

Abstract
An adequate understanding of secretion requires the measurement of exocytosis on the same time scale as that used for second messenger dynamics. To investigate the kinetics of ACh-evoked secretion in pancreatic acinar cells, exocytosis of zymogen granules was quantified by continuous, time-differential analysis of digital images. The validity of this method was confirmed by simultaneous fluorescence imaging of quinacrine-loaded zymogen granules.
Basal rates of exocytosis were low (0.2 events min−1). ACh stimulated a biphasic increase in secretory activity, maximal rates exceeding 20 events min−1 after 10 s of ACh application (10 μm). Over the next 15 s the rate of exocytosis fell to less than 4 events min−1; then began a second phase of secretion that peaked 15 s later at ∼11 events min−1, but subsequently declined in the continued presence of agonist.
Measurements of fura-2 fluorescence demonstrated a biphasic increase in intracellular [Ca2+] ([Ca2+]i). Comparison of the [Ca2+]i records and time-differential analysis revealed that the fall in exocytotic rate following the initial burst occurred despite the fact that [Ca2+]i remained high.
The second phase of secretion depended on both [Ca2+]i and [ACh]. At 10 μm ACh there was a decrease in the steepness of the relationship between [Ca2+]i and exocytosis that led to an enhancement of the slow secretory phase.
We propose that acinar cells contain two pools of secretory vesicles: a small pool of granules that is exocytosed rapidly, but is quickly depleted; and a reserve pool of granules that can be recruited by ACh in a process that is modulated by second messengers other than calcium.
Ever since the classic studies of Palade and colleagues (Jamieson & Palade, 1971; Tartakoff et al. 1975; Palade, 1975), the pancreatic acinar cell has provided a model for the synthesis and export of secretory proteins; nevertheless unlike the mast cell and the chromaffin cell, this cell has not proved particularly amenable to real-time studies of the final step in the secretory pathway, exocytosis. We have sought to remedy this situation by developing a system in which the exocytosis of individual zymogen granules can be directly visualized and quantified in intact acinar cells.
In pancreatic acinar cells a variety of digestive enzymes are packaged in large (∼1 μm diameter) dense-cored secretory vesicles known as zymogen granules. In response to various secretagogues, acinar cells release these digestive enzymes via exocytosis (Palade, 1975) into the lumen of the acinus which ultimately leads to the pancreatic duct. Studies of stimulus-secretion coupling in the exocrine pancreas have largely been carried out by measuring the secretagogue-induced release of amylase, or other secretory products, from large populations of cells, a method that has poor temporal resolution (Pandol et al. 1985; Merritt & Rubin, 1985). A complete understanding of how intracellular signals are coupled to secretory activity can only be obtained by determination of both second messengers and exocytosis with high temporal resolution at the level of the single cell. Several approaches have been developed in other secretory tissues to monitor exocytosis in single cells (Neher & Marty, 1982; Fernandez et al. 1984; Betz & Bewick, 1992), and recently these approaches have also been applied to acinar cells (Maruyama, 1988; Maruyama & Petersen, 1994; Schmid & Schulz, 1996; Giovannucci et al. 1998).
In particular, measurement of membrane capacitance (Cm) has proved a powerful tool for the study of stimulus- secretion coupling (Thomas et al. 1990; Augustine & Neher, 1992; Tse et al. 1993; Heidelberger et al. 1994). When applied to acinar cells this technique has revealed increases in cell surface area in response to Ca2+-mobilizing agonists that have been interpreted as zymogen granule exocytosis (Maruyama, 1988; Maruyama & Petersen, 1994; Giovannucci et al. 1998). Zymogen granules are approximately the same size as mast cell granules and, as such, one would expect to observe clear step-like increases in Cm resulting from the exocytosis of these granules (see for example Fig. 2 of Fernandez et al. 1984). Although some step-like events have been observed (Schmid & Schulz, 1996; Giovannucci et al. 1998; Thomas, 1998), most recordings from acinar cells are largely devoid of such events (see for example Fig. 5 of Maruyama & Petersen, 1994). It has been argued that the lack of step-like events is due to simultaneous exocytotic and endocytotic activity (Giovannucci et al. 1998). In support of this hypothesis, independent measurements of secretory activity using the fluorescent dye FM1-43 (Betz et al. 1996) revealed that much more membrane was inserted into the cell surface than was suggested by the increase in Cm (Giovannucci et al. 1998). Nevertheless, an alternative interpretation is that most of the increase in Cm is due, not to zymogen granule exocytosis, but to the exocytosis of another population of much smaller vesicles (Thomas, 1998). Although measuring the true extent of secretory activity, studies using FM1-43 (just like measurements of Cm), cannot distinguish between different, parallel secretory pathways occurring within the same cell (Ninomiya et al. 1997; Kasai, 1999). An additional problem with Cm measurements is that soluble proteins in the cell escape from the cytosol into the recording electrode, a circumstance that may compromise stimulus-secretion coupling (Burgoyne, 1995; Seward & Nowycky, 1996). To circumvent such ambiguities, we have used a different approach to obtain real-time quantification of zymogen granule exocytosis in intact acinar cells.
Figure 2. ACh-evoked loss of quinacrine fluorescence.
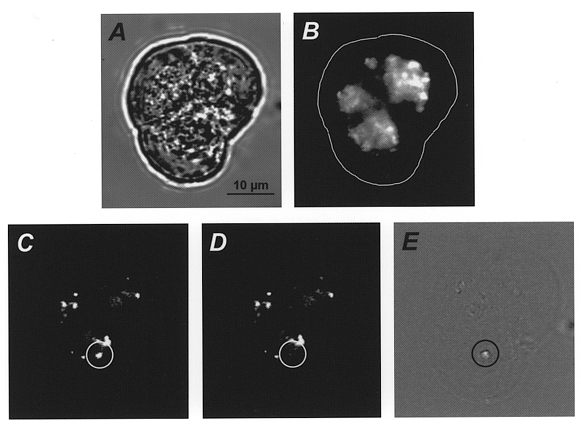
A three-cell acinus loaded with quinacrine before (A and B) and during (C–E) treatment with 10 μm ACh. A, brightfield image at the start of the recording. B, fluorescence image taken at the same time. The continuous white line shows the outline of the acinus made by tracing around the image shown in A. C and D, consecutive fluorescence images captured during application of ACh (note: the image contrast has been adjusted compared with B to allow identification of individual granules, one of which is circled in C). E shows the time-differential image formed by subtraction of the brightfield equivalent of D from the brightfield equivalent of C, exocytosis of a quinacrine-loaded zymogen granule is circled (C-E).
