

Abstract
Evolution has created a large family of different classes of voltage-gated Ca2+ channels and a variety of additional splice variants with different inactivation properties. Inactivation controls the amount of Ca2+ entry during an action potential and is, therefore, believed to play an important role in tissue-specific Ca2+ signalling. Furthermore, mutations in a neuronal Ca2+ channel (Cav2.1) that are associated with the aetiology of neurological disorders such as familial hemiplegic migraine and ataxia cause significant changes in the process of channel inactivation. Ca2+ channels of a given subtype may inactivate by three different conformational changes: a fast and a slow voltage-dependent inactivation process and in some channel types by an additional Ca2+-dependent inactivation mechanism. Inactivation kinetics of Ca2+ channels are determined by the intrinsic properties of their pore-forming α1-subunits and by interactions with other channel subunits. This review focuses on structural determinants of Ca2+ channel inactivation in different parts of Ca2+ channel α1-subunits, including pore-forming transmembrane segments and loops, intracellular domain linkers and the carboxyl terminus. Inactivation is also affected by the interaction of the α1-subunits with auxiliary β-subunits and intracellular regulator proteins. The evidence shows that pore-forming S6 segments and conformational changes in extra- (pore loop) and intracellular linkers connected to pore-forming segments may play a principal role in the modulation of Ca2+ channel inactivation. Structural concepts of Ca2+ channel inactivation are discussed.
Ca2+ entry during an action potential
Ca2+ entry during an action potential initiates and controls multiple cascades of intracellular events which finally result in a variety of cellular functions. These include: impulse generation and propagation, sensory processes, muscle contraction, secretion of hormones and neurotransmitters, cell differentiation and gene expression (Berridge, 1997). Recent interest in the molecular mechanism of voltage-gated Ca2+ entry has been stimulated by the discovery that disorders such as familial hemiplegic migraine (FHM), ataxia type 6 and epilepsy are caused by spontaneous mutations in the pore-forming α1-subunit of neuronal Cav2.1 (class A) Ca2+ channels (see Jen, 1999, for review, and Ertel et al. 2000 for new nomenclature of Ca2+ channels).
Action potentials are initiated by openings of voltage-gated Na+ and Ca2+ channels induced by membrane depolarizations. The resulting influx of Na+ and Ca2+ ions leads to a further membrane depolarization which in turn initiates more channel openings. This positive feedback mechanism underlies the regenerative process forming the ascending phase of an action potential (for review see Hille, 1992). Simultaneously, membrane depolarization initiates channel closures, a process named inactivation (Fig. 1A). The resulting negative feedback restricts the ionic current inflow (jointly with the activation of outward K+ currents) and determines the action potential duration.
Figure 1. Ca2+ channel state transitions during an action potential.
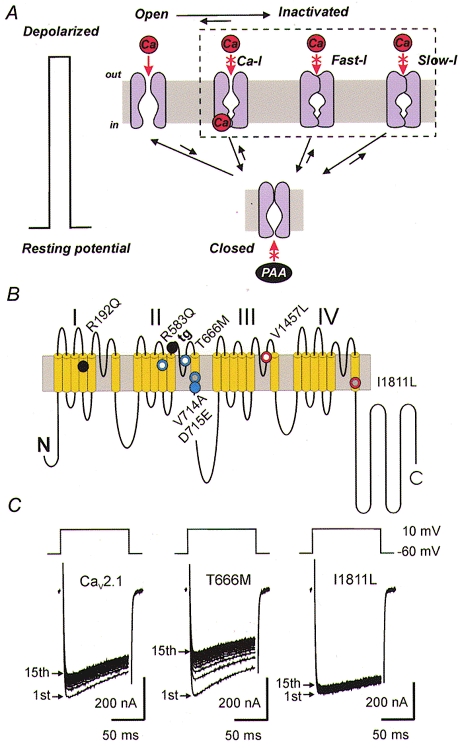
A, Ca2+ channels reside in different conformational populations: closed (resting) states, activated (open) states and several inactivated states. These populations are closely interconnected and the channel distribution between them is altered by changes in the electric field; membrane depolarization causes Ca2+ channels to open and subsequently to close by various inactivation mechanisms. Channel activation (transition from resting to open states) occurs within milliseconds whereas development and recovery from fast (Fast-I) and slow (Slow-I) inactivation occurs within tens of milliseconds or seconds. Ca-I