

Abstract
The accessory β subunits of voltage-dependent Ca2+ channels (VDCCs) have been shown to regulate their biophysical properties and have also been suggested to antagonise the G protein inhibition of N-type (α1B), P/Q-type (α1A) and α1E channels. Here we have examined the voltage-dependent involvement of the four neuronal isoforms (β1b, β2a, β3 and β4) in the process of G protein modulation of α1B Ca2+ channels.
All β subunits hyperpolarised α1B current activation, and all antagonised the G protein-mediated depolarisation of current activation. However, except in the case of β2a, there was no generalised reduction by β subunits in the maximal extent of receptor-mediated inhibition of α1B current.
In addition, all VDCC β subunits enhanced the rate of current facilitation at +100 mV, for both receptor-mediated and tonic modulation. The rank order for enhancement of facilitation rate was β3 > β4 > β1b > β2a. In contrast, the amount of voltage-dependent facilitation during tonic modulation was reduced by β subunit co-expression, despite the fact that the apparent Gβγ dissociation rate at +100 mV was enhanced by β subunits to a similar level as for agonist-induced modulation.
Our data provide evidence that G protein activation antagonises Ca2+-channel β subunit-induced hyperpolarisation of current activation. Conversely, co-expression of all β subunits increases the apparent Gβγ dimer dissociation rate during a depolarising prepulse. This latter feature suggests the co-existence of bound Ca2+-channel β subunits and Gβγ dimers on the α1B subunits. Future work will determine how the interaction between Gβγ dimers and Ca2+-channel β subunits with α1B results in a functional antagonism at the molecular level.
Voltage-dependent Ca2+ channels (VDCCs) are multi-subunit proteins composed of an α1 subunit and a regulatory cytoplasmic β subunit, as well as a largely extracellular α2δ subunit (for review see Dolphin, 1998). Activation of G protein-coupled receptors provides a mechanism for fine tuning of synaptic transmission (Dunlap et al. 1995). Membrane-delimited G protein inhibition of neuronal N-type (α1B), P/Q-type (α1A) and α1E Ca2+ channels has been shown to be mediated by Gβγ dimers (Herlitze et al. 1996; Ikeda, 1996; Shekter et al. 1997). Accessory β subunits regulate the biophysical properties of VDCCs, producing an increase in the current density (in part by recruitment of channels into the membrane) and a hyperpolarizing shift of current activation (De Waard & Campbell, 1995; Brice et al. 1997; Stephens et al. 1997; Jones et al. 1998). Apart from these direct actions on the α1 subunits, a role of VDCC β subunits to produce an apparent antagonism of membrane-delimited G protein inhibition has also been reported in reconstitution studies in Xenopus oocytes (Bourinet et al. 1996; Qin et al. 1998; Roche & Treistman, 1998a). Furthermore, partial depletion of endogenous VDCC β subunits in sensory neurons by the use of antisense DNA was found to enhance GABAB receptor-mediated inhibition of native Ca2+ currents, although not that produced by direct G protein activation (Campbell et al. 1995). This has been interpreted in terms of an interaction at an overlapping binding site (Bourinet et al. 1996, and for review see Dolphin, 1998). This hypothesis is supported by the finding that one of the reported Gβγ binding sites within the I-II loop overlaps with a binding site for β subunits (De Waard et al. 1997). However, there is also an additional Gβγ and β2a binding site on the C-terminus of human α1E channels (Qin et al. 1997), and a VDCC β subunit binding site on the N terminus of α1A (Walker et al. 1999), which is also important for β subunit effects on α1B (Stephens et al. 2000) and overlaps with a site essential for G protein modulation of α1B (Cantíet al. 1999). Since available evidence suggests that only one VDCC β subunit binds per channel (Jones et al. 1998) and only one Gβγ binds per channel, at least as revealed from the re-inhibition kinetics following facilitation (Stephens et al. 1998; Zamponi & Snutch, 1998), it is possible that these three intracellular domains all form part of a complex binding pocket for both VDCC β subunits and Gβγ dimers.
The aim of the present work was to examine the involvement of VDCC β subunits in G protein modulation of α1B currents, by expression studies in Xenopus oocytes. The central strategy was to monitor α1B current activation associated either with the basal, tonic low levels of Gβγ subunits, or with an increase of Gβγ level induced by stimulation of dopamine D2 receptors. Our results provide evidence that VDCC β subunits oppose the Gβγ-mediated depolarising shift of α1B current activation, and that this antagonistic action is facilitated by strong depolarization of the cell membrane. Furthermore, co-expression of all VDCC β subunits results in a dramatic increase in the rate of α1B current facilitation at +100 mV.
METHODS
Molecular biology
The following cDNAs were used: rabbit α1B (GenBank accession number: L15453), rat β1b (X11394), rat β2a (M80545), rat β3 (M88751), rat β4 (LO2315), rat α2δ1 neuronal splice variant (M86621) and rat D2long receptor (X77458, N5→S). Mutant C3,4S-β2a, in which the cysteines at positions 3 and 4 that are substrates for palmitoylation are mutated to serine, was made using standard molecular biology techniques with the forward primer TTC ATG CAG TCC TCC GGG CT, together with the reverse primer TG ACA GGT CAG GTA TCT GG. All cDNAs were subcloned into the expression vector pMT2 (Swick et al. 1992).
Expression of constructs and electrophysiological recording
Adult Xenopus laevis females were anaesthetised by immersion in 0.25% tricaine, and killed by decapitation and pithing. Oocytes were then surgically removed and defolliculated by treatment for 2 h at 21°C with 2 mg ml−1 collagenase type Ia in Ca2+-free ND96 saline containing (mm): 96 NaCl, 2 KCl, 1 MgCl2, 5 Hepes (pH adjusted to 7.4 with NaOH). Plasmid cDNAs (all at 1 ng nl−1) for the α1B subunit plus accessory β and α2δ1 subunits, and D2 receptors were mixed in a ratio of 3:4:1:3 respectively (except when otherwise stated), and 4 nl was injected into the nuclei of stage V and VI oocytes. Injected oocytes were incubated at 18°C for 3–7 days in ND96 saline (as above plus 1.8 mm CaCl2) supplemented with 100 μg ml−1 penicillin and 100 IU ml−1 streptomycin (Life Technologies, Gaithersburg, MD, USA), and 2.5 mm sodium pyruvate. Two electrode voltage clamp recordings from oocytes were performed at room temperature (20–22°C) during continuous superfusion of a chloride-free solution containing (mm): 5 Ba(OH)2, 85 TEA-OH, 2 CsOH, 5 Hepes (pH 7.4 with methanesulfonic acid). Every oocyte was injected with 30–40 nl of a 100 mm solution of K3-1,2-bis(aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid (BAPTA) to suppress endogenous Ca2 +-activated Cl− currents. Electrodes contained 3 M KCl and had resistances of 0.3–2 MΩ. The currents were amplified and low-pass filtered at 1 kHz by means of a Geneclamp 500 amplifier, and digitised through a Digidata 1200 interface using data acquisition software pCLAMP 6.02 (Axon Instruments, Foster City, CA, USA).
Typically the oocytes were held at −100 mV and currents were evoked in sweeps composed of a 100 ms step to the indicated test potential (P1), followed by a 50 ms (unless otherwise stated) depolarising pulse to +100 mV (unless otherwise stated) and a second 100 ms test step (P2). The +100 mV depolarising pulse was separated by 500 ms from P1, and by 20 ms from P2, except when otherwise stated. The interval between sweeps was 15 s. Current amplitude measurements were taken 20 ms after the start of test pulses in order to minimise the influence of time-dependent dissociation of Gβγ subunits during the test pulse, as well as differences due to the inactivation rate. The only exception was the steady-state inactivation protocol, where currents were measured at their peak within 100 ms, since no G protein involvement was studied. All values are means ±s.e.m., and statistical significances were determined by Student's t test or paired t test as appropriate.