Figure 5. [Ca2+]i dependency of the slow phase of secretion.
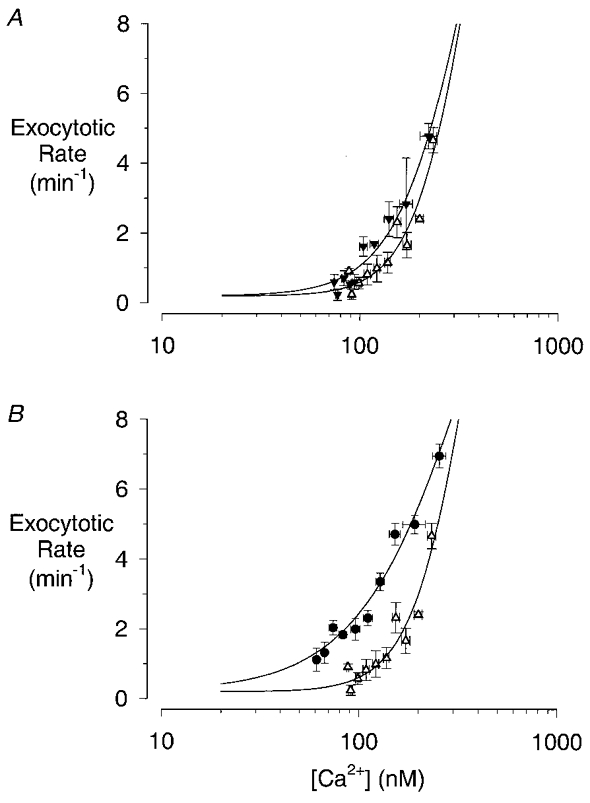
A, plots of the data in Fig. 4Bvs. the data in Fig. 4A for ACh concentrations of 250 nm (▵) and 1 μm (▾). B, plots of the data in Fig. 4Bvs. the data in Fig. 4A for ACh concentrations of 250 nm (▵) and 10 μm (•). Pairs of data points, between 40 and 340 s after the start of ACh application, were ranked in order of [Ca2+] and then divided into bins of three samples each; points represent the mean ±s.e.m. (vertical error bars) or ±s.d. (horizontal error bars). The continuous lines are least-squares fits to eqn (1) with m= 0.2 min−1; a= 19.8 min−1; b= 2.95 (250 nm) or 2.36 (1 μm) or 1.52 (10 μm), and k= 371 nm (250 nm) or 370 nm (1 μm) or 393 nm (10 μm); r2= 0.90 (250 nm), 0.95 (1 μm) and 0.97 (10 μm).
The technique we have used has been pioneered by Terakawa and colleagues (Terakawa et al. 1991; Kamijo et al. 1993) and relies upon the change in optical properties of individual dense-cored vesicles that occurs when the contents of a granule are lost from the lumen during exocytosis. The original methodology used differential interference contrast (DIC) optics; however, we have found that standard brightfield optics are adequate for visualizing the exocytosis of the large zymogen granules of acinar cells. We have used this technique to investigate the relationship between [Ca2+]i and zymogen granule exocytosis, and in particular, to explore the kinetics of release in response to various concentrations of acetylcholine (ACh).
METHODS
Materials
Five- to seven-week-old male Wistar rats (175–225 g) were obtained from the departmental breeding colony. Collagenase type IA, bovine serum albumin fraction V (BSA), soya bean trypsin inhibitor (SBTI), basal medium Eagle’s (BME) amino acids, ACh, polyethyleneimine, quinacrine hydrochloride, DMSO, pluronic F-127 and atropine were from Sigma (Poole, UK). Fura-2 acetoxymethyl ester (AM) and potassium salt were from Molecular Probes (Eugene, OR, USA). All general chemicals were from Sigma; where possible, only cell-culture-tested chemicals were used.
Acinar cell preparation
Acinar cells were obtained by a modification of the procedure of Rogers et al. (1988). Essentially, the pancreas was removed from rats that had been killed by inhalation of CO2. Incubation medium (IM) containing 200 U ml−1 of collagenase type IA (5 ml) was injected into the excised gland. The IM consisted of 120 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 1 mm NaH2PO3, 1 mm NaHCO3, 11 mm glucose, 4 mm D-myo-inositol, 2 mm glutamine, 2 % (v/v) BME amino acids, 25 mm Hepes-NaOH, pH 7.2 plus 0.5 mg ml−1 BSA and 0.5 mg ml−1 SBTI. The injected pancreas was then incubated with shaking for 10 min at 37°C. After incubation the collagenase solution was decanted and the pancreas rinsed twice with IM. The gland was then gently triturated in 5 ml of IM using three plastic Pasteur pipettes of gradually diminishing diameters. After filtering through 200 μm mesh nylon gauze, the dissociated tissue was layered onto two 5 ml cushions of 4 % BSA (in IM) and centrifuged at 150 g for 3 min. The supernatants were carefully removed and the cell pellets washed twice with 5 ml each of IM (and recovered by centrifugation (150 g for 3 min)) before final resuspension in 8–14 ml of IM.
Measurement of exocytosis
Isolated acinar cells were plated on modified, glass-bottomed Petri dishes (Thomas et al. 1996) that had been pre-treated with 0.1 % polyethyleneimine (in 150 mm sodium borate, pH. 8.4). Cells were allowed to attach (at room temperature) for 30 min before use. Dishes were rinsed twice with IM, and then either used for experimentation or kept on ice until needed. Experiments were carried out at 37 ± 2°C using a heated stage (Medical Systems, Greenvale, NY, USA) mounted on a Nikon Diaphot 200 microscope. Various concentrations of ACh were applied to the cells from glass micropipettes (2–5 μm diameter) placed 30–50 μm from the cells using a PicoSpritzer II (General Valve Corp., Fairfield, NJ, USA). To avoid movement artefacts, very low pressure ejection (typically ∼1 p.s.i.) was used and only cells firmly attached to the dish were analysed. Cells were observed using a ×60, 1.4 NA, planapochromat objective lens and the image projected onto a CCD camera (Hamamatsu C4880-80-24 operating in fast mode, i.e. 12 bits) using a ×2.5 projection lens (i.e. final magnification, ×150). Images (40 ms exposure) were captured every 1.25 s using AQM software (Kinetic Imaging, Liverpool, UK). Continuous time-differential analysis was carried out as described by Terakawa and colleagues (Terakawa et al. 1991) using Lucida software (Kinetic Imaging).
