

Abstract
The effects of authentic NO and the NO donor S-nitroso-N-acetylpenicillamine (SNAP) on swelling-activated chloride currents (Iswell) were investigated in freshly dispersed rabbit portal vein smooth muscle cells. Iswell was recorded with the perforated patch configuration of the whole-cell patch clamp technique.
In approximately 50 % of cells NO and SNAP inhibited the amplitude of Iswell by about 45 % in a voltage-independent manner. Iswell was also inhibited by an inhibitor of NO-sensitive guanylate cyclase (1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) and by KT5823, an inhibitor of cGMP-dependent protein kinase.
In other cells both NO and SNAP enhanced Iswell by about 40 % in a voltage-independent manner. A similar increase was produced by application of the cell-permeable cGMP analogue 8-bromo-guanosine 3′,5′-cyclic monophosphate (8-Br-cGMP). However, 8-Br-cGMP had no effect on current amplitude in cells pre-treated with KT5823. In contrast 8-Br-cGMP increased the amplitude of Iswell in cells which had been pre-treated with ODQ.
SNAP also modulated Iswell recorded in the conventional whole-cell configuration with internal solutions containing 10 mm EGTA to rule out any contribution from Ca2+-activated Cl− currents.
These data suggest that the amplitude of Iswell can be enhanced by NO via a cGMP-dependent phosphorylation and inhibited by NO in a cGMP-independent manner.
A membrane chloride current which is activated by cell swelling (Iswell) has been identified in many cell types and is implicated in cell volume regulation, solute transport and also, perhaps, in processes such as cell proliferation (see reviews by Strange et al. 1996; Okada, 1997). Iswell has also been observed in vascular (Yamazaki et al. 1998; Greenwood & Large, 1998) and non-vascular smooth muscle (Xu et al. 1997; Dick et al. 1998). It has been suggested that in vascular smooth muscle activation of Iswell produces depolarisation and contraction because the chloride equilibrium potential (ECl, about −20 to −30 mV) is substantially more positive than the resting membrane potential (Nelson, 1998; Greenwood & Large, 1998). This proposal was based on the observations that the myogenic response in rat cerebral arteries was inhibited by chloride channel blockers (Nelson et al. 1997) and that the sensitivity of this response and Iswell to 4,4′-diisothiocyanatostilbene-2,2′-disulphonic acids (DIDS) was similar (Greenwood & Large, 1998).
Recently evidence has been provided to indicate that NO released from the endothelium regulates smooth muscle chloride conductance. In rat aorta it was shown that reduction of the chloride concentration of the bathing solution (and hence increasing the electrochemical gradient for Cl− ion efflux to produce depolarisation) evoked contraction in de-endothelialised preparations and in aortic segments treated with the NO synthase inhibitor N-nitro-L-arginine but not in aorta with a functionally active endothelium (Lamb & Barna, 1998). It was proposed that the Cl− conductance of smooth muscle cells in intact vessels is low but increases after reducing NO production by the endothelium (Lamb & Barna, 1998). In rat isolated small coronary arteries we demonstrated that inhibition of NO synthesis induces contraction and this response is inhibited by the blockers of Iswell DIDS and tamoxifen (Graves et al. 1998). These results also suggest that NO released spontaneously from the endothelium in coronary arteries may suppress a chloride conductance in the smooth muscle cells, possibly Iswell. In the present study we have investigated the effects of NO and NO donors on Iswell in rabbit portal vein smooth muscle cells, which has been characterised previously (Greenwood & Large, 1998). A preliminary account of this work has been presented to The Physiological Society (Ellershaw et al. 1999).
METHODS
Cell preparation
New Zealand White rabbits (2–3 kg) were killed by injection of a lethal dose of sodium pentobarbitone into the ear vein. Portal veins were excised, cleaned of fat and connective tissue and the exposed muscle sheet was cut into strips which were then immersed in physiological salt solution (PSS) containing 50 μm CaCl2 at 37°C. Single smooth muscle cells were isolated by treating the tissue with protease Type 1 crude or protease Type 14 (0.2–0.3 mg ml−1) for 5 min. After washout of the protease the tissue was immersed in collagenase Type 1A (0.5–1 mg ml−1) for 10 min. Cells were released from the digested tissue by gentle mechanical agitation using a wide-bore Pasteur pipette. Isolated cells were transferred to PSS containing 0.75 mm CaCl2, placed on cover slips for storage at 4°C and used within 6 h of isolation. All experiments were conducted at room temperature (21–23°C).
Electrophysiological recording
Whole-cell membrane currents were recorded with a List LM PCA amplifier using the perforated patch configuration of the whole-cell patch clamp technique. The perforated patch was obtained by adding amphotericin B (200–250 μg ml−1) to the pipette solution from a stock solution of amphotericin B dissolved in dimethyl sulphoxide (DMSO). The stock solution was stored at −10°C and fresh pipette solution was prepared every 2 h. All voltage protocols were generated by the CED (Cambridge, UK) Voltage Clamp program and evoked currents were analysed using the corresponding CED analysis package after filtering at 3 kHz. Further analysis and graphics were produced using Microcal Origin (Northampton, MA, USA). Changes in junction potentials between pipette and bath solutions were minimised by the use of a KCl agar bridge. The voltage-dependent characteristics of the hypotonicity-activated current were investigated by applying voltage ramps every 5 s. This protocol involved stepping the voltage from the holding potential of −50 mV to −100 mV for 50 ms followed by continuously changing the voltage from −100 to +100 mV at a rate of 250 mV s−1 in normal PSS and hypotonic solutions.
