

Abstract
Whole-cell excitatory postsynaptic currents (EPSCs) were recorded from single Purkinje cells (PCs) in rat cerebellar slices in response to alternate activation of two separate sets of parallel fibres (PF1 and PF2). Pairing the stimulation of one input (PF1) with PC depolarisation at 1 Hz for 5 min produced varied effects, including a long-term depression (LTD) of subsequent responses, a medium-term potentiation, or no change relative to baseline levels (n = 14). In all but two cases PF2 responses mirrored those in PF1, in both direction and magnitude even though this second pathway was not specifically activated during pairing.
Increasing the stimulus strength to evoke larger amplitude EPSCs (> 1000 pA) dramatically increased the proportion of cells that underwent LTD in both PF1 and PF2. LTD in both pathways was postsynaptic calcium dependent. PC depolarisation alone (n = 7) or PF1 stimulation paired with PC hyperpolarisation (n = 6) failed to induce LTD at either site.
Pairing PF1 stimulation with climbing fibre (CF) activation at 1 Hz for 5 min produced LTD in the majority of cells regardless of the strength of PF stimulation. LTD under these conditions was not, however, input specific, even at the lowest stimulus strengths.
With EPSCs greater than 1000 pA in amplitude, depression was apparent in both pathways even when the duration of PF1 pairing with depolarisation was limited to 1 min. Full expression of LTD in PF2 required stimulation of this pathway to be resumed within a distinct temporal window of conjunctive pairing with PF1. Introducing a delay of 20 min before resumption of PF2 activation preserved the input specificity of synaptic depression.
We conclude that pairing either PC depolarisation or CF activation with stimulation of a discrete set of PFs produces LTD that spreads to adjacent synapses on the same PC.
Early theories of cerebellar function proposed that learning and memory of motor skills may be encoded by changes in the strength of parallel fibre (PF) to Purkinje cell (PC) synaptic transmission (Marr, 1969; Albus, 1971). A decade later, Ito and co-workers reported a long-term depression (LTD) in the strength of PF to PC synaptic transmission following the repetitive and simultaneous stimulation of climbing fibres (CFs) and PFs (Ito & Kano, 1982; Ito et al. 1982). Cerebellar LTD has since been demonstrated in a number of different in vitro preparations. In acutely prepared cerebellar slices, LTD can be produced following PF stimulation paired with either CF stimulation (Sakurai, 1987) or PC depolarisation (Crepel & Jaillard, 1991). In cultured PCs, pairing iontophoretic application of glutamate with PC depolarisation also leads to a long-lasting form of depression of glutamate responses (Linden et al. 1991).
Experimental evidence obtained from these two, in vitro models has identified three common requirements for the induction of LTD; namely entry of calcium via voltage-dependent calcium channels (VDCCs) together with activation of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and metabotropic glutamate (mGlu)-type receptors (see Linden & Connor, 1995, for a review). Buffering increases in intracellular calcium with either EGTA or BAPTA blocks CF pairing-induced LTD in brain slices (Sakurai, 1990) and depression of glutamate responses induced in cultured PCs (Linden et al. 1991). Fast synaptic transmission at PF to PC synapses in the adult is mediated by AMPA receptors. No functional NMDA receptors are present at this developmental stage (Crepel et al. 1982). mGlu receptors, specifically the mGlu-1 subtype, have also been localised to PC dendrites (Shigemoto et al. 1994). Induction of LTD in cerebellar slices and in cultured PCs requires the activation of both glutamate receptor subtypes (Linden et al. 1991; Aiba et al. 1994; Conquet et al. 1994; Hartell, 1994b; Hemart et al. 1995). In addition to these three basic requirements, protein kinase C (PKC) activity is also necessary for LTD (Crepel & Krupa, 1988; Linden & Connor, 1991). A basic mechanism for LTD has been proposed in which coupling of G-protein-linked mGlu receptor activation to phosphoinositol hydrolysis yields the lipid-soluble messenger diacylglycerol and inositol 1,4,5-tris-phosphate. Diacylglycerol, in the presence of coincident and sufficient increases in intracellular calcium, activates PKC (Nakanishi, 1994; Oancea & Meyer, 1998) leading to a long-term reduction in the postsynaptic sensitivity of AMPA receptors.
This, or a similar associative mechanism, is attractive because it provides an explanation for the dual requirement of PF and CF synaptic inputs for LTD. PFs activate mGlu and AMPA receptors and the CF provides a generalised increase in calcium. Moreover, it provides an explanation for the apparent input specificity that has been reported both in vivo and in vitro (Ekerot & Kano, 1985; Linden, 1994; Chen & Thompson, 1995), whereby depression is confined only to those synapses or receptors that have been specifically activated during the pairing protocol. At the population level, input specificity was preserved at PF pathway separations of over 300 μm (Chen & Thompson, 1995). In cultured neurons, depression of glutamate currents can be restricted to dendritic regions within a single cell, resulting from the synapse-specific activation of PKC produced by mGlu receptor activation (Linden, 1994).
More recently, in cerebellar slices, work from this laboratory has shown that LTD induced by a raised intensity and frequency of PF stimulation alone (LTDPF) can spread to adjacent synapses on the same cell (Hartell, 1996b). The depression that emerges at these distant sites is dependent on NO production. Interestingly, LTD in cultured neurons differs from that observed in cerebellar slices in its lack of requirement for the NO-cGMP cascade (Linden & Connor, 1992; Linden et al. 1995).
In the light of these findings, we have examined the degree of synapse specificity associated with LTD induced by two more conventional protocols commonly used to induce LTD in cerebellar slices. We report that in almost all cases, synaptic depression produced by conjunctive pairing of either PC depolarisation or CF stimulation with activation of one of two distinct PF inputs to a single cell was not input specific. Neither reducing the PF stimulus intensity nor shortening the duration of the pairing protocol revealed input-specific synaptic depression. We did find, however, that there was a tendency towards input specificity when the activation of control PFs was not resumed immediately after the induction protocol. This evidence suggests that there is a distinct temporal window immediately following conjunctive pairing, during which expression of LTD at distant synapses is favoured providing tonic activation of PFs is resumed.
METHODS
Preparation of brain slices
Fourteen- to 21-day-old male Wistar rats were anaesthetised with halothane, decapitated and their brains rapidly removed. Parasaggital slices of approximately 200 μm thickness were prepared from the cerebellar vermis maintained in ice-cold artificial cerebrospinal fluid (ACSF) of the following composition (mm): NaCl, 118; KCl, 4.7; CaCl2.2H2O, 2.5; NaHCO3, 25; KH2PO4, 1.2; MgSO4.7H2O, 1.2; and glucose, 11. Picrotoxin was added to a final concentration of 20 μm and the ACSF was equilibrated with 95 % CO2 and 5 % O2 (pH 7.4). A single brain slice was placed in a tissue chamber, held submerged between two pieces of nylon net, and continually perfused with ACSF at a flow of 2 ml min−1. All experiments were performed at room temperature. Individual PC soma were visualised using a × 40 (0.8 NA) water-immersion lens on an Olympus upright microscope fitted with an infrared-sensitive CCD camera (Hitachi). Infrared video images were captured and stored on a personal computer for the measurement of stimulating electrode separations.
Experimental protocol
Whole-cell patch clamp recordings were made with an Axopatch 200B amplifier (Axon instruments) from individual, visually identified PC soma using pipettes filled with a standard intracellular pipette solution of the following composition (mm): potassium gluconate, 132; NaCl, 8; MgCl2.6H2O, 2; Hepes, 30; Na2ATP, 4; GTP, 0.3; and EGTA, 0.5; the pH was adjusted to 7.3 using KOH. The patch electrodes were fabricated from borosilicate glass and, when filled with standard intracellular solution, had final resistances of between 2 and 4 MΩ. In some recordings the standard intracellular recording solution was replaced with a caesium-based solution of the following composition (mm): caesium methanesulphonate, 132; tetraethylammonium (TEA)Cl, 10; NaCl, 8; MgCl2.6H2O, 2; Hepes, 30; Na2ATP, 4; GTP, 0.3; and EGTA, 0.5; the pH was adjusted to 7.3 using CsOH. Prior to seal formation, two additional, ACSF-filled glass patch electrodes were positioned midway within the molecular layer, either side of the PC soma, in order to stimulate two sets of PF bundles. The linear distance between stimulating electrodes was measured from the calibrated infrared image. In some experiments, a third ACSF-filled glass patch electrode was also positioned within the granule layer to evoke CF responses.
High-resistance seals of at least 3 GΩ were obtained with PC soma before entering the whole-cell configuration. Cells were voltage clamped at –70 mV. Capacitance compensation was applied and series resistance was compensated to at least 70 %. Access resistances were between 3 and 10 MΩ. Membrane and access resistances were monitored at 1 min intervals throughout all recordings by applying a 300 ms duration hyperpolarising voltage step. Data were discarded if access resistances changed by more than 20 % over the course of a recording or if the holding current increased substantially.