Data analysis
Data were analysed using Clampfit (Axon Instruments) and Origin 5.0 (Microcal software, Inc., Northampton, MA, USA). In order to correct for differences in voltage-dependent inactivation, relief of inhibition by a depolarising prepulse was calculated by normalising the P2/P1 ratio in the presence of quinpirole by the P2/P1 ratio in control condition (wash-out after a first application of quinpirole), in an adaptation of the ‘corrected prepulse ratio’ (Simen & Miller, 1998).
Current-voltage (I-V)-derived parameters were obtained by fitting an activation curve of a modified Boltzmann type:
where Vt represents the test potential, Gmax the maximum conductance, Vrev the apparent reversal potential, V50,act the potential for half-activation, and k the potential range required for an e-fold change of current around V50,act. Activation curves were then derived from this.
The steady-state inactivation curves were also fitted by a Boltzmann equation:
where Imax is the peak current value and V50,inact is the potential for half-inactivation, at which there is an e-fold change over k mV.
RESULTS
The currents analysed in the present work were all recorded with the same sequence and timing of protocols. For every cell, complete I-V relationships (−60 to +50 mV) were recorded prior to any application of the D2 receptor agonist quinpirole (100 nm), followed by a time course at the test potential of 0 mV, before, during and after (wash-out) the first perfusion with quinpirole. Subsequently, complete I-V relationships were performed following wash-out and during a second application of 100 nm quinpirole.
Regulation of α1B activation and steady-state inactivation by VDCC β subunits
All known VDCC-β subunit isoforms have been reported consistently to enhance current amplitude and to hyperpolarise the voltage dependence of activation of α1A, α1E and α1C Ca2+ channels. So far, Xenopus oocytes have been shown to endogenously express two VDCC β isoforms, which are highly homologous to the rat brain β3 subunit and have been found to modulate α1E channels in a manner indistinguishable from their mammalian counterpart (Tareilus et al. 1997).
We first assessed the influence of the over-expressed VDCC β subunits on the voltage dependence of activation of α1B currents, recorded over a range of membrane potentials (−60 to +50 mV). In parellel with the enhancement of current amplitude (Fig. 1A), all VDCC β subunits hyperpolarised the mid-point for current activation (V50,act), although β2a was the least effective isoform (Fig. 1A and B). This difference was abolished in a β2a mutant, in which cysteines 3 and 4 are mutated to serine (C3,4S-β2a), which does not become palmitoylated (Qin et al. 1998; Chien & Hosey, 1998) (Fig. 1B).
Figure 1. Regulation of α1B activation and sustained inactivation by VDCC β subunits.

α1B was expressed with α2δ and the dopamine D2 receptor, and either with or without the VDCC β subunits, β1b, β2a, β3, β4 or C3,4S-β2a. All values shown in this figure were obtained before any stimulation of D2 receptors, and are given as means ±s.e.m.A, I-V relationships for the different conditions (symbol key is shown in the box) were obtained by stepping the membrane potential from −60 to +50 mV in 10 mV steps, and current values were fitted to a modified Boltzmann function (see Methods). B, mean V50,act values obtained from A. Compared with α1B/α2δ currents, the co-expression of any VDCC β subunits hyperpolarised significantly the mid-point for current activation (P < 0.01). Numbers of experiments for activation data shown in A and B are as follows: α1B/α2δ, 9; +β1b, 11; +β2a, 20; +C3,4S-β2a, 8; +β3, 12; +β4, 12. C, steady-state inactivation curves were obtained from 100 ms duration test pulses to 0 mV, preceded by a 25 s prepulse to the potential given (−100 to 0 mV). Peak current values were normalised and the fits shown are to a Boltzmann function (see Methods section). The symbols used are the same as in A. D, mean V50,inact values from C. Compared with α1B/α2δ currents, the mid-point for steady-state inactivation was significantly depolarised by co-expression of β2a (P < 0.01), and hyperpolarised by co-expression of the other VDCC β subunits (including non-palmitoylated C3,4S-β2a) (P < 0.01). Numbers of experiments for inactivation data shown in C and D are as follows: α1B/α2δ, 4; +β1b, 5; +β2a, 5; +C3,4S-β2a, 7; +β3, 4; +β4, 4.
β2a, in contrast to other β subunit isoforms, shows the distinctive properties of reducing dramatically the inactivation rate of α1A, α1E and α1C channels, and depolarising rather than hyperpolarising their steady-state inactivation (De Waard & Campbell, 1995; Jones et al. 1998; Cens et al. 1998). We observed a similar shift in the voltage dependence of steady-state inactivation for α1B currents. The potential for 50% inactivation (V50,inact) was hyperpolarised by the co-expression of β1b, β3 or β4 subunits from about −40 mV to −70 mV (Fig. 1C and D). In clear contrast, β2a shifted the voltage dependence of the steady-state inactivation process in the opposite direction by about +10 mV. Again, the palmitoylation-deficient C3,4S-β2a mutant showed a V50,inact value identical to the VDCC β subunits other than β2a (Fig. 1C and D).
Basal G protein modulation of α1B
Membrane-delimited G protein modulation results, at a given potential, in a reduction of current amplitude and a slowing of the activation kinetics of α1A, α1B and α1E Ca2+ channels. The observed slowing of the activation rate is thought to represent a voltage-dependent on-line relief of G protein inhibition, reaching its maximal expression in the fraction of current inhibition that is reversed by a strong depolarising pulse preceding the test pulse, namely prepulse facilitation. Voltage-dependent inhibition defined in this way has been related consistently to Gβγ actions in a number of cell systems (for review see Dolphin, 1998).
We aimed to examine the G protein modulation of α1B in a scenario with a relatively low concentration of activated G proteins, and to compare it with the stronger modulation that occurs upon stimulation of G protein-coupled receptors. We therefore measured the amount of tonic prepulse facilitation under resting conditions, prior to any application of D2 receptor agonist, in oocytes co-expressing the different VDCC β subunits. Figure 2A–F depicts representative currents obtained at 0 mV, before (P1) and after (P2) application of a depolarising prepulse to +100 mV. Traces labelled ‘con’ show that tonic facilitation resulting from the 50 ms prepulse was greatest when no β subunits were co-expressed. Co-expression of any β subunit isoforms markedly reduced the facilitation ratio at this potential (Fig. 3A). However, since a common feature for G protein-mediated effects on calcium currents is that there is a reduction of inhibition at depolarised potentials, the facilitation ratio is also expected to change at different test potentials and this is observed here (Fig. 3A). Comparison of the tonic facilitation ratio at any potential (in the −20 to +30 mV range) resulted in the highest maximal ratio being over 1.3 for the α1B/α2δ combination, whereas the co-expression of any exogenous β subunit reduced the maximal ratio to less than 1.2 (Fig. 3A). Moreover, by lengthening the prepulse to 200 ms, representing a supramaximal duration for facilitation (see Fig. 8A for explanation), the α1B/α2δ currents reached a higher tonic facilitation ratio of ∼1.5 (Fig. 3A), whereas this manipulation had no effect when VDCC β subunits were co-expressed (results not shown).
Figure 2. Current traces in the absence and presence of co-expressed VDCC β subunits, showing tonic facilitation and over-recovery after the initial application of quinpirole.