represents the Ca2+-induced inactivated state. The fast and slow inactivated states are closely interrelated. Transitions to inactivation may occur from the open as well as from the resting state (Patil et al. 1998). Transitions of open and fast inactivated channels to slow inactivation have been reported (Sokolov et al. 2000). Negative shifts of the membrane potential promote transitions of open and/or inactivated channels to the resting state. There is evidence that Ca2+ channel blockers such as phenylalkylamines (PAA) access their receptor from the intracellular site. Inactivation determinants at the inner channel mouth control PAA sensitivity (Hering et al. 1998). B, familial hemiplegic migraine (FHM) mutations affecting inactivation gating. Putative folding structure of a Cav2.1 channel α1-subunit with indicated FHM mutations accelerating (blue) or slowing (red) the development of inactivation. Mutations inducing a delay in recovery from inactivation (using Ba2+ as charge carrier) are marked in white, those accelerating recovery are indicated in grey (see Kraus et al. 1998, 2000). Point mutation R192Q (black) did not affect inactivation (Kraus et al. 1998). The tottering mutation (tg, indicated in black) in the loop between segments IIS5 and IIS6 (P601L; Fletcher et al. 1996) reduces (among other changes in gating) channel inactivation (Wakamori et al. 1998). C, inhibition of Ba2+ currents (IBa) through wild-type Cav2.1 channels and FHM mutants T666M and I1811L (position I1811 in the human sequence corresponds to position I1819 in the rabbit sequence) expressed in Xenopus oocytes during a train of fifteen 100 ms pulses from −60 to 10 mV applied at 1 Hz (reproduced with permission from Kraus et al. 1998). Faster IBa inactivation of mutant T666M was associated with a slower recovery from inactivation resulting in a more pronounced accumulation in an inactivated state during the pulse train. Mutant I1811L displayed a slower inactivation time course. Consequently, less channels accumulated in an inactivated state during the 1 Hz train (compare A).
Ca2+ channels undergo more than one conformational change during inactivation; the existence of a Ca2+-dependent inactivation mechanism was concluded from the finding that inactivation of Ca2+ channels is faster when the charge carrier is Ca2+ (compared with Ba2+ and other divalent or monovalent ions). This type of inactivation was first discovered in the ciliate Paramecium (Brehm & Eckert, 1978), then analysed in Cav1.2 of myocardial and smooth muscle (Lee et al. 1985; Ganitkevich et al. 1987) and later also observed in Cav2.1 (Lee et al. 1999; see also Tareilus et al. 1994). However, Ba2+ currents through Ca2+ channels commonly decay with biexponential kinetics suggesting that these channels additionally close by fast and slow voltage-dependent inactivation processes.
Ca2+ channels dwell for a significantly longer time in an inactivated conformation than in the open state. As illustrated in the schematic diagram in Fig. 1A the channels will have to recover from inactivation before being able to open again. Thus, if the subsequent pulses arrive before recovery from inactivation is complete Ca2+ entry during spiking will be significantly reduced. Changes in the resting potential affect the steady-state fractions of resting and inactivated channels; at more depolarized voltages a larger fraction of Ca2+ channels will reside in one of the inactivated states thereby reducing the number of channels available during an action potential. In summary, Ca2+ entry is determined by the membrane potential (driving force and steady-state fractions in different channel conformations), the kinetics of Ca2+ channel opening, the kinetics of several inactivation processes and last but not least by the kinetics of recovery from inactivation.