The two PF pathways were initially stimulated alternately at 0.2 Hz (200 μs duration stimulus at constant voltages ranging from 2 to 15 V) to evoke excitatory postsynaptic currents (EPSCs). The EPSCs were captured and the peak amplitudes were measured online using a Digidata 1200 series interface and pCLAMP 6 software (Axon Instruments). EPSCs were recorded for at least 10 min, but no longer than 15 min, until a stable baseline was achieved before one of three different LTD induction protocols was applied. When using the standard potassium-based intracellular solution, cells were switched to current clamp mode and held at –70 mV. Activation of one of the two inputs, termed PF1, was then paired simultaneously with either injection of a 500 ms depolarising current to induce firing of calcium spikes, or stimulation of the CF input at 1 Hz for a total duration of 5 min. The other PF pathway (PF2) was not stimulated during this period.
Stable recordings in current clamp were difficult to maintain when using the caesium-based intracellular pipette solution due to the more depolarised membrane potentials resulting from potassium channel blockade. Consequently, cells were held in voltage clamp at –70 mV and PF1 was paired simultaneously with a 500 ms depolarising step to 0 mV at 1 Hz for 5 min. Following this induction procedure, alternate stimulation of PF1 and PF2 was resumed at 0.2 Hz for the duration of the recording. Recordings were discarded if either the membrane resistance or the holding current was found to change permanently relative to the period immediately prior to commencing the induction protocol.
Fluorescence measurements
In some recordings, EGTA was replaced with 0.5 mm calcium green-1 in the intracellular solution in order to examine the spatial elevation of calcium produced by CF activation and somatic depolarisation. Calcium green was excited at 488 nm with a monochromator light source, and the fluorescence emitted at wavelengths greater than 505 nm was captured by a 10-bit cooled CCD camera (Hamamatsu C4880-81). Images were subject to 4 by 4 binning. The relative changes in intracellular calcium between defined dendritic regions of equal area were compared by normalising the change in fluorescence relative to baseline levels measured during a previous control period (δF/F). Image analysis was performed with custom-made software.
Data analysis
Peak EPSC amplitudes were averaged over 1 min intervals and expressed as percentages of the mean peak EPSC amplitude measured during the 10 min baseline period, prior to the induction protocol. Grouped data are expressed as means ± standard error of the mean (s.e.m.). The non-parametric Wilcoxon signed-rank and Mann-Whitney U tests (1- or 2-tailed as stated) were used to test for the level of statistical significance of any changes observed, with P values of less than 0.05 being considered significant.
RESULTS
LTD spreads to adjacent synapses on the same PC
We initially set out to induce LTD in one of two, separate PF pathways to a single Purkinje cell in order to examine the extent to which the resulting depression remained confined to the active PF synapses, or whether it spread to fibres that were not specifically activated during the induction procedure. We initially chose a method of LTD induction whereby one PF input, termed PF1, was activated simultaneously with a 500 ms injection of depolarising current at a rate of 1 Hz for a period of 5 min. The second input, PF2, was not directly activated during the pairing period. Our rationale for choosing this protocol was based upon an earlier report that demonstrated that LTD induced by pairing PF and CF stimulation was optimal under these conditions (Karachot et al. 1995) and because CF activation can be replaced by cell depolarisation (Crepel & Jaillard, 1991). We chose to depolarise cells in preference to activating the CF input for experimental simplicity and because it has been shown that compared to depolarisation, CF activation does not necessarily lead to a uniform increase in calcium throughout the Purkinje cell (Miyakawa et al. 1992).
In a total of 14 recordings obtained from Purkinje cells held at –70 mV in voltage clamp mode, we measured the peak amplitudes of EPSCs in response to alternate activation of PF1 and PF2 inputs at 0.2 Hz prior to and following the LTD induction protocol described above. The mean baseline peak amplitudes of PF1 and PF2 responses were 588.1 ± 33.9 and 562.7 ± 40.1 pA (n = 14), respectively. Following the pairing protocol, PF1 responses increased, decreased or displayed no overall change in amplitude with respect to baseline levels. Although the pairing protocol was confined only to PF1, these changes were mirrored in PF2 in 12 out of the 14 cases, in terms of both the direction and the magnitude of plasticity. The data were therefore grouped according to the three PF1 response outcomes described above (increase, decrease or no change). A fourth group comprised the two examples where PF2 did not mirror PF1. The means and standard errors of PF1 (▪) and PF2 responses (□) measured 30 min after the pairing protocol for each of these groups are summarised in Fig. 1A. Figure 1B shows the time course of the changes of PF1 (•) and PF2 (○) responses for the five cases in which both pathways underwent a depression of synaptic transmission. The level of depression of PF1 responses (73.0 ± 11.4 % of baseline), measured 30 min after pairing was statistically indistinguishable from that in PF2 (73.1 ± 11.1 %; P > 0.05, Wilcoxon signed-rank test, 2-tailed, n = 5). Figure 1C and D provides representative examples of PF1 and PF2 EPSCs recorded 5 min prior to and 30 min after conjunctive pairing. Figure 1E illustrates the waveform associated with pairing PF1 activation with cell depolarisation.
Figure 1. LTD spreads to adjacent synapses on the same PC.
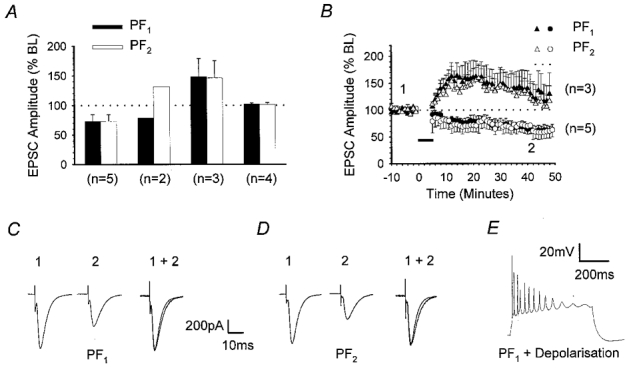
Summary of the effects of pairing one of two, independent PF pathways with PC depolarisation, at 1 Hz for 5 min. Initial EPSC amplitudes of both the paired pathway responses (PF1) and those in a second pathway that was not activated during the pairing process (PF2) ranged between 500 and 600 pA. A, bar chart summarising the mean amplitude of PF1 (▪) and PF2 (□) responses, together with standard errors, measured 30 min after pairing and expressed as percentages of the mean baseline (BL) amplitudes. Data from a total of 14 recordings were subdivided into four groups according to the direction of plasticity of PF1 and PF2 responses; from left to right, these were LTD in both pathways, LTD in PF1 only, long-term potentiation (LTP) in both pathways and no change in both pathways. B, graph displaying the mean and standard errors of PF1 (•, ▴) and PF2 (○, ▵) responses over time for the recordings illustrated in A, where both pathways underwent depression (circles, n = 5) or potentiation (triangles, n = 3) after the period of conjunctive pairing (horizontal filled bar). EPSCs of representative PF1 (C) and PF2 (D) responses that underwent depression, sampled at time points 1 and 2 in B are shown. Responses taken after pairing were normalised to the peak response amplitude prior to pairing (1 + 2). E illustrates the waveform associated with conjunctive pairing in current clamp.
In 3 of the 14 recordings, PF1 amplitude increased after completion of the induction protocol, reaching 148.1 ± 30.8 % of baseline after 30 min and this was accompanied by a similar increase in PF2 response amplitude (146.5 ± 29.1 % at 30 min; Fig. 1A). The time course of the potentiation of PF1 (▴) and PF2 responses (▵) is shown in Fig. 1B. In another four recordings, no change in either PF1 or PF2 responses was observed after 30 min, with peak amplitudes reaching 102.0 ± 2.1 and 101.3 ± 3.6 % of baseline, respectively (Fig. 1A). In the remaining two recordings, however, PF1 responses underwent LTD declining to a mean peak amplitude of 78.6 % whilst PF2 responses increased to 131.4 % of baseline levels after 30 min (n = 2; Fig. 1A).
A thorough analysis of the relative electrode separations, the waveforms associated with the pairing protocol and the kinetic profiles of PF1 and PF2 EPSCs did not reveal any indication as to the mechanism of plasticity or why two examples alone displayed input specificity. Despite the variation in the direction and incidence of plasticity produced by this particular induction protocol, these data reveal that under these conditions, plasticity, either potentiation or depression, is rarely input specific. Moreover, it is noteworthy that depression of PF2 responses never occurred in the absence of LTD in PF1, an observation that might suggest a causative relationship.
The incidence of LTD is dependent upon PF stimulus intensity
We have previously shown that raising the frequency and the intensity of PF stimulation leads to a form of LTD that is also not input specific (Hartell, 1996b). We therefore considered the possibility that the strength of PF activation used in the present model of pairing PF activation with depolarisation might also influence either the incidence of LTD or the degree of input specificity. The baseline PF1 and PF2 responses illustrated in Fig. 1 ranged between 500 and 600 pA. EPSCs with amplitudes within this range are herein described as ‘medium-sized EPSCs’. We next examined the outcome of the above pairing protocol for two further sets of data in which the baseline PF1 and PF2 stimulus intensities were adjusted to yield EPSCs with amplitudes ranging between 1000 and 1300 pA (large EPSCs) and those in which the intensities were selected to produce EPSCs between 200 and 300 pA (small EPSCs).