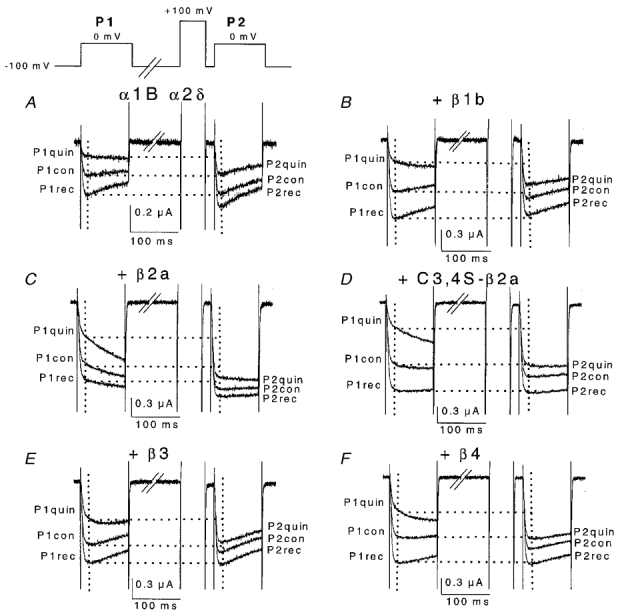
Representative traces are shown for all subunit combinations α1B/α2δ (A), +β1b (B), +β2a (C), + C3,4S-β2a (D), +β3 (E) and +β4 (F). Currents were recorded at 0 mV membrane potential. Top left, voltage waveform used to elicit the depicted currents. P1 indicates test pulse before a 50 ms, +100 mV depolarising prepulse, 20 ms before the P2 test pulse. Control (con) refers to the control condition before application of quinpirole, as explained in the text. The effect of a first application of quinpirole (100 nm, quin) and wash-out of quinpirole, once the current amplitude had stabilised (rec) are shown. The percentage over-recovery is calculated as (1 – P1rec/P1con) × 100. The intersection of the vertical grids (20 ms after the start of the test pulse) with the current traces indicates the points of measurement. Horizontal grids show amount of facilitation in each condition.
Figure 3. Tonic G protein modulation.
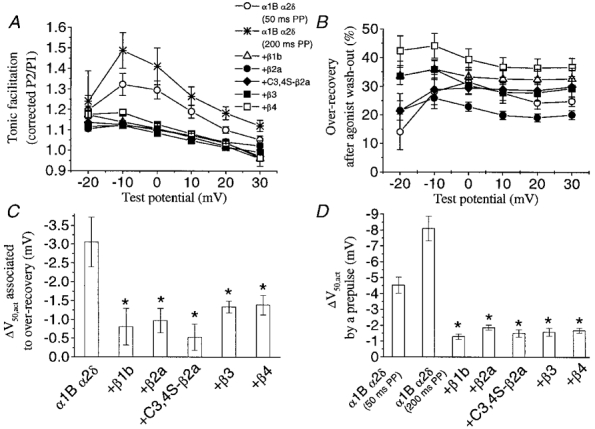
A number of parameters associated with basal G protein modulation have been examined. A, corrected P2/P1 ratios in the −20 to +30 mV range for control currents resulting from a 50 ms prepulse (PP), unless stated in the key, prior to any perfusion of quinpirole. Tonic facilitation was significantly higher in the absence of exogenous VDCC β subunits but the difference decreased at more positive test potentials. ⋆, facilitation values of α1B α2δ currents obtained upon 200 ms prepulses, which we established as a duration producing maximal facilitation in this case. B, percentage over-recovery (P1rec/P1con) during the wash-out, after a first perfusion of the D2 agonist quinpirole (100 nm), at a wide range (−20 to +30 mV) of test pulses. C, hyperpolarisation of V50 for activation associated with removal of tonic inhibition, as the difference between V50,act in control and V50,act in the over-recovered currents. The actual V50,act values for each condition are given in Table 1. D, hyperpolarization of V50 for activation associated with the relief of inhibition by a +100 mV prepulse. V50 values were derived from fitting I-V curves (−60 to +50 mV) to a modified Boltzmann function, as stated in the Methods section. N numbers are as follows: α1B α2δ (50 ms prepulse), 9; α1B α2δ (200 ms prepulse), 4; +β1b, 11; +β2a, 20; + C3,4S-β2a, 8; +β3, 12; +β4, 12. Statistical significance compared with α1B/α2δ for shift in V50,act is indicated by *P < 0.01 (Student's independent t test).
Figure 8. Comparison of the kinetics of facilitation and re-inhibition of α1B currents, in the absence and presence of co-expressed VDCC β3 subunits, during tonic and agonist-induced modulation.
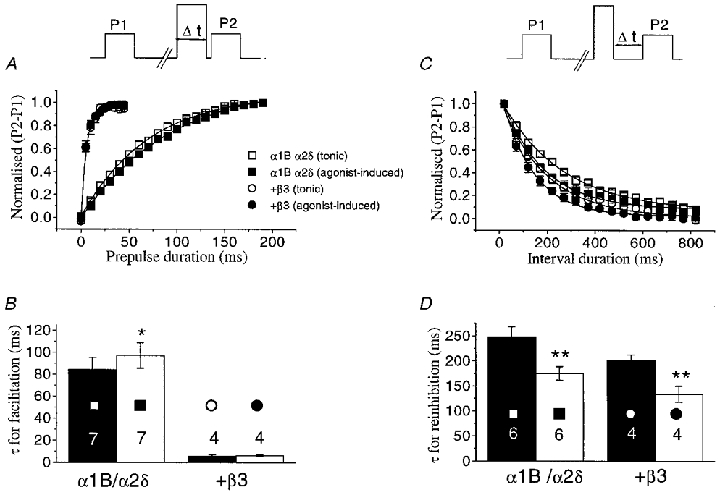
A, top panel, voltage waveform for measurement of facilitation rate. The duration of the prepulse (Δt) was increased in 5 ms (α1B/α2δ co-expressed with VDCC β subunits) or 10 ms steps (α1B/α2δ alone). All data are from −10 mV test pulses. The P1 and P2 currents were then subtracted and the plot of the difference (P2 – P1 values normalised to the maximum) was fitted to a single exponential function. The graph shows the facilitation rate for α1B/α2δ/β3 (circles) and α1B/α2δ currents (squares), both before (tonic modulation, open symbols) and during (agonist-induced modulation, filled symbols) application of quinpirole. Continuous lines are the result of single exponential fits to the data. B, the histogram gives the τ values for facilitation, determined from exponential fits to individual data, for the subunit combinations shown beneath the histogram bars. The filled bars indicate the tonic modulation condition, and the open bars represent agonist-induced modulation. C, top panel, voltage waveform for measurement of re-inhibition rate: following the depolarizing prepulse, the time before the subsequent test pulse (Δt) was progressively increased in 50 ms steps from 20 to 820 ms. Currents were measured and normalised as for the facilitation protocols, and the data were fitted to a single exponential, shown by the continuous lines. All data are from −10 mV test pulses, and data are shown for α1B/α2δ and α1B/α2δ/β3 combinations, using the same symbols as in A. The histogram gives the τ values for re-inhibition, determined from exponential fits to the individual measurements, for the subunit combinations shown beneath the histogram bars. The filled bars indicate the tonic modulation condition, and the open bars represent agonist-induced modulation. Numbers on the histogram bars in both B and D indicate the number of cells for each condition, and statistical significance, of tonic compared to agonist induced modulation, is indicated by *P < 0.05 and **P < 0.01 (Student's paired t test).