The conformationally distinct Ca2+ channel populations (resting, open and inactivated states) are closely interrelated (Fig. 1A). In particular, there is evidence that voltage-dependent fast inactivation (Fast-I) of Ca2+ channels also occurs from the resting state (Patil et al. 1998) and that slow inactivation (Slow-I) in Ca2+ channels occurs not only after entry into the Fast-I state but also to a significant extent directly from the open state (Sokolov et al. 2000).
The molecular mechanism of Ca2+ channel inactivation is much less understood than that of some K+ and Na+ channels. Fast inactivation in Shaker K+ channels (N-type inactivation) involves the occlusion of the inner channel mouth by a ‘ball region’ at the amino (N-) terminus (‘ball and chain mechanism’; Armstrong & Bezanilla, 1977; Hoshi et al. 1990) while fast inactivation of Na+ channels occurs predominantly by occlusion of the channel pore by a ‘hinge lid’ formed by the intracellular linker between domains III and IV of the α1-subunit (IFM-motif; West et al. 1992).
This paper reviews the advances that have been made at the molecular level in the description of the different conformational changes of Ca2+ channels during inactivation. Functional studies on chimeric and mutant Ca2+ channels over recent years have revealed that the determinants of inactivation in this channel type are less localized than in K+ and Na+ channels. Development and recovery from inactivation in Ca2+ channels are affected by numerous structural changes in different parts of the α1-subunit including pore-forming segments, the intracellular loops and the carboxyl (C-) terminus. The inactivation process is also influenced by interactions with intracellular proteins and auxiliary channel subunits, foremost among them being the β-subunit. In view of space constraints, the citations to the literature are selective and focus primarily on general concepts of Ca2+ channel inactivation that are illustrated by examples where structural changes in Ca2+ channel α1-subunits induce major changes in inactivation gating.
Inactivation is a hallmark of Ca2+ channel subtypes
Evolution has not only created a variety of inactivation mechanisms to control Ca2+ entry (Fig. 1A) but additionally has produced a large family of voltage-gated Ca2+ channels with different inactivation kinetics determined by intrinsic properties of their α1-subunits as well as by distinct subunit compositions.
High-voltage-activated Ca2+ channels are hetero-oligomeric complexes formed by α1-, β- and α2/δ-subunits and in some channel classes by an additional γ-subunit.
The α1-subunit is the pore-forming membrane protein consisting of four homologous repeats (I-IV), each of them composed of six transmembrane segments (S1-S6; Fig. 1B). The pore-lining hydrophobic sequence between segments S5 and S6 is called the pore (P-) loop and forms the selectivity filter of the channel (Ellinor et al. 1995). The auxiliary β-, α2/δ- and γ-subunits modulate the activation and inactivation kinetics, expression density, voltage dependence and the pharmacological properties of the α1-subunit (Hofmann et al. 1999). The subunit composition of low-voltage-activated Cav3 (T-type Ca2+ channels) is less clear. The expression density of the α13.1-subunit is regulated by the β1a- (Dolphin et al. 1999) and α2/δ-2-subunits (Gao et al. 2000). A subtype of a γ-subunit has been shown to accelerate T-type activation and inactivation (Klugbauer et al. 2000).
Different classes of voltage-gated Ca2+ channels are encoded by different genes (Cav1.1 (former α1S-subunit), Cav1.2 (α1C), Cav1.3 (α1D), Cav1.4 (α1F), Cav2.1 (α1A), Cav2.2 (α1B), Cav2.3 (α1E), Cav3.1 (α1G), Cav3.2 (α1H), Cav3.3 (α1I)) and can be distinguished with respect to their biophysical and pharmacological properties (see Ertel et al. 2000 for nomenclature).
