

Abstract
In urethane-anaesthetized rats, we assessed the protective effects of glucocorticoids against heatstroke-induced arterial hypotension and ischaemic neuronal damage.
Heatstroke was induced by exposing the animals to an ambient temperature of 42°C. The time at which both the mean arterial pressure (MAP) and local cerebral blood flow (CBF) in the striatum decreased from their peak levels was taken as the onset of heatstroke. Control rats were exposed to a temperature of 24°C.
The values of MAP and CBF after heatstroke onset were all significantly lower than those in control rats. However, the neuronal damage score in the striatum and serum levels of interleukin-1β (IL-1β) were greater.
Systemic pretreatment or treatment with an exogenous glucocorticoid, dexamethasone (4 mg or 6 mg kg−1, i.v.), reduced the heatstroke-induced arterial hypotension, serum IL-1β levels, cerebral ischaemia and neuronal damage, and resulted in prolongation of the time to death (TTD; the interval between the onset of heat stress and cardiac arrest).
Following bilateral adrenalectomy, MAP, CBF and TTD values were found to be significantly lower in the adrenalectomized (ADX) rats than in the sham-ADX rats after heat exposure. These changes were attenuated by dexamethasone.
The data support the argument that glucocorticoids reduce the plasma IL-1β concentration and may provide the neuroprotective effects observed in rat heatstroke.
A clinical diagnosis of heatstroke is strongly suggested when hyperthermia is associated with neurological abnormalities after exposure to high ambient temperature (Austin & Berry, 1956; Hart et al. 1981; Bouchama et al. 1991a; Simon, 1993). There is evidence that cerebral ischaemia (due to arterial hypotension and intracranial hypertension), rather than hyperthermia, is the main reason for the onset of heatstroke (Bouchama et al. 1991a; Lin & Lin, 1992; Lin, 1997). The morbidity and mortality observed in heatstroke may both be related to endotoxaemia and release of interleukin 1 (IL-1) (Bouchama et al. 1991b, 1993). The cerebral ischaemia associated with heatstroke can be attenuated by pretreatment of animals with an IL-1 receptor antagonist (Kao et al. 1994; Lin et al. 1995).
High doses of glucocorticoids have been shown to be of benefit in the treatment of spinal cord injury and experimental cerebral ischaemia (Hall, 1992; Behrmann et al. 1994; De Courten-Myers et al. 1994). This raises the possibility that glucocorticoids may protect against heatstroke-induced cerebral ischaemia and neuronal damage in rats via a decrease in plasma IL-1 levels.
To test this hypothesis we recorded the changes induced in mean arterial pressure (MAP), local cerebral blood flow (CBF), neuronal damage score (NDS) and time to death (TTD; interval between the onset of heat stress and cardiac arrest) following heatstroke in non-adrenalectomized (ADX) rats and in ADX and sham-ADX rats. We also measured changes evoked by heatstroke in the plasma concentration of IL-1β and we tested the effects of an exogenous glucocorticoid, dexamethasone, on the heatstroke-induced cardiovascular, neuropathological and IL-1β responses. Our results suggest that glucocorticoids reduce the IL-1β concentration in plasma, resulting in neuroprotective effects in rat heatstroke.
Methods
Experimental animals
Adult Sprague-Dawley rats (285 ± 18 g) were obtained from the Animal Resource Centre of the National Yang-Ming University (Taipei, Taiwan). The animals were housed individually at a an ambient temperature (Ta) of 22 ± 1°C, with a 12 h light-dark cycle. Pelleted rat chow and tap water were available ad libitum.
All experiments were approved by the Animal Research Committee of the Yang-Ming University School of Medicine. Adequate anaesthesia was maintained to abolish the corneal reflex and pain reflexes induced by tail pinch throughout the course of all experiments (about 8 h) following a single dose of urethane (1.4 g (kg body wt)−1i.p.). At the end of the experiments, control rats and any rats that had survived heatstroke were killed with an overdose of urethane.
Surgery and physiological parameter monitoring
The right femoral artery and vein of rats were cannulated with polyethylene tubing (PE50), under urethane anaesthesia, for blood pressure monitoring, blood sampling (for IL-1β assay) and drug administration. The animals were positioned in a stereotaxic apparatus (Kopf model 1460) to allow insertion of probes for measurement of CBF. Physiological monitoring included colonic temperature (Tco), MAP, heart rate (HR) and CBF values in the corpus striatum. Colon temperature was monitored continuously by a thermocouple.
Control group
Normothermic control rats were exposed to a Ta of 24°C for at least 90 min to reach thermal equilibrium. They were injected with 0.9% saline i.v. (1.0 ml (kg body wt)−1) 0 min before the start of the experiments. Their Tco was maintained at about 36°C using an electric thermal mat before the start of the experiments.
