

Abstract
C-protein is a major component of muscle thick filaments whose function is unknown. We have examined for the first time the role of the regulatory binding domain of C-protein in modulating contraction and intracellular Ca2+ concentration ([Ca2+]i) in intact cardiac myocytes.
Rat ventricular myocytes were reversibly permeabilised with the pore-forming toxin streptolysin O. Myosin S2 (which binds to the regulatory domain of C-protein) was introduced into cells during permeabilisation to compete with the endogenous C-protein-thick filament interaction.
Introduction of S2 into myocytes increased contractility by ∼30%, significantly lengthened the time to peak of the contraction and the time to half-relaxation, but had no effect on [Ca2+]i transient amplitude.
Our data are consistent with increased myofilament Ca2+ sensitivity when there is reduced binding of C-protein to myosin near the head-tail junction.
We propose that the effects of introducing S2 into intact cardiac cells can be equated with the consequences of selectively phosphorylating C-protein in vivo, and that the regulation of contraction by C-protein is mediated by the effects of crossbridge cycling on the Ca2+ affinity of troponin C.
C-protein, also known as myosin-binding protein-C, is a major component of thick filaments in vertebrate striated muscle. Although the function of C-protein is not known, there is compelling evidence for its physiological importance in the heart. C-protein is one of the proteins phosphorylated following b-adrenergic stimulation (Jeacocke & England, 1980) and mutations in the gene which codes for C-protein produce familial hypertrophic cardiomyopathy (FHC) (Carrier et al. 1998).
In the heart, C-protein has been proposed to have both structural and regulatory functions (see Bennett et al. 1999; Winegrad, 1999). A structural role was suggested by its major influence on the 3-dimensional organisation of synthetic thick filaments (Moos et al. 1975). A regulatory role was indicated by an increase in the Ca2+ sensitivity of tension following extraction of the majority of the C-protein from skinned cardiac myocytes (Hofmann et al. 1991), in skinned cardiac muscle from transgenic mice expressing C-terminally truncated C-protein (Yang et al. 1998), and in skinned skeletal muscle exposed to soluble N-terminal fragments of C-protein (Kunst et al. 2000).
C-protein has two myosin-binding sites: a C-terminal site which interacts with light meromyosin and an N-terminal site which binds to myosin subfragment 2 (S2) (Starr & Offer, 1978; Okagaki et al. 1993). S2 is not known to interact with any other protein. In cardiac muscle, the N-terminal binding domain of C-protein (the regulatory domain), which binds to S2 close to the crossbridge (Gruen et al. 1999), has three sites which are phosphorylated following b-adrenergic stimulation (Jeacocke & England, 1980; Hartzell & Glass, 1984; Winegrad, 1999). Evidence suggests that the regulatory domain is of physiological importance as FHC has been shown to be associated with mutations in the S2 region of b-myosin (Vikstrom & Leinwand, 1996).
Many of the experiments carried out in an attempt to elucidate the function of C-protein have been performed in skinned muscle fibres or skinned cells in which soluble proteins and second messengers are lost from the normal milieu of the myofilaments and excitation-contraction coupling does not occur. Transgenic animals have been used (Yang et al. 1998), but because the structural organisation of the myofilaments is changed in these animals it is difficult to separate out the dynamic role of C-protein in regulating contractility.
The present study is the first to investigate the role of C-protein in the intact cardiac cell. Our experiments were performed with permeabilised and resealed ventricular myocytes into which the S2 portion of myosin was introduced. It was our intention to competitively reduce the endogenous C-protein-S2 interaction in order to explore the physiological importance of the binding of C-protein close to the crossbridge.
