaudhary et al. 1999). Bcl-2 expression modulates intracellular signalling and preserves the integrity of the ER Ca2+ pool in cells exposed to various apoptosis-inducing stimuli, including cytotoxic Ca2+ ionophores, TG and reactive oxygen species (Disthelhorst et al. 1996; He et al. 1997). Moreover, it has been shown that Bcl-2 preserves the ER Ca2+ store via an upregulation of calcium pump SERCA gene expression. Bcl-2 may possibly interact with this pump as well (Kuo et al. 1998). Interestingly, the androgen-insensitive DU-145 cells do not express Bcl-2, but rather Bcl-X(L) (Shirahama et al. 1997), which is poorly expressed in LNCaP cells. Wang et al. (1999) have shown that apoptosis induction by TG in DU-145 cells requires an increase in cytoplasmic Ca2+ since it activates the Ca2+-activated protein phosphatase calcineurin that was found to dephosphorylate BAD (proapoptotic member of Bcl-2 family), thus enhancing BAD heterodimerization with Bcl-X(L) (but not with Bcl-2) and promoting apoptosis. Another Ca2+-regulating protein, calreticulin (Krause & Michalak, 1997), has been identified in prostate cells (Zhu & Wang, 1999). The expression of this highly conserved intracellular Ca2+-binding protein in the lumen of the endoplasmic reticulum is regulated by androgen (Zhu et al. 1998). The downregulation of calreticulin by androgen ablation correlates with apoptosis and the upregulation of calreticulin by androgen replacement in castrated rats correlates with proliferation and differentiation of epithelial cells in the prostate (Zhu et al. 1998; Zhu & Wang, 1999). The induction of calreticulin by androgen in prostate organ culture partially resists protein synthesis inhibition, suggesting that calreticulin is a direct androgen-response gene (Zhu et al. 1998). Furthermore, Mery et al. (1996) have shown in a mouse L fibroblast cell line that overexpression of calreticulin increases intracellular Ca2+ storage and decreases store-operated Ca2+ current suggesting an active involvement of calreticulin in intracellular Ca2+ pool refilling regulation. Thus, as calreticulin is a major intracellular Ca2+-binding protein involved in Ca2+ homeostasis and is regulated by androgens, it could be a promising candidate for mediating androgen regulation of intracellular calcium levels in prostate cells. Thus the differences in Ca2+-dependent apoptosis induction between androgen-dependent and androgen-independent prostate cancer cells may be explained by the differential expression of apoptosis-regulating proteins (Bcl-2, Bcl-X(L), calreticulin).
In summary, we have characterized the Ca2+-regulated mechanisms involved in thapsigargin-induced apoptosis in androgen-sensitive human prostate cancer LNCaP cells. We suggest that a decrease in ER calcium is the major factor in apoptosis induction in these cells; however, direct measurement of the Ca2+ concentration in ER lumen would be required to confirm this statement. In contrast to the situation in androgen-insensitive prostate cancer cells, the activation of Istore, responsible for ER refilling, and increasing cytosolic Ca2+ are not required for TG-induced apoptosis. Further studies are needed to identify the precise Ca2+-regulated mechanisms involved in the progression of prostate cancer cells from androgen dependence to androgen independence.
Acknowledgments
The authors gratefully acknowledge the technical assistance of Isabelle Servant and Isabelle Spadone. This work was supported by grants from INSERM, Ministère de l'Education Nationale, ARC (Association pour la Recherche Contre le Cancer), Ligue Nationale Contre le Cancer, ARTP (Association pour le Recherche sur les Tumeurs de la Prostate), France. Y. Shuba was supported by INSERM and University of Science and Technology of Lille International Cooperation Programs.