The mean PF1 and PF2 EPSC baseline amplitudes for the group of nine cells that comprised the large EPSC group were 1180.7 ± 52.4 and 1156.4 ± 29.1 pA, respectively. Within this group, LTD was observed in seven out of nine recordings following pairing with PF1 alone (Fig. 2A). The level of depression in PF1 and PF2 was not statistically different and reached 66.1 ± 4.3 and 64.9 ± 8.6 % of baseline levels, respectively (P > 0.05, Wilcoxon signed-rank test, 2-tailed, n = 7). In the remaining two recordings, no significant change was observed in either PF1 or PF2 responses (107.7 and 90.1 % of baseline at 30 min).
Figure 2. The incidence of LTD produced by PF pairing with depolarisation is dependent on stimulus intensity.
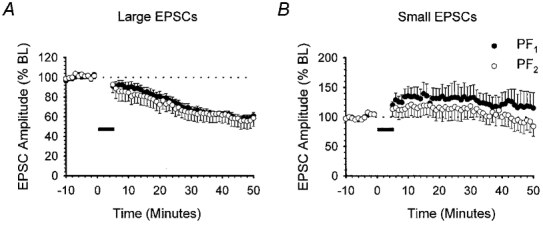
Summary of the effects of pairing PC depolarisation at 1 Hz with PF1 stimulation for 5 min when baseline PF1 (•) and PF2 (○) response amplitudes were either large (1000–1300 pA, n = 7; A) or small (200–300 pA, n = 6; B). Data are presented as in Fig. 1B.
Somewhat surprisingly, reducing the stimulus intensity to yield mean PF1 and PF2 EPSC amplitudes of 288.6 ± 12.0 and 258.9 ± 17.2 pA (n = 6), respectively, failed to produce LTD in either pathway in any of the six recordings. The overall peak amplitude of PF1 and PF2 responses after 30 min was 132.5 ± 20.9 and 118.6 ± 19.0 % of baseline, respectively (Fig. 2B).
From these results, it is apparent that the incidence of LTD produced by conjunctive depolarisation and PF stimulation at 1 Hz is critically dependent on the size of the EPSCs and hence the number of PFs stimulated during the induction procedure. Two recent investigations have both independently reported that LTD can be produced by PF stimulation alone (Hartell, 1996b;Eilers et al. 1997). At a rate of 1 Hz, PF stimulation at intensities that produce excitatory postsynaptic potentials (EPSPs) 9 mV above a holding potential of –70 mV was sufficient to cause a depolarisation sufficient to open VDCCs, leading to a localised influx of calcium and heterosynaptic LTD (Hartell, 1996b). In order to establish whether our EPSCs were sufficiently large to produce LTD on their own, we compared the peak amplitude of EPSCs measured in voltage clamp with the equivalent peak amplitude of EPSPs recorded in current clamp at –70 mV. Figure 3A illustrates the relationship between EPSC and equivalent EPSP amplitudes in 12 different cells and the data reveal that EPSCs larger than 800 pA (indicated by the dotted line) produced EPSPs with peak amplitudes of more than 9 mV. Therefore, we would expect that stimulation of PF1 at 1 Hz, at intensities yielding EPSCs greater than 800 pA, could be sufficient to induce LTD without the need for simultaneous PC depolarisation.
Figure 3. Synaptic requirements for LTD of large-amplitude responses.

A, graph comparing the peak amplitudes of EPSPs recorded in current clamp, elicited at a fixed stimulus intensity against the corresponding EPSCs recorded in voltage clamp, over the range 300 to 1300 pA. Each point represents data from a different Purkinje cell. Linear regression was applied to produce a line of best fit (r = 0.83). EPSCs with amplitudes greater than 800 pA produced EPSPs with amplitudes greater than 9 mV (indicated by the dotted line). B, when baseline PF1 and PF2 EPSC amplitudes exceeded 800 pA, stimulation of PF1 alone for 5 min at 1 Hz (horizontal filled bar) produced LTD of both PF1 (•) and PF2 (○) responses in six out of seven recordings. C, summary of the synaptic requirements for LTD of large-amplitude EPSCs. The bar chart shows the amplitude of PF1 (▪) and PF2 (□) responses 30 min after one of several different stimulus protocols; from left to right, these were PF1 and PF2 stimulation alone at 0.2 Hz (n = 7), 1 Hz stimulation of PF1 alone (n = 6), 1 Hz stimulation of PF1 combined with cell hyperpolarisation (n = 6), 1 Hz depolarisation alone (n = 6), 1 Hz PF1 stimulation paired with depolarisation (n = 7), and 1 Hz PF1 stimulation paired with depolarisation in the presence of 10 mm BAPTA in the patch pipette (n = 6). Asterisks indicate where a significant difference was apparent compared to the effect of 0.2 Hz stimulation alone (P < 0.05, Mann-Whitney U test, 1-tailed). Data are expressed as in Fig. 1A.
To test for this possibility the induction protocol was repeated using large EPSCs but without concurrent PC depolarisation. Prior to 1 Hz PF stimulation, PF1 EPSC amplitude was 1146.4 ± 31.8 pA (corresponding to a peak EPSP of 12.9 ± 1.1 mV; n = 7). In six out of seven recordings, both PF1 (•) and PF2 (○) responses decreased significantly to 70.5 ± 8.2 and 71.5 ± 12.1 % of baseline levels 30 min after 1 Hz stimulation of PF1 (Fig. 3B). In the other recording, PF1 and PF2 responses increased to 134.4 and 128.8 % of baseline levels. The overall decrease in response amplitudes produced by 1 Hz PF stimulation alone was not significantly different from the magnitude of LTD induced by the conjunctive protocol at 30 min (P > 0.05 for both test and control pathways, Mann-Whitney U test, 2-tailed).
We next examined in more detail the precise requirements for the LTD that emerged following 1 Hz PF1 stimulation at intensities that yielded EPSCs greater than 1000 pA in amplitude. The results are summarised in Fig. 3C. Stimulation of PF1 and PF2 at these intensities alternately at a rate of 0.2 Hz throughout failed to produce any significant depression (107 ± 10.8 and 96.7 ± 5.9 % of baseline values, respectively, after 30 min; n = 7). Hyperpolarisation of cells during 1 Hz PF1 stimulation significantly reduced the incidence and the overall extent of depression of PF1 and PF2 responses (97.4 ± 7.0 and 88.5 ± 9.3 %; n = 6). Inclusion of 10 mm BAPTA in the intracellular pipette solution prevented LTD in either pathway following PF1 pairing with depolarisation (106.1 ± 10.4 and 112.4 ± 11.6 %; n = 6). Injection of 500 ms pulses of depolarising current alone at 1 Hz, in the absence of PF stimulation, produced only a slight depression in both pathways to 86.7 ± 5.3 and 88.2 ± 5.0 %, respectively, at 30 min (n = 6). However, this small decrease was not significant when compared with the continuous 0.2 Hz PF stimulation data set (P > 0.05, Mann-Whitney U test, 1-tailed). The results of these experiments accord with our earlier findings (Hartell, 1996b) and lead us to two principle conclusions. First, large-amplitude PF responses alone cannot fulfil the criteria for LTD. The rate of stimulation must be increased to at least 1 Hz. The voltage and calcium dependence of LTD induced under these conditions support the view that if enough PFs are activated, VDCCs are opened. Second, the induction requirements need only be satisfied at PF1 alone for depression to occur in both pathways. This suggests that heterosynaptic LTD is dependent on the production of one or more diffusible messengers at test sites during conjunctive pairing, which then diffuse to adjacent synapses to produce depression. Furthermore, the spread of depression to adjacent synapses occurs regardless of which method is used to induce LTD, either 1 Hz PF stimulation alone or conjunctive pairing with depolarisation.
Heterosynaptic LTD does not reflect pathway overlap or a non-specific increase in potassium conductance
An alternative explanation for the apparent lack of input specificity observed, particularly at higher stimulus intensities, is that a significant number of PFs contributed to both PF1 and PF2 responses. To test for this possibility we used a modified paired-pulse stimulation protocol (Hartell, 1996b) based on the phenomenon known as paired-pulse facilitation (PPF). Figure 4 provides a representative example from a total of six similar recordings. Paired stimulation of PF2, at an interval of 30 ms, resulted in PPF of the second response (Fig. 4A1). PF1 and PF2 electrodes were next stimulated alternately at an interval of 30 ms, over a range of different PF1 stimulus intensities (0–10 V; Fig. 4A2). The intensity of PF2 stimulation was kept constant throughout. This protocol was repeated with PF1 and PF2 electrodes positioned in the molecular layer at separations of 5.6 (Fig. 4A2), 22.0, 57.5 (Fig. 4A3) and 98.8 μm. The resulting PF1 and PF2 peak amplitudes were measured and are plotted in Fig. 4B. At a separation of 5.6 μm an increase in the magnitude of the constant intensity PF2 response was observed when preceded by PF1 activation, suggesting a significant degree of PPF and hence pathway overlap at this separation and over the intensity range used (Fig. 4A2). Pathway overlap was only observed when the stimulating electrodes were closer than 10 μm (Fig. 4B). Since stimulating electrodes were never positioned closer than 35 μm in any of the LTD experiments performed, we can safely conclude that PF1 and PF2 responses derive from entirely separate sets of PF bundles.