METHODS
Reversible cell permeabilisation
Male Wistar rats (200–250 g) were killed by cervical dislocation following stunning and single ventricular myocytes were isolated as described by Frampton et al. (1991). Cells were permeabilised and resealed according to the method of Fawcett et al. (1998). The basis of the permeabilisation procedure is the pore-forming toxin streptolysin O. S2 (100 kDa, 0.11 mg ml−1) was included during the permeabilisation procedure in order to allow it to enter cells. Control experiments were performed with bovine serum albumin (BSA; 67 kDa, 0.074 mg ml−1) included in place of S2. The method of Fawcett et al. (1998) has been shown to permeabilise around 60% of cardiac myocytes. Cells in which contraction and [Ca2+]i were recorded were morphologically normal with a rod-shaped appearance and visible regular striations.
S2 preparation and rhodamine labelling for confocal microscopy
The soluble S2 fragment of myosin was prepared from skeletal muscle of rabbits (killed by an overdose of sodium pentobarbitone, 70 mg kg−1i.v.) according to the method of Quinlan & Stewart (1987). S2 was dialysed overnight into permeabilisation buffer (1.1 mg ml−1). The N-terminal 126 amino acid residues of myosin S2 to which C-protein binds are virtually identical regardless of the myosin isoform or vertebrate species (Gruen et al. 1999). In order to visualise the ability of the S2 molecule to penetrate the cell membrane, tetramethylrhodamine isothiocyanate-labelled S2 (TRITC-S2) was included during permeabilisation. Control cells were permeabilised in the absence of fluorescent S2.
Quiescent cells permeabilised in the presence and absence of TRITC-S2 were imaged by confocal laser microscopy (Leica True Confocal Scanner SP). A krypton laser provided excitation light at 568 nm and emissions were collected between 580 and 620 nm. On-line quantitative analysis software (TCS NT version 1.6.551; Leica) was used to provide a measure of the intensity of the fluorescence of a 2 μm thick section taken through the centre of the cell.
Measurement of contraction and [Ca2+]i
Contraction and [Ca2+]i were measured as described in Calaghan et al. (1998). Cells were electrically stimulated at 0.5 Hz, and cell shortening was monitored using a video edge detection system (Crescent Electronics, UT, USA). [Ca2+]i was measured in cells loaded with fura-2 AM (1.5 μm, Molecular Probes). Experiments were performed at 22–24°C.
Statistical analysis
Data are expressed as means ± s.e.m. of n observations. ANOVA followed by Student's t test with Bonferroni correction was used to test for statistical significance (Bailey, 1977).
RESULTS
Figure 1 shows representative confocal images obtained from rat ventricular myocytes permeabilised in the presence and absence of TRITC-S2. Fluorescence is only visible in the cell permeabilised in the presence of TRITC-S2. Only one of a sample of nine cells permeabilised in the presence of TRITC-S2 showed a level of fluorescence as low as that seen in cells permeabilised in the absence of a fluorescent marker, confirming the presence of S2 in a large majority of cells. These data show that TRITC-S2 was able to enter the cell during permeabilisation. We did not detect a repetitive staining pattern in any of the cells loaded with TRITC-S2.
Figure 1.
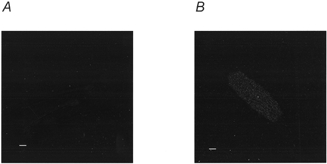
Confocal image of cells permeabilised in the presence and absence of fluorescent S2
Sections were taken through the mid-line plane of representative rat ventricular myocytes following streptolysin O permeabilisation in the absence (A) and in the presence (B) of tetramethylrhodamine isothiocyanate-labelled S2. Permeabilisation and recording of images were performed in parallel. Scale bars represent 10 μm.
Figure 2 shows the effect of permeabilisation in the presence and absence of S2 on contraction in myocytes. Shortening in control, unpermeabilised cells was measured in parallel. Permeabilisation per se had no effect (P > 0.05) on the resting cell length, degree of shortening, time to peak of the contraction, or time to half-relaxation. In cells which were permeabilised in the presence of S2, shortening was significantly greater (P < 0.05) by ∼30% compared with that seen in control permeabilised cells. The time to peak and time to half-relaxation were also longer (P < 0.05) by ∼20% in S2 permeabilised cells compared with control permeabilised cells. By contrast, in a set of experiments in which we examined the effect of including BSA during permeabilisation in place of S2 (at the same molar concentration), we saw no significant difference (P > 0.05) in shortening, time to peak of the contraction or time to half-relaxation between control permeabilised (n = 18) and BSA permeabilised (n = 17) cells (data not shown).