Heatstroke groups
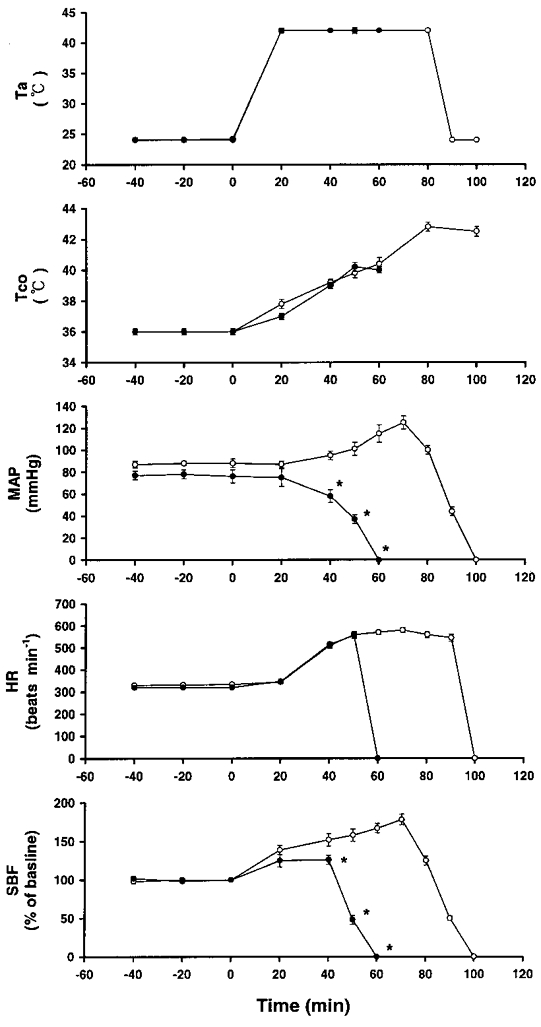
Heatstroke was induced by exposing the animals to a Ta of 42°C (with a relative humidity of 60 % in a temperature-controlled chamber). The time at which both MAP and local CBF in the striatum began to decrease from their peak levels was taken as the onset of heatstroke, as shown in Fig. 1.
Figure 1. Effects of a high ambient temperature (Ta= 42°C) on colonic temperature (Tco), mean arterial pressure (MAP), heart rate (HR) and striatal blood flow (SBF).
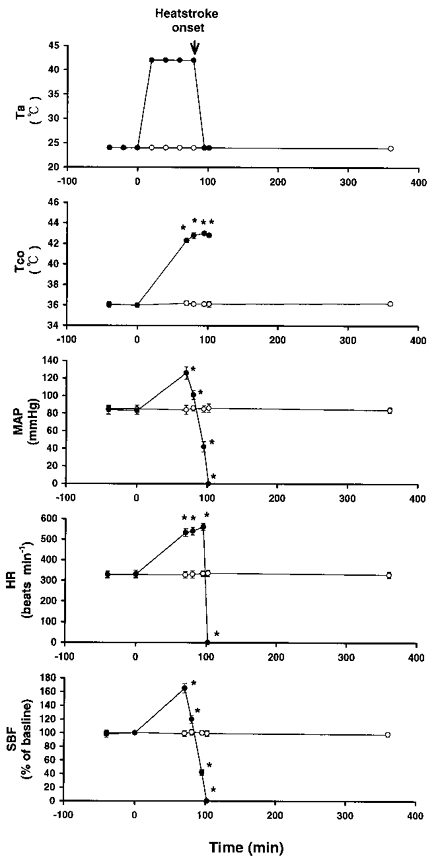
•, values at a Ta of 42°C in 10 rats. Another 8 rats exposed to a Ta of 24°C served as a control (○). The time to death (TTD) value for rats exposed to 42°C was 102 ± 5 min. Points represent means ±s.e.m. * P < 0.05, significantly different from control values (Ta= 24°C group); Student's t test.
Animals were randomly assigned to one of the following three major groups. One group of rats were injected with 0.9 % saline i.v. (1.0 ml (kg body wt)−1) at 0 min before the start of heat exposure (saline pretreated) or immediately after (0 min) the onset of heatstroke (saline treated). The second group were injected with 4 or 6 mg ml−1 (kg body wt)−1 dexamethasone i.v. at 0 min before the start of heat exposure (dexamethasone pretreated) or at 0 min immediately after the onset of heatstroke (dexamethasone treated). The third group were adrenalectomized or sham-operated (see below). These were injected with 4 or 6 mg ml−1 (kg body wt)−1 dexamethasone i.v. or saline (control) at 0 min before the start of heat exposure.
Adrenalectomy
Bilateral adrenalectomy was carried out under general anaesthesia (sodium pentobarbitone, 50 mg kg−1, i.p.). The skin was opened in the dorsal mid-line and the adrenal glands, with some surrounding tissue, were removed through incisions in both left and right flanks. For the sham operation, similar incisions were made and the adrenal gland was located but not removed. At least 10 days were allowed to elapse before experimentation began. During this interval, the rats were allowed free access to 5 % dextrose with 0.9 % saline. At the end of the experiment any rats that had survived heatstroke were killed with an overdose of urethane. Blood was taken from all adrenalectomized and sham-operated rats via cardiac puncture. The plasma from these samples was assayed for the presence of corticosterone (see below). Rats which had very low levels of corticosterone (< 20 ng ml−1) and showed no regeneration of the adrenal glands qualified as adrenalectomized (ADX) rats.
Cerebral blood flow monitoring
Local CBF in the corpus striatum was monitored with a Laserflo BPM2 laser Doppler flowmeter (Vasametics, St Paul, NM, USA). A 24 gauge stainless-steel needle probe (diameter, 0.58 mm; length, 40 mm) was inserted into the right corpus striatum using coordinates: A, interaural 9.7 mm; L, 2.0 mm from the mid-line; and H, 4.5 mm from the top of the skull (Paxinos & Watson, 1982). A representative illustration showing the location of the needle probe placement in the corpus striatum of rat brain is shown in Fig. 2.
Figure 2.
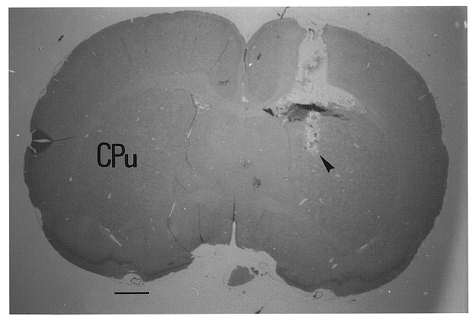
Illustration of the placement of the tip of a needle probe in the caudate-putamen complex (CPu) of rat brain, as indicated by the arrowhead. Scale bar, 1 mm.