R. Skryma and P. Mariot contributed equally to this work.
References
- Berridge MJ. Calcium signalling and cell proliferation. Bioessays. 1995a;17:491–500. doi: 10.1002/bies.950170605. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Capacitative calcium entry. Biochemical Journal. 1995b;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Elementary and global aspects of calcium signalling. The Journal of Physiology. 1997;499:291–306. doi: 10.1113/jphysiol.1997.sp021927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Lipp P. Calcium — a life and death signal. Nature. 1998;935:645–648. doi: 10.1038/27094. [DOI] [PubMed] [Google Scholar]
- Bian X, Hughes FM, Jr, Huang Y, Cidlowski JA, Putney JW., Jr Roles of cytoplasmic Ca2+ and intracellular Ca2+ stores in induction and suppression of apoptosis in S49 cells. American Journal of Physiology. 1997;272:C1241–1249. doi: 10.1152/ajpcell.1997.272.4.C1241. [DOI] [PubMed] [Google Scholar]
- Chaudhary KS, Abel PD, Lalani EN. Role of the bcl-2 gene family in prostate cancer progression and its implications for therapeutic intervention. Environmental Health Perspectives. 1999;107:49–57. doi: 10.1289/ehp.99107s149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disthelhorst CW, Lam M, McCormick TS. Bcl-2 inhibits hydrogen peroxide-induced ER Ca2+ pool depletion. Oncogene. 1996;12:2051–2055. [PubMed] [Google Scholar]
- Dowd DR. Calcium regulation of apoptosis. Advances in Second Messenger and Phosphoprotein Research. 1995;30:255–279. doi: 10.1016/s1040-7952(05)80010-2. [DOI] [PubMed] [Google Scholar]
- Dowd DR, McDonald PN, Komm BS, Haussler MR, Miesfeld R. Stable expression of the calbindin-D28K complementary DNA interferes with the apoptotic pathway in lymphocytes. Molecular Endocrinology. 1992;6:1843–1848. doi: 10.1210/mend.6.11.1336124. [DOI] [PubMed] [Google Scholar]
- Fanger CM, Hoth M, Crabtree GR, Lewis RS. Characterization of T cell mutants with defects in capacitative calcium entry: genetic evidence for the physiological roles of CRAC channels. Journal of Cellular Biology. 1995;131:655–667. doi: 10.1083/jcb.131.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzius D, Hoth M, Penner R. Non-specific effects of calcium entry antagonists in mast cells. Pflügers Archiv. 1994;428:433–438. doi: 10.1007/BF00374562. [DOI] [PubMed] [Google Scholar]
- Furuya Y, Lundmo P, Short AD, Gill DL, Isaacs JT. The role of calcium, pH, and cell proliferation in the programmed (apoptotic) death of androgen-independent prostatic cancer cells induced by thapsigargin. Cancer Research. 1994;54:6167–6175. [PubMed] [Google Scholar]
- Gill DL, Waldron RT, Rys-Sikora KE, Ufret-Vincenty CA, Graber MN, Favre CJ, Alfonso A. Calcium pools, calcium entry, and cell growth. Bioscience Reports. 1996;16:139–157. doi: 10.1007/BF01206203. [DOI] [PubMed] [Google Scholar]
- Gong Y, Blok LJ, Perry JE, Lindzeys JK, Tindall DJ. Calcium regulation of androgen receptor expression in the human prostate cancer cell line LNCaP. Endocrinology. 1995;136:2172–2178. doi: 10.1210/endo.136.5.7720667. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Apoptosis. 1998;281:1309–1316. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Grudt TJ, Usowicz MM, Henderson G. Ca2+ entry following store depletion in SH-SY5Y neuroblastoma cells. Molecular Brain Research. 1996;36:93–100. doi: 10.1016/0169-328x(95)00248-q. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gutiérrez AA, Arias JM, Garcia L, Mas-Oliva J, Guerrero-Hernandez A. Activation of a Ca2+-permeable cation channel by two different inducers of apoptosis in a human prostatic cancer cell line. The Journal of Physiology. 1999;517:95–107. doi: 10.1111/j.1469-7793.1999.0095z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Lam M, McCormick TS, Distelhorst CW. Maintenance of calcium homeostasis in the endoplasmic reticulum by bcl-2. Journal of Cellular Biology. 1997;138:1219–1228. doi: 10.1083/jcb.138.6.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. The Journal of Physiology. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Putney JW. Relationship between intracellular calcium store depletion and calcium release-activated calcium current in a mast cell line (RBL-1) Journal of Biological Chemistry. 1998;273:19554–19559. doi: 10.1074/jbc.273.31.19554. [DOI] [PubMed] [Google Scholar]
- Isaacs JT, Lundmo PI, Berges R, Martikainen P, Kyprianou N, English HF. Androgen regulation of programmed death of normal and malignant prostatic cells. Journal of Andrology. 1992;13:457–464. [PubMed] [Google Scholar]
