

Abstract
The permeability response to acutely applied bradykinin and [des-Arg9]-bradykinin on single cerebral venular capillaries has been investigated using the low molecular mass fluorescent dyes Lucifer Yellow and Sulforhodamine B with the single vessel occlusion technique.
When bradykinin was applied repeatedly for up to 2 h, the permeability increase was small and reversible for concentrations that ranged from 5 nm to 50 μm.
The logEC50 of the permeability response to bradykinin was −5.3 ± 0.15 (logm; mean ± s.e.m.). This was reduced to −6.37 ± 0.24 with the angiotensin-converting enzyme inhibitor captopril, to −6.33 ± 0.19 with the neutral endopeptidase inhibitor phosphoramidon and to −7.3 ± 0.20 with captopril and phosphoramidon combined.
The permeability response to bradykinin was blocked by the bradykinin B2 receptor antagonist HOE 140, by inhibition of the Ca2+-independent phospholipase A2, by the scavenging of free radicals, or by inhibition of both cyclo-oxygenase and lipoxygenase in combination. Block of Ca2+ entry channels with SKF 96365 had no effect on the response.
Application of [des-Arg9]-bradykinin also increased permeability over the concentration range 5 nm to 50 μm, with a logEC50 of −5.6 ± 0.37. This response was not affected by free radical scavenging, but was completely blocked by the histamine H2 receptor blocker cimetidine.
These results imply that the acute permeability response to bradykinin is mediated via the release of arachidonic acid, which is acted on by cyclo-oxygenase and lipoxygenase resulting in the formation of free radicals, and that the response to [des-Arg9]-bradykinin is mediated via histamine.
Traumatic brain injury, stroke, and the active phase of multiple sclerosis result in blood-brain barrier disruption, and sometimes lead to a life-threatening cerebral oedema. The mechanisms of this disruption are still unclear but there is evidence that inflammatory mediators are involved, and it has been suggested recently that bradykinin plays an important role in the cerebral oedema that follows stroke and trauma (Narotam et al. 1998; and see Raidoo & Bhoola, 1998). Furthermore, treatment with a bradykinin B2 receptor antagonist following experimental reversible ischaemia reduced the oedema and infarct volume (Relton et al. 1997), and patients who were operated on for cerebral arterial stenosis made better progress if preoperative cerebral kinin formation was low (Makevnina et al. 1994). The development of non-hydrolysable bradykinin analogues currently under investigation for facilitation of the passage of therapeutic agents into the brain (Jolliet-Riant & Tillement, 1999) additionally highlights the importance of bradykinin in modulating the blood-brain barrier.
Previous experiments on the effects of bradykinin application to the brain have indicated that there is often, but not always, extravasation of marker dyes (e.g. Unterberg et al. 1984; Wahl et al. 1985). It is possible that this dye leakage is secondary to increased vascular pressure, since hypertension itself can result in blood-brain barrier disruption (Mayhan, 1996). The permeability may be estimated independently of pressure by using single microvessel techniques (Easton & Fraser, 1994), an approach pioneered in the brain by Crone & Olesen (1982) who used changes in the electrical cable properties of 500 μm lengths of pial venular capillaries as the basis of the measurement. Results from the frog (Olesen & Crone, 1986), and the rat (Butt, 1995), showed that very high concentrations of bradykinin have only a small permeability-increasing effect. This is apparently at variance with the recent findings that bradykinin is important in the development of oedema following experimental cerebral ischaemia in rats (Kamiya et al. 1993), but it is possible that the resistance measurements underestimate the full permeability response due to uneven permeability along the length of the microvessel itself (see Easton et al. 1997).
We have used a single pial microvessel occlusion technique that allows measurements of permeability change over small regions of microvessels (ca 10 μm in length) in experiments designed to investigate the cerebrovascular permeability effects of bradykinin (which activates B2 receptors) and its active metabolite [des-Arg9]-bradykinin (which activates B1 receptors; see Hall, 1992) on single pial venular capillaries. We have also investigated the intracellular mechanisms that link the activation of the receptors to the permeability changes. Some of the findings reported here have been previously presented in a preliminary form (Sarker & Fraser, 1994, 1995).
METHODS
The method used in this study, and its theoretical basis, has been described fully elsewhere (Fraser & Dallas, 1993; Easton & Fraser, 1994). Briefly, the microcirculation of the surface of the brain of rats (aged 20–30 days) was exposed by removal of the dura and arachnoid. A low molecular mass fluorescent dye, either Lucifer Yellow (457 Da) or Sulforhodamine B (580 Da), was introduced into single venular capillaries via a bolus injection into the carotid artery. Sulforhodamine B has the advantage of requiring a longer wavelength for excitation and results in a more stable preparation. The fluorescence signal, which has been shown to increase linearly with dye concentration, was captured through a microscope, an image-intensifier camera, and analysed through a video-densitometer. Permeability was measured from the rate of loss of dye trapped in a single pial venular capillary by a glass-occluding probe. The fluorescence measurements were made from a small segment 200–300 μm from the open end of the occluded vessel, but not so close to the occluding probe that the vessel diameter was distorted. If the vessel were leaky, a transmural hydrostatic pressure gradient would drive fluid across the wall, which would be replaced by fresh dye-free fluid entering the open end of the occluded portion. The dye concentration in the measured portion will depend on axial flow and diffusion as well as on transmural convection and diffusion. We have shown that when the dye is injected into the arterial blood supplying the pial vasculature the trapped erythrocytes prevent dye diffusion from the open end of the occluded vessel and the axial dye concentration gradient is flat (Easton et al. 1997). Under these circumstances axial diffusion and convection have no effect on the dye concentration in the lumen (Fraser & Dallas, 1993). As in this preparation there is no continuous barrier to free diffusion of dye from the surface of the vessel (Butt et al. 1990), the abluminal dye concentration can be kept at undetectably low levels by maintaining a flow of superfusate of about 1–2 ml min−1. Previous experiments showed that there was no difference in the rate of fall of luminal dye concentration even when the superfusate flow was stopped (Easton, 1993). Permeability was calculated from the rate of decrease in fluorescence under the measured portion of the occluded segment. The diameter of the vessels did not change during the occlusion (Easton & Fraser, 1994). As the rate of fall of the intravascular concentration of a small polar molecule is independent of the hydrostatic pressure (Fraser & Dallas, 1993), the concentration of dye in the vessel early in the occlusion (before axial volume flux distorts the uniform axial concentration of dye in the region of measurement) will be: Ct = C0e−kt, where Ct and C0 are the concentrations at times t and 0, and k = 4P/d, where P is the permeability and d the diameter of the vessel.