In separate experiments, an autoradiographic diffusible tracer technique was used for measuring local CBF (Sakurada et al. 1978). Briefly, at 14 min after heatstroke onset, and at the equivalent time after the injection of saline for the normothermic control rats, approximately 50 μCi of iodo[14C]antipyrine in 1 ml of normal saline was infused at a constant rate through the femoral venous catheter for 1 min, during which time arterial samples were collected on filter paper discs for assay of arterial concentration. At exactly 1 min (i.e. 15 min after the onset of heatstroke), the animal was decapitated, and the brain was removed, frozen, and the 14C concentration assayed by autoradiography. Autoradiographs of sections of rat brain and calibrated plastic 14C standards were used for the determination of the tissue concentration of 14C by densitometry.
Assay for serum interleukin-1β or corticosterone
In separate experiments, blood samples were taken at 0, 40 or 95 min after heat exposure for determination of IL-1β levels. Blood samples were allowed to clot for 2 h at room temperature or overnight at 2–8°C before being centrifuged for 20 min at 2000 g. The serum was quickly removed and assayed immediately for IL-1β or corticosterone. The Quantikine M rat IL-1β immunoassay kit (R&D Systems, Minneapolis, MN, USA) was used for measuring the levels of active rat IL-1β present in serum. This assay employs the quantitative sandwich enzyme immunoassay technique. The assay was based on the competition between rat IL-1β in the test sample and radiolabelled recombinant rat IL-1β (tracer) for binding sites on a specific IL-1β primary antibody. A microplate was pre-coated with an affinity-purified antibody specific for rat IL-1β. Standards, controls and samples were pipetted into the wells and any rat IL-1β present was bound by the immobilized antibody. After washing away any unbound substances, an enzyme-linked polyclonal rat IL-1β antibody was added to the wells. Following a wash to remove any unbound antibody-enzyme reagents, a substrate solution was added to the wells. The enzyme reaction yielded a blue product that turned yellow when the stop solution was added. The intensity of the colour measured is proportional to the amount of rat IL-1β bound in the initial step. The sample values were then read off the standard curve. The minimum detectable dose of rat IL-1β was typically found to be less than 5 pg ml−1. Serum corticosterone concentrations were determined by direct radioimmunoassay (Immu Chem Double Antibody Corticosterone RIA kit, ICN Biomedical Inc., CA, USA).
Neuronal damage score
At the end of the experiment, the brain was removed, fixed in 10 % neutral buffered formalin and embedded in paraffin blocks. Serial (10 μm) sections through the striatum were stained with haematoxylin and eosin for microscopic evaluation. The extent of striatal neuronal damage was scored on a scale of 0–3, modified from the grading system of Pulsinelli et al. (1982), in which 0 is normal, 1 indicates that ∼30 % of the neurons are damaged, 2 that ∼60 % of the neurons are damaged, and 3 that 100 % of the neurons are damaged. Each hemisphere was evaluated independently without the examiner knowing the experimental conditions. Only those areas not invaded by probes were assessed. Our previous results (Kao et al. 1994; Lin et al. 1995) have shown that the striatal, hypothalamic and cortical neurons are susceptible to cerebral ischaemia following heatstroke. However, in the present study, only the striatal regions were chosen for histological examination of neuronal damage.
Statistical analysis
Data are means ±s.e.m. Repeated-measures analysis of variance was used for factorial experiments, whereas Duncan's multiple-range test was used for post hoc multiple comparisons among means. Student's t test was used when only two groups were compared. The criteria for statistical significance was set at P < 0.05.
Results
Figure 1 shows the effects of a high Ta (42°C) on Tco, MAP, HR, striatal blood flow (SBF) and TTD in 10 rats. Another 8 rats maintained at a Ta of 24°C served as controls. Our experiments were terminated 450 min after the start of heat exposure. The values of MAP and SBF after the onset of heatstroke (as indicated by the arrow) were significantly lower than those of the controls. However, the values of Tco and HR were significantly greater. The latency of the onset of heatstroke and the TTD were 80 ± 2 and 100 ± 3 min, respectively. Figure 3 shows the effects of a high Ta (42°C) on Tco, MAP, HR, SBF and TTD in non-adrenalectomized rats that were injected with 0.9 % saline and in others that were injected with dexamethasone (4 or 6 mg kg−1) immediately after the onset of heatstroke. Compared with saline-treated rats, in the heatstroke rats that received dexamethasone, the values of MAP, SBF and TTD after the onset of heatstroke were significantly greater. Figure 4 shows the effects of a high Ta (42°C) on Tco, MAP, HR, SBF and TTD in rats injected with 0.9 % saline and in rats injected with dexamethasone at 0 min before the start of heat exposure. Again, it was found that, in the heatstroke rats injected with dexamethasone, the values of MAP, SBF and TTD following the onset of heatstroke were significantly greater than those of the saline-pretreated rats. The values of Tco and HR obtained after the onset of heatstroke were both similar (not statistically different) in the saline- and dexamethasone-pretreated groups.
Figure 3. Effects of a high ambient temperature (Ta= 42°C) on Tco, MAP, HR and SBF in rats injected with saline or dexamethasone after the onset of heatstroke.