Figure 2.

The effect of permeabilisation in the presence of the S2 fragment of myosin on contraction of rat ventricular myocytes
A, fast time base recordings of representative contractions averaged from 10 recordings obtained in control permeabilised (CP) and S2 permeabilised (S2) myocytes expressed as the change in cell length as a percentage of resting length (CP, 121 μm; S2, 106 μm). B, mean data showing resting cell length (Length), shortening as a percentage of resting cell length (Short), time to peak of the contraction (TTP) and time for half-relaxation (t1/2) in control cells (□; n = 37), streptolysin O-permeabilised cells ( ;n = 60) and cells permeabilised in the presence of the S2 fragment of myosin (
;n = 60) and cells permeabilised in the presence of the S2 fragment of myosin ( ;n = 60). All values are expressed as means +s.e.m. as a percentage of that recorded in control cells. Control values for the parameters were: cell length, 108 ± 3 μm; shortening, 6.9 ± 0.7%; time to peak, 161 ± 7 ms; time to half-relaxation, 85 ± 5 ms. *P < 0.05; **P < 0.01 compared with control permeabilised cells (Student's t test with Bonferroni correction).
;n = 60). All values are expressed as means +s.e.m. as a percentage of that recorded in control cells. Control values for the parameters were: cell length, 108 ± 3 μm; shortening, 6.9 ± 0.7%; time to peak, 161 ± 7 ms; time to half-relaxation, 85 ± 5 ms. *P < 0.05; **P < 0.01 compared with control permeabilised cells (Student's t test with Bonferroni correction).
Figure 3 shows the effect of permeabilisation in the presence and absence of S2 on the [Ca2+]i transient. Permeabilisation per se increased diastolic [Ca2+]i(P < 0.05), reduced the amplitude of the [Ca2+]i transient (P < 0.05) without changing systolic [Ca2+]i, and shortened the time to peak of the transient (P < 0.05). Permeabilisation in the presence of S2 did not significantly alter (P > 0.05) diastolic or systolic [Ca2+]i from that seen in cells permeabilised in the absence of S2. Furthermore, we saw no increase in the amplitude of the [Ca2+]i transient (P > 0.05) which might have accounted for the increase in contractility seen in S2 permeabilised cells. The only significant effect of permeabilisation in the presence of S2 compared with that in its absence was a 20% increase (P < 0.05) in the time to peak of the transient.
Figure 3.
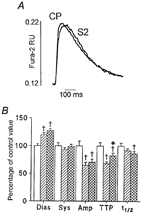
The effect of permeabilisation in the presence of the S2 fragment of myosin on [Ca2+]i transients in fura-2-loaded rat ventricular myocytes
A, fast time base recordings of representative [Ca2+]i transients obtained in control permeabilised (CP) and S2 permeabilised (S2) myocytes. [Ca2+]i is expressed as fura-2 fluorescence ratio units (RU). B, mean data showing diastolic [Ca2+]i (Dias), systolic [Ca2+]i (Sys), amplitude of the [Ca2+]i transient (Amp), time to peak of the transient (TTP) and time to half-recovery of the transient (t1/2) in control cells (□; n = 31), streptolysin O-permeabilised cells ( ; n = 39) and cells permeabilised in the presence of S2 (
; n = 39) and cells permeabilised in the presence of S2 ( ;n = 39). All values are expressed as means +s.e.m. as a percentage of that recorded in control cells. Control values for the parameters were: diastolic [Ca2+]i, 0.111 ± 0.004 RU; systolic [Ca2+]i, 0.216 ± 0.012 RU; [Ca2+]i transient amplitude, 0.105 ± 0.009 RU; time to peak, 94 ± 6 ms; time to half-recovery, 328 ± 18 ms. †P < 0.05 compared with control (unpermeabilised) cells; *P < 0.05 compared with control permeabilised cells (Student's t test with Bonferroni correction).