In some experiments, acinar cells were pre-incubated with 3 μm quinacrine hydrochloride for 10 min at 37°C. Quinacrine fluorescence was excited using 440 nm light from a fast-scanning monochromator using a 12 nm bandpass and set to approximately 40 % of its maximum intensity (Kinetic Imaging). The monochromator was coupled to the microscope using a two-port epifluorescence adapter (T.I.L.L. Photonics, Planegg, Germany) containing a neutral density filter that reflected 50 % of the light to the sample. Excitation light was reflected off a 495 nm dichroic mirror and emitted fluorescence collected through a 500 nm long-pass filter (both from Chroma Technologies, Brattleboro, VT, USA). Pairs of brightfield (100 ms exposure) and fluorescence (500 ms exposure) images were obtained every 2.5 s. Alternate brightfield and fluorescence excitation was achieved by using two high-speed shutters: a VS14 Uniblitz shutter (Vincent Associates, Rochester, NY, USA) in the brightfield illumination path and an 846HP electronic shutter (Newport, Irvine, CA, USA) in the monochromator illumination path. The diameter of quinacrine-loaded granules was also estimated in these experiments. Only those granules undergoing exocytosis (fluorescent spots whose sudden disappearance occurred within two successive frames) were used for these estimates. The diameter was measured (in pixels) along either the horizontal or vertical axis (whichever was the longer). Pixel values were then converted to micrometres by multiplying by 0.165 (effective pixel size, (9.9 μm × 9.9 μm)/magnification; note: in these experiments the ×2.5 projection lens was omitted, and so magnification =× 60).
Measurement of intracellular calcium
For measurement of [Ca2+]i, cells were loaded with 2.5 μm fura-2 AM (0.005 % pluronic F-127, 0.1 % DMSO) for 25 min at 37°C, and then washed free of extracellular dye. Pairs of fluorescence images at 340 nm (700 ms exposure) and 380 nm (500 ms exposure) were obtained every 2 s on the same microscope as described above, except using a ×40, 1.3 NA, CF Fluor objective lens (i.e. final magnification, ×100). Excitation light from the monochromator was reflected off a 400 nm dichroic mirror (Chroma Technologies) and emitted fluorescence was collected through a 500 nm emission filter (70 nm bandpass; Comar, Cambridge, UK). Calibration of the fura-2 signal was carried out using solutions of known [Ca2+] in 20 μm thick microslides (VitroCom Inc., Mountain Lakes, NJ, USA) as described by Thomas et al. (1996).
Except where otherwise stated, all values shown are means ±s.e.m.
RESULTS
Acetylcholine-evoked morphological changes
ACh (10 μm) stimulated morphological alterations in the apical domain of pancreatic acinar cells (Fig. 1). Such changes were immediately apparent when viewed in movie form (especially when viewed at faster than normal speed), but are less striking in still representations (compare Fig. 1E with Fig. 1D). Nevertheless if a difference image is formed by subtraction of one frame from its predecessor (see Fig. 1F), the changes in the apical region become apparent as bright and dark spots (arrows). In previous studies on other cell types, these spots have been interpreted as sites of exocytotic events; the spots appearing either bright or dark depending upon the position of the secretory granule in the focal plane of the objective lens (Segawa et al. 1991; Terakawa et al. 1991; Kamijo et al. 1993).
Figure 1. Continuous, time-differential analysis of ACh-evoked changes in acinar cell morphology.
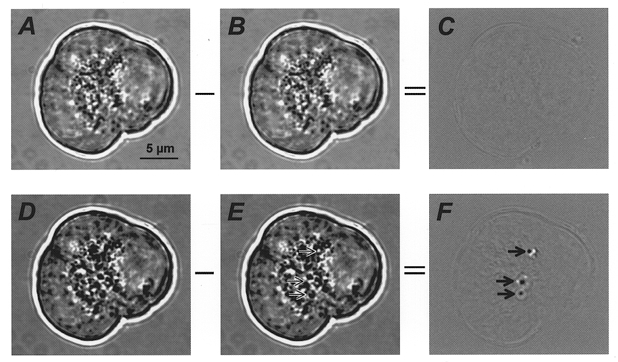
Brightfield images of an acinus consisting of three cells are shown before (A and B) and during (D and E) treatment with 10 μm ACh. Subtraction of one frame from its predecessor (A–B or D–E) creates a time-differential image (C or F, respectively). Three exocytotic events are clearly visible in F (arrows); arrows in E indicate the regions where these exocytotic events occurred.
In our experiments, these spots were rarely seen in cells prior to ACh application (see Fig. 1C which results from subtraction of Fig. 1B from 1A; 0.2 ± 0.03 spots min−1 cell−1; n= 142). The fact that the spots were limited to the apical pole of the acinar cells and that the appearance of the ACh-evoked spots was completely inhibited by atropine (n= 12; 5 μm atropine applied ≤10 min before stimulation with 10 μm ACh) suggests that these events correspond to sites of zymogen granule exocytosis.
To confirm that the spots observed in the time-differential analysis reflect exocytotic activity, we carried out experiments in which we labelled zymogen granules with the fluorescent marker quinacrine. Quinacrine is a weak base that accumulates in acidic compartments such as secretory vesicles (Chiavaroli et al. 1991; Titievsky et al. 1996), and has previously been used to validate the measurement of exocytosis using either complex impedance analysis (Breckenridge & Almers, 1987; Zimmerberg et al. 1987) or voltammetry (Tatham et al. 1991). In acinar cells incubated with quinacrine the apical pole of the cells became brightly fluorescent (Fig. 2B). By carefully adjusting the contrast of the image, individual fluorescent spots can be clearly delineated (Fig. 2C, circled); such spots measured 1.0 ± 0.10 μm in diameter (range, 0.50–2.48 μm; n= 79) which, considering the limited axial resolution of wide-field fluorescence microscopy (Agard et al. 1989), is in reasonable agreement with estimates of the diameter of zymogen granules obtained in electron microscopic studies (diameter ∼0.7 μm, Ermak & Rothman, 1981, 1986; Aughsteen et al. 1996). On stimulation with 10 μm ACh several of these fluorescent spots disappeared (compare circled region in Fig. 2D with that in Fig. 2C). At the same time, and in the same location, spots appeared in the time-differential image (see circled region in Fig. 2E). Disappearance of a fluorescent spot was almost always accompanied, in time and space, by the simultaneous appearance of a spot in the time-differential image; of 79 ‘quinacrine events’ observed, 76 (96 %) were accompanied by a spot in the appropriate time-differential image. This close correspondence clearly demonstrates that the continuous time-differential analysis provides a direct read-out of zymogen granule exocytosis that can be readily quantified (see below).
