

Abstract
β-subunit modulation of slow inactivation of class A calcium (Ca2+) channels was studied with two-microlectrode voltage clamp after expression of the α1A- (BI-2) together with β1a-, β2a-, β3- or β4-subunits in Xenopus oocytes.
On- and off-rates of slow inactivation were estimated from the kinetics of recovery from slow inactivation. Ca2+ channels with an α1A/β-subunit composition inducing the slower rate of fast inactivation displayed the faster rate of slow inactivation. The corresponding order of slow inactivation time constants (τonset) was: α1A/β2a, 33 ± 3 s; α1A/β4, 42 ± 4 s; α1A/β1a, 59 ± 4 s; α1A/β3, 67 ± 5 s (n ≥ 7).
Recovery of class A Ca2+ channels from slow inactivation was voltage dependent and accelerated at hyperpolarized voltages. At a given holding potential recovery kinetics were not significantly modulated by different β-subunits.
Two mutations in segment IIIS6 (IF1612/1613AA) slowed fast inactivation and accelerated the onset of slow inactivation in the resulting mutant (α1A/IF-AA/β3) in a similar manner as coexpression of the β2a-subunit. Recovery from slow inactivation was slightly slowed in the double mutant.
Our data suggest that class A Ca2+ channels enter the ‘slow inactivated’ state more willingly from the open than from the ‘fast inactivated’ state. The rate of slow inactivation is, therefore, indirectly modulated by different β-subunits.
Fast and slow inactivation in class A Ca2+ channels appears to represent structurally independent conformational changes. Fast inactivation is not a prerequisite for slow inactivation.
Ca2+ entry through voltage-gated Ca2+ channels modulates a variety of neuronal functions such as: release of neurotransmitters, generation and propagation of action potentials and gene expression. Nine classes of voltage-gated Ca2+ channels that are coded by at least nine different genes (classes A, B, C, D, E, F, G, H, I) have been identified in neuronal cells (Birnbaumer et al. 1994; Perez-Reyes et al. 1999; Lee et al. 1999).
A hallmark of class A Ca2+ channels is their sensitivity to the funnel web spider venom ω-Aga-IVA (Mintz et al. 1992b). This channel type is widely distributed in the central and peripheral nervous system (Mintz et al. 1992a, b; Stea et al. 1994; Westenbroek et al. 1995) and there is evidence that Ca2+ entry through the α1A-subunit mediates the release of neurotransmitters more efficiently than other neuronal Ca2+ channels (Wu et al. 1999).
Class A Ca2+ channels are hetero-oligomeric protein complexes consisting of a pore-forming α1A-subunit, at least one of four β-subunits (β1-β4) and an α2δ-subunit. The auxiliary β-subunits modulate expression density as well as voltage dependence of channel activation and inactivation kinetics (Stea et al. 1994; Olcese et al. 1994; De Waard & Campbell, 1995). Multiple β-subunits are associated with the α1A-subunit to different extents in different parts of the mammalian brain (Liu et al. 1996; Pichler et al. 1997) suggesting that class A Ca2+ channel properties are modulated by tissue-specific expression of different β-subunits (Tanaka et al. 1995). Missense mutations in α1A are associated with the aetiology of familial hemiplegic migraine (Ophoff et al. 1996; Kraus et al. 1998; Hans et al. 1999), ataxia (Zhuchenko et al. 1997) and epilepsy (Fletcher et al. 1996). Ca2+ influx through class A channels and the sensitivities to the ω-agatoxin IVA are modulated by alternative splicing of the α1A-subunit (see splice variants α1A-a and α1A-b in Bourinet et al. 1999).
Class A Ca2+ channel currents decay under voltage clamp with a biexponential time course suggesting two mechanisms of inactivation. Fast inactivation (corresponding to the transient current decay) is affected by point mutations in different parts of the α1A-subunit (see Hering et al. 1998 for review; Bourinet et al. 1999). The additional slow inactivation is much less understood.
We have, therefore, expressed a neuronal α1A-subunit (BI-2, Mori et al. 1991) together with β1a-, β2a-, β3- or β4-subunits in Xenopus oocytes and analysed the individual effects of various β-subunits on slow inactivation.
We report here that a reduction of fast inactivation by coexpression of α1A- with the β2a-subunit simultaneously accelerates the channel state transitions into the slow inactivated state. Alternatively, slowing fast inactivation of α1A/β3 channels by mutating two inactivation determinants in segment IIIS6 to alanine (IF1612/1613AA) accelerated the onset of slow inactivation in a similar manner. Our data suggest that open class A Ca2+ channels are more willing to enter the slow inactivated state than channels in the fast inactivated conformation. The tissue-specific expression patterns of different β-subunits appear, therefore, as an indirect determinant of slow inactivation in class A Ca2+ channels.
METHODS
Generation of the α1A mutant
The class A Ca2+ channel mutant α1A/IF-AA was constructed by introducing two point mutations (I1612A and F1613A) into α1A cDNA (BI-2 accession number X57477, Mori et al. 1991) by the ‘gene SOEing’ technique (Horton et al. 1989). The point mutations were verified by sequence analysis. All constructs were inserted into the polyadenylating transcription plasmids pNKS2 (a kind gift of Dr O. Pongs, Zentrum für Neurale Signalverarbeitung, Hamburg, Germany).
Electrophysiology
In accordance with national guidelines, female Xenopus laevis (NASCO, USA) were anaesthetized by exposing them for 15 min to a 0.2 % MS-222 (methane sulfonate salt of 3-aminobenzoic acid ethyl ester; Sandoz) solution before surgically removing parts of the ovaries. The frogs were then allowed to recover and returned to their tank. Each frog was reused up to two times and subsequently killed by decapitation under anaesthesia. The interval between the operations was longer than 4 months. Follicle membranes from isolated oocytes were enzymatically digested with 2 mg ml−1 collagenase (Type 1A, Sigma). Preparation of stage V-VI oocytes from Xenopus laevis, synthesis of capped off run-off poly(A+) cRNA transcripts from linearized cDNA templates and injection of cRNA were performed as previously described in detail by Grabner et al. (1996).