Figure 4. Heterosynaptic LTD is not due to pathway overlap or an increase in potassium conductance.
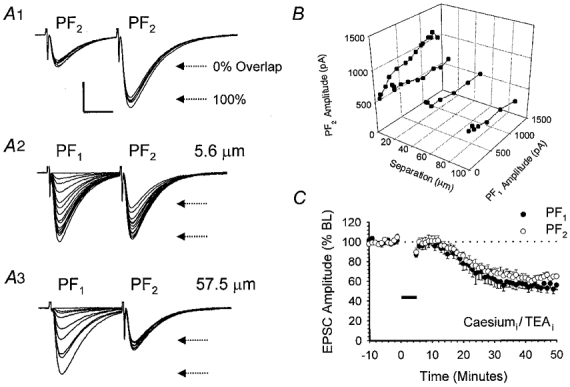
A, illustration of data from a single cell that is representative of a total of six other recordings. A1, paired stimulation of PF2 at an interval of 30 ms produced a facilitation of the second response. A total of five sequential sweeps are shown. A2, the horizontal arrows mark the mean amplitudes of the first and second responses and provide an estimate of the expected PF2 amplitude, at this stimulus strength, under conditions of 0 and 100 % pathway overlap. PF1 and PF2 stimulating electrodes were positioned 5.6 μm apart and activated sequentially at a 30 ms interval over a range of PF1 stimulus intensities. The strength of PF2 stimulation was kept constant throughout. A3, the experiment was repeated with the electrodes repositioned at a further separation of 57.5 μm. B, three-dimensional plot illustrating the relationship between PF1 and PF2 responses for the cell shown in A at inter-electrode separations of 5.6, 22.0, 57.5 and 98.8 μm. C, pooled data from a set of six recordings in which a caesium-based intracellular solution containing TEA was used. PF1 stimulation was paired with depolarisation to 0 mV in voltage clamp for 5 min. Data are expressed as in Fig. 1B.
Voltage- or calcium-dependent changes in membrane conductance produced by the 5 min period of PC depolarisation could alter dendritic filtering, thereby reducing the magnitude of PF responses detected at the soma. In order to eliminate this possibility, we recorded from a total of 10 PCs using a caesium-based intracellular solution that also contained TEA. Of necessity, LTD induction was performed in voltage clamp (see Methods) with PF1 activated in conjunction with depolarising steps to 0 mV at 1 Hz for 5 min. In 6 out of 10 recordings, using the caesium-based solution, both PF1 and PF2 responses underwent LTD, decreasing to 61.7 ± 7.6 and 69.4 ± 4.0 % of baseline levels at 30 min (Fig. 4C). The degree to which PF2 responses were depressed was not significantly different from that observed for PF1 (P > 0.05, Wilcoxon signed-rank test, 2-tailed).
Although the slight change in induction protocol makes a direct comparison of the incidence of LTD in the presence and absence of caesium difficult, these results clearly indicate that potassium channel blockade does not raise the probability of input specificity. However, comparison of the time scale for LTD produced using caesium- versus potassium-based intracellular solutions (Fig. 4CversusFig. 2A) reveals that potassium channel blockade does seemingly delay the onset of depression in both pathways. This suggests that short-lived increases in potassium conductances could account for the immediate decrease in response amplitudes often observed after completion of conjunctive pairing (Konnerth et al. 1992) using potassium-based solutions, but not for the longer term depression.
From these experiments, we can be confident first that only PF1 was specifically activated during the pairing procedure and second, that LTD in PF2 reflects a true reduction in synaptic strength and not a non-specific change in potassium conductance. Therefore, under these conditions of induction, LTD was most commonly heterosynaptic.
Does CF pairing with PFs produce input-specific LTD?
Cerebellar LTD can also be produced by the conjunctive pairing of CFs and PFs at 1 Hz (Sakurai, 1987; Karachot et al. 1995). Since we were unable to induce LTD by pairing small EPSCs with depolarisation, we next examined whether LTD could be more effectively induced, at lower stimulus intensities by pairing PF activation with CF activation. Stimulus intensities were chosen initially to evoke medium-sized PF1 and PF2 responses with peak amplitudes of 591.8 ± 36.5 and 537.6 ± 34.4 pA (n = 6), respectively. For these experiments, a third stimulating electrode was placed within the granule layer to selectively stimulate the CF input to the cell. Once stable baseline PF1 and PF2 responses were established, CF stimulation was paired simultaneously with PF1 activation at 1 Hz for 5 min in current clamp mode.
Under these conditions, LTD of PF1 responses was observed in six out of a total of seven recordings. PF1 (•) and PF2 (○) amplitudes reached 70.9 ± 5.7 and 76.6 ± 5.0 % of baseline levels (n = 6), respectively, after 30 min (Fig. 5A). No statistically significant difference could be detected between PF1 and PF2 response amplitudes (P > 0.05, Wilcoxon signed-rank test, 2-tailed).
Figure 5. CF pairing with PF stimulation also produces heterosynaptic LTD.
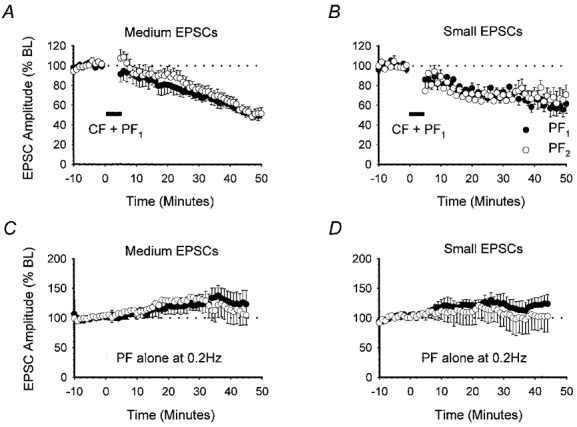
The effects of pairing CF activation with PF1 stimulation at 1 Hz for 5 min. Baseline PF1 (•) and PF2 (○) responses were subdivided into medium (A, n = 6) or small (B, n = 5) groups according to amplitude. The effects of 0.2 Hz stimulation alone for medium (C, n = 6) and small (D, n = 5) PF1 and PF2 responses are also shown. Data are presented as in Fig. 1B.
Figure 6 displays the raw data from one of the six recordings, illustrating the relatively slow onset of depression of both PF1 (•) and PF2 (○) response amplitudes following CF and test PF pairing (Fig. 6A). The CF response remained constant in amplitude throughout the experiment, further illustrating that depression was not due simply to deterioration (Fig. 6C) or to general changes in dendritic filtering.
Figure 6. CF pairing with medium-amplitude responses produces heterosynaptic LTD.
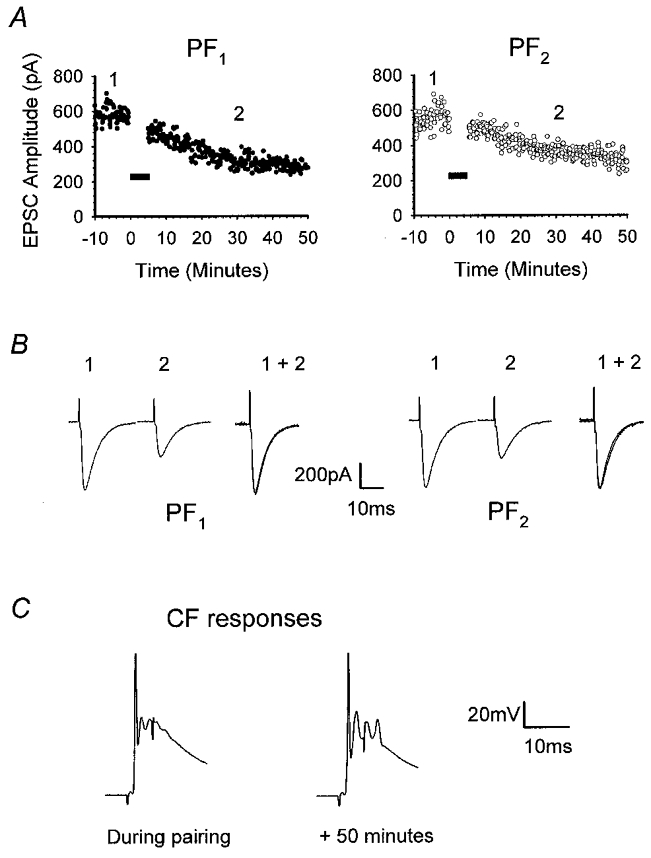
Raw data from one of the six recordings comprising Fig. 5A. A, illustration of the time course of the onset of depression of PF1 (•) and PF2 (○) responses after pairing CF stimulation with activation of PF1. B, representative PF1 and PF2 EPSCs taken at times 1 and 2 illustrated in A. For each pathway, responses measured at time 2 were normalised to the amplitude of those recorded at time 1 (1 + 2). C, CF responses recorded during conjunctive stimulation and 50 min later. Note that the peak amplitudes were identical.