;n = 39). All values are expressed as means +s.e.m. as a percentage of that recorded in control cells. Control values for the parameters were: diastolic [Ca2+]i, 0.111 ± 0.004 RU; systolic [Ca2+]i, 0.216 ± 0.012 RU; [Ca2+]i transient amplitude, 0.105 ± 0.009 RU; time to peak, 94 ± 6 ms; time to half-recovery, 328 ± 18 ms. †P < 0.05 compared with control (unpermeabilised) cells; *P < 0.05 compared with control permeabilised cells (Student's t test with Bonferroni correction).
DISCUSSION
C-protein was first identified some three decades ago and yet the structural or regulatory roles it plays remain unclear. In the present study we used intact cardiac myocytes to examine the effect on contraction and [Ca2+]i of a reduction in the binding of C-protein to endogenous myosin S2.
S2 was introduced into myocytes using the technique of reversible cell permeabilisation, which has been used previously to introduce large molecules into both cultured and cardiac cells. The pores formed by the cholesterol-binding cytolysin streptolysin O have been shown to exceed 30 nm in diameter (Buckingham & Duncan, 1983) and fluorescein isothiocyanate (FITC)-labelled dextrans (148 kDa), as well as FITC-labelled BSA, have been shown to enter permeabilised cardiac myocytes (Fawcett et al. 1998). S2 prepared by the method of Quinlan & Stewart (1987) is a rod 60 nm by 2 nm with a mass of around 100 kDa and the results of our confocal imaging show that TRITC-labelled S2 is also able to enter myocytes during permeabilisation. We did not observe the repetitive staining pattern expected for binding to the C-protein zone with TRITC-S2. This may be because we could image only quiescent cells in which C-protein may bind added S2 weakly, or because excess TRITC-S2 within the cell obscured a specific pattern of binding.
To date, much of the data regarding the role of C-protein in the heart has been collected using skinned skeletal or cardiac muscle. Our experiments are the first to investigate its role in intact cardiac myocytes in which the spatial and temporal interactions between Ca2+ and the myofilaments occur during a twitch contraction. Permeabilisation per se did not affect the contractility of the cardiac cell and, although we saw some changes in the [Ca2+]i transient as a result of the permeabilisation procedure, the normal process of excitation-contraction coupling was in operation. Cells appeared morphologically normal and we did not detect any gross sarcomeric disorganisation as seen in studies using transgenic animals expressing truncated C-protein (Yang et al. 1998). Therefore we ascribe the observed effects of S2 to the regulatory (rather than structural) actions of C-protein.
In cells permeabilised in the presence of S2, contractility was enhanced. We saw a significant increase in the magnitude of shortening, which was accompanied by a slowing of both the time to peak of the contraction and the time to half-relaxation. These effects appear to be specific to S2, as introduction of BSA into cells during permeabilisation had no significant effect on either the amplitude or the kinetics of the twitch.
In cells with added S2 there was no corresponding increase in the magnitude of the [Ca2+]i transient to account for the increase in contractility. However, we saw some changes in the time course of the transient; there was a slight increase in the time to peak and a slight (insignificant) decrease in the time to half-decay of the transient. Taken together, the data are consistent with an increase in the sensitivity of the myofilaments to Ca2+ in cells containing added S2. Interestingly, the changes in the kinetics of the contraction and Ca2+ transient are similar to those seen when myofilament sensitivity is increased through an increase in the affinity of the actin-binding protein troponin C for Ca2+ (see Calaghan & White, 1999).