Calcium channel currents (IBa) were studied 2–7 days after microinjection of approximately equimolar cRNA mixtures of α1A or α1A/IF-AA (0.3 ng (50 nl)−1) with β1a (β2a, β3, β4) (0.1 ng (50 nl)−1) with two-microelectrode voltage clamp in Xenopus oocytes with 40 mm Ba2+ as charge carrier in a bath solution containing (mm): 40 Ba(OH)2, 40 N-methyl-D-glucamine, 10 Hepes, 10 glucose; adjusted to pH 7.4 with methanesulfonic acid as previously described (Grabner et al. 1996). Endogenous chloride currents of the oocytes were suppressed by injecting 20–40 nl of a 0.1 M BAPTA solution 30–240 min before the voltage clamp measurements. Voltage-recording and current-injecting microelectrodes were filled with 2.8 M CsCl, 0.2 M CsOH, 10 mm EGTA and 10 mm Hepes (pH 7.4) and had resistances of 0.3–2 MΩ. The maximum of the activation curve of all α1A/β-subunit combinations studied was in the range of 10 ± 5 mV (n > 5). Only IBa with amplitudes in the range between 0.3 and 1.5 μA were included in the analysis.
Data analysis
Recovery of class A Ca2+ channels from inactivation is sufficiently well described by the equation:
| (1) |
where IN represents the normalized current amplitude, t the recovery time, Afast the fraction of channels that are fast inactivated and Aslow the fraction of channels that are slow inactivated at the end of the inactivating prepulse. 1/τfast gives the rate constant of recovery from the fast inactivated state and 1/τslow the rate constant of recovery from the slow inactivated state (seeAppendix for details).
The time constant of IBa recovery from fast inactivation (τfast) was estimated by means of a conventional double pulse protocol (Fig. 1B, top panel) and the normalized peak current amplitudes were fitted to eqn (1). To enable complete recovery from fast and slow inactivated states we introduced a 1.5 min rest at a holding potential of −120 mV between the subsequently applied double pulses.
Figure 1. Inactivation and recovery kinetics of class A Ca2+ channels with different β-subunit composition.
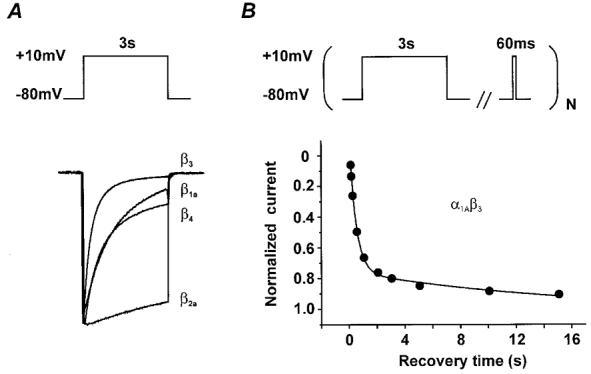
A, IBa through α1A/β1a, α1A/β2a, α1A/β3 and α1A/β4 channels during a 3 s depolarizing step from −80 to 10 mV. Superimposed current traces were scaled to illustrate the different inactivation properties. The IBa decay of α1A/β1a, α1A/β3 and α1A/β4 channels is dominated by fast inactivation; the α1A/β2a channel exhibits slow inactivation kinetics (see also Stea et al. 1994). B, recovery of α1A/β3 Ca2+ channels reveals two components in inactivation. Normalized peak currents are plotted as a function of time (see pulse protocol in the top panel). The smooth line illustrates the two components in IBa recovery corresponding to recovery from fast (τfast) and slow (τslow) inactivation (for estimation of τfast and τslow see Fig. 4).
The onset of slow inactivation (τonset) can be estimated by relating IS, the fraction of channels in the slow inactivated state at the end of a conditioning prepulse, to the prepulse duration (tpre):
| (2) |
(see Appendix for details), which on a logarithmic scale turns to:
where KS = 1/τonset, and C is a constant.
In the experiments described in Fig. 2, the fraction IS and the time constant of recovery from slow inactivation τslow were estimated by fitting the time course of IBa recovery to:
| (3) |
where IN is the normalized current amplitude and t the recovery time.
Figure 2. Estimation of slow inactivation in class A Ca2+ channels by back extrapolation of IBa recovery kinetics.
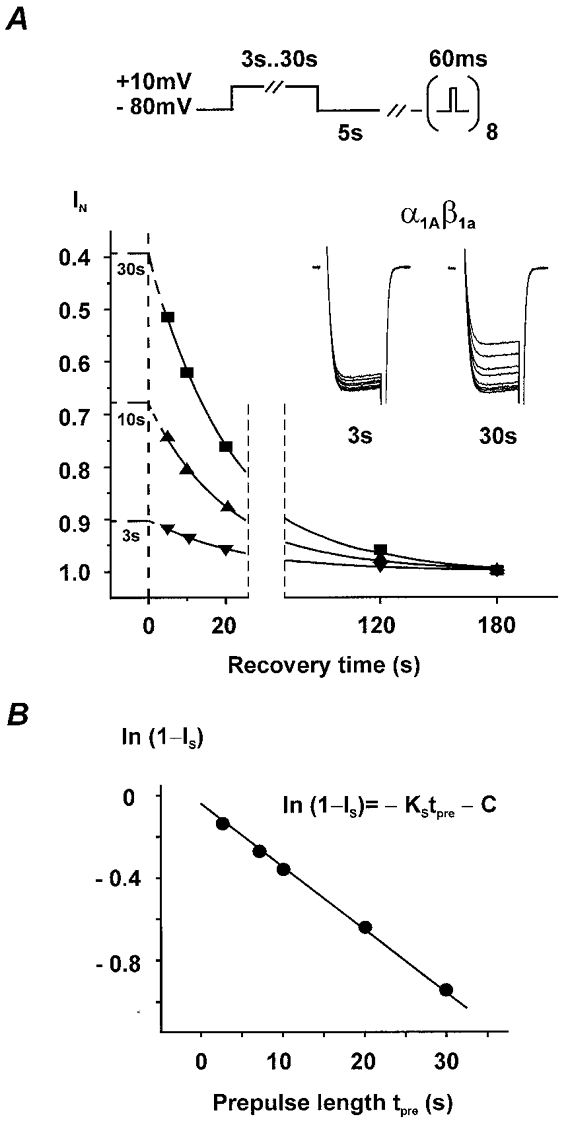
A, recovery from slow inactivation was observed at a holding potential of −80 mV during eight 60 ms test pulses to 10 mV applied 5, 10, 20, 30, 60, 90, 120 and 180 s after conditioning pulses of either 3, 7, 10, 20 or 30 s. Channels recovered completely from fast inactivation during the 5 s rest between the end of the conditioning pulse and the first test pulse (see Fig. 4C for τfast at 80 mV). Current amplitudes were normalized to IBa of the last test pulse and recovery kinetics fitted by an exponential function: IN = 1 –IS exp(-t/τslow), where IN represents the normalized current amplitude, t the recovery time and τslow the time constant of recovery from slow inactivation. τslow was individually estimated for each prepulse length; no significant differences between the mean values for different prepulse lengths were observed. Extrapolation of the mono-exponential recovery to the end of the conditioning pulse (corresponding to time zero) enables the determination of the fraction of channels that have not entered the ‘slow inactivated’ state (1 –IS) during the conditioning pulse. The inset illustrates superimposed 60 ms test pulse currents during recovery from slow inactivation after 3 or 30 s conditioning pulses. B, fractions of non-slow-inactivated channels (1 –IS) after conditioning pulses of different lengths are plotted on a logarithmic scale. The kinetics were fitted by a linear function with the slope KS representing the on-rate of slow inactivation. The coefficient C (y-intercept) was a free parameter of the fit (see Appendix for details).