Animal preparation
The experiments were performed on Wistar rats of either sex and were carried out within guidelines directed by The Animals (Scientific Procedures) Act 1986. The animals were anaesthetized by an intraperitoneal injection of 60 mg (kg body weight)−1 sodium pentobarbitone diluted in water (25 % w/v), supplemented (10% of the original dose) as necessary, as assessed by the foot pinch-withdrawal reflex. At the end of the experiment the animal was killed with an overdose of anaesthetic. Once anaesthetized, a thermal probe was inserted rectally, and the animal was kept on a heating blanket connected via the probe to a feedback circuit (CFP 8105, Harvard Instruments) to maintain the body temperature at 37 ± 1°C. The trachea and the right common carotid artery were cannulated routinely, the arterial cannula being placed orthograde and filled with heparinized saline (100 u ml−1) to prevent clot formation. A section of the frontoparietal bones on the left side, between coronal and lamboid sutures, was thinned with a dental drill. A metal ring (internal diameter, 7 mm; external diameter, 13 mm) was glued onto the cranium surrounding the thinned bone with cyanoacrylate adhesive. The lumen of the ring was filled with artificial cerebrospinal fluid (ACSF; for composition see below), whilst the adhesive set. The thinned cranial surface within the ring was constantly superfused by a pump at a rate of 1–2 ml min−1 with ACSF warmed to 37 ± 0.3°C and delivered through a fine plastic tube, to ensure that a layer of fluid was present at all times. The thinned bone within the ring was then removed carefully avoiding damage to the meninges and cerebral surface. Pial microvessels were exposed by cutting away the overlying meninges, which in these young rats leaves the microvessels free from a continuous layer of overlying tissue (Butt et al. 1990). The rat was then placed on the modified stage of a microscope (ACM, Zeiss Oberkocken) and the exposed cerebral surface illuminated with a 50 W mercury vapour lamp through a ×20 water-immersion objective lens (Cooke; NA, 0.5).
Artificial cerebrospinal fluid and drug application
The cerebral surface was superfused with ACSF at a rate of 0.5–1 ml min−1, warmed to 37 ± 1°C and buffered to pH 7.40. The ACSF contained (mm): NaCl, 110.5; KCl, 4.7; CaCl2, 2.5; KH2PO4, 1.1; MgSO4.7H2O, 1.25; NaHCO3, 25; and Hepes, 15. All chemicals were obtained from BDH (Poole, UK) except MgSO4.7H2O (Fisons, Loughborough, UK) and Hepes (Sigma). The bradykinin B1 receptor agonist [des-Arg9]-bradykinin, the B2 agonist bradykinin, and the B1 antagonist [des-Arg9-Leu8]-bradykinin were generously provided by Dr Martin Perkins of the Novartis Institute for Medical Science. The B2 antagonist HOE 140 was generously provided by Dr Klaus J. Wirth of Hoechst. Possible second messengers were investigated by using the cation channel blocker SKF 96365 (Merritt et al. 1990; generously supplied by SmithKline Beecham Pharmaceuticals), the phospholipase A2 (PLA2) inhibitors aristolochic acid and palmitoyl trifluoromethyl ketone (Calbiochem), the H2 receptor antagonist cimetidine (SmithKline Beecham), the free radical scavengers butylated-hydroxytoluene (BHT) and superoxide dismutase (SOD) with catalase, the cyclo-oxygenase and lipoxygenase blockers indomethacin and nordihydroguaiaretic acid (NDGA), the neutral endopeptidase (NEP) inhibitor phosphoramidon, the H2 receptor agonist dimaprit, and the angiotensin-converting enzyme (ACE) inhibitor captopril (all purchased from Sigma). Bradykinin, [des-Arg9]-bradykinin and other substances were applied abluminally into the pool of ACSF over the exposed cerebral surface, and the concentrations given are the final concentration in the pool.
Protocols
Permeability measurements were carried out on venular capillaries between 10 and 18 μm in diameter in the following manner. The selected dye-containing vessel was occluded, and once a stable level of fluorescence was obtained (after about 30 s) bradykinin or [des-Arg9]-bradykinin, or kinin mixed with other drugs, was applied to the brain surface while the occlusion was maintained. Once the video-densitometer trace had stabilized (between 30 and 60 s) the occlusion was released and the drugs were washed off the brain surface. The order of application of the kinin-drug mixtures was randomized in any series, except with a slowly reversible drug, such as indomethacin, when this was applied towards the end of an experiment. All the experiments, unless otherwise stated, were completed within 2 h of the first measurement. Dose-response curves were constructed by applying all the different concentrations of the drug in pseudo-random order to a single vessel, and at least four vessels from four rats were used for each dose-response curve. The actions of inhibitors and free radical scavengers were investigated by co-application with the stated concentration of bradykinin or [des-Arg9]-bradykinin, and within any series these too were paired experiments.
Statistics
Unless otherwise stated, the results are expressed as the mean ± s.e.m., and the significance of any changes was assessed by a non-parametric one-way analysis of variance test (Kruskal-Wallis) with Dunn's multiple comparison test. Regression lines and sigmoidal dose-response curves were fitted using GraphPad Prism version 3.00 for Windows (GraphPad Software, San Diego, CA, USA).
RESULTS
The present experiments were carried out on pial venular capillaries of at least 200 μm in length, which had negligible or low initial permeabilities, that is from below the limit of detection (nominally 0.2 × 10−6 cm s−1; Easton et al. 1997) up to 2.0 × 10−6 cm s−1. A control series of experiments was carried out in which permeability measurements were made at 5 min intervals on ten venular capillaries from ten rats. There was a very slow spontaneous permeability increase at a rate of 0.09 × 10−6 ± 0.06 × 10−6 cm s−1 h−1 over the first 2 h (see Fig. 1A), which is in contrast with our previous observation on spontaneous permeability increase (Easton et al. 1997), and can be ascribed to improvements in the technique. The effect of repeated bradykinin application was studied by applying it to the cerebral surface for 30–60 s during an occlusion that was released as soon as the new rate of decrease in fluorescence signal was established. The bradykinin was then washed off and a new pair of measurements (control followed by bradykinin) were made 5 min later. This was continued for 150 min (i.e. 30 repetitions of the same concentration), in each of four vessels (from 4 rats) for each concentration of bradykinin from 5 nm to 50 μm. The responses were stable for the first 2 h, and the permeability value returned to the baseline level before a subsequent bradykinin application (Fig. 1B and C). The baseline permeability changed little during this time (rates of change were −0.23 × 10−6 ± 0.08 × 10−6 and 0.11 × 10−6 ± 0.11 × 10−6 cm s−1 h−1 for the 50 nm and 50 μm applications, respectively), although it did change rapidly after this time with the highest concentration of bradykinin. The response to bradykinin also increased dramatically after 2 h, and for these reasons all the subsequent experiments were carried out prior to this. The permeability response to bradykinin was concentration dependent and the dose-response curves obtained were unchanged over the 2 h (see Fig. 1D). The estimated maximal permeability increase was initially 1.25 × 10−6 ± 0.13 × 10−6 cm s−1 with a log median effective concentration (logEC50; units, logm) of −6.34 ± 0.243, and this did not change significantly after 2 h (1.24 × 10−6 ± 0.106 × 10−6 cm s−1 and −6.08 ± 0.188, respectively).