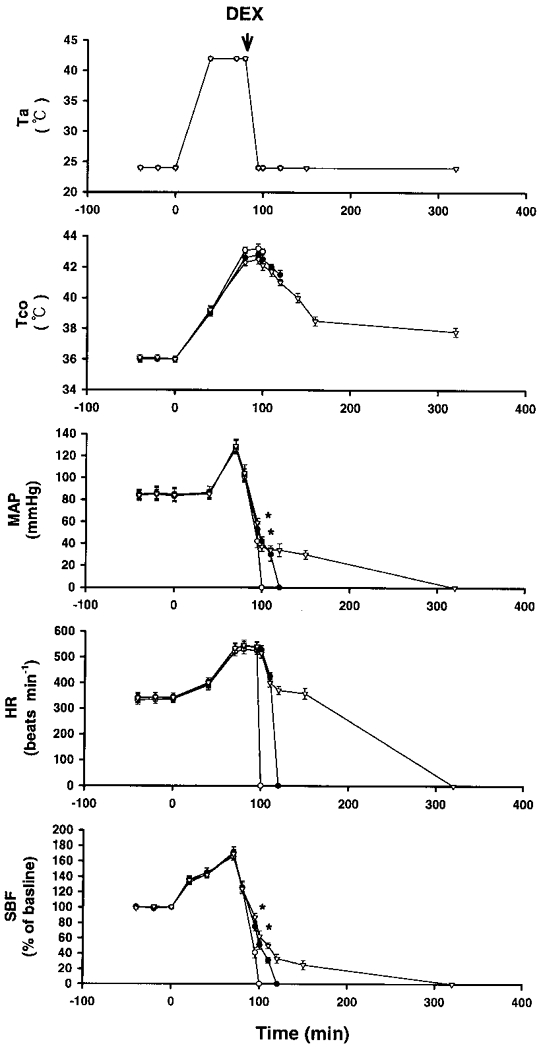
Points represent means ±s.e.m. (10 rats). ○, rats injected with 0.9 % saline; •, rats injected with 4 mg kg−1 dexamethasone (DEX); ▿, rats injected with 6 mg kg−1 dexamethasone. Dexamethasone was injected immediately after the onset of heatstroke, as indicated by the arrow. The TTD values for rats treated with 0.9 % saline, 4 mg kg−1 dexamethasone and 6 mg kg−1 dexamethasone were 100 ± 4, 122 ± 3 and 321 ± 5 min, respectively. The TTD values for the dexamethasone-treated groups were significantly different from control (saline group; P < 0.05). * P < 0.05, significantly different from control (saline group); ANOVA.
Figure 4. Effects of a high ambient temperature (Ta= 42 °C) on Tco, MAP, HR and SBF in rats injected with saline or dexamethasone before heat exposure.
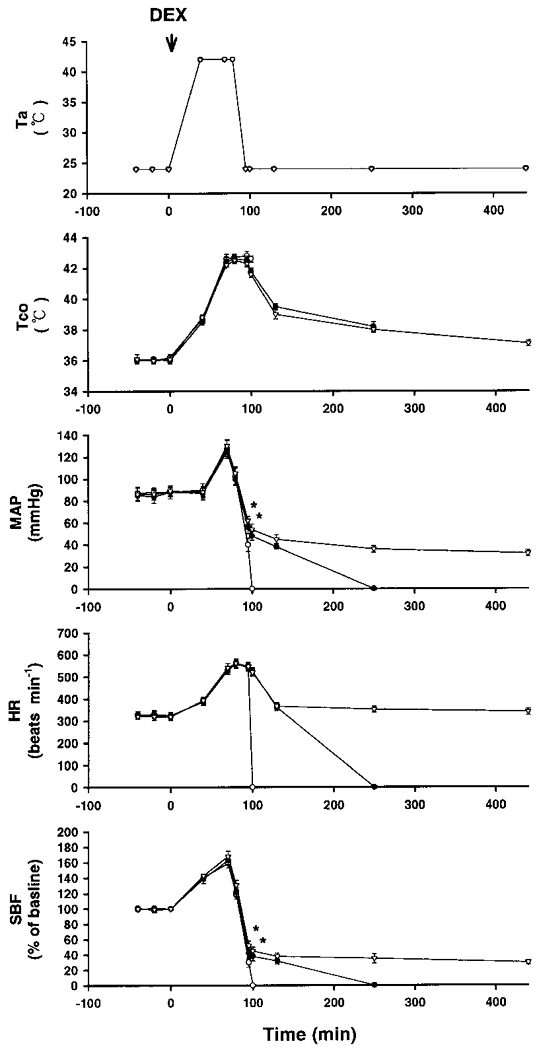
Points represent means ±s.e.m. (10 rats). ○, rats injected with 0.9 % saline; •, rats injected with 4 mg kg−1 dexamethasone (DEX); ▿, rats injected with 6 mg kg−1 dexamethasone. Dexamethasone was injected at ‘0’ min, before the start of heat exposure (arrow). The TTD values for rats pretreated with 0.9 % saline, 4 mg kg−1 dexamethasone and 6 mg kg−1 dexamethasone were 101 ± 3, 250 ± 9 and > 450 min, respectively. TTD values for the dexamethasone-pretreated groups were significantly different from control (P < 0.05). * P < 0.05, significantly different from control (saline group); ANOVA.
In separate studies, absolute CBF and the NDS were estimated using the autoradiography diffusable tracer technique and histological techniques, respectively. As shown in Table 1, compared with normothermic, control rats, the rats without dexamethasone treatment had a high NDS value accompanied by a lower value of striatal CBF 15 min after the onset of heatstroke. However, heatstroke-induced neuronal injury and cerebral ischaemia were attenuated by dexamethasone treatment either 0 min before the start of heat exposure or 80 min after the start of heat exposure (or immediately after the onset of heatstroke). Figure 5 shows that the heatstroke-induced cell body shrinkage, pyknosis of the nucleus, and loss of Nissl substance in the striatum (B) were attenuated after dexamethasone treatment (C).