Kunst et al. (2000) recently showed that the addition of soluble fragments of C-protein containing the regulatory domain (C1C2) to skeletal muscle increased Ca2+ sensitivity and decreased active force. In skinned cardiac muscle from transgenic animals expressing C-terminally truncated C-protein, an increase in myofilament sensitivity has also been observed (Yang et al. 1998). These data have been interpreted as evidence that the binding of the regulatory domain of C-protein to S2 alone is sufficient to cause changes in myofilaments which may increase their Ca2+ sensitivity. However, there are data which cannot be explained in this way. Extraction of the entire C-protein molecule from skinned cardiac cells has been shown to increase myofilament Ca2+ sensitivity (Hofmann et al. 1991). Furthermore, in the present study, introduction of S2 into the intact cardiac cell to compete with the endogenous C-protein-S2 interaction in the thick filament also increased myofilament sensitivity. Taken together, these data suggest that the binding of endogenous C-protein to myosin S2 of the thick filament acts to reduce the Ca2+ sensitivity, and that any treatment which removes this interaction will therefore result in an increase in Ca2+ sensitivity. Whether this interaction is blocked by extraction of the entire C-protein molecule, by expression of C-terminally truncated C-protein fragments, by addition of soluble C1C2 fragments which block the S2 binding site, or by addition of S2 which interacts with the N-terminus of C-protein, the consequence should be the same. The tethering actions of C-protein linking S2 of one myosin molecule with the light meromyosin portion of another may be of importance in this effect. This proposed impact of C-protein modifications on crossbridge behaviour is summarised schematically in Fig. 4.
Figure 4.
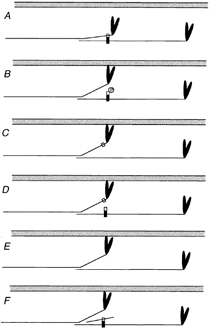
Unifying model for the impact of C-protein modifications on crossbridge behaviour
Each panel shows a schematic representation of a thin filament (grey) and a pair of myosin molecules from a thick filament that are axially staggered appropriately to be crosslinked by a C-protein molecule (vertical rectangle). The C-terminal part of the C-protein (black) binds to the light meromyosin part of the tail, the N-terminal part (white) to S2. C-protein is too short (∼40 nm) to span the distance (> 50 nm) between the binding sites in the S2 and light meromyosin portions of a single myosin molecule, therefore it crosslinks molecules. A shows the quiescent state, in which we propose that S2 is tethered by C-protein. In B, phosphorylation of C-protein releases the S2 part (Weisberg & Winegrad, 1996), allowing the myosin heads greater ease of interaction with the thin filament, thereby promoting thin filament activation. C, C-protein, truncated at the C-terminus (Yang et al. 1998) cannot crosslink. D, exogenous C1C2 domains compete with endogenous C-protein, releasing the S2 part, but do not compete if phosphorylated (Kunst et al. 2000). E, extraction of C-protein (Hofmann et al. 1991) removes the tether. F, exogenous S2 can compete with the S2 part of myosin (this work), releasing the tether. C–F thus all mimic B. The model predicts that weakening of the interactions between myosin and C-protein caused by mutations in the S2 part of myosin heavy chain will have effects similar to these other interventions.
The regulatory domain of C-protein binds to the myosin tail close to the crossbridge, and it has been suggested that the phosphorylation of C-protein regulates contractile function through an effect on crossbridge cycling (Winegrad, 1999). In isolated thick filaments from cardiac muscle, phosphorylation of C-protein extends the crossbridges from the backbone of the filaments (Weisberg & Winegrad, 1996). This may alter the rate of attachment or detachment of crossbridges.
When C-protein is phosphorylated, evidence suggests that the interaction between the regulatory domain and S2 does not occur. In vitro, trisphosphorylation of the C1C2 fragment of C-protein abolishes its interaction with myosin S2 (Gruen et al. 1999) and the effects of C1C2 on contractile force and Ca2+ sensitivity in skinned skeletal muscle are abolished by phosphorylation (Kunst et al. 2000). Thus in vivo, the effects of C-protein phosphorylation and addition of S2 may be similar, and result in increased myofilament Ca2+ sensitivity (see Fig. 4).