Figure 1.
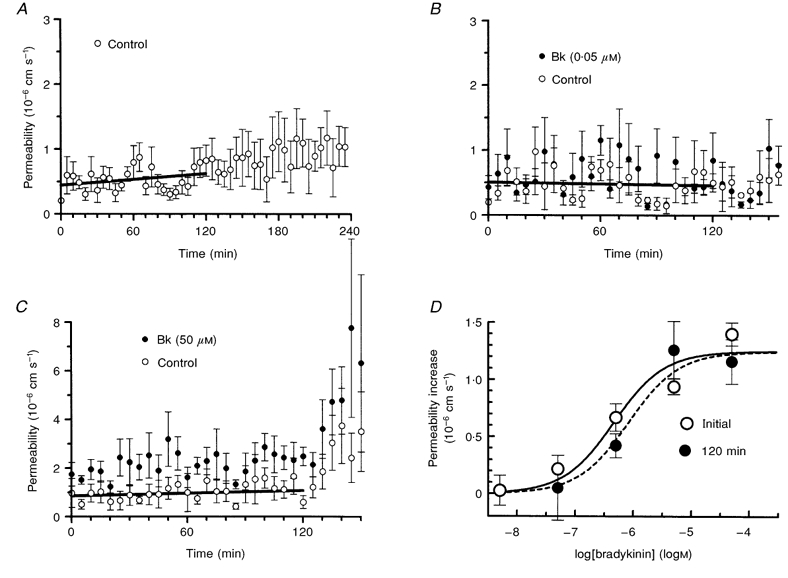
The stability of the permeability response to acute topical application of bradykinin
A, permeability measured with no addition of bradykinin at 5 min intervals in ten venular capillaries to assess the stability of the preparation. B and C, examples of the permeability response to bradykinin application to the brain surface at concentrations of 0.05 and 50 μm, respectively, at 5 min intervals. The permeability was measured immediately before, and from 30 to 60 s during, each application. Each concentration was repeated on four microvessels. The regression lines through the control values, shown for the first 2 h, do not increase significantly. D, dose-response curve for the permeability increase due to bradykinin application, estimated from initial values and those obtained 2 h later.
Bradykinin-destroying enzymes
The log EC50 value for the permeability response to bradykinin in the above experiments was much higher than would be predicted from the KD for bradykinin on the B2 receptor (see Hall, 1992). We investigated the possibility that bradykinin-destroying enzymes are present on the cerebral microvessels (Brecher et al. 1981; Brownson et al. 1994) by constructing dose-response curves for bradykinin alone, and in the presence of the ACE inhibitor captopril or the NEP inhibitor phosphoramidon, or both enzyme inhibitors together (Fig. 2). The logEC50 of the response to bradykinin was −5.30 ± 0.15 (rather higher than in the earlier series of experiments) and this decreased to −6.37 ± 0.24 with captopril, to −6.33 ± 0.19 with phosphoramidon, and to −7.3 ± 0.20 with captopril and phosphoramidon combined. One-way analysis of variance showed that this decrease was significant when captopril and phosphoramidon were applied separately (P < 0.05), and when they were applied together (P < 0.001), as assessed by Dunn's correction. There was no change in the estimated maximum permeability increase with any of the enzyme inhibitors, and the apparent increase in the maximum response when compared with that in Fig. 1D (i.e. 1.82 × 10−6 ± 0.13 × 10−6vs. 1.25 × 10−6 ± 0.29 × 10−6 cm s−1) was also not significant.
Figure 2.
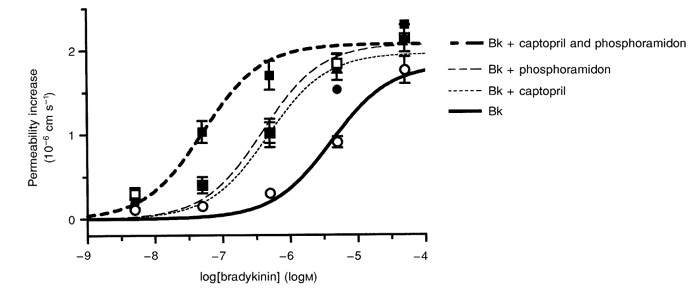
The effect of inhibiting bradykinin-destroying enzymes on the permeability response to bradykinin
Dose-response curves for bradykinin (Bk; 5 nm to 50 μm) when applied to the brain surface for 30 to 60 s, alone (○) or in combination with the neutral endopeptidase inhibitor phosphoramidon (□; 200 μm), the angiotensin-converting enzyme inhibitor captopril (•; 1 μm) or with both inhibitors in combination (▪). Four vessels were used for each dose-response curve.
Bradykinin and [des-Arg9]-bradykinin receptor antagonists
Bradykinin is rapidly converted to [des-Arg9]-bradykinin by the action of kininase I (see Bhoola et al. 1992), and when [des-Arg9]-bradykinin was applied in a similar way to bradykinin there was also a permeability increase. The dose-response curve is illustrated in Fig. 3; the estimated logEC50 of −5.6 ± 0.37 was similar to that of bradykinin, although the maximal permeability increase of 4.1 × 10−6 ± 0.82 × 10−6 cm s−1 was higher.
Figure 3.
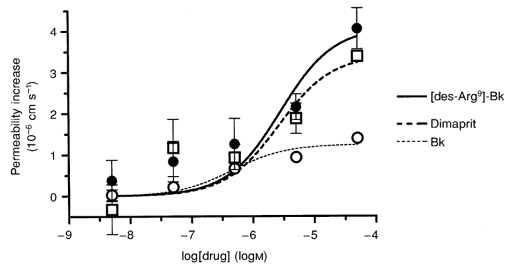
Permeability dose-response curve for [des-Arg9]-bradykinin compared with those for bradykinin and dimaprit
The bradykinin B1 receptor agonist [des-Arg9]-bradykinin (•) was applied to the brain surface over a range of concentrations (from 5 nm to 50 μm) and the permeability response measured in eight vessels. The dose-response curves for bradykinin (○) and dimaprit (□; histamine H2 receptor agonist) are shown for comparison, the data being taken from Fig. 1D and Sarker et al. (1998), respectively.
It is possible that all the permeability response is mediated via the B1 receptor. However, when the specific subtype receptor inhibitors ([des-Arg9-Leu8]-bradykinin for the B1 receptor and HOE 140 for the B2 receptor) were co-applied with bradykinin and [des-Arg9]-bradykinin, there was no evidence of such cross-reactivity (see Fig. 4). The permeability increase in response to bradykinin was abolished by HOE 140 (from 1.6 × 10−6 ± 0.42 × 10−6 to 0.02 × 10−6 ± 0.21 × 10−6 cm s−1), and was not significantly affected by B1 blockade (1.0 × 10−6 ± 0.28 × 10−6 cm s−1). Blocking the B1 receptor with [des-Arg9-Leu8]-bradykinin (0.5 μm) abolished the response to 5 μm [des-Arg9]-bradykinin (1.2 × 10−6 ± 0.38 × 10−6 to 0.0 × 10−6 ± 0.14 × 10−6 cm s−1). B2 receptor blockade with HOE 140 did not affect the response to [des-Arg9]-bradykinin ([des-Arg9]-bradykinin with HOE 140: 1.1 × 10−6 ± 0.17 × 10−6 cm s−1).