After assessing the tonic facilitation values, quinpirole (100 nm) was then applied. As we have shown in previous studies (Page et al. 1998; Cantíet al. 1999), D2 receptor stimulation inhibited the amplitude and slowed the activation rate of α1B currents, both effects being partially relieved by a depolarising prepulse (Fig. 2). In all cases the action of quinpirole reached a peak within 0.5–1 min after the start of its application. After 2 min, quinpirole was washed out for 3–5 min, resulting in over-recovery compared to initial control values (traces labelled P1rec in Fig. 2). The over-recovery phenomenon has been related to removal of basal modulation (Roche & Treistman, 1998b). The amount of over-recovery also provides, by subtraction, a different estimate of tonic inhibition, and we investigated its evolution along the I-V relationship. In all cases the peak potential for over-recovery was around −10 mV, although the values showed little or no decline as the test pulse was depolarised in the −20 to +30 mV range (Fig. 3B). The α1B/α2δ combination was maximally tonically inhibited by 31.5%, which was not significantly different from the maximal tonic inhibition in the presence of any of the β subunits. However, there were significant differences between different β subunits, such that the tonic inhibition by α1B/α2δ/β4 was consistently elevated compared with α1B/α2δ/β2a over the whole voltage range (P < 0.01).
Together with the effects on current amplitude and kinetics, G protein modulation depolarises the voltage dependence of activation of Ca2+ channel currents (for review see Dolphin, 1998). This also proved true for the tonically modulated α1B currents recorded under our experimental conditions, and the effect was countered by co-expression of VDCC β subunits (Fig. 3C and Table 1). Indeed, in parallel to the facilitation of current amplitude, application of a depolarising prepulse induced a negative shift of the I-V relationship, which was reduced by co-expression of VDCC β subunits where the activation was already hyperpolarised (Fig. 3D and Table 1). Thus, although no antagonism was found in terms of current inhibition (except for β2a from the over-recovery measure of tonic inhibition), VDCC β subunits opposed the depolarising shift in current activation resulting from basally active G proteins.
Table 1.
V50,act (in mV) values for the different experimental conditions used
| Control | Overrecovery | Quinpirole (2nd application) | |||||
|---|---|---|---|---|---|---|---|
| Condition | n | P1 | P2 | P1 | P2 | P1 | P2 |
| α1B/α2δ (50 ms PP) | 9 | +1.0 ± 1.0 | −3.6 ± 0.9 | −1.7 ± 0.7 | −3.3 ± 0.8 | +8.5 ± 0.7 | +1.6 ± 1.1 |
| α1B/α2δ (200 ms PP) | 4 | +3.7 ± 0.1 | −4.6 ± 1.0 | +0.8 ± 0.9 | −4.6 ± 0.6 | +10.3 ± 0.4 | −2.6 ± 0.8 |
| +β1b | 11 | −14.9 ± 0.8 | −16.2 ± 0.9 | −15.4 ± 0.8 | −16.0 ± 1.1 | −9.2 ± 0.9 | −16.9 ± 0.8 |
| +β2a | 20 | −9.8 ± 0.7 | −11.6 ± 0.7 | −11.3 ± 0.6 | −12.8 ± 0.7 | −6.6 ± 0.7 | −11.8 ± 0.7 |
| + C3,4Sβ2a | 8 | −15.2 ± 1.2 | −16.7 ± 1.3 | −15.7 ± 1.2 | −16.9 ± 1.3 | −10.5 ± 1.1 | −16.5 ± 1.2 |
| +β3 | 12 | −13.5 ± 0.8 | −15.0 ± 0.8 | −15.3 ± 0.7 | −16.5 ± 0.7 | −8.4 ± 0.6 | −15.6 ± 0.6 |
| +β4 | 12 | −11.6 ± 0.8 | −13.3 ± 0.7 | −13.2 ± 0.8 | −14.1 ± 0.8 | −7.7 ± 0.8 | −13.4 ± 0.9 |
P1 refers to before, and P2 after, a prepulse to +100 mV. The prepulse (PP) duration was 50 ms except where otherwise indicated. See Figs 3C and D and 6D for statistical significance of differences between the values. The number of cells for each condition is given by n.
Modulation of α1B by stimulation of the D2 receptor
Over-recovery after the first application of quinpirole was useful not only for assessment of tonic inhibition, but also because in the process the Ba2+ currents recovered from tonic inhibition and therefore constituted a better starting point for studying agonist-mediated inhibition, which otherwise could be largely occluded. Nonetheless, over-recovered currents were not completely devoid of tonic inhibition as shown by the traces of α1B/α2δ and α1B/α2δ/β2a currents depicted in Fig. 2 (labelled Prec), which reveal a residual facilitation at 0 mV. This feature was not unique to these subunit combinations, since significant residual facilitation was also present for the other combinations including VDCC β subunits at more negative potentials (not shown).
Following over-recovery, the effect of a second application of 100 nm quinpirole was examined (designated quin(2), Fig. 4). Regardless of the co-expression of VDCC β subunits, agonist-mediated inhibition of α1B currents reached a peak at potentials before the V50 for activation and then decayed steeply (Fig. 5A). As the I-V relationships peaked at different potentials, depending on the co-expression of VDCC β subunits (Fig. 1, Fig. 6A–C), it was of interest to compare the maximal values for inhibition irrespective of the membrane potential. For α1B/α2δ in the absence of co-expressed β subunits, a peak inhibition of 69.6% was obtained at −10 mV test pulses. Co-expression of most VDCC β subunits hardly attenuated the inhibitory effect: β1b did not diminish the current inhibition, with 70.4% inhibition at −20 mV, followed by β3 (62.2% inhibition at −20 mV) and β4 (59.0% inhibition at −20 mV) (Fig. 5A). In contrast, β2a was the only β subunit to significantly and consistently result in a reduced inhibition (50.9% inhibition at −10 mV, P < 0.01) (Fig. 5B). This effect of β2a was largely reversed at all potentials in the C3,4S-β2a mutant (61.8% inhibition at −20 mV; Fig. 5B).
Figure 4. Current traces in the absence and presence of co-expressed VDCC β subunits, showing inhibition by a second application of quinpirole and reversal by a depolarizing prepulse.

A–F, membrane currents elicited upon 0 mV test pulses as in Fig. 2 (waveform shown top left). Currents following wash-out of the first application of quinpirole (rec) were challenged with quinpirole (100 nm) for a second time (quin(2)). The percentage inhibition is calculated as (1 – Pquin/Pcon) × 100, either before (P1) or after (P2) a depolarising prepulse. The displayed traces are from the same cells whose current traces are shown in Fig. 2.
Figure 5. Inhibition by quinpirole of α1B currents, in the presence or absence of co-expressed VDCC β subunits.
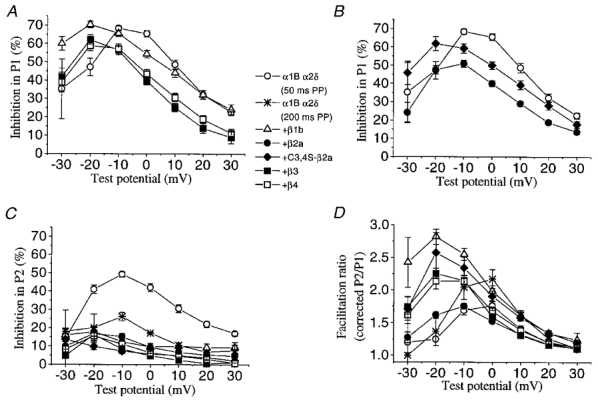
A, the percentage inhibition by quinpirole in P1 for currents formed by α1B/α2δ without co-expressed VDCC β subunits, or with the VDCC β1b, β3 or β4 subunits, as shown in the key. Inhibition was measured over the −30 to +30 mV range. The control values for the 50 and 200 ms prepulse (PP) data were not significantly different and have been combined (○). B, the percentage inhibition by quinpirole in P1 for currents formed by α1B/α2δ without co-expressed VDCC β subunits, or with the VDCC β2a, or C3,4S-β2a subunits, symbols as shown in the key. The controls for the 50 and 200 ms prepulse (PP) data were not significantly different and have been combined (○). C, the percentage inhibition in P2, after a depolarizing prepulse of 50 or 200 ms duration, as stated, for the same combinations of subunits as in A and B. D, facilitation ratio (as corrected P2/P1, see Methods) for the different subunit combinations (see key), between −30 and +30 mV. The numbers of experiments are as follows for both A and B:α1B α2δ (50 ms prepulse), 9; α1B α2δ (200 ms prepulse), 4; +β1b, 11; +β2a, 20; + C3,4S-β2a, 8; +β3, 12; +β4, 12.