Physiologically, b-adrenergic stimulation results in phosphorylation of several myocardial proteins including the Ca2+ channel, phospholamban, troponin I and C-protein. It is difficult to dissect out the contributions of the various proteins to the consequences of b-adrenergic stimulation. In the present study, by adding S2 to the intact myocyte, we can see for the first time an effect equivalent to selective phosphorylation of endogenous C-protein in vivo. The increase in myofilament sensitivity observed with added S2 could be ascribed to an increase in the affinity of troponin C (TnC) for Ca2+. Evidence suggests that the effects of added S2 on myofilament sensitivity are likely to be due to a change in crossbridge cycling. If myosin heads spend a greater fraction of their cycle attached to actin, an increase in TnC affinity for Ca2+ would be expected, as strong-binding heads increase TnC affinity (Rice et al. 1999). This would result from changes in troponin conformation brought about by tropomyosin movement, induced by strong-binding crossbridges (Vibert et al. 1997). Thus this may be the mechanism by which myofilament Ca2+ sensitivity is increased in the cardiac myocyte when the regulatory domain of C-protein releases the myosin tail.
Acknowledgments
This work was sponsored by the British Heart Foundation.
References
- Bailey BJR. Tables of the Bonferroni t statistic. Journal of the Americal Statistical Association. 1977;72:469–478. [Google Scholar]
- Bennett PM, Fürst DO, Gautel M. The C-protein (myosin binding protein C) family: Regulators of contraction and sarcomere formation? Reviews of Physiology Biochemistry and Pharmacology. 1999;138:203–234. doi: 10.1007/BFb0119628. [DOI] [PubMed] [Google Scholar]
- Buckingham L, Duncan JL. Approximate dimensions of membrane lesions produced by streptolysin-S and streptolysin-O. Biochimica et Biophysica Acta. 1983;729:115–122. doi: 10.1016/0005-2736(83)90462-5. [DOI] [PubMed] [Google Scholar]
- Calaghan SC, White E. The role of calcium in the response of cardiac muscle to stretch. Progress in Biophysics and Molecular Biology. 1999;71:59–90. doi: 10.1016/s0079-6107(98)00037-6. [DOI] [PubMed] [Google Scholar]
- Calaghan SC, White E, Colyer J. Co-ordinated changes in cAMPphosphorylated phospholamban, Ca2+ and contraction following b-adrenergic stimulation of rat heart. Pflugers Archiv. 1998;436:948–956. doi: 10.1007/s004240050728. [DOI] [PubMed] [Google Scholar]
- Carrier L, Bonne G, Schwartz K. Cardiac myosin-binding protein C and hypertrophic cardiomyopathy. Trends in Cardiovascular Medicine. 1998;8:151–157. doi: 10.1016/S1050-1738(97)00144-8. [DOI] [PubMed] [Google Scholar]
- Fawcett JM, Harrison SM, Orchard CH. A method for reversible permeabilisation of isolated rat ventricular myocytes. Experimental Physiology. 1998;83:293–303. doi: 10.1113/expphysiol.1998.sp004114. [DOI] [PubMed] [Google Scholar]
- Frampton JE, Orchard CH, Boyett MR. Diastolicsystolic and sarcoplasmic reticulum [Ca2+] during inotropic interventions in isolated rat ventricular myocytes. Journal of Physiology. 1991;437:351–375. doi: 10.1113/jphysiol.1991.sp018600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruen M, Prinz H, Gautel M. cAPK-phosphorylation controls the interaction of the regulatory domain of cardiac myosin-binding protein C with myosin S2 in an on-off fashion. FEBS Letters. 1999;453:254–259. doi: 10.1016/s0014-5793(99)00727-9. [DOI] [PubMed] [Google Scholar]