Table 1.
Effects of heat exposure (HE) on striatal cerebral blood flow (CBF) and neuronal damage score
| Treatment | CBF (ml (100 g)−1 min−1) | Neuronal damage score (0–3) |
|---|---|---|
| Normothermic control rats pretreated with 0.9% saline | 161 ± 12 | 0 |
| Saline-pretreated rats 0 min before the start of HE0 | 48 ± 7 * | 2.10 ± 0.05 * |
| Dexamethasone (6 mg kg−1, i.v.)-pretreated rats 0 min before the start of HE | 147 ± 9 *† | 0.30 ± 0.05 *† |
| Salinetreated rats 80 min after the start of HE | 44 ± 3 * | 2.00 ± 0.05 * |
| Dexamethasone (6 mg kg−1, i.v.)-treated rats 80 min after the start of HE | 115 ± 11 *† | 0.70 ± 0.05 *† |
Values are means ±s.e.m.of 10 rats per group. For the determination of CBF (autoradiographic diffusible tracer technique) and neuronal damage score animals were killed 15 min after the onset of heatstroke or at the equivalent time after injection of saline for the normothermic controls.
P < 0.05, significantly different from corresponding values (normothermia, control rats); ANOVA.
P < 0.05, significantly different from corresponding values (salinepretreated or salinetreated rats); ANOVA.
Figure 5. Histological examination of neuronal damage.
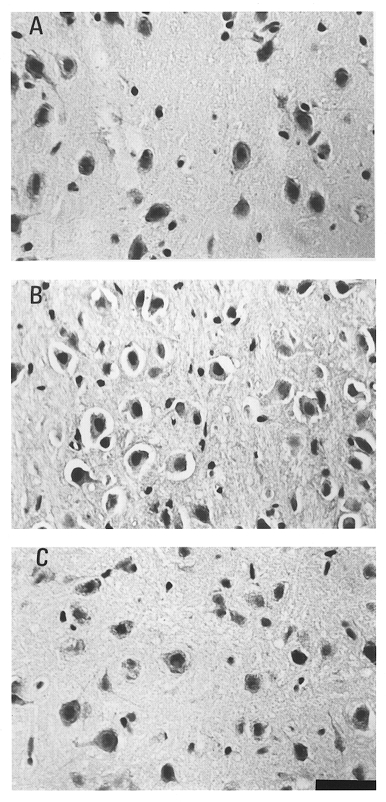
A, photomicrographs of the striatum of a control normothermic rat injected with 0.9% saline. B, striatum of a heatstroke rat injected with 0.9% saline at 0 min before the onset of heat exposure. C, striatum of a heatstroke rat injected with 6 mg kg−1 dexamethasone at 0 min before the onset of heat exposure. Fifteen minutes after the onset of heatstroke, the striatum of the rat not injected with dexamethasone showed cell shrinkage, pyknosis of the nucleus and loss of Nissl substance (B). However, with dexamethasone injection, neuroprotection was induced, as shown in C. Scale bar, 50 μm.
In another series of experiments, we assessed the effects of heat exposure on plasma IL-1β levels in saline-pretreated rats and compared them with the effects in dexamethasone-pretreated rats. As shown in Table 2, the evoked increase in IL-1β levels in serum 15 min after the onset of heatstroke was significantly attenuated by pretreatment of intact rats with dexamethasone at ‘0’ min before the start of heat exposure (42°C).
Table 2.
Effects of heat exposure (HE; 42 °C) on Tco, MAP, striatal CBF and serum IL-1βlevels in saline-and dexamethasone-pretreated rats
| Tco(°C) | MAP (mmHg) | CBF (% of baseline) | Serum [IL-1β] (pg ml−1) | |
|---|---|---|---|---|
| Salinepretreated rats | ||||
| 0 min after HE | 36.0 ± 0.2 | 85 ± 4 | 100 | 250 ± 17 |
| 40 min after HE | 39.0 ± 0.3* | 86 ± 5 | 148 ± 8* | 452 ± 22* |
| 95 min after HE (or 15 min after heatstroke onset) | 43.0 ± 0.4* | 38 ± 3* | 36 ± 4* | 1250 ± 61* |
| Dexamethasonepretreated rats | ||||
| 0 min after HE | 36.1 ± 0.2 | 83 ± 3 | 100 | 252 ± 15 |
| 40 min after HE | 38.5 ± 0.2* | 88 ± 4 | 150 ± 7* | 456 ± 18* |
| 95 min after HE (or 15 min after heatstroke onset) | 42.6 ± 0.2* | 52 ± 5*† | 50 ± 6*† | 487 ± 56*† |
Values are means ±s.e.m.of 8 rats per group. An i.v. dose of 4 mg kg−1 dexamethasone, or 0.9% saline, was administered at [0] min before the start of heat exposure.
P < 0.05, significantly different from corresponding control values (0 min); ANOVA.
P < 0.05, significantly different from corresponding control values (saline-treated); ANOVA.
Figure 6 shows the effects of a high Ta (42°C) on Tco, MAP, HR, SBF and TTD in sham-ADX and ADX rats. The values of MAP, SBF and TTD in ADX rats after heat stress were significantly lower than those in sham-ADX rats. However, the values of both Tco and HR were indistinguishable between the sham-ADX group and ADX group at 50 min after the start of heat stress.
Figure 6. Effects of a high ambient temperature (Ta= 42°C) on Tco, MAP, HR and SBF in ADX and sham-ADX rats.