Figure 4.
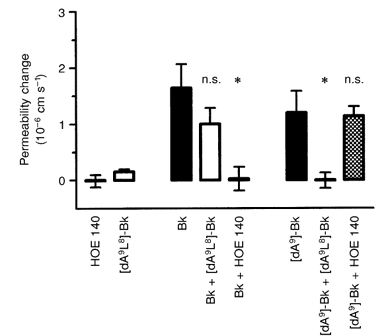
The effect of bradykinin receptor antagonists on the permeability responses to bradykinin and [des-Arg9]-bradykinin
The permeability-increasing effects of 5 μm bradykinin and 5 μm[des-Arg9]-bradykinin ([dA9]-Bk) were tested against the bradykinin B1 and B2 receptor antagonists [des-Arg9-Leu8]-bradykinin ([dA9L8]-Bk; 0.5 μm) and HOE 140 (0.5 μm), respectively. The bradykinin-induced permeability increase was completely blocked by HOE 140 (*P < 0.05, Kruskal-Wallis test, Dunn's correction), but not by the B1 receptor antagonist [des-Arg9-Leu8]-bradykinin. Similarly, [des-Arg9-Leu8]-bradykinin blocked the effect of [des-Arg9]-bradykinin (*P < 0.05), but HOE 140 had no effect. [des-Arg9-Leu8]-Bradykinin and HOE 140 alone had no effect on permeability. These paired experiments were carried out on two groups of four vessels.
Second messengers that mediate the permeability response to bradykinin and [des-Arg9]-bradykinin
Raised cytosolic Ca2+ in the endothelium has been implicated as having an important role in the rapid permeability response to a variety of agonists (see Crone, 1986), and we have found previously that the permeability response to histamine in pial venular capillaries was attenuated by the Ca2+ channel blocker SKF 96365 (Sarker et al. 1998). Co-application of SKF 96365 (10 μm) with bradykinin (5 μm), however, did not alter the permeability response (2.3 × 10−6 ± 0.12 × 10−6 cm s−1 with bradykinin alone and 2.5 × 10−6 ± 0.05 × 10−6 cm s−1 with SKF 96365 in 4 vessels; see Fig. 5). On the other hand, the response to [des-Arg9]-bradykinin (5 μm) was much reduced by SKF 96365 ([des-Arg9]-bradykinin alone, 3.8 × 10−6 ± 0.12 × 10−6 cm s−1; with SKF 96365, 0.89 × 10−6 ± 0.10 × 10−6 cm s−1 in 4 vessels).
Figure 5.
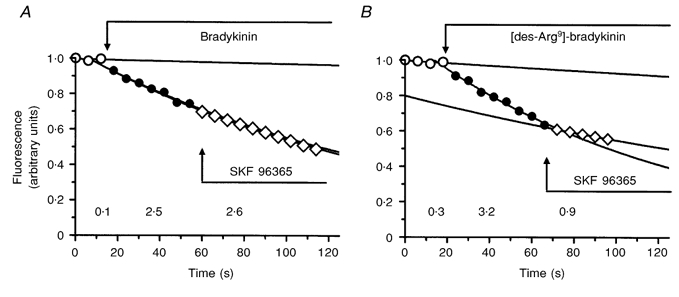
Differences in the effects of Ca2+ channel block on the permeability responses to bradykinin and [des-Arg9]-bradykinin
A and B, representative occlusion experiment carried out on single venular capillaries with bradykinin (5 μm; A) and [des-Arg9]-bradykinin (5 μm; B) applied to the brain surface. Permeability (as measured from the rate of fall of fluorescence signal) increased with both agonists. The Ca2+ channel blocker SKF 96365 (10 μm) had no effect on the increase induced by bradykinin, while it reduced that to [des-Arg9]-bradykinin. The permeability values (shown below the relevant portions of the traces and expressed as 10−6 cm s−1) were calculated from the rate constants of mono-exponentials fitted to the fluorescence signals from the occluded microvessel segments.
Free radicals
Free radicals released as a consequence of arachidonic acid metabolism have been shown to increase the permeability of these vessels (Easton & Fraser, 1998), and the possibility that they are involved in the bradykinin-mediated increase in permeability was tested by applying known scavengers. A series of experiments was carried out on four vessels from four rats. Figure 6A shows that co-application of bradykinin with a combination of SOD (100 U ml−1) and catalase (100 U ml−1) was required to block the permeability response to bradykinin (bradykinin alone, 1.52 × 10−6 ± 0.26 × 10−6 cm s−1; with SOD and catalase, −0.07 × 10−6 ± 0.09 × 10−6 cm s−1). In a further series of four experiments BHT (1 μm), a lipid peroxidation chain blocker, also prevented the response to 5 μm bradykinin, but not that to 5 μm [des-Arg9]-bradykinin (bradykinin alone, 2.2 × 10−6 ± 0.67 × 10−6 cm s−1; with BHT, 0.11 × 10−6 ± 0.26 × 10−6 cm s−1; [des-Arg9]-bradykinin alone, 1.47 × 10−6 ± 0.10 × 10−6 cm s−1; with BHT, 2.07 × 10−6 ± 0.28 × 10−6 cm s−1; Fig. 6B).
Figure 6.
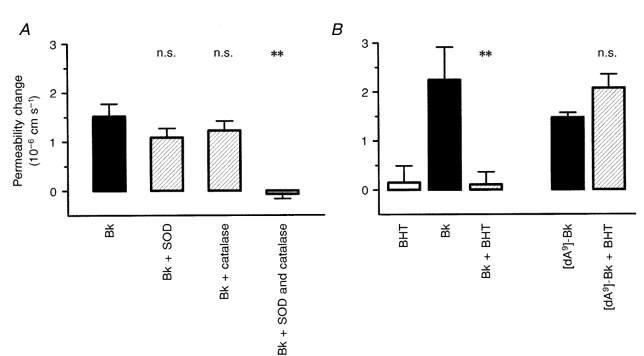
The effects of antioxidants on the permeability response to kinins
A, superoxide dismutase (SOD) and catalase (each 100 U ml−1) applied separately to the brain surface had little effect on the permeability response to 5 μm bradykinin, but completely blocked it when used in combination (**P < 0.01, Kruskal-Wallis test, Dunn's correction). B, the lipid peroxidation chain blocker butylated-hydroxytoluene (BHT; 1 μm) also inhibited the permeability response to bradykinin (5 μm; **P < 0.01), but had no effect on that to [des-Arg9]-bradykinin. Each of the two groups of experiments was carried out on four vessels.