- Hartzell C, Glass D. Phosphorylation of purified cardiac muscle C-protein by purified cAMP-dependent and endogenous Ca2+-calmodulin-dependent protein kinases. Journal of Biological Chemistry. 1984;259:15587–15596. [PubMed] [Google Scholar]
- Hofmann PA, Hartzell HC, Moss RL. Alterations in Ca sensitive tension due to partial extraction of C protein from rat skinned cardiac myocytes and rabbit skeletal muscle fibres. Journal of General Physiology. 1991;97:1141–1163. doi: 10.1085/jgp.97.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeacocke SA, England PJ. Phosphorylation of a myofibrillar protein of Mr 150 000 in perfused rat heartand the tentative identification of this as C-protein. FEBS Letters. 1980;22:129–132. doi: 10.1016/0014-5793(80)80418-2. [DOI] [PubMed] [Google Scholar]
- Kunst G, Kress KR, Gruen M, Uttenweiler D, Gautel M, Fink RHA. Myosin binding protein Ca phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circulation Research. 2000;86:51–58. doi: 10.1161/01.res.86.1.51. [DOI] [PubMed] [Google Scholar]
- Moos C, Offer G, Starr R, Bennett P. Interaction of C-protein with myosinmyosin rod and light meromyosin. Journal of Molecular Biology. 1975;97:1–9. doi: 10.1016/s0022-2836(75)80017-9. [DOI] [PubMed] [Google Scholar]
- Okagaki T, Weber FE, Fischman DA, Vaughan KT, Mikawa T, Reinach FC. The major myosin-binding domain of skeletal muscle MyBP-C (C protein) resides in the COOH-terminalimmunoglobulin C2 motif. Journal of Cell Biology. 1993;123:619–626. doi: 10.1083/jcb.123.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan RA, Stewart M. Crystalline tubes of myosin subfragment-2 showing the coiled-coil and molecular interaction geometry. Journal of Cell Biology. 1987;105:403–415. doi: 10.1083/jcb.105.1.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice JJ, Winslow RL, Hunter WC. Comparison of putative co-operative mechanisms in cardiac muscle: length dependence and dynamic responses. American Journal of Physiology. 1999;276:H1734–1754. doi: 10.1152/ajpheart.1999.276.5.H1734. [DOI] [PubMed] [Google Scholar]
- Starr R, Offer G. The interaction of C-protein with heavy meromyosin and subfragment-2. Biochemical Journal. 1978;171:813–816. doi: 10.1042/bj1710813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vibert P, Craig R, Lehman W. Steric-model for activation of muscle thin filaments. Journal of Molecular Biology. 1997;266:8–14. doi: 10.1006/jmbi.1996.0800. [DOI] [PubMed] [Google Scholar]
- Vikstrom KL, Leinwand LA. Contractile protein mutations and heart disease. Current Opinion in Cell Biology. 1996;8:87–105. doi: 10.1016/s0955-0674(96)80053-6. [DOI] [PubMed] [Google Scholar]
- Weisberg S, Winegrad S. Alteration of myosin cross bridges by phosphorylation of myosin-binding protein C in cardiac muscle. Proceedings of the National Academy of Sciences of the USA. 1996;93:8999–9003. doi: 10.1073/pnas.93.17.8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winegrad S. Cardiac myosin binding protein C. Circulation Research. 1999;84:1117–1126. doi: 10.1161/01.res.84.10.1117. [DOI] [PubMed] [Google Scholar]
- Yang Q, Sanbe A, Osinka H, Hewett TE, Klevitsky R, Robbins J. A mouse model of myosin binding protein C human familial hypertrophic cardiomyopathy. Journal of Clinical Investigation. 1998;102:1292–1300. doi: 10.1172/JCI3880. [DOI] [PMC free article] [PubMed] [Google Scholar]