Figure 6. Facilitation of α1B/α2δ currents in the absence and presence of over-expressed β subunits.
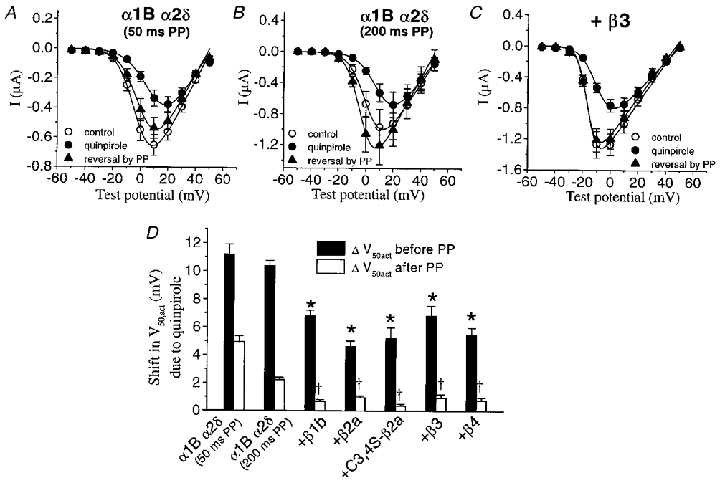
A, I-V relationships for α1B/α2δ currents: for control (over-recovery), during the second quinpirole application, and after a 50 ms depolarizing prepulse (PP) in the presence of the agonist. B, I-V relationships for α1B/α2δ currents as in A, from a different set of cells, where a prepulse of 200 ms duration was applied. C, I-V relationships for α1B/α2δ/β3 currents, showing the effect of quinpirole and reversal by a 50 ms depolarizing prepulse. D, mean depolarization of V50,act values from I-V relationships as in A-C, for all different subunit combinations. Depolarization of the V50,act due to quinpirole is partially antagonised by co-expression of any VDCC β subunits (filled bars). When preceded by a 50 or 200 ms prepulse (as stated, open bars), the depolarization of the V50,act is reduced. Numbers of experiments are as follows: α1B/α2δ (50 ms prepulse), 9; α1B α2δ (200 ms prepulse), 4; +β1b, 11; +β2a, 20; + C3,4S-β2a, 8; +β3, 12; +β4, 12. Statistical significance compared to α1B/α2δ (200 ms data) are given by *P < 0.01 for shift in V50,act before PP and †P < 0.01 for the shift in V50,act after PP (Student's independent t test).
We also measured the degree of quinpirole-induced inhibition for test pulses applied 20 ms after a 50 ms depolarising pulse to +100 mV, termed P2 pulses. In this condition all groups showed a decrease of inhibition, though the co-expression of any VDCC β subunits strongly enhanced such voltage-dependent relief, in remarkable contrast with the effects seen on basal modulation. For α1B/α2δ in the absence of β subunits, the agonist-induced inhibition in P2 after a 50 ms prepulse was 49.2% (at −10 mV) and this was lowered to 17.8% (at −20 mV, P < 0.01) for β1b, 16.9% (at −20 mV, P < 0.01) for β2a, 16.3% (at −20 mV, P < 0.01) for β3, 15.6% (at −20 mV, P < 0.01) for β4 and 13.6% (at −30 mV, P < 0.01) for C3,4S-β2a (Fig. 5C). However, application of a 200 ms prepulse reduced the maximal residual inhibition in P2 for the α1B/α2δ combination to a level approaching that obtained with co-expression of VDCC β subunits (26.2% at −10 mV, P < 0.01) (Fig. 5C).
In all cases the prepulse facilitation ratio peaked at the same potential as that at which the P1 inhibition was maximal (Fig. 5D). Although α1B/α2δ expressed alone displayed a much higher P2 inhibition, its maximal facilitation ratio for a 50 ms prepulse was not different from that in the presence of β2a (1.8 at 0 mV). The other VDCC β isoforms gave higher values, being 2.2 (β4), 2.3 (β3) and 2.6 (β1b, C3,4S-β2a), all at −20 mV test pulses (Fig. 5D). Nevertheless, application of a supramaximal 200 ms prepulse enhanced the facilitation of α1B/α2δ currents up to a level similar to the co-expression of the β3 and β4 isoforms (∼2.2) (Fig. 5D).
We also analysed the role of VDCC β subunits in the quinpirole-mediated depolarisation of α1B current activation. As shown in Fig. 1, VDCC β subunits strongly hyperpolarise the control I-V relationship of α1B currents. The agonist-induced depolarisation of the V50 for activation in all experimental groups is shown in Fig. 6D (filled bars) and the actual V50,act values are given in Table 1. In the absence of exogenous VDCC β subunits, quinpirole caused a +11.2 mV shift of the α1B current-voltage relationship. Co-expression of all VDCC β isoforms consistently opposed this depolarising action, the values being reduced between +4.6 and +6.8 mV (P < 0.01). The antagonism by VDCC β subunits of G protein-mediated depolarisation of current activation was augmented by application of +100 mV steps before the test pulses (Fig. 6D, open bars). Thus a 50 ms depolarising prepulse reversed the G protein effects on current activation, and the degree of facilitation was enhanced by co-expression of all β subtypes from a partial to a complete reversal of inhibition. However, for α1B/α2δ an increase in the reversal of a quinpirole-mediated shift in V50,act was achieved by increasing the depolarising prepulse to 200 ms duration (Fig. 6D), while there was no significant effect of lengthening the prepulse duration for the combinations containing β subunits (not shown).
Kinetics of facilitation and re-inhibition for the α1B-Gβγ interaction
Because of the reduced ability of a 50 ms prepulse to reverse the quinpirole-induced inhibition in the absence of co-expressed VDCC β subunits (Fig. 4A, compared with Fig. 4B–F), we examined the dependence of facilitation in the presence of quinpirole on prepulse duration (Fig. 7A). The rate of current facilitation, which can be measured by varying the length of the depolarising prepulse, is believed to reflect the apparent dissociation rate of Gβγ from the open α1B Ca2+ channels. In all cases the facilitation rate could be fitted with a single exponential (Fig. 7A). Co-expression of any VDCC β subunit caused a significant increase of the facilitation rate during a +100 mV prepulse, compared with the α1B/α2δ currents (Fig. 7A and B). In the absence of expressed VDCC β subunits, the facilitation time constant (τfacil) was 88.7 ms. The most dramatic reduction in τfacil (to 4.5 ms) occurred with expression of the β3 subunit. The rank order for facilitation rate was β3 > β4 (6.9 ms) > β1b (10.3 ms) > C3,4S-β2a (11.9 ms) > β2a (17.5 ms) (Fig. 7B). For α1B/α2δ co-expressed with VDCC β3, the voltage for 50% facilitation by a 50 ms prepulse was +12.8 mV, and saturation occurred at +80 mV. In the absence of co-expressed VDCC β subunits, the equivalent value was +30.9 mV (results not shown). Thus significant facilitation will occur during test steps, particularly in the presence of VDCC β subunits.