The kinetics of inactivation differ significantly between different Ca2+ channel classes. Heterologous expression experiments enabled the kinetics of Ca2+ channels of similar subunit composition to be compared. Such experiments suggest that Ba2+ currents through the Cav3 channel family (Cav3.1-Cav3.3, also known as T-type channels; Perez-Reyes et al. 1999) display the fastest time course of voltage-gated inactivation, followed by Cav2.3 (possibly R-type; Zhang et al. 1994; Spaetgens & Zamponi, 1999), Cav2.2 (N-type; Hans et al. 1999b) and Cav2.1 (P/Q-type; Sather et al. 1993) channels. Ba2+ currents through Cav1 channels (various L-type channels; Williams et al. 1992; see Hofmann et al. 1999 for review) display the slowest time course of inactivation. More recently, different splice variants of the α11.2- (Soldatov et al. 1998) and α12.1-subunits (Bourinet et al. 1999) with surprisingly different inactivation kinetics have been discovered. The data of Forsythe et al. (1998) suggest that different inactivation properties of neuronal Cav2.1 could be important determinants of synaptic efficacy.
Evidence regarding the molecular mechanism of Ca2+ channel inactivation has come from various sources: diseased states associated with altered Ca2+ channel inactivation (e.g. FHM mutations, Kraus et al. 1998, 2000; and ataxia, Wakamori et al. 1998), gain-of-function chimeras, channel splice variants with different inactivation properties, mutational studies directed towards the identification of drug-binding determinants and functional studies analysing the role of auxiliary Ca2+ channel subunits and other intracellular proteins.
Mutations associated with familial hemiplegic migraine and ataxia induce changes in inactivation gating
Cav2.1 channels at presynaptic terminals mediate the release of neurotransmitters (Wu et al. 1999). Missense mutations and deletions in the pore-forming α12.1-subunit of human Cav2.1 channels are associated with the aetiology of FHM (Ophoff et al. 1996; Kraus et al. 1998; Hans et al. 1999a), ataxia type 6 and epilepsy (Fletcher et al. 1996; Zhuchenko et al. 1997; see Jen, 1999, for review). Moreover, Cav2.1-deficient mice develop specific characteristics of ataxia and dystonia before dying about 3–4 weeks after birth (Jun et al. 1999).
Kraus et al. (1998) reported that the introduction of FHM mutations into the rabbit α12.1-subunit induced significant changes in inactivation gating. Three out of four point mutations either accelerated (T666M, V714A) or slowed (I1811L, corresponds to I1819L in the rabbit sequence) the inactivation of the corresponding Ba2+ currents (IBa). Recovery from fast inactivation was slowed in mutant T666M and accelerated in V714A and I1811L (Fig. 1B). Some of the possible pathophysiological consequences of these FHM mutations can be deduced from Fig. 1A. On the one hand, an accelerated channel inactivation reduces Ca2+ entry during a depolarization (e.g. prominent for T666M, Fig. 1C). On the other hand, a delay or facilitation in recovery from an inactivated state will either reduce (T666M) or enhance (I1811L) Ca2+ entry during a train of consecutive pulses (see Fig. 1C for typical IBa of mutant channels). Almost identical changes in inactivation gating were later reported by Hans et al. (1999a) who introduced the same FHM mutations into the human α12.1-subunit.
Most surprising in our initial study on the functional impact of FHM mutations in Xenopus oocytes (Kraus et al. 1998) was the finding that changes in the accumulation of inactivated Ca2+ channels could be monitored during pulse trains applied at frequencies as low as 1 pulse s−1 (Fig. 1C). This is far below the spiking frequency in neuronal networks in mammals and more pronounced effects might be expected in patients with FHM. However, the Ca2+‘pulses’ during action potentials in the central nervous system at 37°C are orders of magnitude shorter than the voltage clamp steps illustrated in Fig. 1C. The pathophysiological impact of the changed inactivation properties (compared to changes in Cav2.1 expression level, threshold of activation and single channel conductance; Kraus et al. 1998, 2000; Hans et al. 1999a) remains to be elucidated under more physiological conditions in mammalian cells.