Pairing CF stimulation with test PF stimulation was clearly a more effective method of inducing LTD of smaller responses. We therefore used this new induction protocol to test whether stimulation of fewer PFs could produce input-specific LTD. The stimulus intensity was reduced to evoke small mean PF1 and PF2 response amplitudes of 296.2 ± 15.3 and 275.7 ± 13.4 pA, respectively (n = 6). This is equivalent to activation of approximately 15–20 PFs (Barbour, 1993). Surprisingly, pairing small EPSCs with CF activation not only led to a clear LTD in five out of six recordings, but also caused a depression in PF2 responses. At 30 min, PF1 and PF2 responses were depressed to 73.6 ± 4.9 and 70.6 ± 7.2 % of baseline levels, respectively (Fig. 5B). No statistically significant difference could be found in the level of depression observed in the two pathways at 30 min (P > 0.05, Wilcoxon signed-rank test, 2-tailed).
The long-term stability of medium- and small-amplitude PF responses evoked alternately at 0.2 Hz, without CF pairing, was monitored in two additional sets of experiments in order to eliminate the possibility that smaller PF responses may have simply deteriorated with time. The mean and standard errors of the peak amplitudes for both pathways, at each set of intensities, are displayed in Fig. 5C and D. PF response amplitudes, whether medium or small, failed to show any degree of time-dependent deterioration comparable to the depression seen after CF pairing. On the contrary, at the equivalent time point to LTD experiments (30 min), medium-amplitude responses increased to 132.6 ± 20.6 and 123.5 ± 16.3 % (n = 6) of baseline, which is significantly greater than those measured in both pathways after CF pairing (Fig. 5A and C; P > 0.05, Mann-Whitney U test, 1-tailed). Similarly, small-amplitude PF responses increased slightly to 123.5 ± 16.4 % in PF1 and to 108.0 ± 22.9 % (n = 5) of baseline values in PF2, at the 30 min time point (Fig. 5D).
Although cell depolarisation and CF stimulation are ostensibly considered to be interchangeable, these results indicate that CF activation contributes an additional factor that facilitates the incidence of LTD when fewer PFs are activated. Nevertheless, this increased incidence at lower stimulus intensities did not uncover input-specific LTD.
Does somatic depolarisation elevate intracellular calcium at distal sites?
One possibility for the low incidence of LTD produced by PF pairing with depolarisation for PF responses less than 800 pA in amplitude may simply have been insufficient elevation of intracellular calcium at distal sites. We therefore examined calcium changes during LTD induction to establish whether there were clear differences in calcium mobilisation under the two induction conditions. In four cells, the single wavelength calcium indicator calcium green was included in the intracellular patch solution at a concentration of 500 μm and changes in fluorescence were measured before, during and after each of the LTD induction protocols. Imaging was commenced after a delay period of at least 20 min after gaining whole-cell access to ensure a reasonably uniform level of filling.
Figure 7 illustrates data from a representative experiment, in which a single set of PFs was first paired with CF stimulation at 1 Hz for 5 min, followed 25 min later by pairing of the same PF pathway with cell depolarisation for 5 min. Fluorescence measurements were made from nine separate regions of interest of similar size. CF activation produced relatively small increases in calcium yet LTD was clearly induced. The peak changes in fluorescence produced by somatic depolarisation were typically much larger in all regions (Fig. 7C). In proximal dendrites, fluorescence levels tended to decline with distance from the soma. In some distal dendrites, similar graded effects were observed. In other regions much larger changes were apparent that presumably resulted from the generation of regenerative spikes in spatially restricted regions. Similar patterns of calcium change produced by somatic depolarisation have previously been reported (Ross & Werman, 1987; Ross et al. 1990; Miyakawa et al. 1992).
Figure 7. Somatic depolarisation elevates intracellular calcium throughout the dendritic tree.
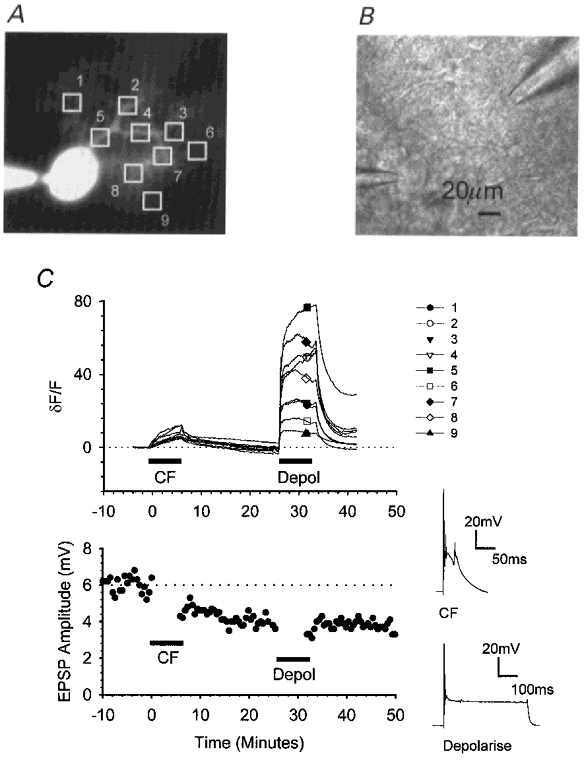
In cells injected with the calcium indicator calcium green-1, fluorescence measurements over time were combined with electrophysiological measurements of the synaptic changes resulting from pairing PF stimulation in current clamp with CF activation followed by cell depolarisation. A, fluorescence image illustrating the positions of nine identically sized regions of interest. B, bright-field image illustrating the positions of the recording electrode (bottom left) and a single stimulating pipette (top right). C, upper graph, changes in the mean fluorescence intensities within each of the regions of interest were measured over time. For each region, intensities were normalised to the initial fluorescence level and expressed as a percentage change (δF/F). After 10 min, the CF input was activated conjunctively with PF stimulation for 5 min at 1 Hz; 25 min later, PF activation was paired with cell depolarisation. The accompanying PF responses are shown in the lower graph.
These results indicate that the low incidence of LTD that we observed following the depolarising protocol, at lower stimulus intensities, is unlikely to be due to a failure to elevate intracellular calcium at distal regions of the dendritic tree. This suggests that under these experimental conditions there are insufficient levels of an additional factor, provided by CFs, for which increasing the stimulus intensity to the PF pathway compensates.
The temporal and spatial constraints to the occurrence of heterosynaptic LTD
LTD at PF2 consistently accompanied LTD at PF1 even without direct activation of this second pathway during the induction protocol (Figs 1, 2 and 5). Input-specific depression was typically not observed at electrode separations ranging between 35.7 and 109.2 μm. At the lower stimulus intensities afforded by pairing PF1 and CF stimulation, input specificity was still not observed even at electrode separations as wide as 107.2 μm (Fig. 5B).
We next considered whether input specificity was favoured by a shorter pairing duration. In six recordings, large-amplitude responses were paired with PC depolarisation at 1 Hz for only 1 min. This protocol effectively induced depression of PF1 responses to 64.4 ± 6.6 % (n = 6) of baseline at 30 min. Depression also occurred in PF2 to a similar magnitude (61.6 ± 9.8 %, n = 6; Fig. 8A) that was not significantly different from that in PF1 (P > 0.05, Wilcoxon signed-rank test, 2-tailed).
Figure 8. Temporal constraints to the induction of heterosynaptic LTD.
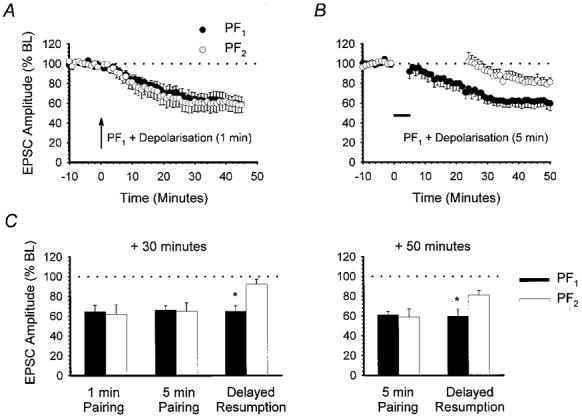
A, the effect of conjunctive pairing of PF1 responses with depolarisation at 1 Hz for 1 min (arrow). Large-amplitude baseline PF responses were used (n = 6). B, using large-amplitude baseline PF responses, PF1 stimulation was paired with depolarisation at 1 Hz for 5 min (horizontal filled bar). PF2 was not activated during pairing and stimulation was not resumed for a further 20 min (n = 6). C, bar chart comparing PF1 (▪) and PF2 (□) responses measured 30 min after (from left to right): 1 min conjunctive pairing of PF1 with depolarisation, 5 min conjunctive pairing with immediate resumption of both PF1 and PF2 stimulation, and 5 min conjunctive pairing with delayed PF2 stimulation. Measurements after 50 min for the last two of these three experiments are shown on the right. Asterisks indicate a significant difference between PF1 and PF2 responses (P < 0.05, Wilcoxon signed-rank test, 2-tailed).