Figure 7. The effects of co-expressed VDCC β subunits on kinetics of facilitation and re-inhibition of α1B currents in the presence of quinpirole.
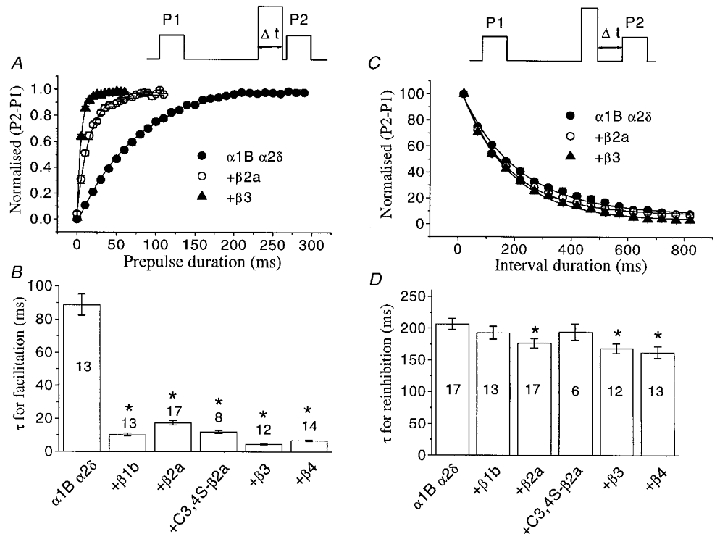
A, top panel, voltage waveform for measurement of facilitation rate in the presence of quinpirole. The duration of the prepulse (Δt) was increased in 5 ms (α1B/α2δ co-expressed with VDCC β subunits) or 10 ms steps (α1B/α2δ alone). The P1 and P2 currents (measured at −10 mV) were then subtracted and the plot of the differences (P2 – P1 values normalised to the maximum) was fitted to a single exponential function. The graph shows examples of the facilitation rate for α1B/α2δ/β3, α1B/α2δ/β2a and α1B/α2δ currents. Continuous lines are the result of exponential fits to the data. B, the histogram of the resulting τ values for facilitation, determined as in A, for all subunit combinations, as shown beneath the histogram bars. C, top panel, voltage waveform for measurement of re-inhibition: following the depolarizing prepulse, the time before the subsequent test pulse (Δt) was progressively increased in 50 ms steps from 20 to 820 ms. Currents were measured and normalised as for the facilitation protocols, and the data were fitted to a single exponential, shown by the continuous lines. All data are from −10 mV test pulses, and data are only shown for α1B/α2δ, α1B/α2δ/β2a and α1B/α2δ/β3. D, histogram of the resulting τ values for re-inhibition, determined as in C, for all subunit combinations, as shown beneath the histogram bars. Numbers on the histogram bars indicate the number of cells for each condition, and statistical significance, compared with α1B/α2δ alone, is indicated by *P < 0.01 (Student's t test).
The rate of re-inhibition of α1B channels at the holding potential, following the depolarising prepulse, which is thought to represent the net rate of re-binding of Gβγ subunits, can be assessed by varying the duration of the interval between the depolarising step and the subsequent test pulse, P2. It has been shown previously that the rate of re-inhibition following facilitation is strongly influenced by the concentration of free Gβγ dimers (Stephens et al. 1998; Delmas et al. 1998). Moreover, a direct relationship was found between the rate of re-inhibition and the concentration of Gβγ, fitting a bimolecular reaction model (Zamponi & Snutch, 1998). In contrast, in the present study, we observed that co-expression of α1B/α2δ with any of the VDCC β subunits did not cause a major change in the rate of re-inhibition (Fig. 7C and D). For the α1B/α2δ combination alone, the time constant for re-inhibition (τreinhib) was 207.0 ms, not significantly different from that with β1b (τreinhib, 193.1 ms) or C3,4S-β2a (τreinhib, 194.5 ms). The rest of the β subunit isoforms induced a small, yet statistically significant, increase in re-inhibition rate, the τreinhib being 176.8 ms for β2a, 168.7 ms for β3 and 162.4 ms for β4.
These results show that a higher facilitation ratio for 50 ms prepulses is primarily associated with a faster facilitation rate, consistent with a faster unbinding rate for the Gβγ dimers. The curves for facilitation demonstrate that by increasing the time of the depolarisation, facilitation reaches a maximum and reverses most of the inhibition. Whereas this process is much slower in the absence of exogenously expressed VDCC β subunits, it also eventually reaches maximal reversal (Figs 6B and 7A).
Comparison of the kinetics of facilitation and re-inhibition of α1B currents, in the absence and presence of co-expressed VDCC β3 subunits, during tonic and agonist-induced modulation
To complement the experiments described above, we have also examined the properties of prepulse facilitation under resting conditions, where there is a low tonic basal level of Gβγ subunits, prior to any application of D2 receptor agonist, to examine whether the facilitation rate at +100 mV is dependent on Gβγ concentration. Currents were examined at −10 mV, before and after application of a depolarising prepulse to +100 mV for the two extreme combinations: α1B/α2δ and α1B/α2δ/β3. The data in Fig. 8A show a comparison, in which facilitation rate was measured before and during quinpirole application in the same cells, expressing either α1B/α2δ or α1B/α2δ/β3. For the α1B/α2δ/β3 combination, there was no difference in the τfacil at +100 mV, in these two conditions, whereas for the α1B/α2δ combination, the facilitation rate was slightly but significantly faster during tonic facilitation, compared with agonist-induced facilitation (Fig. 8B). The marked difference in rate between the presence and absence of over-expressed VDCC β3 subunits was observed under both these conditions (Fig. 8A and B). As expected, the rate of re-inhibition at −100 mV following a prepulse was markedly slower for tonic inhibition (filled bars), than for quinpirole-mediated inhibition (open bars), again compared in the same cells (Fig. 8C and D). Furthermore, a small but consistently faster rate of re-inhibition was observed in the presence, compared to the absence, of co-expressed VDCC β3 subunits, for both tonic and agonist-induced modulation (Fig. 8C and D).
DISCUSSION
An important regulatory feature of VDCC β subunits is to hyperpolarise the activation of Ca2+ channels. Even though this has a partial contribution related to removal of G protein-mediated basal inhibition, such a change in the activation voltage is the result of a direct regulatory action of VDCC β subunits on the α1B Ca2+ channels, since it also applies to α1B constructs insensitive to G proteins (see Page et al. 1998). Conversely, a hallmark for G protein-mediated effects is a depolarisation of current activation. There is currently a broad consensus that the voltage-dependent relief of inhibition resulting from a large depolarising prepulse is partially the result of voltage-dependent unbinding of Gβγ subunits from Ca2+ channels (see Dolphin, 1998 for review). Our data suggest that the actions of the VDCC β and Gβγ subunits are not only opposite, but they also antagonise each other in a voltage-dependent fashion. Furthermore, strong depolarisation will remove the influence of Gβγ, hence facilitating the hyperpolarising effects of VDCC β subunits on the activation of α1B channels. In contrast, the rate of facilitation during a prepulse is dependent on the levels of VDCC β subunits. It is possible that this is because the α1B channels attached to β subunits will adopt a conformation making the Gβγ subunits more susceptible to voltage-dependent unbinding.