Solutions
Normal PSS used for dissection contained (mm): NaCl 126, KCl 6, MgCl2 1.2, CaCl2 1.5, Hepes 10 and glucose 11, and was adjusted to pH 7.2 with NaOH. Experiments were performed in K+-free conditions to remove contaminating K+ currents and the normal K+-free extracellular solution had the following composition (mm): NaCl 126, MgCl2 1.2, CaCl2 1.5, Hepes 10 and glucose 11, and was adjusted to pH 7.2 with NaOH. In all experiments voltage-dependent Ca2+ currents were blocked by the inclusion of 5 μm nicardipine in the bathing solution. In all perforated patch experiments the K+-free pipette solution contained (mm): CsCl 126, MgCl2 1.2, Hepes 10, glucose 11 and EGTA 0.1, and the pH was adjusted to 7.2 with CsOH. However, in a series of experiments Iswell was recorded using the conventional whole-cell configuration and the K+-free pipette solution contained 10 mm EGTA to eliminate any contamination from Ca2+-activated Cl− currents. Iswell was evoked by bathing the cell in a hypotonic external solution in which the NaCl concentration was reduced to 60 mm (see Greenwood & Large, 1998, for further details).
Chemicals
NO solution was prepared by a method described by Trepakova et al. (1999). Briefly, NO gas (BDH, Poole, Dorset, UK) was bubbled through distilled water containing an anion exchange resin (Bio-Rad) to mop up possible nitrites and nitrates formed by NO reacting with oxygen. The resulting 3 mm solution was stored at 4°C and used within 1 week. Dilutions were made by drawing off NO solution with a syringe and adding it directly to the external solution in the perfusion reservoir immediate to the recording chamber. With this method we estimated that the concentration of NO which reached the smooth muscle cells was about 1 μm. All enzymes, amphotericin B and DIDS were purchased from Sigma (Poole, Dorset, UK). S-nitroso-N-acetylpenicillamine (SNAP), 8-bromo-guanosine 3′,5′-cyclic monophosphate (8-Br-cGMP) and KT5823 were purchased from Calbiochem (La Jolla, CA, USA). 1H-[1,2,4]Oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) was purchased from Tocris (Avonmouth, Bristol, UK). Reagents were dissolved in either DMSO or ethanol, which at the highest concentration used (0.1 %) had no effect on Iswell. None of the agents investigated had any effect on the degree of cell swelling produced by bathing in hypotonic solutions. Maximum cell width was measured by a graticule located within the optical pathway and in isotonic conditions was 12 ± 2 μm (n = 30). Perfusion with hypotonic external solution produced cell swelling and increased cell width to 20 ± 3 μm. In the continued presence of hypotonic solution the maximum width in the presence of 100 μm 8-Br-cGMP, 10 μm SNAP and NO was 19 ± 3, 21 ± 3 and 20 ± 2 μm, respectively (n = 6–8).
Statistics
All data are presented as the mean ± s.e.m. of n cells. Student's t test was used to compare mean values and statistical significance was set at P < 0.05.
RESULTS
Inhibition of Iswell by NO and SNAP
We have shown previously that application of hypotonic solutions to rabbit portal vein myocytes caused significant cell swelling and concomitant activation of a Cl− current that was designated Iswell (Greenwood & Large, 1998). In the present study, changing the external solution from one containing 126 mm NaCl to one with 60 mm NaCl evoked a current that reversed at −5 ± 1 mV (n = 50), close to the predicted ECl as calculated by the Nernst equation. This current exhibited slight outward rectification and was inhibited by DIDS in a voltage-dependent manner consistent with Iswell in this cell type (Greenwood & Large, 1998). Figure 1A shows that after Iswell had reached a plateau the current was well maintained and persisted in the continued presence of the hypotonic solution as reported previously (Greenwood & Large, 1998). Similar well-sustained currents were observed in five other control cells bathed in hypotonic solution for 30 min. The addition of NO (approximate concentration was 1 μm) to the bathing solution reduced the amplitude of Iswell in five out of 11 cells. The maximal inhibition was produced after approximately 500 s exposure to NO and a typical record is shown in Fig. 1B. The effect of NO was not voltage dependent and the maximum inhibition at −50 and +100 mV was 49 ± 3 % (n = 5) and 42 ± 2 % (n = 5 cells from 4 different animals), respectively (Fig. 1C). Application of 10 μm SNAP to the bathing solution also reduced Iswell in 15 out of 30 cells isolated from 22 different animals and the maximal inhibition was observed after about 500 s (Fig. 2A). The SNAP-induced inhibition was also voltage independent and the maximum inhibition at −50 and +100 mV was 50 ± 7 and 57 ± 7 %, respectively (n = 15), which was qualitatively similar to that produced by NO (Fig. 2B). SNAP had no effect on the resting conductance recorded under isotonic conditions (n = 9, 3 different animals).