The possibility that arachidonic acid is the source for the free radicals was investigated by co-applying bradykinin with the Ca2+-dependent PLA2 inhibitor aristolochic acid (Rosenthal et al. 1989) and the Ca2+-independent PLA2 inhibitor palmitoyl trifluoromethyl ketone (PaF3MeK; Liu, 1999). Figure 7A shows that PaF3MeK (50 μm) completely blocked the bradykinin-mediated permeability response (bradykinin alone 1.65 × 10−6 ± 0.30 × 10−6 cm s−1, and with PaF3MeK 0.27 × 10−6 ± 0.23 × 10−6 cm s−1), whereas aristolochic acid had no effect on the response (bradykinin with aristolochic acid, 1.46 × 10−6 ± 0.42 × 10−6 cm s−1). The permeability increase in response to dimaprit, a histamine H2 agonist previously shown to act via SKF 96365-sensitive Ca2+ entry (Sarker et al. 1998), was unaffected by PaF3MeK.
Figure 7.
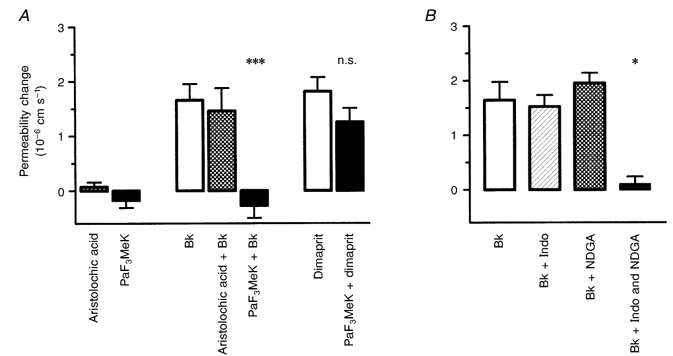
Arachidonic acid mediates the bradykinin-induced permeability increase
The idea that arachidonic acid release following acute bradykinin (5 μm) application leads to free radical generation was tested by co-application of PLA2 antagonists, and blockers of lipoxygenase and cyclo-oxygenase. A, the antagonist of the Ca2+-dependent PLA2 (aristolochic acid; 5 μm) had no effect, but that of the Ca2+-independent PLA2 (palmitoyl trifluoromethyl ketone, PaF3MeK; 50 μm) completely blocked the permeability increase. PaF3MeK did not affect the response to the histamine H2 agonist dimaprit (50 μm) (***P < 0.001, Kruskal-Wallis test, Dunn's correction). B, the permeability response to bradykinin was also abolished by a combination of indomethacin (Indo) and nordihydroguaiaretic acid (NDGA) (1 μm of each), but not when they were applied separately (*P < 0.05, Kruskal-Wallis test, Dunn's correction). The two groups of experiments were carried out on five vessels each.
Free radicals are formed during the metabolism of arachidonic acid via either cyclo-oxygenase or lipoxygenase, so inhibition of both these enzymes should abolish the permeability response to bradykinin. This was tested by co-applying bradykinin with indomethacin and NDGA, inhibitors of cyclo-oxygenases and lipoxygenases, respectively, separately and in combination. Figure 7B shows that only combined application of the inhibitors prevented the permeability increase (5 μm bradykinin alone, 1.64 × 10−6 ± 0.33 × 10−6 cm s−1; with 1 μm indomethacin, 1.52 × 10−6 ± 0.21 × 10−6 cm s−1; with 1 μm NDGA, 1.96 × 10−6 ± 0.19 × 10−6 cm s−1; with both, 0.10 × 10−6 ± 0.14 × 10−6 cm s−1). These results indicate that bradykinin stimulates the production of arachidonic acid and that free radicals are generated via its metabolism through cyclo-oxygenase and lipoxygenase pathways.
There is evidence that kinins are able to release histamine from mast cells (see Hall, 1992), which raises the possibility that histamine has a role in the permeability response. This was examined by blocking the H2 receptor (responsible for cerebrovascular permeability increase; Sarker et al. 1998) during bradykinin and [des-Arg9]-bradykinin application. Figure 8 shows that 2 nm cimetidine blocked the response to 5 μm [des-Arg9]-bradykinin ([des-Arg9]-bradykinin alone, 1.15 × 10−6 ± 0.22 × 10−6 cm s−1; with cimetidine, 0.03 × 10−6 ± 0.03 × 10−6 cm s−1), but had no effect on that to 5 μm bradykinin (bradykinin alone, 1.75 × 10−6 ± 0.31 × 10−6 cm s−1; with cimetidine, 1.59 × 10−6 ± 0.46 × 10−6 cm s−1).
Figure 8.
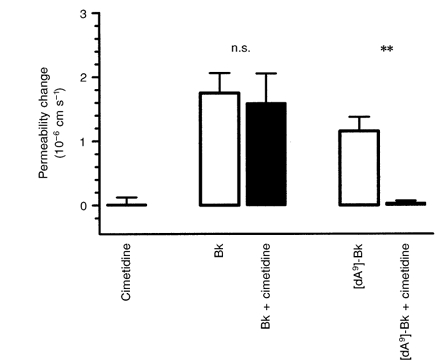
The [des-Arg9]-bradykinin-induced permeability increase is blocked by cimetidine
The histamine H2 receptor antagonist cimetidine (2 nm) blocked the permeability response to [des-Arg9]-bradykinin (5 μm), but not that to bradykinin (5 μm; **P < 0.01, Kruskal-Wallis test, Dunn's correction).
DISCUSSION
The present study shows that brief applications of bradykinin that last no longer than a minute can increase cerebrovascular permeability to a limited extent (about half of that to histamine; Sarker et al. 1998), but only at relatively high concentrations. Longer lasting applications can give rise to complicated interactions with the release of other inflammatory mediators. For example, a 10 min application resulted in an irreversible permeability increase due to the release of interleukin-1β (Hu & Fraser, 1997). Applications for 30–60 s at 5 min intervals, on the other hand, produced fully reversible responses which were stable for 2 h even with a bradykinin concentration as high as 50 μm.
This lack of sensitivity (high EC50) can be explained by the activity of the bradykinin-destroying enzymes ACE and NEP, which are widely distributed on microvessels and in many tissues including the brain (Bhoola et al. 1992). We found that inhibition of either of these enzymes shifted the dose-response curve to the left by an order of magnitude, and when both were blocked the EC50 was much closer to the reported KD of bradykinin on the B2 receptor (see Hall, 1992). A similar enhancement by a combination of captopril and phosphoramidon on the effect of bradykinin on permeability was also observed in hamster cheek pouch (Yong et al. 1992). It is possible that the kinin-destroying enzymes ACE and NEP have a physiological role in protecting the blood-brain barrier from circulating bradykinin. It has been reported that the expression of these enzymes can vary between individual animals (Winkler et al. 1998) and this may be at the root of the differences in the observed logEC50 for the two series of experiments shown in Figs 1 and 2.