Points represent means ±s.e.m.○, 8 sham-operated rats; •, 8 adrenalectomized rats. The TTD values for sham-operated and adrenalectomized rats were 100 ± 4 and 62 ± 3 min, respectively. These values were significantly different (P < 0.05). * P < 0.05, significantly different from control values (sham-operated group); Student's t test.
Table 3 summarizes the effects of a high Ta (42°C) on Tco, MAP, striatal CBF and TTD in ADX rats pretreated with 0.9 % saline or dexamethasone. In the ADX rats injected with dexamethasone, the values of MAP and striatal CBF at 40–60 min after the start of heat exposure were significantly greater than those of ADX rats injected with 0.09% saline. In addition, the TTD values of the ADX rats injected with dexamethasone were also significantly greater than those of the ADX rats injected with 0.9 % saline.
Table 3.
Effects of heat exposure (HE; 42 °C) on Tco, MAP, striatal CBF and time to death (TTD) in ADX rats injected with 0.9% saline or dexamethasone at ‘0’ min before the start of heat exposure.
| Tco(°C) | MAP(mmHg) | CBF (% of base line) | TTD(min) | |
|---|---|---|---|---|
| Salinepretreated rats | — | — | — | 62 ± 2 |
| 0 min after HE | 36.0 ± 0.2 | 78 ± 2 | 100 | — |
| 40 min after HE | 39.4 ± 0.3 | 58 ± 5 | 106 ± 7 | — |
| 60 min after HE | 41.6 ± 0.3 | 21 ± 3 | 30 ± 4 | — |
| Dexamethasone (4 mg kg−1)-pretreated rats | — | — | — | 91 ± 4* |
| 0 min after HE | 36.0 ± 0.3 | 80 ± 3 | 100 | — |
| 40 min after HE | 39.1 ± 0.3 | 78 ± 2* | 129 ± 4* | — |
| 60 min after HE | 41.8 ± 0.2 | 94 ± 4* | 115 ± 3* | — |
| Dexamethasone (6 mg kg−1)-pretreated rats | — | — | — | 122 ± 6* |
| 0 min after HE | 36.1 ± 0.3 | 79 ± 3 | 100 | — |
| 40 min after HE | 38.9 ± 0.2 | 96 ± 4* | 123 ± 3* | — |
| 60 min after HE | 41.4 ± 0.3 | 103 ± 5* | 132 ± 6* | — |
Values are means ±s.e.m.of 8 rats per group.
P < 0.05, significantly different from corresponding control values (saline group); ANOVA.
Discussion
The present study has shown that heatstroke evoked arterial hypotension, cerebral ischaemia and neuronal damage, and elevated IL-1β levels in anaesthetized rats. These results are in good agreement with those of Bouchama et al. (1991b, 1993) and Lin et al. (1994, 1997), who have also shown that the concentration of circulating endotoxins and pyrogenic cytokines is elevated in heatstroke patients or animals. Increases in the concentration of these pyrogenic cytokines can cause increased production of hypothalamic arachidonic acid metabolites and thereby raise the hypothalamic set-point (Dinarello et al. 1988). Animals injected intravenously with an IL-1 receptor antagonist at the time of heatstroke onset were protected from some of the cardiovascular effects of heatstroke, such as decreased ventricular depolarization, decreased stroke volume, decreased cardiac output and arterial hypotension (Lin et al. 1994, 1997). Furthermore, the haemodynamic changes associated with heatstroke can be mimicked by IL-1β administration (Lin et al. 1997). These observations suggest that a selective decline in stroke volume resulting from increased plasma levels of IL-1β may be an important mechanism signalling arterial hypotension, cerebral ischaemia and neuronal damage in experimental heatstroke.
In the present study, systemic pretreatment or treatment with an exogenous glucocorticoid, dexamethasone, attenuated the heatstroke-induced arterial hypotension, elevated IL-1β levels in plasma and ischaemic neuronal injury, and resulted in prolongation of survival of heatstroke rats. Furthermore, after bilateral adrenalectomy, most of the syndromes associated with heatstroke were enhanced or exacerbated and, therefore, the time to death of the rats with heatstroke was reduced. This exacerbation after adrenalectomy was attenuated by dexamethasone pretreatment. These observations suggest that endogenous glucocorticoids are an important modulator of cerebrocardiovascular and cytokine responses induced in rat heatstroke. In addition, the data demonstrate that an exogenous glucocorticoid can provide neuroprotective effects in rat heatstroke. In fact, this contention is supported by many previous findings. Glucocorticoids are known to be potent inhibitors of cytokine production and to exert a protective effect against lipopolysaccharide (LPS)-induced death (Staruch & Wood, 1985; Zuckerman et al. 1989). Steroidal anti-inflammatory drugs such as dexamethasone have been shown to decrease the generation of leukotrienes and prostaglandins by inhibiting the secretion of phospholipase A2 and the release of arachidonic acid (Simpson, 1990).
The present results show that the depressor response evoked by heatstroke was enhanced in ADX rats. This exacerbated arterial hypotension in ADX rats with heatstroke may result from a lack of the normal permissive action of glucocorticoids on the cardiovascular response to catecholamines and other hormones (Granner, 1979). The finding that the depressor response to heatstroke was exacerbated after adrenalectomy may be taken to imply that adrenal hormones are important in counteracting the depressor response. Since the exacerbated depressor response to heatstroke in ADX rats was reversed by pretreatment with dexamethasone, it is likely that glucocorticoids, by their permissive action, are the adrenal hormones required for antagonizing the full expression of the heatstroke-induced depressor effect. As described previously, heatstroke increases the plasma levels of IL-1β and results in decreases in both stroke volume and arterial blood pressure in rats (Lin et al. 1997). Pretreatment of animals with glucocorticoids protects against the cardiovascular collapse elicited by LPS (Altura & Altura, 1974; Szabo et al. 1993). The present results further show that the evoked increases in the plasma levels of IL-1β and decreases in blood pressure in rat heatstroke were attenuated by dexamethasone treatment. Thus, it appears that glucocorticoids reduce the IL-1β concentration in plasma and result in attenuation of the evoked arterial hypotension or cerebral ischaemia in rat heatstroke.