The pCLAMP software package (version 6.0, Axon Instruments, Inc.) was used for data acquisition and preliminary analysis. Microcal Origin 5.0 was employed for analysis and curve fitting. Data are given as means ±s.e.m. Statistical significance was calculated according to Student's unpaired t test (P < 0.05).
RESULTS
Estimation of slow inactivation in class A Ca2+ channels
To evaluate the effects of different β-subunits on slow inactivation of class A Ca2+ channels we expressed the α1A-subunit together with β1a-, β2a-, β3- or β4-subunits in Xenopus oocytes for later voltage clamp experiments with the two-microelectrode technique. Negligible run-down during two-microelectrode voltage clamp makes this channel type particular suitable for long-lasting experiments as required for kinetic studies of slow inactivation.
As shown in Fig. 1A, each subunit combination produced Ca2+ channels with distinct kinetic phenotypes (see also Stea et al. 1994; De Waard & Campbell, 1995). Since slow inactivation in class A Ca2+ channels with different β-subunit composition (except the α1A/β2a channel) is masked by fast inactivation, this process is difficult to observe from the kinetics of IBa decay.
However, as illustrated in Fig. 1B, IBa of class A Ca2+ channels recovered from inactivation with a biexponential time course. The fraction of channels in the slow inactivated state can be estimated by extrapolating the slow recovery component to the end of the inactivating prepulse (Khodorov et al. 1976; see Appendix).
This approach facilitated the estimation of the on-rate of slow inactivation by means of a ‘back-extrapolation technique’ illustrated in Fig. 2. Conditioning pulses ranging from 3 to 30 s were applied from a holding potential of −80 mV to 10 mV and recovery from slow inactivation was observed at the same holding potential (−80 mV) during a series of short (60 ms) test pulses to 10 mV (Fig. 2A). A 5 s rest between the end of the inactivating prepulse and the first test pulse enabled complete recovery from fast inactivation (see Fig. 4C and Table 1 for corresponding time constants of recovery from fast inactivation). The recovery time constant from slow inactivation (τslow) was estimated by fitting IBa recovery to a mono-exponential function. The fraction of Ca2+ channels in the slow inactivated state (IS, see eqn (3) in Data analysis) was subsequently calculated by extrapolating the exponential function to the end of the inactivating prepulse (Fig. 2A). A conventional double pulse protocol, where single test pulses were applied at various intervals after the conditioning pulse, yielded similar results ensuring that inactivation during the 60 ms test pulse train did not affect the time course of recovery from slow inactivation (data not shown).
Figure 4. Voltage dependence of recovery from fast and slow inactivation of class A Ca2+ channels with different β-subunit composition.
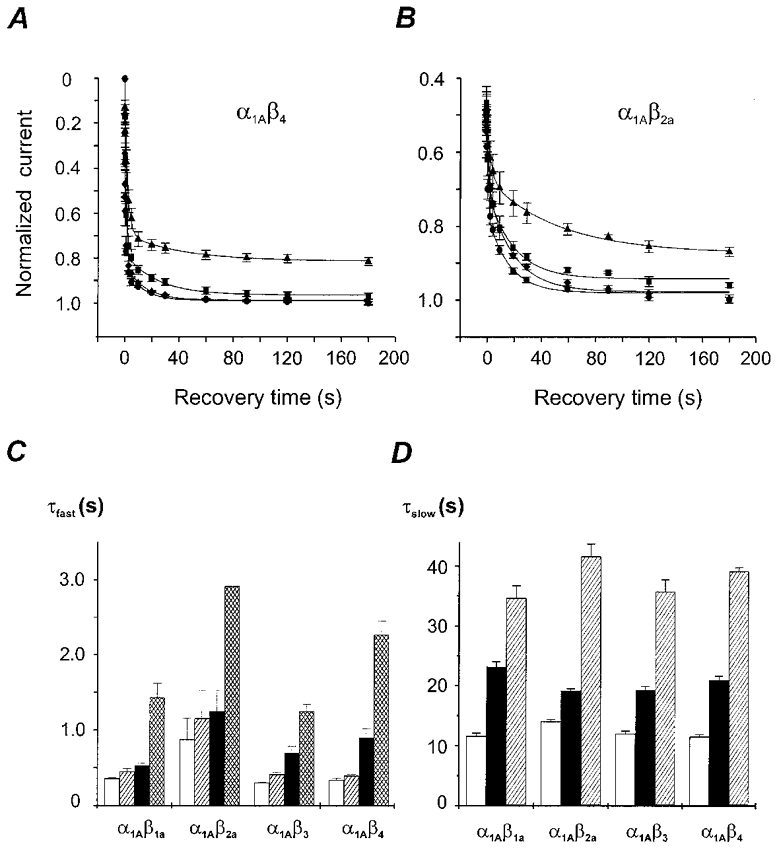
A and B, IBa recovery from inactivation after 10 s conditioning pulses to 10 mV measured by a conventional double pulse protocol (similar to Fig. 1B, top panel). The recovery potentials were −120 mV (•), −100 mV (♦), −80 mV (▪) or −60 mV (▴). The holding potential was −120 mV in all experiments. Mean recovery time courses of α1A/β4 and α1A/β2a channels are shown. Data points are fitted by a biexponential function. Fit parameters are displayed in Table 1. C, time constants of recovery from fast inactivation of class A Ca2+ channels with different β-subunit composition at −120 mV (□), −100 mV ( ) −80 mV (▪) and −60 mV (▪). D, time constants of recovery from slow inactivation at −120 mV (□), −80 mV (▪) and −60 mV (
) −80 mV (▪) and −60 mV (▪). D, time constants of recovery from slow inactivation at −120 mV (□), −80 mV (▪) and −60 mV ( ) were estimated as described in Fig. 2A (n > 20). No significant differences with τslow estimated from double pulse experiments (Table 1) were observed (P > 0.05). The mean values for different subunit combinations at the corresponding holding potentials were not significantly different (P > 0.05).