Quantification of zymogen granule exocytosis
To determine the time course of exocytotic activity in response to ACh, we counted the number of exocytotic events occurring in 10 s bins throughout a 10 min period, in the course of which we made a 6 min application of 10 μm ACh (see Fig. 3A). The time course shows very low levels of exocytosis before ACh addition (0.3 ± 0.05 events min−1 cell−1, n= 50) followed by a burst of activity within 20 s of agonist application (peak rate, 12.2 ± 3.68 events min−1 cell−1; n= 50). This high rate of activity soon subsided to a much lower level, albeit still significantly higher than baseline (1.8 ± 0.30 events min−1 cell−1 measured over a 60 s period beginning 4 min after the start of ACh application; n= 50; P < 0.05, pairwise comparison by Student’s t test).
Figure 3. Time course of ACh-stimulated changes in [Ca2+]i and zymogen granule exocytosis.
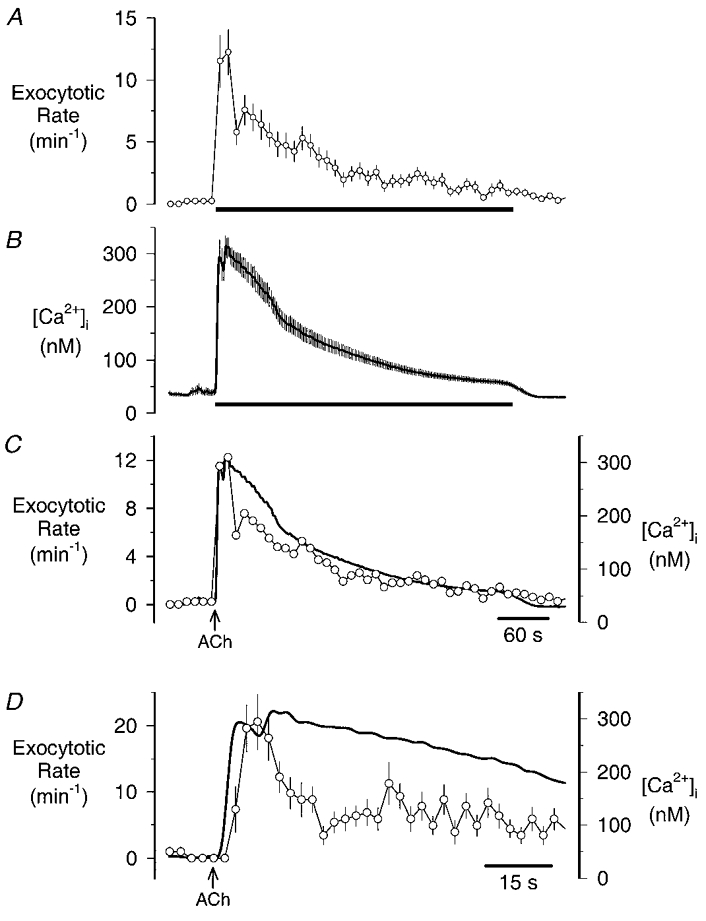
A, changes in the rate of exocytosis (measured every 1.25 s and analysed in 10 s bins) evoked by a 6 min application (horizontal bar) of 10 μm ACh; points represent the mean ±s.e.m. determined in 50 cells. B, changes in [Ca2+]i elicited by a 6 min application (horizontal bar) of 10 μm ACh; points represent the mean ±s.e.m. determined in 19 cells. C, traces from A and B superimposed (thick continuous trace, [Ca2+]i; ○, exocytosis), error bars omitted for clarity. D, as in C except the rate of exocytosis is analysed in 2.5 s bins and error bars are shown. Arrows in C and D indicate the start of the ACh application.
In parallel experiments, examination of the [Ca2+]i responses of cells (from the same preparations) revealed a similar time course to that observed for exocytosis (Fig. 3B); an initial peak followed by a plateau phase. To examine the relationship more closely the two time courses are superimposed in Fig. 3C. One thing is apparent from this comparison: that the rate of exocytosis following the peak falls significantly despite the fact that [Ca2+]i remains elevated. This observation is more clearly seen in Fig. 3D where we have reanalysed the data with higher temporal resolution (2.5 s bins) and expanded the portion of the recordings around the start of the ACh application. (Note: analysis at 2 times the sampling frequency gave optimal results; when rates were estimated frame-by-frame the results were too noisy for meaningful interpretation.) This analysis reveals that the burst of exocytosis begins soon after the rise in [Ca2+]i. At its peak, this burst of activity reached 20.6 ± 4.14 events min−1, but then the rate of exocytosis fell precipitously even though [Ca2+]i was still high (> 250 nm); the rate of secretion decreasing to 3.4 ± 1.4 min−1 after 25 s of treatment. However, secretory activity then recovered slightly to reach a secondary peak (11.3 ± 3.22 events min−1) about 40 s after the start of ACh application.
Dose response to acetylcholine
Figure 4A reveals the [Ca2+]i responses evoked by different concentrations of ACh. The lowest dose of ACh that we used, 10 nm, elicited no measurable response, whereas 50 nm gave a small monophasic increase in [Ca2+]i. At higher doses (250 nm, 1 μm and 10 μm) ACh gave rise to very similar responses with a well-defined peak that was of similar magnitude with each concentration of ACh. The subsequent plateau phase however, was more varied. Curiously with increasing ACh concentrations (≤ 250 nm), the height of the [Ca2+]i plateau appeared to decrease. In contrast, the slower phase of secretion that we observed above appeared to be much greater with 10 μm than with 250 nm or 1 μm ACh. Examination of Fig. 4B shows that the burst of exocytosis, just like the initial peak in [Ca2+]i, was very similar for [ACh]≤ 250 nm; however, the rate of zymogen granule exocytosis at later times was higher with 10 μm ACh than with 250 nm or 1 μm. This is more clearly seen in the integral of the time courses, i.e. the cumulative secretory response, shown in Fig. 4C. The integral (Fig. 4C) also shows the remarkable similarity between the exocytotic responses obtained with 250 nm and 1 μm ACh.