Figure 1.
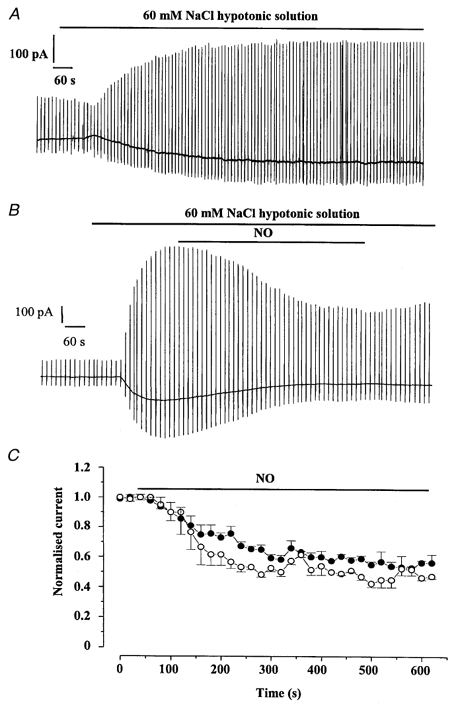
Inhibitory effect of NO on Iswell
A, representative trace showing a control cell where Iswell developed following exposure of a single portal vein smooth muscle cell to hypotonic solution. Note that the current is sustained throughout the application of hypotonic solution. B, the inhibitory effect of NO on Iswell. The concentration of NO was about 1 μm (see Methods) and in A and B large deflections represent the ramp protocol from a holding potential of −50 mV. C, the mean time dependence of the NO-induced inhibition shown at −50 mV (○) and +100 mV (•). Currents were normalised to peak Iswell prior to application of NO. Each point is the mean ± s.e.m. of 5 cells.
Figure 2.
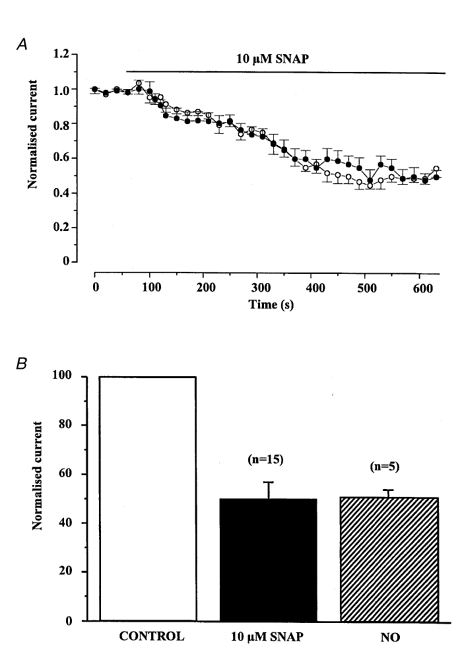
Inhibitory effect of SNAP on Iswell
A, the mean time dependence of the SNAP-induced inhibition of Iswell shown at −50 mV (○) and +100 mV (•). Each point is the mean ±s.e.m. of 15 cells. B, comparison of the maximum SNAP- and NO-induced inhibition of Iswell recorded at −50 mV. Currents were normalised to peak Iswell prior to application of SNAP or NO.
Enhancement of Iswell by NO and SNAP
However, in nine cells isolated from seven different animals SNAP produced an increase in the current amplitude. An example of the SNAP-induced increase in Iswell is illustrated in Fig. 3A and it can be seen that this effect was reversible. The enhancement had a slow time course similar to the inhibitory effect (cf. Fig. 3B and Fig. 2A). In these cells SNAP increased Iswell at −50 mV by 39 ± 4 % and at +100 mV by 40 ± 6 %. A similar increase in the amplitude of Iswell was observed in six cells when NO was added to the bathing solution. Thus, NO increased Iswell at −50 and +100 mV by 41 ± 5 and 43 ± 5 %, respectively, which was not significantly different to the enhancement produced by 10 μm SNAP (P < 0.05, Fig. 4C). Therefore, it is apparent that NO and SNAP can increase or decrease Iswell evoked in vascular myocytes but have no effect on the resting conductance recorded under isotonic conditions. In six cells (4 different animals) SNAP had no effect on Iswell.
Figure 3.
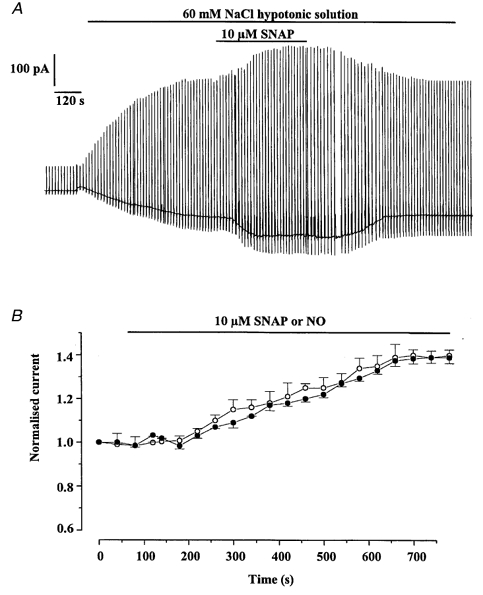
Cells in which SNAP and NO increased Iswell
A, typical cell showing the increase of Iswell by 10 μm SNAP. B, time dependence of the potentiating effect of SNAP (○) and NO (•) on Iswell recorded at −50 mV. Each point is the mean ±s.e.m. of 9 cells for SNAP and 6 cells for NO.