The effect of adrenalectomy on the heatstroke-induced cerebrovascular dysfunction may be attributed not only to a lack of glucocorticoids but also to the removal of aldosterone, adrenaline and other hormones. Indeed, IL-1β has been shown to stimulate the secretion of the mineralocorticoid aldosterone (Berkenbosch et al. 1989; Bataillard et al. 1992; Hashimoto et al. 1993). Since aldosterone and adrenaline are well known to have pronounced effects on cerebrovascular functions, it would be essential to determine the relative roles of these hormones in the heatstroke-induced arterial hypotension and cerebral ischaemia. However, as ADX rats with free access to salt water have similar plasma osmolality to sham-ADX rats (Watanabe et al. 1995), a change in plasma osmolality is unlikely to be responsible for the cerebrovascular responses observed in the ADX rats in our study.
In agreement with the present results, recent studies (Hall, 1992; Behrmann et al. 1994; De Courten-Myers et al. 1994) have shown that glucocorticoids are of benefit in the treatment of spinal cord injury and experimental cerebral ischaemia. In addition, evidence has accumulated to indicate that glucocorticoids provide protection against hypoxic-ischaemic damage by decreasing basal metabolic energy requirements and/or increasing the availability or efficiency of use of energy substrates (Tuor, 1997), which may be responsible for the neuroprotective effect exerted by dexamethasone in rat heatstroke.
In summary, our findings show that heatstroke onset induced arterial hypotension, cerebral ischaemia and neuronal damage, increased serum levels of IL-1β and decreased the survival time in intact rats. Most of these effects were greatly attenuated in dexamethasone-treated rats. After bilateral adrenalectomy, the heatstroke-induced arterial hypotension, cerebral ischaemia and reduced survival time were significantly exacerbated. These changes could be attenuated by dexamethasone. This may suggest that glucocorticoids have two distinct actions. First, endogenous glucocorticoids may be important for the development of protective effects in heatstroke. Second, injection of exogenous glucocorticoids may reduce the heatstroke syndromes by affecting the release of IL-1β, responsible for its mediation, in the plasma.
Acknowledgments
We are grateful to Dr Paul S. K. Wang for his kind help in performing the assay of corticosterone levels in serum. This work was supported by grants from the National Science Council of the Republic of China (NSC 89-2316-B-010-014) and the Veterans' General Hospital-National Yang-Ming University joint research program (VTY 88-P5-43), Tsou's Foundation, and the Ministry of Education of the Republic of China (89-B-FA 22-1-4-02). M.-T. Lin received a Medical Research and Advancement Foundation award in Memory of Dr Chi-Shuen Tsou.
References
- Altura BM, Altura BT. Peripheral vascular actions of glucocorticoids and their relationship to protection in circulatory shock. Journal of Pharmacology and Experimental Therapeutics. 1974;190:300–315. [PubMed] [Google Scholar]
- Austin MG, Berry JW. Observations on one hundred cases of heatstroke. Journal of the American Medical Association. 1956;161:1525–1529. doi: 10.1001/jama.1956.02970160005002. [DOI] [PubMed] [Google Scholar]
- Bataillard A, Del Rey A, Klusman I, Monge-Arditi G, Besedovsky HO. Interleukin-1 stimulates aldosterone secretion: involvement of renin, ACTH, and prostaglandins. American Journal of Physiology. 1992;263:R884–888. doi: 10.1152/ajpregu.1992.263.4.R840. [DOI] [PubMed] [Google Scholar]
- Behrmann DL, Bresnahan JC, Beattie MS. Modeling of acute spinal cord injury in the rat: Neuroprotection and enhanced recovery with methylprednisolone, U-74006F and YM-14673. Experimental Neurology. 1994;126:61–75. doi: 10.1006/exnr.1994.1042. [DOI] [PubMed] [Google Scholar]
- Berkenbosch F, de Goeij DEC, Sel Rey A, Besedovsky HO. Neuroendocrine, sympathetic and metabolic responses induced by interleukin-1. Neuroendocrinology. 1989;50:570–576. doi: 10.1159/000125283. [DOI] [PubMed] [Google Scholar]
- Bouchama A, Al-Sedairy S, Siddiqui S, Shail E, Rezeig M. Elevated pyrogenic cytokines in heatstroke. Chest. 1993;104:1498–1502. doi: 10.1378/chest.104.5.1498. [DOI] [PubMed] [Google Scholar]
- Bouchama A, Cafege A, Devol EB, Labdi O, El-Assil K, Seraj M. Ineffectiveness of dantrolene sodium in the treatment of heatstroke. Critical Care Medicine. 1991a;19:176–186. doi: 10.1097/00003246-199102000-00011. [DOI] [PubMed] [Google Scholar]
- Bouchama A, Parhar RS, El-Yazigi A, Sheth K, Al-Sedairy S. Endotoxemia and release of tumor necrosis factor and interleukin-1α in acute heatstroke. Journal of Applied Physiology. 1991b;70:2640–2644. doi: 10.1152/jappl.1991.70.6.2640. [DOI] [PubMed] [Google Scholar]