- Jiang S, Chow SC, Nicotera P, Orremius S. Intracellular Ca2+ signals activate apoptosis in thymocytes: studies using the Ca2+-ATPase inhibitor thapsigargin. Experimental Cellular Research. 1998;212:84–92. doi: 10.1006/excr.1994.1121. [DOI] [PubMed] [Google Scholar]
- Juin P, Pelletier M, Oliver L, Tremblais K, Grégoire M, Meflah K, Vallette FM. Induction of a caspase-3-like activity by calcium in normal cytosolic extracts triggers nuclear apoptosis in a cell-free system. Journal of Biological Chemistry. 1998;273:17559–17564. doi: 10.1074/jbc.273.28.17559. [DOI] [PubMed] [Google Scholar]
- Koch GLE. The endoplasmic reticulum and calcium storage. Bioessays. 1990;12:527–531. doi: 10.1002/bies.950121105. [DOI] [PubMed] [Google Scholar]
- Kohn EC, Reed E, Sarosy G, Christian M, Link CJ, Cole K, Figg WD, Davis PA, Jaco Goldspiel B, Liotta LA. Clinical investigation of a cyanotic calcium influx inhibitor in patients with refractory cancers. Cancer Research. 1996;56:569–573. [PubMed] [Google Scholar]
- Krause KH, Michalak M. Calreticulin. Cell. 1997;88:439–443. doi: 10.1016/s0092-8674(00)81884-x. [DOI] [PubMed] [Google Scholar]
- Kuo TH, Kim HRC, Zhu L, Yu Y, Lin HM, Tsang W. Modulation of endoplasmic reticulum calcium pump by bcl-2. Oncogen. 1998;17:1903–1910. doi: 10.1038/sj.onc.1202110. [DOI] [PubMed] [Google Scholar]
- Kuznetsov G, Brostrom MA, Brostrom CO. Role of endoplasmic reticular calcium in oligosaccharide processing of alpha 1-antitrypsin. Journal of Biological Chemistry. 1992;268:2001–2008. [PubMed] [Google Scholar]
- Lückhoff A, Clapham DE. Calcium channels activated by depletion of internal calcium stores in A431 cells. Biophysical Journal. 1994;67:177–182. doi: 10.1016/S0006-3495(94)80467-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConkey DJ, Nicotera P, Hartzell P, Belloms G, Wyllie AM, Orremius S. Glucocorticoids activate a suicide process in thymocytes through an elevation of cytosolic Ca2+ concentration. Archives of Biochemistry and Biophysics. 1989;269:365–370. doi: 10.1016/0003-9861(89)90119-7. [DOI] [PubMed] [Google Scholar]
- McConkey DJ, Orrenius S. The role of calcium in the regulation of apoptosis. Biochemical and Biophysical Research Communications. 1997;239:357–366. doi: 10.1006/bbrc.1997.7409. [DOI] [PubMed] [Google Scholar]
- Marin MC, Fernandez A, Bick RJ, Brisbay S, Buja LM, Snuggs M, McConkey DJ, Von Eschenbach AC, Keating MJ, McDonnell TJ. Apoptosis suppression by bcl-2 is correlated with the regulation of nuclear and cytosolic Ca2+ Oncogen. 1996;12:2259–2266. [PubMed] [Google Scholar]
- Martikainen P, Kyprianou N, Tucker RW, Isaacs JT. Programmed death of nonproliferating androgen independent prostatic cancer cells. Cancer Research. 1991;51:4693–4700. [PubMed] [Google Scholar]
- Mason MJ, Garcia-Rodriguez C, Grinstein S. Coupling between intracellular Ca2+ stores and the Ca2+ permeability of the plasma membrane. Comparison of the effects of thapsigargin, 2,5-di-(tert-butyl)-1,4-hydroquinone, and cyclopiazonic acid in rat thymic lymphocytes. Journal of Biological Chemistry. 1991;266:20856–20862. [PubMed] [Google Scholar]
- Mery L, Mesaeli N, Michalak M, Opas M, Lew DP, Krause KH. Overexpression of calreticulin increases intracellular Ca2+ storage and decreases store-operated Ca2+ influx. Journal of Biological Chemistry. 1996;271:9332–9339. doi: 10.1074/jbc.271.16.9332. [DOI] [PubMed] [Google Scholar]
- Montironi R, Magi-Galluzzi C, Muzzunigro G, Prete E, Polito M, Fabris G. Effects of combination endocrine treatment on normal prostate, prostatic intraepithelial neoplasia, and prostatic adenocarcinoma. Journal of Clinical Pathology. 1994;47:906–913. doi: 10.1136/jcp.47.10.906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Activation of store-operated calcium influx at resting InsP3 levels by sensitization of the InsP3 receptor in rat basophilic leukaemia cells. The Journal of Physiology. 1995;489:377–382. doi: 10.1113/jphysiol.1995.sp021058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Regulation of store-operated calcium currents in mast cells. Organellar Ion Channels and Transporters. 1996;51:231–239. [PubMed] [Google Scholar]
- Parekh AB, Penner R. Store depletion and calcium influx. Physiological Reviews. 1997;77:901–929. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Parker SL, Tong T, Balden S, Wingo PA. Cancer statistics. CA Cancer Journal for Clinicians. 1997;47:5–27. doi: 10.3322/canjclin.47.1.5. [DOI] [PubMed] [Google Scholar]
- Premack BA, McDonald TV, Gardner P. Activation of Ca2+ current in Jurkat T cells following the depletion of Ca2+ stores by microsomal Ca2+-ATPase inhibitors. Journal of Immunology. 1994;152:5226–5240. [PubMed] [Google Scholar]