Figure 4.
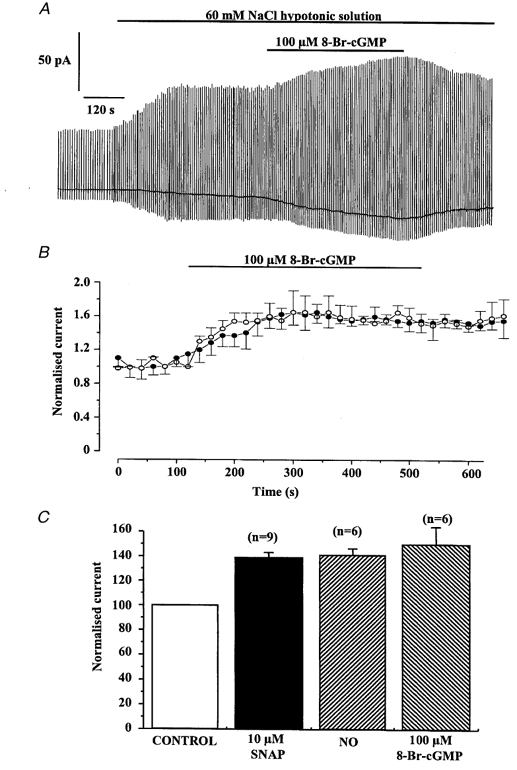
Effect of 8-Br-cGMP on Iswell
A, record illustrating the excitatory effect of 100 μm 8-Br-cGMP on Iswell. B, time dependence of the 100 μm 8-Br-cGMP-induced increase shown at −50 mV (○) and +100 mV (•). Each time point is the mean ±s.e.m. of 6 cells. C, comparison of the potentiating effect of SNAP, NO and 8-Br-cGMP on Iswell recorded at −50 mV. Currents were normalised to peak Iswell prior to application of SNAP, NO or 8-Br-cGMP and the number of cells is shown in parentheses.
Effect of SNAP on Iswell recorded from cells with the whole-cell configuration using pipette solutions containing 10 mm EGTA
Since Ca2+-activated Cl− currents (ICl(Ca)) are readily recorded with the perforated patch configuration it is possible that the variable effects of SNAP and NO may be due to an effect on ICl(Ca) as well as Iswell. Consequently, we performed experiments using the conventional whole-cell configuration with a pipette solution containing 10 mm EGTA to chelate any increase in [Ca2+]i evoked by cell swelling and therefore prevent activation of ICl(Ca). Under these conditions bathing the cell in hypotonic solution evoked Iswell that was indistinguishable from Iswell recorded with the perforated patch technique. The current exhibited outward rectification, reversed at +5 ± 2 mV (close to the theoretical ECl,n = 12) and was well maintained in the continued presence of the hypotonic solution. Application of SNAP had similar variable effects on Iswell recorded under these conditions to those described above. Thus in five cells from four different rabbits SNAP inhibited Iswell at −50 and +100 mV by 50 ± 8 and 51 ± 8 %, respectively. However, in five other cells (5 animals) SNAP enhanced Iswell at −50 and +100 mV by 60 ± 4 and 55 ± 7 %, respectively. SNAP had no effect on Iswell in four cells isolated from four different animals. The magnitudes of the changes produced by SNAP under these conditions were similar to those observed for Iswell recorded with the perforated patch technique. These data show that the effects of NO and SNAP are due to an action on Iswell and not on ICl(Ca).
Reversal potential of Iswell when Na+ was replaced by N-methyl-d-glucamine
We were concerned that a cation conductance may contribute to Iswell which may give rise to the variability of the data with NO and SNAP. To test this possibility we replaced the external Na+ ions with the less permeant cation N-methyl-d-glucamine (NMDG). The reversal potential of Iswell with 75 mm NaCl was +5 ± 0.6 mV (n = 7, 6 rabbits) and with 75 mm NMDG-Cl− it was +7 ± 0.7 mV (n = 8, 6 rabbits). Since there was no change in the reversal potential it is concluded that in this preparation Iswell is purely an anion current with no contribution from a cation conductance.