) were estimated as described in Fig. 2A (n > 20). No significant differences with τslow estimated from double pulse experiments (Table 1) were observed (P > 0.05). The mean values for different subunit combinations at the corresponding holding potentials were not significantly different (P > 0.05).
Table 1.
Parameters of the biexponential fit to recovery from inactivation at various voltages
| Channel | Vh (mV) | τfast (s) | Afast | τslow (s) | Aslow | Offset | n |
|---|---|---|---|---|---|---|---|
| α1A/β1a | −120 | 0.14 ± 0.01 | 0.81 ± 0.02 | 8.2 ± 0.7 | 0.18 ± 0.01 | 1.01 ± 0.01 | 4 |
| −100 | 0.24 ± 0.04 | 0.82 ± 0.03 | 16 ± 3 | 0.17 ± 0.03 | 0.99 ± 0.01 | 4 | |
| −80 | 0.32 ± 0.04 | 0.69 ± 0.03 | 21 ± 2 | 0.19 ± 0.02 | 0.95 ± 0.01 | 5 | |
| −60 | 1.3 ± 0.2 | 0.68 ± 0.02 | 45 ± 6 | 0.20 ± 0.01 | 0.87 ± 0.01 | 4 | |
| α1A/β2a | −120 | 0.7 ± 0.3 | 0.24 ± 0.03 | 16 ± 2 | 0.26 ± 0.02 | 1.00 ± 0.01 | 4 |
| −100 | 1.0 ± 0.4 | 0.21 ± 0.02 | 20 ± 3 | 0.29 ± 0.02 | 1.01 ± 0.01 | 4 | |
| −80 | 1.1 ± 0.3 | 0.23 ± 0.02 | 19 ± 3 | 0.25 ± 0.02 | 0.97 ± 0.01 | 5 | |
| −60 | 2.9 ± 0.3 | 0.19 ± 0.01 | 47 ± 9 | 0.21 ± 0.01 | 0.90 ± 0.01 | 3 | |
| α1A/β3 | −120 | 0.08 ± 0.01 | 0.86 ± 0.02 | 8 ± 1 | 0.12 ± 0.03 | 1.00 ± 0.01 | 5 |
| −100 | 0.20 ± 0.03 | 0.85 ± 0.04 | 10 ± 1 | 0.15 ± 0.03 | 1.01 ± 0.01 | 4 | |
| −80 | 0.5 ± 0.1 | 0.76 ± 0.06 | 19 ± 2 | 0.10 ± 0.01 | 0.93 ± 0.01 | 8 | |
| −60 | 1.1 ± 0.1 | 0.77 ± 0.02 | 35 ± 5 | 0.15 ± 0.01 | 0.69 ± 0.01 | 8 | |
| α1A/β4 | −120 | 0.12 ± 0.02 | 0.83 ± 0.02 | 14 ± 1 | 0.17 ± 0.01 | 1.01 ± 0.01 | 4 |
| −100 | 0.18 ± 0.02 | 0.83 ± 0.02 | 16 ± 2 | 0.16 ± 0.02 | 1.01 ± 0.01 | 4 | |
| −80 | 0.72 ± 0.13 | 0.64 ± 0.03 | 23 ± 5 | 0.19 ± 0.02 | 0.97 ± 0.01 | 4 | |
| −60 | 2.2 ± 0.2 | 0.57 ± 0.02 | 41 ± 11 | 0.12 ± 0.02 | 0.82 ± 0.01 | 4 |
In all experiments the holding potential (Vh) between the double pulses was −120 mV.
As demonstrated in Fig. 2A, the fraction of channels in the slow inactivated state increased with the prolongation of the conditioning prepulse. On a logarithmic scale the time course of the onset of slow inactivation could be approximated by a linear function:
(Fig. 2B) with the slope KS = 1/τonset representing the on-rate of slow inactivation, tpre the length of the inactivating prepulse, IS the fraction of channels in the slow inactivated state and C the y-intercept. The interpretation of the y-intercept is given in Discussion.
Slow inactivation of class A Ca2+ channels with different β-subunit composition
The ‘back-extrapolation’ technique described above enabled the estimation of the on- and off-rates of slow inactivation in class A Ca2+ channels with different β-subunit composition. In line with previous observations, the β3-subunit induced the fastest, β2a- the slowest and the β1a- and β4-subunits intermediate rates of fast inactivation (see also Stea et al. 1994; De Waard & Campbell, 1995; Fig. 1A). The reversed order was, however, observed for the kinetics of slow inactivation. During a test pulse to 10 mV α1A/β2a channels entered the slow inactivated state at an about 2-fold higher rate than rapidly inactivating α1A/β3 channels (Fig. 3A, see Fig. 3B for a comparison of the slow inactivation rates of all β-subunit combinations).
Figure 3. Onset of slow inactivation is modulated by β-subunit composition.
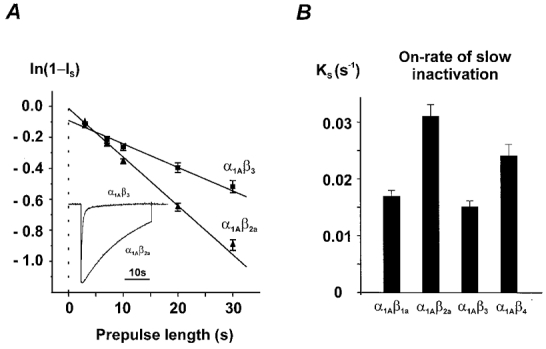
A, slow inactivation was estimated by using conditioning pulses of different length (Fig. 2A). The rate constants were calculated as described in Fig. 2B. A comparison of IBa inactivation in α1A/β2a and α1A/β3 channels during a 30 s test pulse from −80 to 10 mV is illustrated in the inset. B, on-rate constants of slow inactivation of the four different α1A/β-subunit combinations at 10 mV. The corresponding time constants (τonset = 1/KS) are α1A/β1a: 59 ± 4 s, n = 7; α1A/β2a: 33 ± 3 s, n = 12; α1A/β3: 67 ± 5 s, n = 10; α1A/β4: 42 ± 4 s, n = 7.