- Prevarskaya N, Skryma R, Vacher P, Daniel N, Djiane J, Dufy B. Rôle of tyrosine phosphorylation in potassium channel activation. Journal of Biological Chemistry. 1995;270:24292–24299. doi: 10.1074/jbc.270.41.24292. [DOI] [PubMed] [Google Scholar]
- Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Putney JW., Jr Capacitative calcium entry revisited. Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- Raffo AJ, Perlman H, Chen MW, Day ML, Streitman JS, Buttyan R. Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Research. 1995;55:4438–4445. [PubMed] [Google Scholar]
- Sambrook JF. The involvement of calcium in transport of secretary proteins from the endoplasmic reticulum. Cell. 1990;61:197–199. doi: 10.1016/0092-8674(90)90798-j. [DOI] [PubMed] [Google Scholar]
- Santella L. The role of calcium in the cell cycle: facts and hypotheses. Biochemical and Biophysical Research Communications. 1998;244:317–324. doi: 10.1006/bbrc.1998.8086. [DOI] [PubMed] [Google Scholar]
- Schlegel W, Mollard P, Demaurex N, Theler JM, Chiavaroli C, Guérineau N, Vacher P, Mayr G, Krause KH, Wollheim CB, Lew PD. Calcium signalling: comparison of the role of Ca2+ influx in excitable endocrine and non-excitable myeloid cells. Advances in Second Messenger and Phosphoprotein Research. 1993;28:142–152. [PubMed] [Google Scholar]
- Shirahama T, Sakakura C, Sweeney EA, Ozawa M, Takemoto M, Nishiyama K, Ohi Y, Igarashi Y. Sphingosine induces apoptosis in androgen-independent human prostatic carcinoma DU-145 cells by suppression of bcl-X(L) gene expression. FEBS Letters. 1997;407:97–100. doi: 10.1016/s0014-5793(97)00304-9. [DOI] [PubMed] [Google Scholar]
- Skryma R, Prevarskaya N, Vacher P, Dufy B. Voltage-dependent ionic conductances in Chinese hamster ovary cells. American Journal of Physiology. 1994;267:544–553. doi: 10.1152/ajpcell.1994.267.2.C544. [DOI] [PubMed] [Google Scholar]
- Skryma RN, Prevarskaya NB, Dufy-Barbe L, Odessa MF, Audin J, Dufy B. Potassium conductance in the androgen-sensitive prostate cancer cell line, LNCaP: involvement in cell proliferation. The Prostate. 1997;33:112–122. doi: 10.1002/(sici)1097-0045(19971001)33:2<112::aid-pros5>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Skryma RN, Van Coppenolle F, Dufy-Barbe L, Dufy B, Prevarskaya N. Characterization of Ca2+-inhibited potassium channels in the LNCaP human prostate cancer cell line. Receptors and Channels. 1999;6:241–253. [PubMed] [Google Scholar]
- Spielberg H, June CH, Blair OC, Nystrom-Rosander C, Cereb N, Deeg HJ. UV irradiation of lymphocytes triggers an increase in intracellular Ca2+ and prevents lectin-stimulated Ca2+ mobilization: evidence for UV- and nifedipine-sensitive Ca2+ channels. Experimental Hematology. 1991;19:742–748. [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin a tumor promoter discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proceedings of the National Academy of Sciences of the USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaca L, Kunze DL. Depletion of intracellular Ca2+ stores activates a Ca2+-selective channel in vascular endothelium. American Journal of Physiology. 1995;269:C733–738. doi: 10.1152/ajpcell.1994.267.4.C920. [DOI] [PubMed] [Google Scholar]
- Wang H-G, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- Wei H, Wei W, Bredesen DE, Perry DC. Bcl-2 protects against apoptosis in neuronal cell line caused by thapsigargin-induced depletion of intracellular calcium stores. Journal of Neurochemistry. 1998;70:2305–2314. doi: 10.1046/j.1471-4159.1998.70062305.x. [DOI] [PubMed] [Google Scholar]
- Woolf SH. Screening for prostate cancer with prostate specific antigen. An examination of the evidence. New England Journal of Medicine. 1995;333:1401–1405. doi: 10.1056/NEJM199511233332107. [DOI] [PubMed] [Google Scholar]
- Zhu N, Wang Z. Calreticulin expression is associated with androgen regulation of the sensitivity to calcium ionophore-induced apoptosis in LNCaP prostate cancer cells. Cancer Research. 1999;59:1896–1902. [PubMed] [Google Scholar]
- Zhu N, Pewitt EB, Cai X, Cohn EB, Lang S, Chen R, Wangs Z. Calreticulin: an intracellular Ca2+-binding protein abundantly expressed and regulated by androgen in prostatic epithelial cells. Endocrinology. 1998;139:4337–4344. doi: 10.1210/endo.139.10.6242. [DOI] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proceedings of the National Academy of Sciences of the USA. 1993;90:6295–6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]