Modulation of Iswell by cGMP and cGMP-dependent protein kinase
Many of the effects of NO are due to the activation of guanylate cyclase and subsequent increase in cGMP (McDonald & Murad, 1996) and therefore we investigated the effect of cGMP on Iswell to delineate the effects of NO on this conductance. In six cells isolated from five different animals application of the cell-permeable cGMP analogue 8-Br-cGMP (100 μm) to the bathing solution during activation of Iswell produced a reversible increase in the amplitude of the current (Fig. 4A), although in two cells 8-Br-cGMP had no effect on Iswell. The enhancement of Iswell by 8-Br-cGMP reached a peak after about 300 s (Fig. 4B) and the mean increase at −50 and +100 mV was 52 ± 14 and 50 ± 8 % (n = 6 cells, 5 animals), respectively. The magnitude of the enhancement produced by 8-Br-cGMP was similar to that produced by NO or SNAP (Fig. 4C). A further series of experiments was performed to investigate the effect of a selective inhibitor of NO-sensitive guanylate cyclase, ODQ, on Iswell. In five cells (4 animals) application of 10 μm ODQ to the bath solution caused inhibition of Iswell. A typical cell is shown in Fig. 5A. The mean reduction of Iswell by ODQ was 34 ± 5 and 22 ± 5 % at −50 and +100 (n = 5 cells). Application of either ODQ or 8-Br-cGMP for 8 min had no effect on the resting conductance recorded under isotonic conditions (n = 4 cells from 4 different animals for each agent). These data suggest that an increase in cGMP levels is insufficient to activate Iswell but cGMP augments Iswell when it has been activated by hypotonic solution. Consequently increases in cGMP levels in portal vein myocytes appear to enhance the activation of Iswell.
Figure 5.
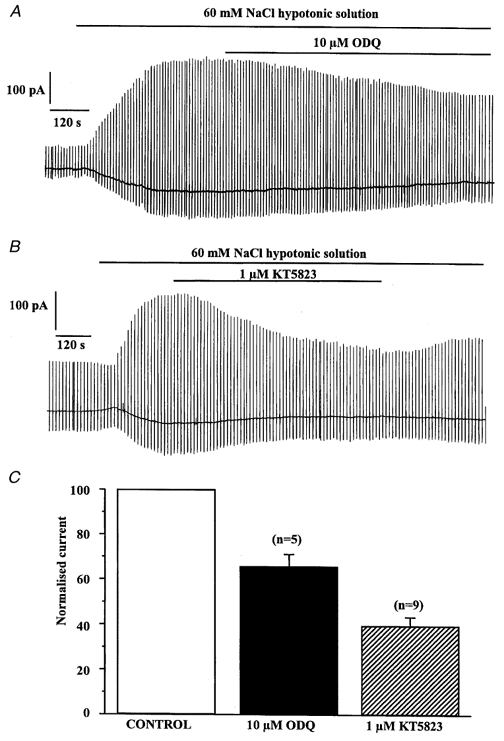
Effect of ODQ and KT5823 on Iswell
A, the inhibitory effect of 10 μm ODQ on Iswell. B, the inhibitory effect of 1 μm KT5823 on Iswell. C, comparison of ODQ- and KT5823-induced inhibition of Iswell recorded at −50 mV. Currents were normalised to peak Iswell prior to application of ODQ or KT5823.
Most of the cellular effects of cGMP are mediated by a cGMP-dependent protein kinase (McDonald & Murad, 1996). Consequently we studied the effect of the selective cGMP-dependent protein kinase inhibitor KT5823 on evoked Iswell. This agent has a Ki for inhibition of protein kinase G of 234 nm compared to Ki values against protein kinase C and protein kinase A of 4 and 10 μm, respectively (Ito & Karachot, 1990). Consequently we used KT5823 at a concentration of 1 μm where it is relatively selective for cGMP-dependent protein kinase. In nine cells isolated from eight animals addition of 1 μm KT5823 to the bathing solution produced a slow, voltage-independent inhibition of Iswell. A typical cell is shown in Fig. 5B. The degree of inhibition was 60 ± 3 and 69 ± 7 % at −50 and +100 mV, respectively (n = 9, Fig. 5C), which was significantly greater than the inhibition produced by ODQ (P < 0.05). Removal of KT5823 from the bathing solution caused a partial reversal of the inhibition (Fig. 5B). In four cells (4 animals) KT5823 was without effect, but it is worth noting that neither KT5823 nor ODQ increased Iswell in any cell tested. Consequently, when Iswell has been activated the intracellular concentration of cGMP is capable of stimulating a cGMP-dependent protein kinase that in turn enhances Iswell. When 8-Br-cGMP was applied in the presence of KT5823 there was no increase in Iswell (Fig. 6A); similar results were observed in six cells from four different rabbits. Consequently, the stimulatory effect of 8-Br-cGMP appears to be mediated by a cGMP-dependent protein kinase. In comparison, application of 8-Br-cGMP in the continued presence of ODQ caused an increase in the amplitude of Iswell. Thus, application of 10 μm ODQ inhibited Iswell by 37 ± 4 % at −50 mV (similar to that reported above, n = 3) and after application of 8-Br-cGMP in the presence of ODQ the current was increased by 40 ± 10 % (n = 3), which is similar to the increase of Iswell by 8-Br-cGMP in the absence of ODQ (about 50 %, see above).
Figure 6.