In contrast to previous studies, we have found that co-expression of VDCC β subunits caused no generalised reduction of dopamine D2 receptor-mediated inhibition of α1B currents, which maximally reached ∼20% smaller inhibition for β2a, compared with the greater degree of antagonism reported for α1E or α1A subunits (Bourinet et al. 1996; Qin et al. 1998; Roche & Treistman, 1998a). Although this may indicate real differences in the sensitivity of the different α1 subunits to the influence of VDCC β and Gβγ subunits, in previous studies isopotential measurements were made, not taking into account the hyperpolarisation of activation resulting from co-expression of VDCC β subunits. Furthermore, current measurements were generally taken at 50 or 100 ms, instead of 20 ms used in the present study, allowing more time for Gβγ dissociation to occur during the test pulse, and we have found a much higher apparent Gβγ dissociation rate associated with VDCC β subunit co-expression (Fig. 7). For example, measuring currents at the end of the 100 ms pulse instead of at 20 ms would almost double the apparent reduction of inhibition due to β2a at 0 mV in our experiments.
From facilitation rate to facilitation ratio; basal modulation versus agonist-induced modulation
For agonist-mediated inhibitory modulation of α1B, the final amount of facilitation is not correlated with the rate of facilitation, provided the prepulse duration is maximal (compare Figs 5D and 7B). Thus, VDCC β subunits enhance the facilitation rate, but not the maximal facilitation ratio itself. Another important point is that under conditions of tonic modulation, associated with a low tonic level of activated G proteins, the exogenous expression of VDCC β subunits halts voltage-dependent facilitation. Such an apparent contradiction is not due to a distinct dissociation rate for the low level Gβγ subunits present tonically, since we have found that the main difference between tonic and agonist-induced facilitation is a slowing of the re-inhibition rate under tonic modulation conditions (Fig. 8A and B), in good agreement with data from COS-7 cells (Stephens et al. 1998). A possible explanation comes from representation of the Gβγ binding to α1B subunits as a bi-molecular reaction:
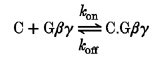 |
where C is a closed state of the calcium channel α1B subunit, kon is the association time constant and koff is the dissociation time constant, both of them likely to have intrinsic voltage dependencies.
At equilibrium, by the law of mass action:
where [Gβγ] and [C.Gβγ] are the concentrations of free and channel-bound Gβγ, respectively, at −100 mV.
| (1) |
thus an increase in Gβγ concentration (agonist-induced compared with basal modulation) will result in a decrease in the time constant for re-inhibition, as previously reported (Stephens et al. 1998; Fig. 8A and B in the present work).
| (2) |
and
| (3) |
therefore the facilitation ratio
| (4) |
As formulated, the facilitation ratio will approach unity (P2/P1 ≈ 1) when the when [Gβγ] is low (basal modulation) and koff is high (by the co-expression of VDCC β subunits). In contrast, P2 > P1 either in the absence of co-expressed VDCC β subunits (low koff value) or in a situation of agonist-induced modulation (high [Gβγ]). Furthermore, according to eqn (4), the co-expression of VDCC β subunits will result in a lower agonist-induced facilitation ratio by increasing koff, unless this effect is voltage dependent, and τreinhib will be smaller under these conditions, for the same reason. These predictions fulfil our experimental observations.
It is also of interest that while both over-recovery and tonic voltage-dependent facilitation provide estimates of tonic G protein-mediated inhibition, they are not identical. Notably, over-recovery does not diminish over the voltage range measured. Over-recovery probably represents a reduction in the tonic level of free Gβγ, associated with the recovery from agonist stimulation. This is supported by the fact that over-expression of either Gαi (Roche & Treistman, 1998b) or Gαo (C. Cantí & A. C. Dolphin, unpublished results) in combination with α1B subunits, induces a complete removal of tonic modulation (both tonic facilitation and over-recovery features) together with an enhancement of the agonist-mediated inhibition. This observation is consistent with the assumption that G protein α subunits act as a sink for tonically free Gβγ, and thus recruit more Gαβγ heterotrimers, which are able to dissociate upon receptor activation. In contrast to over-recovery, tonic facilitation represents the ability of a depolarising prepulse to overcome the modulation induced by the tonic low level of Gβγ dimers, and as such is also reduced at large test depolarisations.
An alternative hypothesis to explain why co-expression of VDCC β subunits diminishes tonic facilitation, despite enhancing the facilitation rate, would be if α1B/α2δ currents were significantly more tonically inhibited than α1B/α2δ/β currents, as suggested by a higher residual facilitation at 0 mV in the absence of β subunits (Fig. 2, traces labelled Prec). This explanation would imply that the agonist-mediated inhibition is underestimated specifically in the absence of β subunits. However, it should be noted that: (1) residual inhibition is voltage dependent, and it is present for all β subunit combinations at more negative test potentials, (2) the percentage over-recovery does not vary significantly between α1B/α2δ and α1B/α2δ/β currents, and (3) Roche & Treistman (1998b) found that co-expression of Gαi2 subunits enhanced agonist-mediated inhibition of the over-recovered α1B currents to a slightly smaller extent than for α1B/β3 currents.
Differences between VDCC β subtypes
The VDCC β2a subunit behaves in a strikingly different manner from the other VDCC β subunit isoforms, in that it depolarises rather than hyperpolarises the steady-state inactivation. These unique properties are also linked to a clear antagonism of G protein-mediated inhibition and to an apparently lower ability to promote the voltage-dependent relief of G protein-mediated inhibition by a strong positive prepulse. Intriguingly, N terminal palmitoylation is responsible for all differential features of β2a, as the β2a mutant in which cysteines 3 and 4 are mutated to serines, and which is not palmitoylated (Qin et al. 1998), shows characteristics close to the other β subunit isoforms, and in particular very close to β1b. At present the molecular mechanism for such differences remains unknown (but see Cens et al. 1999). As palmitoyl residues allow non-transmembrane proteins to anchor to the membrane, a plausible explanation would be that β2a, unlike the other β subunit isoforms, could have the ability to bind to additional domains on the α1B channel by virtue of its palmitate tether.
Conclusions
Our data suggest that receptor-activated high levels of Gβγ subunits result in a functional interference (either direct or allosteric) with the ability of VDCC β subunits to hyperpolarise the voltage-dependent activation of α1B Ca2+ channels. The Gβγ-mediated inhibition of the α1B channels is voltage sensitive, and the effect of VDCC β subunit on the rate of loss of G protein regulation is also dependent on the membrane potential, being observed only during strong depolarisation.
One of the implications arising from such a model is the necessity of the presence of VDCC β subunits for voltage-dependent facilitation and reciprocal inhibition to occur. With respect to this point, we have observed in another study that voltage-dependent G protein-mediated inhibition of α1B channels is absent from COS-7 cells unless a VDCC β subunit is co-expressed (Meir et al. 2000). The endogenous level of VDCC β subunit protein appears to be negligible in COS-7 cells (Meir et al. 2000), whereas an endogenous β3 is present in Xenopus oocytes (Tareilus et al. 1997). Future work will determine how the interaction between Gβγ dimers and Ca2+ channel β subunits with α1B results in functional antagonism at the molecular level.
Acknowledgments
We thank the following for generous gifts of cDNAs: T. Snutch (UBC, Vancouver, Canada), rat β1b; H. Chin (NIH, Bethesda, USA), rat α2δ-1; Y. Mori (Seriken, Okazaki, Japan), rabbit α1B; E. Perez-Reyes (University of Virginia, USA), rat β2a, β3 and β4; P. G. Strange (Reading, UK), rat D2 receptor; Genetics Institute (Cambridge, MA, USA), pMT2. We also gratefully acknowledge financial support from The Wellcome Trust, the European Community (Marie Curie Fellowship to C.C.) and the MRC (UK). We thank N. Balaguero for technical assistance.