Figure 4. ACh dose dependency of [Ca2+]i and exocytosis in acinar cells.

A, changes in [Ca2+]i in acinar cells treated with various concentrations of ACh for 6 min: 10 nm, n= 19; 50 nm, n= 27; 250 nm, n= 17; 1 μm, n= 26; and 10 μm, n= 19. B, changes in the rate of exocytosis in acinar cells treated with various concentrations of ACh for 6 min: 10 nm, n= 8; 50 nm, n= 22; 250 nm, n= 24; 1 μm, n= 33; and 10 μm, n= 50. C, cumulative secretory response (i.e. the time integral of the exocytotic responses shown in B). The period of ACh application is indicated by the horizontal bars in A and B. The arrow in C indicates the start of ACh application.
The reason for the apparent discrepancy between the [Ca2+]i signal and the later phase of secretion at higher doses of ACh was examined by plotting the rate of exocytosis measured in a 5 min period starting 40 s after the start of the ACh application versus the [Ca2+]i measured at the same time in the parallel experiments (Fig. 5). It is apparent from this analysis that even though the rate of exocytosis in the later phase of secretion was dependent upon [Ca2+]i, it was also dramatically affected by the agonist concentration. To make quantitative comparisons we fitted all the data to the Hill equation:
| (1) |
where E is the rate of exocytosis; m, minimum rate; a, maximum rate −m; b, Hill coefficient; Ca, [Ca2+]i; and k, EC50 for [Ca2+]i.
To obtain estimates of the EC50 and Hill coefficient for each dose of ACh, we assumed that m was equal to the basal rates that we measured in the absence of agonist (0.2 events min−1) and that a was equal to the highest rates we measured during the exocytotic burst (20 events min−1) −m (0.2 min−1), i.e. 19.8 min−1. Comparisons of the data obtained with 250 nm ACh to that obtained with either 1 μm (Fig. 5A) or 10 μm (Fig. 5B) show no differences in the EC50 of the slow phase for [Ca2+]i (250 nm ACh, 371 nm; 1 μm ACh, 370 nm; 10 μm ACh, 393 nm), and yet, cumulative exocytosis is clearly greater with 10 μm ACh than with the other two doses (see Fig. 4C). It would seem that 10 μm ACh has effects other than an increase in Ca2+ sensitivity. One potential explanation of the enhanced secretory response with 10 μm ACh suggested by this analysis is that the steepness of the relationship between the exocytotic rate and [Ca2+]i is markedly reduced at the higher [ACh], the Hill coefficient being decreased from 2.95 at 250 nm ACh to 1.52 at 10 μm ACh (2.36 at 1 μm ACh).
DISCUSSION
Previous studies of pancreatic acini have proposed that the secretory response to Ca2+-mobilizing agonists is divided into two phases: an early phase that correlates well with the increase in [Ca2+]i during the first 5 min of treatment; followed by a slower sustained phase of secretion that continues as long as agonist is applied (15–30 min) even though [Ca2+]i has returned to baseline levels (Pandol et al. 1985). We have now investigated this initial, Ca2+-dependent phase of secretion using a technique with much better temporal resolution than used in previous studies. Our results clearly show that the early phase can itself be subdivided into an initial exocytotic burst followed by a slower phase that is dependent both on [Ca2+]i and agonist concentration.
Quantitative comparisons of our work with previous studies using amylase release assays are difficult. Nevertheless, morphometric analysis of rat pancreatic acinar cells has suggested that the average cell in a fasted rat contains 343 zymogen granules (Aughsteen et al. 1996). On feeding, the number of zymogen granules decreases by about 35 % (Ermak & Rothman, 1981); so in the fed rats that we have used, one would expect an average cell to contain ∼220 granules. Thus, after 6 min of treatment with 10 μm ACh the exocytosis of 21 granules (see Fig. 4C) would constitute almost 10 % of the total granule pool, this amount is in very good agreement with amylase release studies using rat pancreatic acini; for example, Fig. 1 of Halenda & Rubin (1982) shows release of ∼11 % of cellular amylase in 6 min of treatment with 10 μm carbachol. Furthermore in the present study, the five granules (see Fig. 4C) exocytosed in the initial burst would be equivalent to ∼2 % of the granule pool.
Studies on permeabilized acini have also revealed a biphasic secretory response (Padfield & Panesar, 1997, 1998). In these studies an initial Ca2+-stimulated, MgATP-independent phase of amylase secretion was complete at the earliest time studied, 2.5 min, and was then followed by a second phase of release that was dependent upon the presence of MgATP. In the study of Padfield & Panesar (1997) the ‘primed’ granules, i.e. those secreted in the absence of MgATP, released ∼5 % of the total amylase content of the cell. From the comparison above, this would suggest that primed granules represent about half of the secretory response we measured in 6 min of treatment with 10 μm ACh. However, it is clear that the proportion of primed granules declines with time after permeabilization (Padfield & Panesar, 1997), and so we cannot rule out the possibility that in intact acinar cells the secretory response we have measured is due solely to the exocytosis of primed zymogen granules.
Considering that the exocytotic burst we measured represents the exocytosis of 2 % of the total granule pool, our results imply that < 50 % of the primed zymogen granules are exocytosed during the exocytotic burst, and that this sub-population of granules is more ready for release than the rest of the primed pool. This observation is similar to those made on neuroendocrine cells which have also suggested that primed secretory vesicles are found in various states of readiness for release (Thomas et al. 1993; Heinemann et al. 1994). Indeed, in both melanotrophs and chromaffin cells the release-ready pool of secretory vesicles constitutes only about 10 % of the primed vesicles (Thomas et al. 1993; Parsons et al. 1995). Despite these similarities, the exocytotic burst in acinar cells probably does not represent truly ‘release-ready’ granules. Acinar cells secrete much more slowly than neuroendocrine cells, for example, rates of agonist-evoked secretion in gonadotrophs (Thomas & Waring, 1997) and chromaffin cells (Augustine & Neher, 1992; Mollard et al. 1995), even at room temperature, can exceed 25 vesicles s−1; such rates are more than 70-fold faster than the highest rates measured in this study at 37°C. It is likely that even the zymogen granules exocytosed during the exocytotic burst must undergo further modifications after the rise in [Ca2+]i before finally being exocytosed.