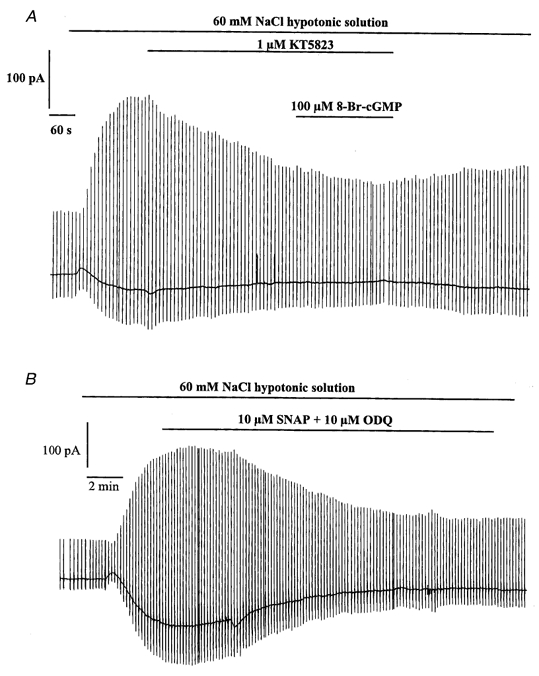
Effect of KT5823 on the modulation of Iswell by 8-Br-cGMP and effect of ODQ on the action of SNAP
A, representative trace showing the inhibitory effect of 1 μm KT5823. Application of 8-Br-cGMP had no effect on Iswell in the continued presence of KT5823. B, representative trace showing the combined inhibitory effect of 10 μm ODQ and 10 μm SNAP.
Since NO can either decrease or increase the amplitude of Iswell, we investigated whether these actions occurred simultaneously in the same cell. For these experiments SNAP was applied simultaneously with ODQ, which would be expected to inhibit the NO-sensitive guanylate cyclase-mediated increase in Iswell in response to SNAP. A typical experiment is shown in Fig. 6B where 10 μm SNAP applied with 10 μm ODQ decreased Iswell by about 60 %. In five cells (3 animals) the mean reduction of Iswell produced by SNAP and ODQ was 52 ± 5 % at −50 mV. This value is not significantly different to the decrease in Iswell produced by SNAP in the absence of ODQ (see Fig. 2B). If SNAP was producing simultaneous excitatory and inhibitory effects on Iswell, it might have been expected that after the removal of the facilitatory action with ODQ SNAP would produce a larger inhibitory effect than observed with SNAP alone.
DISCUSSION
The major finding of this study is that authentic NO and the NO donor SNAP modulate Iswell recorded in rabbit portal vein smooth muscle cells. In approximately half of the cells these agents produced marked voltage-independent inhibition of Iswell. Both the magnitude and the time course of the inhibition produced by NO and SNAP were similar suggesting a common mechanism. Furthermore, the rate of inhibition was markedly slower than that of voltage-dependent inhibition produced by blockers such as DIDS (see Greenwood & Large, 1998) suggesting that NO and SNAP were not acting as direct channel blockers but were probably affecting regulatory mechanisms of the conductance. In other cells, NO and SNAP produced a significant increase of Iswell similar to that produced by the application of a cell-permeable analogue of cGMP, 8-Br-cGMP. Similar effects were observed when Iswell was recorded with pipette solutions containing 10 mm EGTA to suppress Ca2+-activated Cl− currents. These results indicate that neither the increase nor the decrease of Iswell produced by NO was due to an effect on contaminating ICl(Ca) recorded simultaneously with Iswell. In support of this conclusion, SNAP and NO at concentrations up to 1 mm and 1 μm, respectively, had no effect on whole-cell ICl(Ca) in tracheal smooth muscle (Waniishi et al. 1998) or ICl(Ca) in excised patches from aortic smooth muscle (Hirakawa et al. 1999). Consequently NO regulates Iswell without a direct effect on ICl(Ca).
Mechanisms involved in the increase in Iswell by NO and NO donors
In some portal vein myocytes NO, SNAP and 8-Br-cGMP enhanced Iswell. Furthermore, inhibition of both NO-dependent guanylate cyclase and cGMP-dependent phosphorylation by ODQ and KT5823, respectively, inhibited Iswell. These data suggest that NO and SNAP stimulate guanylate cyclase to generate sufficient cGMP to activate cGMP-dependent protein kinase, which enhances Iswell. However, 8-Br-cGMP did not activate Iswell under isotonic conditions but enhanced Iswell once it had been activated. This result suggests that 8-Br-cGMP does not directly activate the Cl− channel but accentuates channel activity once opened. In addition, the stimulatory effect of 8-Br-cGMP on Iswell was not apparent when the cell had been pre-treated with the selective cGMP-dependent protein kinase inhibitor KT5823, and the NO-sensitive guanylate cyclase inhibitor ODQ also reduced Iswell. These data suggest that during activation of Iswell there is a tonic generation of cGMP that activates sufficient KT5823-sensitive kinase to enhance the amplitude of Iswell. Since the inhibitory effect of KT5823 was greater than that of ODQ it is possible that during activation of Iswell cGMP is produced by both NO-sensitive and NO-insensitive guanylate cyclase. Interestingly, in contraction studies NO and sodium nitroprusside contracted the opossum oesophagus by a cGMP-dependent mechanism (Saha et al. 1993). Overall the data of the present study suggest that in vascular smooth muscle cells isolated from rabbit portal vein the activity of Iswell is enhanced by a cGMP-dependent protein kinase.