References
- Bourinet E, Soong TW, Stea A, Snutch TP. Determinants of the G protein-dependent opioid modulation of neuronal calcium channels. Proceedings of the National Academy of Sciences of the USA. 1996;93:1486–1491. doi: 10.1073/pnas.93.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brice NL, Berrow NS, Campbell V, Page KM, Brickley K, Tedder I, Dolphin AC. Importance of the different β subunits in the membrane expression of the α1A and α2 calcium channel subunits: studies using a depolarisation-sensitive α1A antibody. European Journal of Neuroscience. 1997;9:749–759. doi: 10.1111/j.1460-9568.1997.tb01423.x. [DOI] [PubMed] [Google Scholar]
- Campbell V, Berrow N, Brickley K, Page K, Wade R, Dolphin AC. Voltage-dependent calcium channel β-subunits in combination with α1 subunits have a GTPase activating effect to promote hydrolysis of GTP by Gαo in rat frontal cortex. FEBS Letters. 1995;370:135–140. doi: 10.1016/0014-5793(95)00813-o. [DOI] [PubMed] [Google Scholar]
- Cantí C, Page KM, Stephens GJ, Dolphin AC. Identification of residues in the N-terminus of α1B critical for inhibition of the voltage-dependent calcium channel by Gβγ. Journal of Neuroscience. 1999;19:6855–6864. doi: 10.1523/JNEUROSCI.19-16-06855.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cens T, Restituito S, Galas S, Charnet P. Voltage and calcium use the same molecular determinants to inactivate calcium channels. Journal of Biological Chemistry. 1999;274:5483–5490. doi: 10.1074/jbc.274.9.5483. [DOI] [PubMed] [Google Scholar]
- Cens T, Restituito S, Vallentin A, Charnet P. Promotion and inhibition of L-type Ca2+ channel facilitation by distinct domains of the subunit. Journal of Biological Chemistry. 1998;273:18308–18315. doi: 10.1074/jbc.273.29.18308. [DOI] [PubMed] [Google Scholar]
- Chien AJ, Hosey MM. Post-translational modifications of β subunits of voltage-dependent calcium channels. Journal of Bioenergetics and Biomembranes. 1998;30:377–386. doi: 10.1023/a:1021941706726. [DOI] [PubMed] [Google Scholar]
- De Waard M, Campbell KP. Subunit regulation of the neuronal α1A Ca2+ channel expressed in Xenopus oocytes. The Journal of Physiology. 1995;485:619–634. doi: 10.1113/jphysiol.1995.sp020757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Waard M, Liu HY, Walker D, Scott VES, Gurnett CA, Campbell KP. Direct binding of G protein βγ complex to voltage-dependent calcium channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- Delmas P, Brown DA, Dayrell M, Abogadie FC, Caulfield MP, Buckley NJ. On the role of endogenous G protein βγ subunits in N-type Ca2+ current inhibition by neurotransmitters in rat sympathetic neurones. The Journal of Physiology. 1998;506:319–329. doi: 10.1111/j.1469-7793.1998.319bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. Mechanisms of modulation of voltage-dependent calcium channels by G proteins. The Journal of Physiology. 1998;506:3–11. doi: 10.1111/j.1469-7793.1998.003bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends in Neurosciences. 1995;18:89–98. [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G protein βγ subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G protein βgamma subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Jones LP, Wei SK, Yue DT. Mechanism of auxiliary subunit modulation of neuronal α1E calcium channels. Journal of General Physiology. 1998;112:125–143. doi: 10.1085/jgp.112.2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meir A, Bell DC, Stephens GJ, Page KM, Dolphin AC. Calcium channel β subunit promotes voltage-dependent modulation of α1B by Gβγ. Biophysical Journal. 2000 doi: 10.1016/S0006-3495(00)76331-4. (in the Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page KM, Cantí C, Stephens GJ, Berrow NS, Dolphin AC. Identification of the amino terminus of neuronal Ca2+ channel α1 subunits α1B and α1E as an essential determinant of G protein modulation. Journal of Neuroscience. 1998;18:4815–4824. doi: 10.1523/JNEUROSCI.18-13-04815.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin N, Platano D, Olcese R, Costantin JL, Stefani E, Birnbaumer L. Unique regulatory properties of the type 2a Ca2+ channel β subunit caused by palmitoylation. Proceedings of the National Academy of Sciences of the USA. 1998;95:4690–4695. doi: 10.1073/pnas.95.8.4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin N, Platano D, Olcese R, Stefani E, Birnbaumer L. Direct interaction of Gβγ with a C terminal Gβgamma binding domain of the calcium channel α1 subunit is responsible for channel inhibition by G protein coupled receptors. Proceedings of the National Academy of Sciences of the USA. 1997;94:8866–8871. doi: 10.1073/pnas.94.16.8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche JP, Treistman SN. The Ca2+ channel β3 subunit differentially modulates G protein sensitivity of α1A and α1B Ca2+ channels. Journal of Neuroscience. 1998a;18:878–886. doi: 10.1523/JNEUROSCI.18-03-00878.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche JP, Treistman SN. The calcium channel β3 subunit enhances voltage-dependent relief of G protein inhibition induced by muscarinic receptor activation and Gβγ. Journal of Neuroscience. 1998b;18:4883–4890. doi: 10.1523/JNEUROSCI.18-13-04883.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekter LR, Taussig R, Gillard SE, Miller RJ. Regulation of human neuronal calcium channels by G protein βgamma subunits expressed in human embryonic kidney 293 cells. Molecular Pharmacology. 1997;52:282–291. doi: 10.1124/mol.52.2.282. [DOI] [PubMed] [Google Scholar]
- Simen AA, Miller RJ. Structural features determining differential receptor regulation of neuronal Ca2+ channels. Journal of Neuroscience. 1998;18:3689–3698. doi: 10.1523/JNEUROSCI.18-10-03689.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens GJ, Brice NL, Berrow NS, Dolphin AC. Facilitation of rabbit α1B calcium channels: involvement of endogenous Gβγ subunits. The Journal of Physiology. 1998;509:15–27. doi: 10.1111/j.1469-7793.1998.015bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens GJ, Page KM, Burley JR, Berrow NS, Dolphin AC. Functional expression of rat brain cloned α1E calcium channels in COS-7 cells. Pflügers Archiv. 1997;433:523–532. doi: 10.1007/s004240050308. [DOI] [PubMed] [Google Scholar]
- Swick AG, Janicot M, Cheneval-Kastelic T, McLenithan JC, Lane DM. Promoter-cDNA-directed heterologous protein expression in Xenopus laevis oocytes. Proceedings of the National Academy of Sciences of the USA. 1992;89:1812–1816. doi: 10.1073/pnas.89.5.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tareilus E, Roux M, Qin N, Olcese R, Zhou JM, Stefani E, Birnbaumer L. A Xenopus oocyte β subunit: Evidence for a role in the assembly/expression of voltage-gated calcium channels that is separate from its role as a regulatory subunit. Proceedings of the National Academy of Sciences of the USA. 1997;94:1703–1708. doi: 10.1073/pnas.94.5.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker D, Bichet D, Geib S, Mori E, Cornet V, Snutch TP, Mori Y, De Waard M. A new-β subtype-specific interaction in α1A subunit controls P/Q type Ca2+ channel activation. Journal of Biological Chemistry. 1999;274:12383–12390. doi: 10.1074/jbc.274.18.12383. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Snutch TP. Decay of prepulse facilitation of N type calcium channels during G protein inhibition is consistent with binding of a single Gβγ subunit. Proceedings of the National Academy of Sciences of the USA. 1998;95:4035–4039. doi: 10.1073/pnas.95.7.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]