Recovery of class A Ca2+ channels from the slow inactivated state was voltage dependent and occurred in all four α1/β-subunit combinations at a significantly faster rate if the membrane was held at more negative voltages (Fig. 4D). However, at a given holding potential (−120, −80 or −60 mV) channels with different β-subunit composition recovered from slow inactivation with similar kinetics.
Voltage dependence of recovery from fast inactivation in class A Ca2+ channels with different β-subunit composition
IBa recovery from fast inactivation was examined by means of a conventional double pulse protocol (similar to Fig. 1B, top panel) over a period of 180 s at −120, −100, −80 and −60 mV. The holding potential was −120 mV in all experiments. As illustrated in Fig. 4A and B, recovery of rapidly inactivating channels (e.g. α1A/β4) was dominated by recovery from fast inactivation whereas α1A/β2a channels generally recovered at a slower rate and with an apparent smaller impact of fast inactivation. Coexpression of α1A with β2a did not totally prevent but substantially diminished the fast component of IBa inactivation. α1A/β2a channels recovered with significantly slower kinetics from fast inactivation (e.g. at −60 mV: τfast,α1A/β2a = 2.9 ± 0.3 s, n = 3vs.τfast,α1A/β1a = 1.3 ± 0.2 s, n = 4) suggesting a direct effect of the β2a-subunit on the ‘fast inactivated’ channel conformation.
The time constants of recovery from fast (τfast) and slow (τslow) inactivation of class A channels with different α1A/β-subunit composition at different recovery potentials and corresponding amplitude coefficients are summarized in Fig. 4C and D, and Table 1. Recovery from both fast and slow inactivation was voltage dependent and occurred at a significantly faster rate at more negative voltages. Note that at −120 and −100 mV, where recovery from inactivation is complete, amplitude coefficients of fast and slow components in recovery represent the fractions of fast- and slow-inactivated channels at the end of the depolarizing pulse and are invariant of recovery voltage. However, at −80 and −60 mV normalized IBa did not recover to unity and amplitude coefficients of biexponential fit did not represent such fractions.
Point mutations at the inner mouth of segment IIIS6 reduce fast inactivation and enhance the rate of slow inactivation
The data shown in Fig. 3 indicate that class A channels with α1A/β-subunit composition inducing a slower rate of fast inactivation, enter the slow inactivated state at a significantly higher rate (KS: α1A/β2a > α1A/β4 > α1A/β1a≈α1A/β3). It was interesting to investigate if this correlation reflects a direct β-subunit effect on the slow inactivated channel conformation or, instead, if the β-subunit effect on slow inactivation is secondary to the changes in fast inactivation.
We have, therefore, constructed an α1A mutant with a slower rate of fast inactivation than wild-type by substituting two amino acids in IIIS6 of α1A by alanine (IF1612/1613AA, α1A numbering). Both amino acids at the inner mouth of segment IIIS6 have previously been shown to play an essential role in inactivation of class A/L-type channel chimeras (see IFV-motif in Hering et al. 1997, 1998). As illustrated in Fig. 5A, the resulting mutant (α1A/IF-AA/β3) displayed dramatically slower fast-inactivation kinetics. Furthermore, α1A/IF-AA/β3 channels entered the slow inactivated state at a faster rate compared with α1A/β3 channels (τonset,α1A/IF-AA/β3 = 38 ± 5 s (n = 6) vs.τonset,α1A/β3 = 67 ± 5 s, n = 10,P < 0.01; Fig. 5A).
Figure 5. Point mutations in segment IIIS6 of α1A (IF1612,1613AA) affect the on-rate of slow inactivation and recovery.
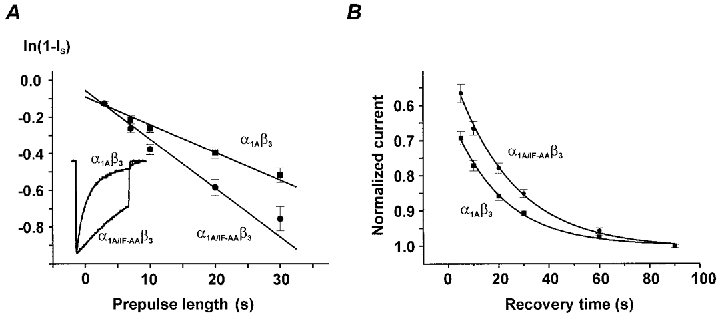
A, onset of slow inactivation in α1A/β3 and α1A/IF-AA/β3 channels (estimated as described in Fig. 2). Slow inactivation time constants (τonset) of α1A/β3 (67 ± 5 s, n = 10) and α1A/IF-AA/β3 channels (38 ± 5 s, n = 6) were significantly different (P < 0.01). Representative normalized currents of both subunit combinations during a 3 s pulse from −80 mV to 10 mV are illustrated in the inset. Note the slower fast-inactivation rate in the double mutant α1A/IF-AA/β3. B, recovery of α1A/β3 and α1A/IF-AA/β3 channels from the slow inactivated state after 30 s conditioning pulse to +10 mV (holding potential −80 mV). Recovery time constants (τslow) of α1A/β3 (19.2 ± 0.6 s, n = 47) and α1A/IF-AA/β3 channels (23.8 ± 0.8 s, n = 26) were significantly different (P < 0.01).
However, we also observed a small but significant difference in recovery of α1A/IF-AA/β3 from slow inactivation (24 ± 1 s in α1A/IF-AA/β3vs. 19 ± 1 s in α1A/β3 at −80 mV, P < 0.01,Fig. 5B).
DISCUSSION
Class A Ca2+ channels affect synaptic function in the mammalian nervous system by mediating the release of neurotransmitters. It is believed that the fine tuning of voltage-dependent Ca2+ entry into neurons is mediated by the β-subunit composition of class A Ca2+ channels. In the present study we expressed an α1A-subunit (BI-2) together with β1a-, β2a-, β3- or β4-subunits in Xenopus oocytes and estimated the on- and off-rate constants of slow inactivation in channels with different β-subunit composition.