Mechanisms involved in the decrease of Iswell by NO and NO donors
Our experiments did not reveal the mechanism by which NO inhibits Iswell. The present study indicates that the inhibitory effect of NO on Iswell is not likely to be mediated via the well-known action of NO to stimulate NO-sensitive guanylate cyclase to produce cGMP. As discussed above, this mechanism enhances Iswell. It is worth re-iterating that NO did not reduce Iswell by an action of cGMP independent of cGMP-dependent protein kinase since cGMP had no effect on Iswell when the cells were pre-treated with the kinase inhibitor KT5823. Thus, NO may inhibit Iswell by an unidentified transduction mechanism. Alternatively, NO has been shown to activate directly Ca2+-sensitive K+ channels in vascular and tracheal smooth muscle (Bolotina et al. 1994; Abderrahmane et al. 1998) via modulation of redox-sensitive amino acid residues (Beckman & Koppenol, 1996). Consequently it is possible that the NO-induced inhibition of Iswell may be due to NO, either directly or via a reactive intermediate such as peroxynitrite, modifying the chloride channel protein.
Variable effects of NO and SNAP on Iswell
The data of the present study show that Iswell is regulated by NO via a cGMP-dependent pathway to increase Iswell and by a cGMP-independent mechanism to decrease Iswell. However, the net effect of NO and SNAP differed between populations of cells and in some cases NO, SNAP and 8-Br-cGMP had no effect. Similar variability has been observed in contraction studies on opossum oesophageal smooth muscle (Saha et al. 1993) and human detrusor smooth muscle (A. Moon, personal communication) where authentic NO and NO donors elicit both contraction and relaxation responses over the same concentration ranges. In the present study SNAP also produced similar effects on Iswell recorded with pipette solutions containing 10 mm EGTA to prevent any rise in [Ca2+]i that would activate ICl(Ca). Consequently, the different effects of SNAP and NO are not due to variable modulation of ICl(Ca). The present work indicates that cGMP-dependent and -independent mechanisms modulate Iswell but there is also strong evidence that other kinases modulate Iswell in other cell types. Thus, activation of protein kinase C by phorbol esters inhibits Iswell in NIH3T3 cells transfected with ClC-3 isolated from canine atrial muscle (Duan et al. 1997, 1999) and native Iswell in canine colonic myocytes (Dick et al. 1998). However, protein kinase C and tyrosine kinases have been implicated in the activation of Iswell in other cell types (Lepple-Wienhues et al. 1998; Du & Sorota, 1999). Consequently, the observed variable responses in portal vein myocytes may reflect the balance of various regulatory mechanisms in the cells used. In summary, the data of the present study suggest that NO regulates Iswell in rabbit portal vein myocytes by a cGMP-dependent pathway to increase Iswell and by a cGMP-independent mechanism to decrease Iswell.
Physiological implications
If the inhibitory effect of NO on Iswell was to prevail in physiological conditions this interaction may have a profound effect on blood flow. Since opening of Cl− channels in smooth muscle produces depolarisation and contraction, the inhibitory effect of NO on Iswell may contribute to the vasorelaxant effect of NO. In both vascular and non-vascular smooth muscle it has been shown that Iswell is active in isotonic conditions (Greenwood & Large, 1998; Dick et al. 1998). Moreover it has been proposed that Iswell may be involved in the myogenic response (Nelson, 1998). Therefore tonic release of NO from the endothelium may suppress Iswell in vascular smooth muscle cells. Reduction of NO release, for example by damage to the endothelium, may lead to depolarisation and increased contractility of the vascular smooth muscle cell. This mechanism would explain the results of both Lamb & Barna (1998) and Graves et al. (1998), which were outlined in the Introduction. Hence, NO-induced modulation of Iswell in vascular smooth muscle may play an important role in controlling blood flow in physiological and pathophysiological conditions.
Acknowledgments
This work was funded by The British Heart Foundation. I.A.G. is a Wellcome Trust Research Fellow. The helpful comments of Dr Jon Graves were much appreciated.