A relatively stable, low-level disruption of the blood-brain barrier may account for the observations of workers who found that bradykinin was unable to open the barrier to proteins or other macromolecules (Gabbiani et al. 1970; Saria et al. 1983; Unterberg et al. 1984). The values obtained in the present series of experiments are similar to those obtained with previous single vessel studies. Thus bradykinin (5 μm) acutely applied to frog pial microvessels reduced the specific resistance of the vessel wall from 1500 to 280 Ω cm2 (equivalent to a Lucifer Yellow permeability of 1 × 10−6 cm s−1; Olesen & Crone, 1986), but no lower. Likewise, the rat pial microvascular specific resistance fell to a mean of about 550 Ω cm2 (ca 0.5 × 10−6 cm s−1) even after 30 min superfusion with 100 μm bradykinin (Butt, 1995).
The finding that [des-Arg9]-bradykinin also produced a reversible permeability increase was unexpected, as the bradykinin B1 receptor is usually expressed only in response to an inflammatory event subsequent to interleukin-1β release (see Marceau et al. 1998). The specific receptor subtype inhibitors for the B1 and B2 receptors, [des-Arg9-Leu8]-bradykinin and HOE 140, respectively, showed no cross-reactivity for the natural agonists, [des-Arg9]-bradykinin and bradykinin. As cimetidine blocked the permeability-increasing effect of [des-Arg9]-bradykinin, it is likely that the B1 receptors are not located on the endothelium, but on a histamine-secreting cell. For instance, histaminergic neurones are distributed widely and have nerve endings close to most small blood vessels of the brain (Takagi et al. 1986). This idea is supported by the similarity between the dimaprit and [des-Arg9]-bradykinin dose-response curves (see Fig. 3), and is consistent with the finding that B2, and not B1, ligand binding sites were found on isolated rat and pig cerebral microvessels (Homayoun & Harik, 1991).
Signal transduction linked to bradykinin receptor
In the present study we found that the cation channel blocker SKF 96365 failed to inhibit the permeability increase due to bradykinin. This was surprising as in cultured and large vessel endothelium, bradykinin is known to raise [Ca2+]i via a phospholipase C pathway. The IP3 so formed mediates release of Ca2+ from Ca2+ stores, and their depletion results in the opening of SKF 96365-sensitive Ca2+ channels (Schilling et al. 1992). Increased [Ca2+]i is often linked to a rapid increase in microvascular permeability both in vivo (He & Curry, 1993) and in cell culture (Wysolmerski & Lagunoff, 1991), and we have previously shown that the H2 receptor on these vessels operates via such channels (Sarker et al. 1998). The resulting endothelial nitric oxide synthase (eNOS) activation and subsequent stimulation of soluble guanylyl cyclase to form cGMP are thought to be key steps in increasing microvascular permeability (He et al. 1998), and bradykinin has been shown to be a very potent mediator of vascular relaxation mediated by eNOS activation (Cherry et al. 1982). In many in vivo studies little attempt has been made to separate the effects of vasodilatation from those of permeability (Mayhan, 1992), but where this was done (in the hamster cheek pouch preparation: Félétou et al. 1996; and the rat knee: Cambridge & Brain, 1995) bradykinin application resulted in nitric oxide-dependent vasodilatation, but nitric oxide-independent macromolecular extravasation.
There are other instances where the effects of bradykinin have been shown to be mediated via a free radical-generating pathway. Thus, bradykinin-mediated cerebral arteriolar dilatation was blocked by either scavenging free radicals (Navari et al. 1979; Kontos et al. 1984; Rosenblum, 1987) or by blocking their production by chelating iron or haem with desferrioxamine (Kontos et al. 1990).
We found that the permeability response to bradykinin in cerebral microvessels is linked to free radical formation as it was completely blocked by either a combination of SOD and catalase, or by inhibiting lipid peroxidation with BHT. Bradykinin has been shown to release arachidonic acid by activating PLA2 in cultured endothelium (Briand et al. 1996) and the present experiments indicate that bradykinin activates a Ca2+-independent PLA2, one variety of which has recently been shown to be strongly expressed in all regions of rat brain (Molloy et al. 1998). The increase in cerebral microvascular permeability induced by direct application of arachidonic acid was blocked by scavenging free radicals (Wei et al. 1986) that were formed from both cyclo-oxygenase and lipoxygenase (Easton & Fraser, 1998). Thus the observation that a combination of low concentrations of indomethacin and NDGA was effective in blocking the acute response to bradykinin further supports the idea that the B2 receptor activates PLA2. This is of particular interest as arachidonic acid release from endothelium is one of the earliest events in cerebral ischaemia (Bazan & Rodriguez de Turco, 1980). It has been pointed out to us that the relatively low concentrations of indomethacin and NDGA used in the present experiments may allow arachidonic acid to be shunted into a cytochrome P450 or 12 or 15 lipoxygenase pathway, which may be protective of inflammation (Conrad, 1999), and thus may not necessarily reflect a reduction of free radical production. This is an interesting possibility, but as these pathways would still result in free radical formation we think that it is more likely that the direct effect is the inhibition of their generation from the actions of both lipoxygenase and cyclooxygenase.
In conclusion we have found that bradykinin and [des-Arg9]-bradykinin applied to the brain surface for no more than 60 s results in a small reversible opening of the blood-brain barrier, which is mediated via B2 and B1 receptors, respectively. The B1 receptors do not appear to be located on the endothelium, but on some histamine-releasing structure, possibly on the histaminergic nerve endings. The B2 receptor appears to activate a Ca2+-independent PLA2 which releases arachidonic acid, which in turn is the substrate for cyclo-oxygenase and lipoxygenase. The permeability increase results from the free radicals that are formed during the metabolism of arachidonic acid, possibly as a consequence of Ca2+ entry through areas of lipid that have undergone peroxidation.
Acknowledgments
This work was supported by the Wellcome Trust and The Novartis Institute for Medical Science. M.H.S. was a Commonwealth Staff Scholar when some of this work was carried out.