Regardless of the induction method used, the LTD we observed in both pathways was always slow in onset (Figs 1, 2 and 5). Additionally, for heterosynaptic depression to occur, activation of control PFs during the induction protocol was not found to be necessary. We therefore considered whether expression of heterosynaptic LTD required control PF activation to be resumed during the slow declining phase, immediately following pairing. Experiments were performed using large-amplitude EPSCs. PC depolarisation was paired with PF1 stimulation for 5 min as before; however, only stimulation of PF1 at 0.2 Hz was resumed. Activation of PF2 was delayed for a further 20 min. In six out of a total of nine recordings, PF1 was depressed to 64.7 ± 5.7 % of baseline levels at 30 min. However, at the same time point PF2 response amplitude was only slightly depressed to 92.4 ± 4.7 % of baseline levels, which was significantly different from PF1 (n = 6,P < 0.05, Wilcoxon signed-rank test, 2-tailed; Fig. 8C). After a further 20 min, however, there was a further small decline in the PF2 response to 81.2 ± 4.4 % at 50 min (Fig. 8B). Nevertheless, the level of depression in the two pathways at 50 min was still significantly different (P < 0.05, Wilcoxon signed-rank test, 2-tailed; Fig. 8C). These results suggest that expression of heterosynaptic LTD does indeed require PF2 stimulation, although not conjunctively with the depolarising signal. More specifically, expression of heterosynaptic LTD appears to require PF stimulation to be resumed soon after completion of the induction protocol. Delaying PF2 stimulation for a further 20 min tended to favour input-specific depression.
DISCUSSION
LTD induced in cerebellar slices lacks input specificity
The experiments described above were designed to evaluate whether LTD, induced in cerebellar slices by pairing PF activation with either cell depolarisation or CF activation, remains input specific at the single cell level. We report three principal findings. First, synaptic depression resulting from repeated pairing of either PC depolarisation or CF activation with 1 Hz PF stimulation does not remain confined to the active synapses but spreads upwards of 100 μm to spatially distant synapses on the same cell. Reducing the stimulus intensity to activate as few as 20 PFs or decreasing the duration of the pairing protocol did not enhance the degree of input specificity. Second, the incidence of synaptic depression produced by depolarisation paired with PF stimulation was strongly dependent on the strength of PF activation, i.e. the more PFs activated, the greater the incidence of LTD. Pairing CF with PF stimulation effectively produced LTD regardless of the strength of PF activation. Third, expression of heterosynaptic LTD was dependent upon the resumption of PF stimulation within a distinct temporal window after the pairing procedure. Depression in the distant pathway was attenuated if PF stimulation was delayed for 20 min after pairing.
Is heterosynaptic LTD genuine?
Cerebellar LTD is classically considered to be associative by virtue of its requirement for conjunctive stimulation of the two morphologically distinct inputs, PFs and CFs (Ito & Kano, 1982; Sakurai, 1987). At the cellular level, LTD induction requires the activation of both AMPA and mGlu receptors in combination with an increase in intracellular calcium (Linden & Connor, 1995). Physiologically, repetitive activation of the PF is thought to satisfy the first two of these requirements (Konnerth et al. 1990; Batchelor et al. 1994). CF activation, which causes cell depolarisation and consequently calcium influx through VDCCs (Ross & Werman, 1987), satisfies the final requirement. Given that CF activation (or cell depolarisation) results in a fairly global calcium influx (Fig. 7), the above model predicts that input specificity must be determined by the PF.
Such a model fully provides for the depression that we observed at PF1. However, it cannot explain the heterosynaptic spread of depression that we routinely observed since PF2 was not specifically activated. One obvious explanation is that the depression we observed merely reflected a pre- or postsynaptic deterioration of the recording conditions. Several pieces of evidence indicate that this is not the case. First, depression was not accompanied by a significant reduction in membrane resistance in any example included in this study. Second, heterosynaptic LTD was also evident in the presence of internal TEA and caesium. Third, LTD was effectively blocked by postsynaptic inclusion of 10 mm BAPTA. Together, these data demonstrate that the loss of input specificity does not reflect a postsynaptic change in dendritic filtering either as a consequence of general deterioration or through a change in voltage- and/or calcium-dependent potassium conductances. Depression was not observed in either pathway following low-frequency PF stimulation alone, regardless of the intensity, indicating that presynaptic recording conditions were also stable for the duration of the recordings.
The loss of input specificity could, alternatively, simply reflect a significant overlap of fibres between PF1 and PF2 pathways, such that both inputs were activated during the induction protocol. Experiments using a modified paired-pulse protocol revealed that within the range of stimulus intensities used, significant pathway overlap was only encountered when stimulating electrodes were positioned closer than 10 μm (Fig. 4B). As stimulating electrodes were never closer than 35 μm apart in any of the LTD experiments, we can be confident that the PF1 pathway was selectively activated during the induction protocol, even in experiments where large EPSCs were recorded.
On the basis of these results, it is reasonable to assume that the heterosynaptic LTD that we observed is a genuine depression of synaptic activity. How then can LTD take place at distant synapses without concurrent PF activity during depolarisation or CF activation? One possibility is that basal levels of glutamate release at PF2 during the induction process might be sufficient to satisfy the associative requirements of LTD. In agreement with previous reports, low-frequency PF activation was not, on its own, capable of producing synaptic depression in either pathway, regardless of stimulus strength (Fig. 5C and D). Since PC depolarisation can open calcium-activated potassium channels, it is conceivable that basal glutamate release is non-specifically enhanced around the Purkinje cell as a consequence of depolarisation-induced potassium extrusion. Indeed, increases in extracellular potassium can be detected using potassium-sensitive microelectrodes following molecular layer and white matter stimulation in cerebellar slices (Shibuki & Okada, 1990). Although PC depolarisation alone, without concurrent PF stimulation, produced a small depression in both pathways, perhaps suggesting enhanced glutamate release during depolarisation, the level of depression was significantly less than that produced by conjunctive pairing with PF1 activation (Fig. 3C). Therefore, unless LTD is fully induced at PF1, any non-specific increase in basal glutamate is insufficient to induce full LTD at PF2. Moreover, heterosynaptic LTD was still apparent in the presence of TEA and internal caesium, which would have significantly reduced the likelihood of potassium extrusion (Fig. 4D). Therefore, indirect activation of distant PF terminals by increases in extracellular potassium is unlikely to account for the spread of LTD.
Irrespective of the induction protocol used, LTD at PF2 was never observed unless depression took place at PF1, i.e. the site that was specifically activated during induction. Assuming that heterosynaptic LTD is a genuine phenomenon, conjunctive stimulation at PF1 must in some way facilitate depression at distant sites such that LTD can occur via less stringent associative mechanisms or by an entirely different mechanism.
LTD at both sites was prevented by the inclusion of intracellular BAPTA (Fig. 3C). In this respect, the LTD resembles that previously described in slices (Sakurai, 1990; Crepel & Jaillard, 1991) and in culture (Linden et al. 1991). As LTD at PF2 was never observed on its own, we cannot distinguish whether LTD at distant sites is itself calcium dependent or whether it merely depends upon the calcium dependence of LTD at PF1.
One way in which LTD might spread is through the production of a diffusible messenger, such as nitric oxide (NO). NO synthase (NOS) is present in granule cells (Bredt et al. 1990), and NO has been linked to LTD in cerebellar slices (Daniel et al. 1993; Hartell, 1994a, 1996a; Lev Ram et al. 1995). Pairing caged release of NO with cell depolarisation induces a form of LTD (Lev Ram et al. 1995). Since high-frequency stimulation of the molecular layer causes the production of NO, it is entirely conceivable that the combination of NO release through PF activation and calcium influx through cell depolarisation or CF activation contributed to the LTD that we observed. If so, one would predict that the degree of input specificity would depend upon the diffusion radius of NO. Indeed, inhibition of NOS prevents the spread of LTD that follows raised intensity and frequency PF activation (Hartell, 1996b).
Alternative or additional candidates that could mediate the spread of depression include arachidonic acid. Inhibition of phospholipase A2, which releases arachidonic acid from membrane phospholipids, produces only a short-term depression lasting less than 30 min (Linden, 1995). Arachidonic acid could be produced through conjunctive activation of both AMPA and mGlu receptors (Dumuis et al. 1990). The possible roles of either NO or arachidonic acid in heterosynaptic LTD are currently the subject of investigation.