A back-extrapolation technique for estimation of slow inactivation
The ‘back-extrapolation’ technique described in Fig. 2 enabled the estimation of the on- and off-rate constants of slow inactivation in class A Ca2+ channels. The first detailed study of slow inactivation in Ca2+ channels was performed by Boyett et al. (1994) who focused on an analysis of the kinetics of current decay of L-type channels during long-lasting membrane depolarizations. Slow inactivation is, however, often masked by fast and/or Ca2+-dependent inactivation (see McMorn et al. 1995 for studies in L-type channels). A comparable picture was observed for class A Ca2+ channels where a transient component in IBa inactivation of α1A/β1a, α1A/β3 and α1A/β4 channels prevented a straight estimation of transitions to the slow inactivated state from the current shape (Fig. 1A). Moreover, transitions from fast to slow inactivation cannot be observed from the kinetics of current decay.
The ‘back-extrapolation’ technique accounts for all channel state transitions to the slow inactivated state including those that are camouflaged by fast or Ca2+-dependent inactivation and requires shorter depolarizing pulses.
Role of β-subunits in slow inactivation of class A Ca2+ channels
The rate of slow inactivation is crucially dependent on subunit composition. Slowly inactivating α1A/β2a channels (Fig. 1A) displayed the fastest rate of slow inactivation. Our results demonstrate that a β2a-subunit-induced reduction in fast inactivation does not prevent but even accelerates state transitions to the slow inactivated state (Fig. 3). There is evidence that the properties of α1A proteins are regulated by interaction with more than a single β-subunit (Burgess et al. 1999). Tissue-specific expression patterns of different β-subunits or changes in the subunit assembly during development (Tanaka et al. 1995) appear, therefore, as significant determinants of slow inactivation in class A Ca2+ channels.
The kinetics of class A channel recovery from the slow inactivated state were independent of the length of the applied prepulses (Fig. 2A). The latter finding indicates that class A Ca2+ channels recover, unlike NaII and NaIIA channels (Toib et al. 1998), in a non-duration-of-activity-dependent manner.
Class A Ca2+ channels are more willing to enter slow inactivation from the open state
To answer the question if enhanced slow inactivation in α1A/β2a channels (Fig. 3) reflects a specific β-subunit effect or, alternatively, occurs at a higher rate from the open state, we designed a class A Ca2+ channel mutant with slower inactivation kinetics. In previous studies on class A/L-type chimeras we have identified a hot spot of inactivation determinants at the inner channel mouth in segment IIIS6 (IFV-motif, Hering et al. 1998) and suggested an inactivation model where fast inactivation is substantially determined by non-covalent interaction of S6 segements (Hering et al. 1996, 1998). Here we demonstrate for the first time that two amino acids of a homologous motif at the inner mouth of segment IIIS6 in α1A (I1612, F1613) play an essential role in fast inactivation of class A Ca2+ channels (Fig. 5). The faster on-rate of slow inactivation in the slowly inactivating double mutant α1A/IF-AA/β3 supports the hypothesis that the faster rate of slow inactivation in α1A/IF-AA/β3 and α1A/β2a channels is more probably caused by reduced fast inactivation than by a direct β-subunit effect on the ‘slow inactivated’ state. However, a small but significant increase of the slow recovery time constant in α1A/IF-AA/β3 compared with α1A/β3 channels (Fig. 5B) does not allow an unequivocal conclusion. Hence, the apparent faster on-rate demonstrated in Fig. 5A could also partially result from an additional stabilization of the slow inactivated conformation by mutations IF1612/1613AA.
A higher rate of slow inactivation from the open to the slow inactivated state is also suggested by the y-intercept of the linear function fitted to the log of slow inactivation in α1A/β3 channels compared with α1A/β2a (Fig. 3A). α1A/β3 channels enter the slow inactivated state during the first few seconds of the inactivating prepulse predominantly from the open state and later exclusively from the ‘fast inactivated’ state. Due to the much slower current decay α1A/β2a channels enter the slow inactivated state during the prepulse from both open and ‘fast inactivated’ states. The y-intercept in α1A/β3 channels can, therefore, be explained by an initially higher transition rate of open channels to the slow inactivated state causing a somewhat steeper slope during the first few seconds whereas later during the pulse the slope is determined mainly by the slower rate of redistribution between fast and slow inactivation. α1A/β2a channels enter the slow inactivated state during the conditioning pulse from both states with an obviously smaller impact of the redistribution rate between fast and the slow inactivated state resulting in the smaller y-intercept. For a detailed mathematical description of the y-intercept see Appendix.
Possible implications for synaptic function
Ca2+ channels that have opened during an action potential appear to be more willing to enter the slow inactivated state than channels in the fast inactivated conformation. It is tempting to speculate, that, due to slow recovery (Fig. 4D), a certain fraction of class A Ca2+ channels will inevitably accumulate in the slow inactivated state during action potential trains. Such a mechanism would be particularly important for the slowly inactivating α1A-b splice variant (Bourinet et al. 1999). Slow inactivation in class A channels could, therefore, play an important role in regulating the voltage-dependent Ca2+ entry into neurons.
Direct evidence for a role of class A Ca2+ channel inactivation in post-tetanic depression of excitatory postsynaptic currents was recently presented by Forsythe et al. (1998). Presynaptic P-type channels in a brainstem giant synapse (calyx of Held) recover with biexponential kinetics from inactivation. Forsythe et al. (1998) suggest that accumulation of the channels during high frequency stimulation in inactivation represents a fail safe mechanism that would reduce Ca2+ entry and thereby slow or prevent presynaptic vesicle depletion. The mean time constant of about 50 s for slow P-type channel recovery in the presynaptic membrane of the calyx of Held is comparable to the values of class A Ca2+ channel recovery from slow inactivation (Fig. 4D). The β-subunit composition of class A Ca2+ channels may, therefore, represent a significant determinant of short-term synaptic plasticity.
This assumption is supported by our finding that only the on-rate was β-subunit-modulated whereas recovery from slow inactivation at a given membrane potential was not affected. However, voltage clamp experiments with faster temporal resolution on mammalian cells are required to compare the relative impacts of slow inactivation, fast inactivation of open channels and closed-state inactivation (Patil et al. 1998) during trains of brief action potential waveforms.