- De Courten-Myers GM, Kleinholz M, Wagner KR, Xi G, Myers RE. Efficacious experimental stroke treatment with high-dose methylprednisolone. Stroke. 1994;25:487–492. doi: 10.1161/01.str.25.2.487. [DOI] [PubMed] [Google Scholar]
- Dinarello CA, Cannon JG, Wolff SM. New concepts on the pathogenesis of fever. Reviews of Infectious Diseases. 1988;10:168–189. doi: 10.1093/clinids/10.1.168. [DOI] [PubMed] [Google Scholar]
- Granner DK. The role of glucocorticoid hormones as biological amplifiers. In: Baxter JD, Rousseau GG, editors. Glucocorticoid Hormone Action. New York: Springer-Verlag; 1979. pp. 593–611. [DOI] [PubMed] [Google Scholar]
- Hall ED. The neuroprotective pharmacology of methylprednisolone. Journal of Neurosurgery. 1992;76:13–22. doi: 10.3171/jns.1992.76.1.0013. [DOI] [PubMed] [Google Scholar]
- Hart GR, Anderson RJ, Crumpler CP, Shulikin A, Reed G, Knochel JP. Epidemic classical heatstroke: Clinical characteristics and course of 28 patients. Medicine (Baltimore) 1981;61:189–197. [PubMed] [Google Scholar]
- Hashimoto K, Hirasawa R, Makino S. Comparison of the effects of intra-third ventricular administration of interleukin-1 or platelet activating factor on ACTH secretion and the sympathetic-adrenomedullary system in conscious rats. Acta Medica Okayama. 1993;47:1–6. doi: 10.18926/AMO/31608. [DOI] [PubMed] [Google Scholar]
- Kao TY, Chio CC, Lin MT. Hypothalamic dopamine release and local cerebral flow during onset of heatstroke in rats. Stroke. 1994;25:2483–2487. doi: 10.1161/01.str.25.12.2483. [DOI] [PubMed] [Google Scholar]
- Lin MT. Heatstroke-induced cerebral ischemia and neuronal damage. Involvement of cytokines and monoamines. Annals of the New York Academy of Sciences. 1997;813:572–580. doi: 10.1111/j.1749-6632.1997.tb51748.x. [DOI] [PubMed] [Google Scholar]
- Lin MT, Kao TY, Jin YT, Chen CF. Interleukin-1 receptor antagonist attenuates the heat stroke-induced neuronal damage by reducing the cerebral ischemia in rats. Brain Research Bulletin. 1995;37:595–598. doi: 10.1016/0361-9230(95)00046-h. [DOI] [PubMed] [Google Scholar]
- Lin MT, Kao TY, Su CF, Hsu SSF. Interleukin-1β production during the onset of heatstroke in rabbits. Neuroscience Letters. 1994;174:17–20. doi: 10.1016/0304-3940(94)90108-2. [DOI] [PubMed] [Google Scholar]
- Lin MT, Lin SZ. Cerebral ischemia is the main cause for the onset of heat stroke syndrome in rabbits. Experientia. 1992;48:225–227. doi: 10.1007/BF01930459. [DOI] [PubMed] [Google Scholar]
- Lin MT, Liu HH, Yang YL. Involvement of interleukin-1 receptor mechanisms in development of arterial hypotension in rat heatstroke. American Journal of Physiology. 1997;273:H2072–2077. doi: 10.1152/ajpheart.1997.273.4.H2072. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. New York: Academic Press; 1982. [Google Scholar]
- Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Annals of Neurology. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- Sakurada O, Kennedy C, Jehle J, Brown JD, Clabon GL, Sokoloff L. Measurement of local cerebral blood flow with iodo-[14C]-antipyrine. American Journal of Physiology. 1978;234:H59–66. doi: 10.1152/ajpheart.1978.234.1.H59. [DOI] [PubMed] [Google Scholar]
- Simon HB. Hyperthermia. New England Journal of Medicine. 1993;329:483–487. doi: 10.1056/NEJM199308123290708. [DOI] [PubMed] [Google Scholar]
- Simpson PJ. Arachidonic acid metabolites. In: Zelenock GB, editor. Clinical Ischemic Syndromes. St Louis, USA: CV Mosby; 1990. pp. 277–285. [Google Scholar]
- Staruch MJ, Wood DD. Reduction of serum interleukin-1-like activity after treatment with dexamethasone. Journal of Leukocyte Biology. 1985;37:193–207. doi: 10.1002/jlb.37.2.193. [DOI] [PubMed] [Google Scholar]
- Szabo C, Thiemermann C, Vane JR. Inhibition of the production of nitric oxide and vasodilator prostaglandins attenuates the cardiovascular response to bacterial endotoxin in adrenalectomized rats. Proceedings of the Royal Society B. 1993;253:233–238. doi: 10.1098/rspb.1993.0108. [DOI] [PubMed] [Google Scholar]
- Tuor UI. Glucocorticoids and the prevention of hypoxic-ischemic brain damage. Neuroscience and Biobehavioral Reviews. 1997;21:175–179. doi: 10.1016/s0149-7634(96)00007-3. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Makisumi T, Macari M, Tan N, Nakamori T, Nakamura S, Murakami N. Febrile responses induced in adrenalectomized rats by administration of interleukin-1β or prostaglandin E2. The Journal of Physiology. 1995;484:767–775. doi: 10.1113/jphysiol.1995.sp020702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerman SH, Shellhaas J, Butler LD. Differential regulation of lipopolysaccharide-induced interleukin-1 and tumor necrosis factor synthesis: Effects of endogenous glucocorticoids and the role of the pituitary adrenal axis. European Journal of Pharmacology. 1989;19:301–305. doi: 10.1002/eji.1830190213. [DOI] [PubMed] [Google Scholar]