References
- Bazan NG, Rodriguez de Turco EB. Membrane lipids in the pathogenesis of brain edema: phospholipids and arachidonic acid, the earliest membrane components changed at the onset of ischemia. Advances in Neurology. 1980;28:197–205. [PubMed] [Google Scholar]
- Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacological Reviews. 1992;44:1–80. [PubMed] [Google Scholar]
- Brecher P, Hingorani V, Reininga K, Chobanian AV. Distribution and properties of angiotensin-converting enzyme in cerebral microvessels of the rabbit and rat. Clinical Science. 1981;61:249s–251s. doi: 10.1042/cs061249s. [DOI] [PubMed] [Google Scholar]
- Briand SI, Bernier SG, Guillemette G. Calcium-calmodulin plays a major role in bradykinin-induced arachidonic acid release by bovine aortic endothelial cells. Journal of Cellular Biochemistry. 1996;63:292–301. doi: 10.1002/(SICI)1097-4644(19961201)63:3%3C292::AID-JCB4%3E3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Brownson EA, Abbruscato TJ, Gillespie TJ, Hruby VJ, Davis TP. Effect of peptidases at the blood brain barrier on the permeability of enkephalin. Journal of Pharmacology and Experimental Therapeutics. 1994;270:675–680. [PubMed] [Google Scholar]
- Butt AM. Effect of inflammatory agents on electrical resistance across the blood-brain barrier in pial microvessels of anaesthetized rats. Brain Research. 1995;696:145–150. doi: 10.1016/0006-8993(95)00811-4. [DOI] [PubMed] [Google Scholar]
- Butt AM, Jones HC, Abbott NJ. Electrical resistance across the blood-brain barrier in anaesthetized rats: a developmental study. The Journal of Physiology. 1990;429:47–62. doi: 10.1113/jphysiol.1990.sp018243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambridge H, Brain SD. Mechanism of bradykinin-induced plasma extravasation in the rat knee joint. British Journal of Pharmacology. 1995;115:641–647. doi: 10.1111/j.1476-5381.1995.tb14980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry PD, Furchgott RF, Zawadzki JV, Jothianandan D. Role of endothelial cells in relaxation of isolated arteries by bradykinin. Proceedings of the National Academy of Sciences of the USA. 1982;79:2106–2110. doi: 10.1073/pnas.79.6.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DJ. The arachidonate 12/15 lipoxygenases. A review of tissue expression and biologic function. Clinical Reviews in Allergy and Immunology. 1999;17:71–89. doi: 10.1007/BF02737598. [DOI] [PubMed] [Google Scholar]
- Crone C. Modulation of solute permeability in microvascular endothelium. Federation Proceedings. 1986;45:77–83. [PubMed] [Google Scholar]
- Crone C, Olesen S-P. Electrical resistance of brain microvascular endothelium. Brain Research. 1982;241:49–55. doi: 10.1016/0006-8993(82)91227-6. [DOI] [PubMed] [Google Scholar]
- Easton AS. 1993. The characteristics of blood-brain barrier disruption studied in single microvessels in vivo. PhD Thesis, University of London. [Google Scholar]
- Easton AS, Fraser PA. Variable restriction of albumin diffusion across inflamed cerebral microvessels of the anaesthetized rat. The Journal of Physiology. 1994;475:147–157. doi: 10.1113/jphysiol.1994.sp020056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton AS, Fraser PA. Arachidonic acid increases cerebral microvascular permeability by free radicals in single pial microvessels of the anaesthetized rat. The Journal of Physiology. 1998;507:541–547. doi: 10.1111/j.1469-7793.1998.541bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton AS, Sarker MH, Fraser PA. Two components of blood-brain barrier disruption in the rat. The Journal of Physiology. 1997;503:613–623. doi: 10.1111/j.1469-7793.1997.613bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félétou M, Bonnardel E, Canet E. Bradykinin and changes in microvascular permeability in the hamster cheek pouch: the role of nitric oxide. British Journal of Pharmacology. 1996;118:1371–1376. doi: 10.1111/j.1476-5381.1996.tb15547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser PA, Dallas AD. Permeability of disrupted cerebral microvessels in the frog. The Journal of Physiology. 1993;461:619–632. doi: 10.1113/jphysiol.1993.sp019532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbiani G, Badonnel MC, Majno G. Intra-arterial injections of histamine, serotonin, or bradykinin: a topographic study of vascular leakage. Proceedings of the Society for Experimental Biology and Medicine. 1970;135:447–452. doi: 10.3181/00379727-135-35072. [DOI] [PubMed] [Google Scholar]
- Hall JM. Bradykinin receptors: pharmacological properties and biological roles. Pharmacology and Therapeutics. 1992;56:131–190. doi: 10.1016/0163-7258(92)90016-s. [DOI] [PubMed] [Google Scholar]
- He P, Curry FE. Albumin modulation of capillary permeability: Role of endothelial cell [Ca2+]i. American Journal of Physiology. 1993;265:H74–82. doi: 10.1152/ajpheart.1993.265.1.H74. [DOI] [PubMed] [Google Scholar]
- He P, Zeng M, Curry FE. cGMP modulates basal and activated microvessel permeability independently of [Ca2+]i. American Journal of Physiology. 1998;274:H1865–1874. doi: 10.1152/ajpheart.1998.274.6.H1865. [DOI] [PubMed] [Google Scholar]
- Homayoun P, Harik SI. Bradykinin receptors of cerebral microvessels stimulate phosphoinositide turnover. Journal of Cerebral Blood Flow and Metabolism. 1991;11:557–566. doi: 10.1038/jcbfm.1991.104. [DOI] [PubMed] [Google Scholar]
- Hu D-E, Fraser PA. Evidence for interleukin-1β mediating enhanced permeability responses to bradykinin in single pial venular capillaries of anaesthetized rats. The Journal of Physiology. 1997;505.P:53. [Google Scholar]
- Jolliet-Riant P, Tillement JP. Drug transfer across the blood-brain barrier and improvement of brain delivery. Fundamental and Clinical Pharmacology. 1999;13:16–26. doi: 10.1111/j.1472-8206.1999.tb00316.x. [DOI] [PubMed] [Google Scholar]
- Kamiya T, Katayama Y, Kashiwagi F, Terashi A. The role of bradykinin in mediating ischemic brain edema in rats. Stroke. 1993;24:571–575. doi: 10.1161/01.str.24.4.571. [DOI] [PubMed] [Google Scholar]
- Kontos HA, Wei EP, Kukreja RC, Ellis EF, Hess ML. Differences in endothelium-dependent cerebral dilation by bradykinin and acetylcholine. American Journal of Physiology. 1990;258:H1261–1266. doi: 10.1152/ajpheart.1990.258.5.H1261. [DOI] [PubMed] [Google Scholar]
- Kontos HA, Wei EP, Povlishock JT, Christman CW. Oxygen radicals mediate the cerebral arteriolar dilation from arachidonate and bradykinin in cats. Circulation Research. 1984;55:295–303. doi: 10.1161/01.res.55.3.295. [DOI] [PubMed] [Google Scholar]