Exocytosis of this small population of granules is followed by a second, slower phase of secretion, the extent of which varies according to the concentration of ACh used. The rate of secretion at times after 30 s of treatment is clearly dependent upon the [Ca2+]i (see Fig. 4A and B); however, it appears that the steepness of this relationship changes according to the agonist concentration. Comparing 1 μm ACh to 250 nm, the change is quite small (Hill coefficient, 2.4 vs. 3.0), but at 10 μm there is an approximately 2-fold reduction in steepness (Hill coefficient, 1.5). This has the effect of increasing the amount of exocytosis at [Ca2+]i levels below the EC50 value, but decreasing the response at higher [Ca2+]i. At the low [Ca2+]i levels during the plateau phase of the response, it is the former effect that has most influence on the slow secretory activity. Over the time period studied, the small effect on the steepness of the Ca2+ dependency of exocytosis observed at 1 μm ACh is just sufficient to maintain the slow phase of secretion at the same levels obtained at lower [ACh], i.e. 250 nm; however, the larger effect seen with 10 μm ACh clearly leads to an enhancement of the second phase of secretion (see Fig. 4C). The similarity of the secretory response obtained with 1 μm and 250 nm ACh seems contradictory in the light of the response with 10 μm. Previous studies of amylase release would suggest that the response with 1 μm ACh would be intermediate between 250 nm and 10 μm ACh (Merritt & Rubin, 1985). However, determination of the dose dependency of amylase release has generally been carried out using longer periods of incubation (≤ 15 min) than we have used. It is possible that when agonist application is continued for longer than 6 min, [Ca2+]i falls to pre-stimulus levels and secretion then enters a phase essentially independent of [Ca2+]i (Pandol et al. 1985) during which secretion at 1 μm ACh might exceed that obtained at 250 nm.
Which second messenger pathways contribute to the second phase of release will be the focus of future studies, but this phase may involve changes in the actin cytoskeleton (Muallem et al. 1995; Valentijn et al. 1999) brought about by the diacylglycerol-mediated activation of protein kinase C (PKC). PKC may act directly on the cytoskeleton (Vitale et al. 1995) or indirectly via activation of the p38 mitogen-activated protein kinase pathway (Schäfer et al. 1998). Alternatively, the enhancement of the slow secretory phase observed with higher doses of ACh might reflect the agonist-dependent activation of a GTP-binding protein such as rab3 (Jena et al. 1994) or Gq/11 (Ohnishi et al. 1997).
As discussed above, there is good quantitative agreement between our results and biochemical measurements of amylase release (Halenda & Rubin, 1982; Padfield & Panesar, 1997); nevertheless, there seem to be some discrepancies compared with the results of Giovannucci et al. (1998) who monitored secretion either electrophysiologically (Cm measurements) or optically (with FM1-43). These workers calculated that in their experiments ∼50 zymogen granules were exocytosed in a 90 s period following treatment with 10 μm carbachol (5 times more than we have observed in the first 90 s of ACh application). However, their Cm traces were noticeably free of the ‘steps’ one would expect to see on exocytosis of large zymogen granules (see Introduction). They argued that this was due to concomitant endocytotic activity, and by using FM1-43 measurements they demonstrated that much more exocytotic activity had occurred than was apparent from the Cm recordings. Additionally, they observed many more steps in the Cm recordings when endocytosis was inhibited by including GTP-γ-S in the recording electrode. Nonetheless, examination of Fig. 1B of Giovannucci et al. (1998) shows only six steps during a 30 s application of carbachol (in the presence of intracellular GTP-γ-S) together with three further steps after agonist application had ceased. And yet, the Cm increased by over 1000 fF in the same period; considering the mean granule Cm of 22 fF in this study, nine granules would only increase the cellular Cm by 198 fF. Likewise, Fig. 1C shows only 21 steps detected in two cells treated with carbachol (+ GTP-γ-S), presumably in a 90 s period. Thus the frequency of step-like events observed by Giovannucci et al. (10.5 steps in 90 s), is very similar to the frequency of exocytotic events that we have measured by continuous, time-differential analysis (11 events in the first 90 s of treatment). We would argue, therefore, that the steps observed by Giovannucci and colleagues are a good measure of zymogen granule exocytosis, and that the major component of secretion measured as a ‘smooth’ increase in Cm (see also Maruyama (1988) and Maruyama & Petersen (1994)), or with FM1-43, is not due to zymogen granule exocytosis, but reflects the activity of a parallel secretory pathway (Ninomiya et al. 1997; Kasai, 1999) involving an, as-yet, uncharacterized population of smaller vesicles (Thomas, 1998).
In conclusion, we propose that secretion in acinar cells consists of three phases. First, an initial exocytotic burst that involves, on average, five granules in each acinar cell (or ∼2 % of the total granule pool). Second, an intermediate phase that is dependent on both [Ca2+]i and other second messenger pathways, and contributes approximately three times the number of granules exocytosed in the initial burst. Third, a phase of release, which we have not studied here, but which occurs on prolonged stimulation (> 5 min), and which may be Ca2+ independent and PKC dependent (Pandol et al. 1985; Muallem et al. 1995).
Acknowledgments
This work was supported by Grant No. 8/C11044 from the Biotechnology and Biological Sciences Research Council. P.T. is the recipient a Department of Pharmacology Fellowship. We would like to thank Dr Alois Hodel for critical reading of the manuscript.