LTD induced with depolarisation depends upon the number of PFs activated
When paired with cell depolarisation LTD was only effectively induced at levels of PF activation that gave rise to EPSCs greater than 1000 pA in amplitude. This is equivalent to EPSPs with amplitudes greater than 9 mV (Fig. 3A). It is important to note that these levels of PF activation are comparable to and in many cases smaller than those used in the vast majority of investigations in which LTD was produced in cerebellar slices by pairing PF stimulation with PC depolarisation where EPSPs with peak magnitudes as large as 15–20 mV were used (see for example Crepel & Jaillard, 1991; Hemart et al. 1995; Boxall & Garthwaite, 1996). Although PF responses are typically superimposed on hyperpolarising steps introduced ostensibly to allow measurements of membrane resistance, these hyperpolarising steps may have reduced the likelihood of VDCC activation before and after induction but not during induction itself. Interestingly, in none of these aforementioned studies were the effects of 1 Hz PF stimulation alone specifically tested.
At lower stimulus strengths, pairing PF activation with depolarisation was relatively ineffectual compared to that which ensued pairing with CF activation. This was not simply due to a lack of calcium influx during cell depolarisation. Fluorescence measurements revealed that depolarisation was far more effective in mobilising a global calcium increase than was CF activation (Fig. 7). This suggests, therefore, that the CF may contribute something more to LTD than merely calcium influx. One possibility, recently proposed, is that CFs release the peptide corticotropin releasing factor, which facilitates LTD induction by enhancing activation of PKC (Miyata et al. 1999), one of the intracellular messengers essential for LTD induction in both cultures (Linden & Connor, 1991) and slices (Crepel & Krupa, 1988). Raised levels of PF activation appear then to compensate for the deficiency of a CF-derived factor that facilitates LTD induction. In cerebellar slices, increasing the stimulus intensity within the molecular layer produces a proportional increase in the amount of NO release (Shibuki & Kimura, 1997). Therefore, the relative contribution of NO to the induction mechanism may increase with PF stimulus intensity. NO is thought to then activate guanylate cyclase within Purkinje cells to produce cGMP (Daniel et al. 1993; Boxall & Garthwaite, 1996), which in turn activates protein kinase G (PKG; Ito & Karachot, 1992; Hartell, 1994a) and G-substrate. If G-substrate behaves as a phosphatase inhibitor, as has been suggested (Ajima & Ito, 1995), the NO–cGMP–PKG–G-substrate cascade could compensate for insufficient activation of PKC at higher PF stimulus intensities. Although pairing with CF activation permitted LTD induction at lower PF stimulus intensities, we still failed to observe input-specific LTD.
Expression of LTD requires PF stimulation
Synaptic depression in pathway PF2 was not immediate after PF1 pairing with depolarisation but it emerged gradually only when low-frequency stimulation of PF2 was resumed. If PF2 stimulation was delayed for a further 20 min, LTD was not immediately apparent and, subsequently, only a partial depression occurred. The onset of heterosynaptic LTD is therefore not simply time dependent; it requires low-frequency PF activation for expression. These findings further support the view that AMPA receptors must be activated for LTD induction (Linden et al. 1993; Hemart et al. 1995) and are similar to the LTD that was induced after injection of the phosphatase inhibitor calyculin A (Ajima & Ito, 1995), where delaying the onset of PF stimulation after injection delayed the onset of depression. Moreover, after the induction of LTD at PF1, one or more intracellular signals must persist for at least 20 min to allow LTD at PF2 once stimulation is resumed. Therefore, a temporal window of up to 20 min exists during which heterosynaptic LTD is favoured. The molecular switch is unlikely to be calcium since increases in intracellular calcium do not persist for this long (Fig. 7).
Physiological importance of input specificity
We conclude from this study that LTD produced at a single set of PF synapses spreads to adjacent synapses on the same PC. The signals or molecular switches that are set by the induction protocol are capable of modifying synaptic transmission at distant synapses providing PF activation is resumed within 20 min of completion of the induction protocol. These findings differ from earlier studies using models of LTD performed in culture where input-specific LTD was conferred by the receptor-specific activation of PKC (Linden, 1994). However, the models of LTD expressed in culture contrast with those in cerebellar slices in that they neither involve nor require the NO–cGMP cascade (Linden & Connor, 1992; Linden et al. 1995). We have previously shown that LTD resulting from PF stimulation alone in cerebellar slices lacked input specificity at the single cell level and that the heterosynaptic depression was dependent on the generation of NO (Hartell, 1996b). Although a recent study reported that input-specific LTD can be induced in slices following the repetitive localised release of caged IP3 within PCs (Finch & Augustine, 1998), this form of depression does not involve or require PF activity in conjunction with caged IP3 release. We suggest, therefore, that the degree of input specificity in slices is largely governed by the extent of spread of PF-mediated NO release.
At the PC population level, LTD induced both in vivo and in vitro is input specific (Ekerot & Kano, 1985; Chen & Thompson, 1995). LTD of PF-mediated extracellular field potentials recorded from cerebellar slices remains input specific when stimulating electrodes are positioned 300 μm apart (Chen & Thompson, 1995). This distance is at least three times greater than the maximum stimulating electrode separations used in our study. When this evidence is considered alongside the single cell studies, the extent of spread of LTD appears to be limited to PF synapses on the same PC. The original theories of cerebellar function suggest that learning and memory of motor skills at the cerebellar level are achieved through the selective modification of subsets of active PF synapses (Marr, 1969; Albus, 1971). At the individual PC level we have demonstrated that LTD spreads to adjacent synapses. This finding apparently contradicts earlier theories, in so far as changes occur at PF synapses that are not stimulated during PC depolarisation or CF activation. However, we did find that the expression of heterosynaptic LTD only occurred on resuming PF2 stimulation. In this respect, the LTD we observed was indeed confined only to active PFs.
Acknowledgments
This study was supported by grants from the Medical Research Council, the BBSRC, the Royal Society of Great Britain and the Nuffield Foundation.
References
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- Ajima A, Ito M. A unique role of protein phosphatases in cerebellar long-term depression. NeuroReport. 1995;6:297–300. doi: 10.1097/00001756-199501000-00018. [DOI] [PubMed] [Google Scholar]
- Albus JS. A theory of cerebellar function. Mathematical Biosciences. 1971;28:167–171. [Google Scholar]
- Barbour B. Synaptic currents evoked in Purkinje cells by stimulating individual granule cells. Neuron. 1993;11:759–769. doi: 10.1016/0896-6273(93)90085-6. [DOI] [PubMed] [Google Scholar]
- Batchelor AM, Madge DJ, Garthwaite J. Synaptic activation of metabotropic glutamate receptors in the parallel fibre-Purkinje cell pathway in rat cerebellar slices. Neuroscience. 1994;63:911–915. doi: 10.1016/0306-4522(94)90558-4. [DOI] [PubMed] [Google Scholar]
- Boxall AR, Garthwaite J. Long-term depression in rat cerebellum requires both NO synthase and NO-sensitive guanylyl cyclase. European Journal of Neuroscience. 1996;8:2209–2212. doi: 10.1111/j.1460-9568.1996.tb00743.x. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Hwang PM, Snyder SH. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347:3768–3770. doi: 10.1038/347768a0. [DOI] [PubMed] [Google Scholar]
- Chen C, Thompson RF. Temporal specificity of long-term depression in parallel fiber-Purkinje synapses in rat cerebellar slice. Learning and Memory. 1995;2:185–198. doi: 10.1101/lm.2.3-4.185. [DOI] [PubMed] [Google Scholar]
- Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, Franz-Bacon K, Reggiani A, Matarese V, Conde F, Collingridge GL, Crepel F. Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature. 1994;372:237–243. doi: 10.1038/372237a0. [DOI] [PubMed] [Google Scholar]
- Crepel F, Dhanjal SS, Sears TA. Effect of glutamate, aspartate and related derivatives on cerebellar Purkinje-cell dendrites in the rat — an in vitro study. The Journal of Physiology. 1982;329:297–317. doi: 10.1113/jphysiol.1982.sp014304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crepel F, Jaillard D. Pairing of pre- and postsynaptic responses in cerebellar Purkinje cells induces long-term changes in synaptic efficacy in vitro. The Journal of Physiology. 1991;432:123–141. doi: 10.1113/jphysiol.1991.sp018380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crepel F, Krupa M. Activation of protein kinase C induces a long-term depression of glutamate sensitivity of cerebellar Purkinje cells. An in vitro study. Brain Research. 1988;458:397–401. doi: 10.1016/0006-8993(88)90486-6. [DOI] [PubMed] [Google Scholar]