- Liu L. Regulation of lung surfactant secretion by phospholipase A2. Journal of Cellular Biochemistry. 1999;72:103–110. [PubMed] [Google Scholar]
- Makevnina LG, Lomova IP, Zubkov Yu N, Semenyutin VB. Kininogen consumption in cerebral circulation of humans during brain ischemia and postischemic reperfusion. Brazilian Journal of Medical and Biological Research. 1994;27:1955–1963. [PubMed] [Google Scholar]
- Marceau F, Hess JF, Bachvarov DR. The B1 receptors for kinins. Pharmacological Reviews. 1998;50:357–386. [PubMed] [Google Scholar]
- Mayhan WG. Role of nitric oxide in modulating permeability of hamster cheek pouch in response to adenosine 5′-diphosphate and bradykinin. Inflammation. 1992;16:295–305. doi: 10.1007/BF00917622. [DOI] [PubMed] [Google Scholar]
- Mayhan WG. Role of activation of bradykinin B2 receptors in disruption of the blood-brain barrier during acute hypertension. Brain Research. 1996;738:337–341. doi: 10.1016/s0006-8993(96)01000-1. [DOI] [PubMed] [Google Scholar]
- Merritt JE, Armstrong WP, Benham CD, Hallam TJ, Jacob R, Jaxa-Chamiec A, Leigh BK, McCarthy SA, Moores KE, Rink TJ. SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochemical Journal. 1990;15:515–522. doi: 10.1042/bj2710515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy GY, Rattray M, Williams RJ. Genes encoding multiple forms of phospholipase A2 are expressed in rat brain. Neuroscience Letters. 1998;258:139–142. doi: 10.1016/s0304-3940(98)00838-6. [DOI] [PubMed] [Google Scholar]
- Narotam PK, Rodell TC, Nadvi SS, Bhoola KD, Troha JM, Parbhoosingh R, van Dellen JR. Traumatic brain contusions: a clinical role for the kinin antagonist CP-0127. Acta Neurochirurgica. 1998;140:793–802. doi: 10.1007/s007010050181. [DOI] [PubMed] [Google Scholar]
- Navari RM, Wei EP, Kontos HA, Patterson JL., Jr Oxygen consumption of pial arteries. American Journal of Physiology. 1979;236:H151–156. doi: 10.1152/ajpheart.1979.236.1.H151. [DOI] [PubMed] [Google Scholar]
- Olesen S-P, Crone C. Substances that rapidly augment ionic conductance of endothelium in cerebral venules. Acta Physiologica Scandinavica. 1986;127:233–241. doi: 10.1111/j.1748-1716.1986.tb07898.x. [DOI] [PubMed] [Google Scholar]
- Raidoo DM, Bhoola KD. Pathophysiology of the kallikrein-kinin system in mammalian nervous tissue. Pharmacology and Therapeutics. 1998;79:105–127. doi: 10.1016/s0163-7258(98)00011-4. [DOI] [PubMed] [Google Scholar]
- Relton JK, Beckey VE, Hanson WL, Whalley ET. CP-0597, a selective bradykinin B2 receptor antagonist, inhibits brain injury in a rat model of reversible middle cerebral artery occlusion. Stroke. 1997;28:1430–1436. doi: 10.1161/01.str.28.7.1430. [DOI] [PubMed] [Google Scholar]
- Rosenblum WI. Hydroxyl radical mediates the endothelium-dependent relaxation produced by bradykinin in mouse cerebral arterioles. Circulation Research. 1987;61:601–603. doi: 10.1161/01.res.61.4.601. [DOI] [PubMed] [Google Scholar]
- Rosenthal MD, Vishwanath BS, Franson RC. Effects of aristolochic acid on phospholipase A2 activity and arachidonate metabolism of human neutrophils. Biochimica et Biophysica Acta. 1989;1001:1–8. doi: 10.1016/0005-2760(89)90299-3. [DOI] [PubMed] [Google Scholar]
- Saria A, Lundberg JM, Skofitsch G, Lembeck F. Vascular protein linkage in various tissue induced by substance P, capsaicin, bradykinin, serotonin, histamine and by antigen challenge. Naunyn-Schmiedeberg's Archives of Pharmacology. 1983;324:212–218. doi: 10.1007/BF00503897. [DOI] [PubMed] [Google Scholar]
- Sarker MH, Easton AS, Fraser PA. Regulation of cerebral microvascular permeability by histamine in the anaesthetized rat. The Journal of Physiology. 1998;507:909–918. doi: 10.1111/j.1469-7793.1998.909bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarker MH, Fraser PA. Evidence that bradykinin increases permeability of single cerebral microvessels via free radicals in the rat. The Journal of Physiology. 1994;479.P:36. [Google Scholar]
- Sarker MH, Fraser PA. Bradykinin and des-Arg9-bradykinin increase permeability of single cerebral microvessels by different mechanisms in the anaesthetized rat. The Journal of Physiology. 1995;483.P:141. [Google Scholar]
- Schilling WP, Cabello OA, Rajan L. Depletion of the inositol 1,4,5-trisphosphate-sensitive intracellular Ca2+ store in vascular endothelial cells activates the agonist-sensitive Ca2+-influx pathway. Biochemical Journal. 1992;284:521–530. doi: 10.1042/bj2840521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi H, Morishima Y, Matsuyama T, Hayashi H, Watanabe T, Wada H. Histaminergic axons in the neostriatum and cerebral cortex of the rat: a correlated light and electron microscopic immunocytochemical study using histidine decarboxylase as a marker. Brain Research. 1986;364:114–123. doi: 10.1016/0006-8993(86)90992-3. [DOI] [PubMed] [Google Scholar]
- Unterberg A, Wahl M, Baethmann A. Effects of bradykinin on permeability and diameter of pial vessels in vivo. Journal of Cerebral Blood Flow and Metabolism. 1984;4:574–585. doi: 10.1038/jcbfm.1984.82. [DOI] [PubMed] [Google Scholar]
- Wahl M, Unterberg A, Baethmann A. Intravital fluorescence microscopy for the study of blood-brain-barrier function. International Journal of Microcirculation: Clinical and Experimental. 1985;4:3–18. [PubMed] [Google Scholar]
- Wei EP, Ellison MD, Kontos HA, Povlishock JT. O2 radicals in arachidonate-induced increased blood-brain barrier permeability to proteins. American Journal of Physiology. 1986;251:H693–699. doi: 10.1152/ajpheart.1986.251.4.H693. [DOI] [PubMed] [Google Scholar]
- Winkler A, Rottmann M, Heder G, Hyytia P, Siems WE, Melzig MF. Gene expression and activity of specific opioid-degrading enzymes in different brain regions of the AA and ANA lines of rats. Biochimica et Biophysica Acta. 1998;1406:219–227. doi: 10.1016/s0925-4439(97)00041-0. [DOI] [PubMed] [Google Scholar]
- Wysolmerski RB, Lagunoff D. Regulation of permeabilized endothelial cell retraction by myosin phosphorylation. American Journal of Physiology. 1991;261:C32–40. doi: 10.1152/ajpcell.1991.261.1.C32. [DOI] [PubMed] [Google Scholar]
- Yong T, Gao XP, Koizumi S, Conlon JM, Rennard SI, Mayhan WG, Rubinstein I. Role of peptidases in bradykinin-induced increase in vascular permeability in vivo. Circulation Research. 1992;70:952–959. doi: 10.1161/01.res.70.5.952. [DOI] [PubMed] [Google Scholar]