References
- Agard DA, Hiraoka Y, Shaw P, Sedat JW. Fluorescence microscopy in three dimensions. Methods in Cell Biology. 1989;30:353–377. doi: 10.1016/s0091-679x(08)60986-3. [DOI] [PubMed] [Google Scholar]
- Aughsteen AA, Kataoka K, Shair SA. Correlative morphometric and biochemical study on pancreatic amylase in normal and streptozotocin-diabetic rats. Pancreas. 1996;13:295–303. doi: 10.1097/00006676-199610000-00013. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Neher E. Calcium requirements for secretion in bovine chromaffin cells. The Journal of Physiology. 1992;450:247–271. doi: 10.1113/jphysiol.1992.sp019126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz W, Mao F, Smith CB. Imaging exocytosis and endocytosis. Current Opinion in Neurobiology. 1996;6:365–371. doi: 10.1016/s0959-4388(96)80121-8. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science. 1992;255:200–203. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- Breckenridge LJ, Almers W. Final steps in exocytosis observed in a cell with giant secretory granules. Proceedings of the National Academy of Sciences of the USA. 1987;84:1945–1949. doi: 10.1073/pnas.84.7.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne RD. Fast exocytosis and endocytosis triggered by depolarisation in single adrenal chromaffin cells before rapid Ca2+ current run-down. Pflügers Archiv. 1995;430:213–219. doi: 10.1007/BF00374652. [DOI] [PubMed] [Google Scholar]
- Chiavaroli C, Vacher P, Vecsey A, Mons N, Letari O, Pralong W, Lagnaux Y, Whelan R, Schlegel W. Simultaneous monitoring of cytosolic free calcium and exocytosis at the single cell level. Journal of Neuroendocrinology. 1991;3:253–260. doi: 10.1111/j.1365-2826.1991.tb00272.x. [DOI] [PubMed] [Google Scholar]
- Ermak TH, Rothman SS. Zymogen granules of pancreas decrease in size in response to feeding. Cell and Tissue Research. 1981;214:51–66. doi: 10.1007/BF00235144. [DOI] [PubMed] [Google Scholar]
- Ermak TH, Rothman SS. Zymogen granule size in pancreas of nursing rats. Journal of Morphology. 1986;187:289–299. doi: 10.1002/jmor.1051870303. [DOI] [PubMed] [Google Scholar]
- Fernandez JM, Neher E, Gomperts BD. Capacitance measurements reveal stepwise fusion events in degranulating mast cells. Nature. 1984;312:453–455. doi: 10.1038/312453a0. [DOI] [PubMed] [Google Scholar]
- Giovannucci DR, Yule DI, Stuenkel EL. Optical measurement of stimulus-evoked membrane dynamics in single pancreatic acinar cells. American Journal of Physiology. 1998;275:C732–739. doi: 10.1152/ajpcell.1998.275.3.C732. [DOI] [PubMed] [Google Scholar]
- Halenda SP, Rubin RP. Phospholipid turnover in isolated rat pancreatic acini. Biochemical Journal. 1982;208:713–721. doi: 10.1042/bj2080713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger R, Heinemann C, Neher E, Matthews G. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature. 1994;371:513–515. doi: 10.1038/371513a0. [DOI] [PubMed] [Google Scholar]
- Heinemann C, Chow RH, Neher E, Zucker RS. Kinetics of the secretory response in bovine chromaffin cells following flash photolysis of caged Ca2+ Biophysical Journal. 1994;67:2546–2557. doi: 10.1016/S0006-3495(94)80744-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson JD, Palade GE. Synthesis, intracellular transport, and discharge of secretory proteins in stimulated pancreatic exocrine cells. Journal of Cell Biology. 1971;50:135–158. doi: 10.1083/jcb.50.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jena BP, Gumkowski FD, Konieczko EM, Fischer von Mollard G, Jahn R, Jamieson JD. Redistribution of a rab3-like GTP-binding protein from secretory granules to the Golgi complex in pancreatic acinar cells during regulated exocytosis. Journal of Cell Biology. 1994;124:43–53. doi: 10.1083/jcb.124.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo A, Terakawa S, Hisamatsu K. Neurotransmitter-induced exocytosis in goblet and acinar cells of rat nasal mucosa studied by video microscopy. American Journal of Physiology. 1993;265:L200–209. doi: 10.1152/ajplung.1993.265.2.L200. [DOI] [PubMed] [Google Scholar]
- Kasai H. Comparative biology of Ca2+-dependent exocytosis: implications of kinetic diversity for secretory function. Trends in Neurosciences. 1999;22:88–93. doi: 10.1016/s0166-2236(98)01293-4. [DOI] [PubMed] [Google Scholar]
- Maruyama Y. Agonist-induced changes in cell membrane capacitance and conductance in dialysed pancreatic acinar cells of rats. The Journal of Physiology. 1988;406:299–313. doi: 10.1113/jphysiol.1988.sp017381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama Y, Petersen OH. Delay in granular fusion evoked by repetitive cytosolic Ca2+ spikes in mouse pancreatic acinar cells. Cell Calcium. 1994;16:419–430. doi: 10.1016/0143-4160(94)90035-3. [DOI] [PubMed] [Google Scholar]
- Merritt JE, Rubin RP. Pancreatic amylase secretion and cytoplasmic free calcium. Biochemical Journal. 1985;230:151–159. doi: 10.1042/bj2300151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollard P, Seward EP, Nowycky MC. Activation of nicotinic receptors triggers exocytosis from bovine chromaffin cells in the absence of membrane depolarization. Proceedings of the National Academy of Sciences of the USA. 1995;92:3065–3069. doi: 10.1073/pnas.92.7.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muallem S, Kwiatkowska K, Xu X, Yin HL. Actin filament disassembly is a sufficient final trigger for exocytosis in nonexcitable cells. Journal of Cell Biology. 1995;128:589–598. doi: 10.1083/jcb.128.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Marty A. Discrete changes of cell membrane capacitance observed under condition of enhanced secretion in bovine adrenal chromaffin cells. Proceedings of the National Academy of Sciences of the USA. 1982;79:6712–6716. doi: 10.1073/pnas.79.21.6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya Y, Kishimoto T, Yamazawa T, Ikeda H, Miyashita Y, Kasai H. Kinetic diversity in the fusion of exocytotic vesicles. EMBO Journal. 1997;16:929–934. doi: 10.1093/emboj/16.5.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi H, Ernst SA, Yule DI, Baker CW, Williams JA. Heterotrimeric G-protein Gq/11 localized on pancreatic zymogen granules is involved in calcium-regulated amylase secretion. Journal of Biological Chemistry. 1997;272:16056–16061. doi: 10.1074/jbc.272.25.16056. [DOI] [PubMed] [Google Scholar]