- Daniel H, Hemart N, Jaillard D, Crepel F. Long-term depression requires nitric oxide and guanosine 3′:5′cyclic monophosphate production in rat cerebellar Purkinje cells. European Journal of Neuroscience. 1993;5:1079–1082. doi: 10.1111/j.1460-9568.1993.tb00961.x. [DOI] [PubMed] [Google Scholar]
- Dumuis A, Pin JP, Oomagari K, Sebben M, Bockaert J. Arachidonic-acid released from striatal neurons by joint stimulation of ionotropic and metabotropic quisqualate receptors. Nature. 1990;347:182–184. doi: 10.1038/347182a0. [DOI] [PubMed] [Google Scholar]
- Eilers J, Takechi H, Finch EA, Augustine GJ, Konnerth A. Local dendritic Ca2+ signaling induces cerebellar long-term depression. Learning and Memory. 1997;4:159–168. doi: 10.1101/lm.4.1.159. [DOI] [PubMed] [Google Scholar]
- Ekerot CF, Kano M. Long-term depression of parallel fiber synapses following stimulation of climbing fibers. Brain Research. 1985;342:357–360. doi: 10.1016/0006-8993(85)91136-9. [DOI] [PubMed] [Google Scholar]
- Finch EA, Augustine GJ. Local calcium signalling by inositol-1,4,5-trisphosphate in Purkinje cell dendrites. Nature. 1998;396:753–756. doi: 10.1038/25541. [DOI] [PubMed] [Google Scholar]
- Hartell NA. cGMP acts within cerebellar Purkinje cells to produce long term depression via mechanisms involving PKC and PKG. NeuroReport. 1994a;5:833–836. doi: 10.1097/00001756-199403000-00024. [DOI] [PubMed] [Google Scholar]
- Hartell NA. Induction of cerebellar long term depression requires activation of glutamate metabotropic receptors. NeuroReport. 1994b;5:913–916. doi: 10.1097/00001756-199404000-00015. [DOI] [PubMed] [Google Scholar]
- Hartell NA. Inhibition of cGMP breakdown promotes the induction of cerebellar long-term depression. Journal of Neuroscience. 1996a;16:2881–2890. doi: 10.1523/JNEUROSCI.16-09-02881.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartell NA. Strong activation of parallel fibers produces localized calcium transients and a form of LTD that spreads to distant synapses. Neuron. 1996b;16:601–610. doi: 10.1016/s0896-6273(00)80079-3. [DOI] [PubMed] [Google Scholar]
- Hemart N, Daniel H, Jaillard D, Crepel F. Receptors and second messengers involved in long-term depression in rat cerebellar slices in vitro: a reappraisal. European Journal of Neuroscience. 1995;7:45–53. doi: 10.1111/j.1460-9568.1995.tb01019.x. [DOI] [PubMed] [Google Scholar]
- Ito M, Kano M. Long-lasting depression of parallel fiber-Purjkinje cell transmission induced by conjunctive stimulation of parallel fibres and climbing fibers in the cerebellar cortex. Neuroscience Letters. 1982;33:253–258. doi: 10.1016/0304-3940(82)90380-9. [DOI] [PubMed] [Google Scholar]
- Ito M, Karachot L. Protein kinases and phosphatase inhibitors mediating long-term desensitization of glutamate receptors in cerebellar Purkinje cells. Neuroscience. 1992;14:27–38. doi: 10.1016/s0168-0102(05)80004-5. [DOI] [PubMed] [Google Scholar]
- Ito M, Sakurai M, Tongroach P. Climbing fibre induced depression of both mossy fibre responsiveness and glutamate sensitivity of cerebellar Purkinje cells. The Journal of Physiology. 1982;324:113–134. doi: 10.1113/jphysiol.1982.sp014103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karachot L, Kado RT, Ito M. Stimulus parameters for induction of long-term depression in in vitro rat Purkinje cells. Neuroscience Research. 1995;21:161–168. doi: 10.1016/0168-0102(94)90158-9. [DOI] [PubMed] [Google Scholar]
- Konnerth A, Dreessen J, Augustine GJ. Brief dendritic calcium signals initiate long-lasting synaptic depression in cerebellar Purkinje cells. Proceedings of the National Academy of Sciences of the USA. 1992;89:7051–7055. doi: 10.1073/pnas.89.15.7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konnerth A, Llano I, Armstrong CM. Synaptic currents in cerebellar Purkinje cells. Proceedings of the National Academy of Sciences of the USA. 1990;87:2662–2665. doi: 10.1073/pnas.87.7.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev Ram V, Makings LR, Keitz PF, Kao JY, Tsien RY. Long-term depression in cerebellar Purkinje neurons results from coincidence of nitric oxide and depolarization-induced Ca2+ transients. Neuron. 1995;15:407–415. doi: 10.1016/0896-6273(95)90044-6. [DOI] [PubMed] [Google Scholar]
- Linden DJ. Input-specific induction of cerebellar long-term depression does not require presynaptic alteration. Learning and Memory. 1994;1:121–128. [PubMed] [Google Scholar]
- Linden DJ. Phospholipase A2 controls the induction of short-term versus long-term depression in the cerebellar Purkinje neuron in culture. Neuron. 1995;15:1393–1401. doi: 10.1016/0896-6273(95)90017-9. [DOI] [PubMed] [Google Scholar]
- Linden DJ, Connor JA. Participation of postsynaptic PKC in cerebellar long-term depression in culture. Science. 1991;254:1656–1659. doi: 10.1126/science.1721243. [DOI] [PubMed] [Google Scholar]
- Linden DJ, Connor JA. Long-term depression of glutamate currents in cultured cerebellar Purkinje neurons does not require nitric oxide signalling. European Journal of Neuroscience. 1992;4:10–15. doi: 10.1111/j.1460-9568.1992.tb00104.x. [DOI] [PubMed] [Google Scholar]
- Linden DJ, Connor JA. Long-term synaptic depression. Annual Review Of Neuroscience. 1995;18:319–357. doi: 10.1146/annurev.ne.18.030195.001535. [DOI] [PubMed] [Google Scholar]
- Linden DJ, Dawson TM, Dawson VL. An evaluation of the nitric oxide/cGMP-dependent protein kinase cascade in the induction of cerebellar long-term depression in culture. Journal of Neuroscience. 1995;15:5098–5105. doi: 10.1523/JNEUROSCI.15-07-05098.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DJ, Dickenson MH, Smeyne M, Connor JA. A long-term depression of AMPA currents in cultured cerebellar Purkinje neurons. Neuron. 1991;7:81–89. doi: 10.1016/0896-6273(91)90076-c. [DOI] [PubMed] [Google Scholar]
- Linden DJ, Smeyne M, Connor JA. Induction of cerebellar long-term depression in culture requires postsynaptic action of sodium ions. Neuron. 1993;11:1093–1110. doi: 10.1016/0896-6273(93)90222-d. [DOI] [PubMed] [Google Scholar]
- Marr D. A theory of cerebellar cortex. The Journal of Physiology. 1969;202:437–470. doi: 10.1113/jphysiol.1969.sp008820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa H, Lev Ram V, Lasser-Ross N, Ross WN. Calcium transients evoked by climbing fiber and parallel fiber synaptic inputs in guinea pig cerebellar Purkinje neurons. Journal of Neurophysiology. 1992;68:1178–1189. doi: 10.1152/jn.1992.68.4.1178. [DOI] [PubMed] [Google Scholar]
- Miyata M, Okada D, Hashimoto K, Kano M, Ito M. Corticotropin-releasing factor plays a permissive role in cerebellar long-term depression. Neuron. 1999;22:763–775. doi: 10.1016/s0896-6273(00)80735-7. [DOI] [PubMed] [Google Scholar]
- Nakanishi S. Metabotropic glutamate receptors — synaptic transmission, modulation, and plasticity. Neuron. 1994;13:1031–1037. doi: 10.1016/0896-6273(94)90043-4. [DOI] [PubMed] [Google Scholar]
- Oancea E, Meyer T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 1998;95:307–318. doi: 10.1016/s0092-8674(00)81763-8. [DOI] [PubMed] [Google Scholar]
- Ross WN, Lasser-Ross N, Werman R. Spatial and temporal analysis of calcium-dependent electrical activity in guinea pig Purkinje cell dendrites. Proceedings of the Royal Society B. 1990;240:173–185. doi: 10.1098/rspb.1990.0032. [DOI] [PubMed] [Google Scholar]
- Ross WN, Werman R. Mapping calcium transients in the dendrites of Purkinje cells from the guinea-pig cerebellum in vitro. The Journal of Physiology. 1987;389:319–336. doi: 10.1113/jphysiol.1987.sp016659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai M. Synaptic modification of parallel fibre-Purkinje cell transmission in in vitro guinea-pig cerebellar slices. The Journal of Physiology. 1987;394:463–480. doi: 10.1113/jphysiol.1987.sp016881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai M. Calcium is an intracellular mediator of the climbing fibre induction of cerebellar long-term depression. Proceedings of the National Academy of Sciences of the USA. 1990;87:3383–3385. doi: 10.1073/pnas.87.9.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuki K, Kimura S. Dynamic properties of nitric oxide release from parallel fibres in rat cerebellar slices. The Journal of Physiology. 1997;498:443–452. doi: 10.1113/jphysiol.1997.sp021870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuki K, Okada D. Long-term synaptic changes in rat cerebellar slices reflected in extracellular K+ activity. Neuroscience. 1990;113:34–39. doi: 10.1016/0304-3940(90)90490-z. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Abe T, Nomura S, Nakanishi S, Hirano T. Antibodies inactivating mGluR1 metabotropic glutamate receptor block long-term depression in cultured Purkinje cells. Neuron. 1994;12:1245–1255. doi: 10.1016/0896-6273(94)90441-3. [DOI] [PubMed] [Google Scholar]