Slow inactivation of class A Ca2+ channels may also represent an important mechanism to prevent Ca2+ overload during long-lasting depolarizations in neuronal tissue during hypoxic or ischaemic events (Kristián & Siesjö, 1996).
Taken together, we demonstrate a technique that enabled a detailed description of the on- and off-rate constants of slow inactivation in Ca2+ channels. Class A Ca2+ channels enter the ‘slow inactivated’ state from both open and ‘fast inactivated’ conformations. Open channels appear, however, to be more willing to enter the slow inactivated state.
The β-subunit regulation of fast inactivation has an indirect effect on the rate of slow inactivation which may play an important role in fine tuning of voltage-dependent Ca2+ entry and help to maintain the neuronal Ca2+ homeostasis.
Acknowledgments
We thank Professor H. Glossmann for continuous support of our work and Drs S. Berjukow and J. Mitterdorfer for comments on the manuscript. We also thank Drs Y. Mori and K. Imoto for the gift of the α1A cDNA, Dr Schwartz for providing the α2/δ cDNA and Dr Birnbaumer for providing the β2a, β3 and β4 cDNAs. This work was supported by grants from the Fonds zur Förderung der Wissenschaftlichen Forschung P12649 (S.H.), a grant from the Else Kröner Fresenius Stiftung (S.H.) and a grant from the Austrian National Bank (S.H.) and is part of the thesis of S.S.
APPENDIX
Our data are consistent with a state diagram where class A Ca2+ channels enter the slow inactivation state (S) either directly from the open (O) or via the fast inactivated state (F). Possible state transitions during a membrane depolarization are illustrated in the reaction scheme:
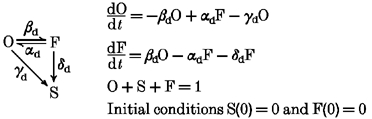 |
Index d indicates rate constants at a depolarizing potential.
The exact solution for this system of differential equations
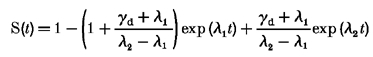 |
incorporates the characteristic roots of the system:
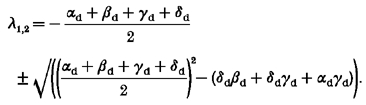 |
Assuming that slow inactivation develops at a much slower rate than fast inactivation (γd,δd << αd,βd), this process can be approximated by:
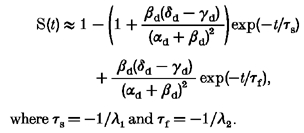 |
where τs =−1/λ1 and τf =−1/λ2.
During the slow phase of onset of inactivation (when fast inactivation is completed):
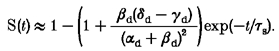 |
If this function is extrapolated to zero time we obtain an expression for the experimentally observed y-intercept:
If transitions to the slow inactivated state occur predominantly via the fast inactivated state O → F → S, γd < δd and the y-intercept is expected to have a negative value whereas predominantly parallel transition (from O directly to S, i.e. δd < γd) will result in a positive y-intercept, as observed in our experiments.
Recovery from inactivation at negative membrane potentials occurs in accordance with the following scheme:
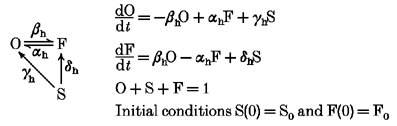 |
Index h indicates rate constants at a hyperpolarizing potential.
Solving this set of equations we obtain:
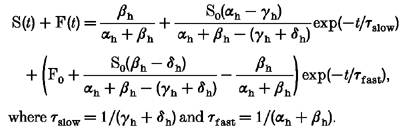 |
where τslow = 1/(γh+δh) and τfast = 1/(αh+βh).
Under the assumptions that the recovery from fast inactivation occurs at a much faster rate than recovery from slow inactivation (αh >> γh, αh >> δh, αh >> βh) this equation can be simplified to:
This expression demonstrates that under conditions where the fast and slow kinetic components of recovery are well separated from each other, the amplitude coefficient of the slow component reflects the fraction of slow-inactivated Ca2+ channels at the end of the conditioning prepulse.
References
- Birnbaumer L, Campbell KP, Catterall WA, Harpold MM, Hofmann F, Horne WA, Mori Y, Schwartz A, Snutch TP, Tanabe T. The naming of voltage-gated calcium channels. Neuron. 1994;13:505–506. doi: 10.1016/0896-6273(94)90021-3. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Sutton K, Slaymaker S, Mathews E, Monteil A, Zamponi GW, Nargeot J, Snutch TP. Splicing of α1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nature Neuroscience. 1999;5:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- Boyett MR, Honjo H, Harrison SM, Zang W-J, Kirby MS. Ultra-slow voltage-dependent inactivation of the calcium current in guinea-pig and ferret ventricular myocytes. Pflügers Archiv. 1994;428:39–50. doi: 10.1007/BF00374750. [DOI] [PubMed] [Google Scholar]
- Burgess DL, Biddlecome GH, McDonough SI, Diaz ME, Zilinski CA, Bean BP, Campbell KP, Noebels JL. β-subunit reshuffling modifies N- and P/Q-type Ca2+ channel subunit compositions in lethargic mouse brain. Molecular and Cellular Neuroscience. 1999;13:293–311. doi: 10.1006/mcne.1999.0748. [DOI] [PubMed] [Google Scholar]
- De Waard M, Campbell KP. Subunit regulation of the neuronal α1A Ca2+ channel expressed in Xenopus oocytes. The Journal of Physiology. 1995;485:619–634. doi: 10.1113/jphysiol.1995.sp020757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher CF, Lutz CM, O'Sullivan TN, Shaughnessy JD, Jr, Hawkes R, Frankel WN, Copeland NG, Jenkins NA. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87:607–617. doi: 10.1016/s0092-8674(00)81381-1. [DOI] [PubMed] [Google Scholar]
- Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. [DOI] [PubMed] [Google Scholar]
- Grabner M, Wang Z, Hering S, Striessnig J, Glossmann H. Transfer of 1,4-dihydropyridine sensitivity from L-type to class A (BI) calcium channels. Neuron. 1996;16:207–218. doi: 10.1016/s0896-6273(00)80037-9. [DOI] [PubMed] [Google Scholar]