- Padfield PJ, Panesar N. MgATP acts before Ca2+ to prime amylase secretion from permeabilized rat pancreatic acinar acini. American Journal of Physiology. 1997;273:G655–660. doi: 10.1152/ajpgi.1997.273.3.G655. [DOI] [PubMed] [Google Scholar]
- Padfield PJ, Panesar N. The two phases of regulated exocytosis in permeabilized pancreatic acini are modulated differently by heterotrimeric G-proteins. Biochemical and Biophysical Research Communications. 1998;245:332–336. doi: 10.1006/bbrc.1998.8350. [DOI] [PubMed] [Google Scholar]
- Palade G. Intracellular aspects of the process of protein synthesis. Science. 1975;189:347–358. doi: 10.1126/science.1096303. [DOI] [PubMed] [Google Scholar]
- Pandol SJ, Schoeffield MS, Sachs G, Muallem S. Role of free cytosolic calcium in secretagogue-stimulated amylase release from dispersed acini from guinea pig pancreas. Journal of Biological Chemistry. 1985;260:10081–10086. [PubMed] [Google Scholar]
- Parsons TD, Coorssen JR, Horstmann H, Almers W. Docked granules, the exocytic burst, and the need for ATP hydrolysis in endocrine cells. Neuron. 1995;15:1085–1096. doi: 10.1016/0896-6273(95)90097-7. [DOI] [PubMed] [Google Scholar]
- Rogers J, Hughes RG, Matthews EK. Cyclic GMP inhibits protein kinase C-mediated secretion in rat pancreatic acini. Journal of Biological Chemistry. 1988;263:3713–3719. [PubMed] [Google Scholar]
- Schäfer C, Ross SE, Bragado JM, Groblewski GE, Ernst SA. A role for the p38 mitogen-activated protein kinase/HSP27 pathway in cholecystokinin-induced changes in the actin cytoskeleton in rat pancreatic acini. Journal of Biological Chemistry. 1998;273:24173–24180. doi: 10.1074/jbc.273.37.24173. [DOI] [PubMed] [Google Scholar]
- Schmid A, Schulz I. Different time courses of GTP[γ-S]-induced exocytosis and current oscillations in isolated mouse pancreatic acinar cells. Pflügers Archiv. 1996;432:876–884. doi: 10.1007/s004240050211. [DOI] [PubMed] [Google Scholar]
- Segawa A, Terakawa S, Yamashina S, Hopkins CR. Exocytosis in living salivary glands: direct visualization by video-enhanced microscopy and confocal laser microscopy. European Journal of Cell Biology. 1991;54:322–330. [PubMed] [Google Scholar]
- Seward EP, Nowycky MC. Kinetics of stimulus-coupled secretion in dialyzed bovine chromaffin cells in response to trains of depolarizing pulses. Journal of Neuroscience. 1996;16:553–562. doi: 10.1523/JNEUROSCI.16-02-00553.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartakoff AM, Jamieson JD, Scheele GA, Palade GE. Studies on the pancreas of the guinea pig. Parallel processing and discharge of exocrine proteins. Journal of Biological Chemistry. 1975;250:2671–2677. [PubMed] [Google Scholar]
- Tatham PER, Duchen MR, Millar J. Monitoring exocytosis from single mast cells by fast voltammetry. Pflügers Archiv. 1991;419:409–414. doi: 10.1007/BF00371124. [DOI] [PubMed] [Google Scholar]
- Terakawa S, Fan J-H, Kumakura K, Imaizumi-Ohara M. Quantitative analysis of exocytosis directly visualized in living chromaffin cells. Neuroscience Letters. 1991;123:82–86. doi: 10.1016/0304-3940(91)90163-n. [DOI] [PubMed] [Google Scholar]
- Thomas P. Two types of exocytosis observed in single, rat pancreatic acinar cells. The Journal of Physiology. 1998;509.P:186. [Google Scholar]
- Thomas P, Mellon PL, Turgeon JL, Waring DW. The LβT2 clonal gonadotrope: a model for single cell studies of endocrine cell secretion. Endocrinology. 1996;137:2979–2989. doi: 10.1210/endo.137.7.8770922. [DOI] [PubMed] [Google Scholar]
- Thomas P, Surprenant A, Almers W. Cytosolic Ca2+, exocytosis and endocytosis in single melanotrophs of the rat pituitary. Neuron. 1990;5:723–733. doi: 10.1016/0896-6273(90)90226-6. [DOI] [PubMed] [Google Scholar]
- Thomas P, Waring DW. Modulation of stimulus- secretion coupling in single rat gonadotrophs. The Journal of Physiology. 1997;504:705–719. doi: 10.1111/j.1469-7793.1997.705bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P, Wong JG, Lee AK, Almers W. A low affinity Ca2+ receptor controls the final steps in peptide secretion from pituitary melanotrophs. Neuron. 1993;11:93–104. doi: 10.1016/0896-6273(93)90274-u. [DOI] [PubMed] [Google Scholar]
- Titievsky AV, Takeo T, Tepikin AV, Petersen OH. Decrease of acidity inside zymogen granules inhibits acetylcholine- or inositol trisphosphate-evoked cytosolic Ca2+ spiking in pancreatic acinar cells. Pflügers Archiv. 1996;432:938–940. doi: 10.1007/s004240050218. [DOI] [PubMed] [Google Scholar]
- Tse A, Tse FW, Almers W, Hille B. Rhythmic exocytosis stimulated by GnRH-induced calcium oscillations in rat gonadotropes. Science. 1993;260:82–84. doi: 10.1126/science.8385366. [DOI] [PubMed] [Google Scholar]
- Valentijn K, Valentijn JA, Jamieson JD. Role of actin in regulated exocytosis and compensatory membrane retrieval: insights from an old acquaintance. Biochemical and Biophysical Research Communications. 1999;266:652–661. doi: 10.1006/bbrc.1999.1883. [DOI] [PubMed] [Google Scholar]
- Vitale ML, Seward EP, Trifaró JM. Chromaffin cell cortical actin network dynamics control the size of the release-ready vesicle pool and the initial rate of exocytosis. Neuron. 1995;14:353–363. doi: 10.1016/0896-6273(95)90291-0. [DOI] [PubMed] [Google Scholar]
- Zimmerberg J, Curran M, Cohen FS, Brodwick M. Simultaneous electrical and optical measurements show that membrane fusion precedes secretory granule swelling during exocytosis of beige mouse mast cells. Proceedings of the National Academy of Sciences of the USA. 1987;84:1585–1589. doi: 10.1073/pnas.84.6.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]