References
- Abderrahmane A, Salvail D, Dumoulin M, Garon J, Cadieux A, Rousseau E. Direct activation of a KCa channel in airway smooth muscle by nitric oxide: involvement of a nitrosylation mechanism? American Journal of Respiratory Cell and Molecular Biology. 1998;18:1–13. doi: 10.1165/ajrcmb.19.3.2996. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and the ugly. American Journal of Physiology. 1996;271:C1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- Dick GM, Bradley KK, Horowitz B, Hume JR, Sanders KM. Functional and molecular identification of a novel chloride conductance in canine colonic smooth muscle. American Journal of Physiology. 1998;275:C940–950. doi: 10.1152/ajpcell.1998.275.4.C940. [DOI] [PubMed] [Google Scholar]
- Du X-Y, Sorota S. Protein kinase C stimulates swelling-induced chloride current in canine atrial cells. Pflügers Archiv. 1999;437:227–234. doi: 10.1007/s004240050773. [DOI] [PubMed] [Google Scholar]
- Duan D, Cowley S, Horowitz B, Hume JR. A serine residue in ClC-3 links phosphorylation-dephosphorylation to chloride channel regulation by cell volume. Journal of General Physiology. 1999;113:57–70. doi: 10.1085/jgp.113.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Winter C, Cowley S, Hume JR, Horowitz B. Molecular identification of a volume-regulated chloride channel. Nature. 1997;390:417–421. doi: 10.1038/37151. [DOI] [PubMed] [Google Scholar]
- Ellershaw DC, Greenwood IA, Large WA. The effects of NO and NO donors on the swell-activated chloride currents in rabbit portal vein myocytes. The Journal of Physiology. 1999;521.P:59P. doi: 10.1111/j.1469-7793.2000.00015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves JE, Greenwood IA, Large WA. Effect of chloride current inhibitors on vascular tone in rat isolated coronary arteries. The Journal of Physiology. 1998;511.P:170. P. [Google Scholar]
- Greenwood IA, Large WA. Properties of a Cl− current activated by cell swelling in rabbit portal vein vascular smooth muscle cells. American Journal of Physiology. 1998;275:H1524–1532. doi: 10.1152/ajpheart.1998.275.5.H1524. [DOI] [PubMed] [Google Scholar]
- Hirakawa Y, Gericke M, Cohen R, Bolotina VM. Ca2+-dependent Cl− channels in mouse and rabbit aortic smooth muscle cells: regulation by intracellular Ca2+ and NO. American Journal of Physiology. 1999;277:H1732–1744. doi: 10.1152/ajpheart.1999.277.5.H1732. [DOI] [PubMed] [Google Scholar]
- Ito M, Karachot L. Messengers mediating long-term desensitization in cerebellar Purkinje cells. NeuroReport. 1990;1:129–132. doi: 10.1097/00001756-199010000-00012. [DOI] [PubMed] [Google Scholar]
- Lamb FS, Barna TJ. The endothelium modulates the contribution of chloride currents to norepinephrine-induced vascular contraction. American Journal of Physiology. 1998;275:H161–168. doi: 10.1152/ajpheart.1998.275.1.H161. [DOI] [PubMed] [Google Scholar]
- Lepple-Wienhues A, Szabò I, Laun T, Kaba NK, Gulbins E, Lang F. The tyrosine kinase p56lck mediates activation of swelling-induced chloride channels in lymphocytes. Journal of Cell Biology. 1998;141:281–286. doi: 10.1083/jcb.141.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald LJ, Murad F. Nitric oxide and cGMP signalling. Proceedings of the Society for Experimental Biology and Medicine. 1996;211:1–6. doi: 10.3181/00379727-211-43950a. [DOI] [PubMed] [Google Scholar]
- Nelson MT. Bayliss, myogenic tone and volume-regulated chloride channels in arterial smooth muscle. The Journal of Physiology. 1998;507:629. doi: 10.1111/j.1469-7793.1998.629bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Conway MA, Knot HJ, Brayden JE. Chloride channel blockers inhibit myogenic tone in rat cerebral arteries. The Journal of Physiology. 1997;502:259–264. doi: 10.1111/j.1469-7793.1997.259bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y. Volume expansion-sensing outward rectifier Cl− channel: fresh start to the molecular identity and volume sensor. American Journal of Physiology. 1997;273:C755–789. doi: 10.1152/ajpcell.1997.273.3.C755. [DOI] [PubMed] [Google Scholar]
- Saha JK, Hirano I, Goyal RK. Biphasic effect of SNP on opossum esophageal longitudinal muscle: involvement of cGMP and eicosanoids. American Journal of Physiology. 1993;265:G403–407. doi: 10.1152/ajpgi.1993.265.2.G403. [DOI] [PubMed] [Google Scholar]
- Strange K, Emma F, Jackson PS. Cellular and molecular physiology of volume-sensitive anion channels. American Journal of Physiology. 1996;270:C711–730. doi: 10.1152/ajpcell.1996.270.3.C711. [DOI] [PubMed] [Google Scholar]
- Trepakova ES, Cohen RA, Bolotina VM. Nitric oxide inhibits capacitative cation influx in human platelets by promoting sarcoplasmic/endoplasmic reticulum Ca2+-ATPase-dependent refilling of Ca2+ stores. Circulation Research. 1999;84:201–209. doi: 10.1161/01.res.84.2.201. [DOI] [PubMed] [Google Scholar]
- Waniishi Y, Inoue R, Morita H, Teramoto N, Abe K, Ito Y. Cyclic GMP-dependent but G-kinase-independent inhibition of Ca2+-dependent Cl− currents by NO donors in cat tracheal smooth muscle. The Journal of Physiology. 1998;511:719–731. doi: 10.1111/j.1469-7793.1998.719bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu WX, Kim SJ, So I, Kang TM, Rhee JC, Kim KW. Volume-sensitive chloride current activated by hyposmotic swelling in antral gastric myocytes of the guinea-pig. Pflügers Archiv. 1997;435:9–19. doi: 10.1007/s004240050478. [DOI] [PubMed] [Google Scholar]
- Yamazaki J, Duan D, Janiak R, Kuenzli K, Horowitz B, Hume JR. Functional and molecular expression of volume-regulated chloride channels in canine vascular smooth muscle cells. The Journal of Physiology. 1998;507:729–736. doi: 10.1111/j.1469-7793.1998.729bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]