- Hans M, Luvisetto S, Williams ME, Spagnolo M, Urrutia A, Tottene A, Brust PF, Johnson EC, Harpold MM, Stauderman KA, Pietrobon D. Functional consequences of mutations in the human alpha1A calcium channel subunit linked to familial hemiplegic migraine. Journal of Neuroscience. 1999;19:1610–1619. doi: 10.1523/JNEUROSCI.19-05-01610.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering S, Aczel S, Grabner M, Doring F, Berjukow S, Mitterdorfer J, Sinnegger MJ, Striessnig J, Degtiar VE, Wang Z, Glossmann H. Transfer of high sensitivity for benzothiazepines from L-type to class A (BI) calcium channels. Journal of Biological Chemistry. 1996;271:24471–24475. doi: 10.1074/jbc.271.40.24471. [DOI] [PubMed] [Google Scholar]
- Hering S, Aczel S, Kraus RL, Berjukow S, Striessnig J, Timin EN. Molecular mechanism of use-dependent calcium channel block by phenylalkylamines: role of inactivation. Proceedings of the National Academy of Sciences of the USA. 1997;94:13323–13328. doi: 10.1073/pnas.94.24.13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering S, Berjukow S, Aczél S, Timin EN. Calcium channel block and inactivation: common molecular determinants. Trends in Pharmacological Sciences. 1998;19:439–443. doi: 10.1016/s0165-6147(98)01258-9. [DOI] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Khodorov B, Shishkova L, Peganov E, Revenko S. Inhibition of sodium currents in frog Ranvier node treated with local anesthetics. Role of slow sodium inactivation. Biochimica et Biophysica Acta. 1976;433:409–435. [Google Scholar]
- Kraus RL, Sinnegger MJ, Glossmann H, Hering S, Striessnig J. Familial hemiplegic migraine mutations change alpha1A Ca2+ channel kinetics. Journal of Biological Chemistry. 1998;273:5586–5590. doi: 10.1074/jbc.273.10.5586. [DOI] [PubMed] [Google Scholar]
- Kristián T, Siesjö BK. Calcium-related damage in ischemia. Life Sciences. 1996;59:357–367. doi: 10.1016/0024-3205(96)00314-1. [DOI] [PubMed] [Google Scholar]
- Lee JH, Daud AN, Cribbs LL, Lacerda AE, Pereverzev A, Klockner U, Schneider T, Perez-Reyes E. Cloning and expression of a novel member of the low voltage-activated T-type calcium channel family. Journal of Neuroscience. 1999;19:1912–1921. doi: 10.1523/JNEUROSCI.19-06-01912.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, De Waard M, Scott VES, Gurnett CA, Lennon VA, Campbell KP. Identification of three subunits of the high affinity omega-conotoxin MVIIC-sensitive Ca2+ channel. Journal of Biological Chemistry. 1996;271:13804–13810. [PubMed] [Google Scholar]
- McMorn SO, Harrison SM, Zang W-J, Boyett MR. Comparison of ultra-slow voltage-dependent inactivation of the cardiac L-type Ca2+ channel with Ca2+ or Ba2+ as the charge carrier in ferret ventricular myocytes. Experimental Physiology. 1995;80:565–575. doi: 10.1113/expphysiol.1996.sp003959. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Adams ME, Bean BP. P-type calcium channels in rat central and peripheral neurons. Neuron. 1992a;9:85–95. doi: 10.1016/0896-6273(92)90223-z. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Venema VJ, Swiderek KM, Lee TD, Bean BP, Adams ME. P-type calcium channels blocked by the spider toxin omegAga-IVA. Nature. 1992b;355:827–829. doi: 10.1038/355827a0. [DOI] [PubMed] [Google Scholar]
- Mori Y, Friedrich T, Kim MS, Mikami A, Nakai J, Ruth P, Bosse E, Hofmann F, Flockerzi V, Furuichi T. Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature. 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- Olcese R, Qin N, Schneider T, Neely A, Wie X, Stefani E, Birnbaumer L. The amino terminus of a calcium channel beta subunit sets rates of channel inactivation independently of the subunit's effect on activation. Neuron. 1994;13:1433–1438. doi: 10.1016/0896-6273(94)90428-6. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- Patil PG, Brody DL, Yue DT. Preferential closed-state inactivation of neuronal calcium channels. Neuron. 1998;20:1027–1038. doi: 10.1016/s0896-6273(00)80483-3. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E, Lee JH, Cribbs L-L. Molecular characterization of two members of the T-type calcium channel family. Annals of the New York Academy of Sciences. 1999;868:131–143. doi: 10.1111/j.1749-6632.1999.tb11283.x. [DOI] [PubMed] [Google Scholar]
- Pichler M, Cassidy TN, Reimer D, Haase H, Kraus R, Ostler D, Striessnig J. β Subunit heterogeneity in neuronal L-type Ca2+ channels. Journal of Biological Chemistry. 1997;272:13877–13882. doi: 10.1074/jbc.272.21.13877. [DOI] [PubMed] [Google Scholar]
- Stea A, Tomlinson WJ, Soong TW, Bourinet E, Dubel SJ, Vincent SR, Snutch TP. Localization and functional properties of a rat brain α1A calcium channel reflect similarities to neuronal Q- and P-type channels. Proceedings of the National Academy of Sciences of the USA. 1994;91:10576–10580. doi: 10.1073/pnas.91.22.10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka O, Sakagami H, Kondo H. Localization of mRNAs of voltage-dependent Ca2+-channels: four subtypes of α1- and β-subunits in developing and mature rat brain. Molecular Brain Research. 1995;30:1–16. doi: 10.1016/0169-328x(94)00265-g. [DOI] [PubMed] [Google Scholar]
- Toib A, Lyakhov V, Marom S. Interaction between duration of activity and time course of recovery from slow inactivation in mammalian brain Na+ channels. Journal of Neuroscience. 1998;18:1893–1903. doi: 10.1523/JNEUROSCI.18-05-01893.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Sakurai T, Elliott EM, Hell JW, Starr TV, Snutch TP, Catterall WA. Immunochemical identification and subcellular distribution of the α1A subunits of brain calcium channels. Journal of Neuroscience. 1995;15:6403–6418. doi: 10.1523/JNEUROSCI.15-10-06403.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Westenbroek RE, Borst JGG, Catterall WA, Sakmann B. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. Journal of Neuroscience. 1999;19:726–736. doi: 10.1523/JNEUROSCI.19-02-00726.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α1A-voltage-dependent calcium channel. Nature Genetics. 1997;15:62–69. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]