The general concept that FHM mutations modulate Ca2+ entry through Cav2.1 channels by modulating channel inactivation (Fig. 1A) was recently confirmed by Kraus et al. (2000) who observed similar effects for three newly identified FHM mutations (R583Q, D715E and V1457L (corresponds to V1465L in the rabbit sequence); Fig. 1B).
Except for the two mutations in S4 segments, all of the FHM mutations analysed so far are located either in the putative pore-forming S5 and S6 segments or the connecting pore loops (Fig. 1B). The charge neutralization in the voltage sensor IIS4 by R583Q also accelerates the time course of channel inactivation and simultaneously delays recovery from inactivation. The pathophysiological mechanism by which FHM results from point mutations that cause either enhanced or reduced Ca2+ entry (Fig. 1C) remains unclear.
A mutation associated with the pathological state of ataxia (the tottering mutation tg, P601L; Fig. 1B) is also located in the extracellular linker between segments IIS5 and IIS6. Wakamori et al. (1998) reported for Purkinje cells of homozygous ataxic mice carrying the tg mutation a reduction in the Cav2.1 current density, an about 10 mV shift in the threshold of channel activation and inactivation to more negative voltages and also distinct changes in the inactivation kinetics of Ca2+ channel currents. Recent work of Plomp et al. (2000) provided the first hints as to how changes in Ca2+ channel gating might be associated with ataxia. Plomp and colleagues described an increased Cav2.1-mediated release of acetylcholine at neuromuscular junctions of homo- and heterozygote tg-mutant mice associated with an increased influx of Ca2+ at rest. The observed changes in neurotransmitter release could be associated with the leftward shift in the Cav2.1 activation threshold and/or reduced channel inactivation.
Studying inactivation in gain-of-function chimeras
Different kinetics in different channel classes implied that it should be possible to transfer inactivation determinants from one Ca2+ channel class to another. In line with this, Zhang et al. (1994) reported the successful transfer of the transient inactivation properties from a marine ray (doe-1) Cav2.3 channel to a slower inactivating Cav2.1 channel by transfer of segments IS6 and adjacent intra- and extracellular sequence stretches (Fig. 2A).
Figure 2. Chimeric channels as tools for studying structure and function of Ca2+ channel inactivation.
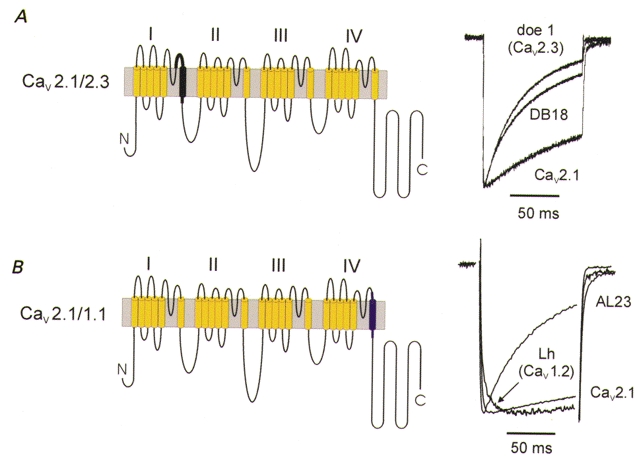
A, a chimeric channel consisting of α12.1 sequence (yellow) and segment IS6 and adjacent extra- and intracellular sequence stretches from Cav2.3 (doe-1, black) demonstrates a crucial role of this sequence in inactivation gating (chimera DB18 and current traces (right panel) are reproduced from Zhang et al. 1994 with permission from Nature; http://www.nature.com/). B, Cav2.1/1.1 chimera consisting of α12.1 sequence (yellow) and segment IVS6 of L-type sequence (purple, α11.1 sequence from carp skeletal muscle; Grabner et al. 1996). The chimera AL23 inactivated more rapidly than Cav2.1 and Cav1.m channels. A trace of the Cav1.2 construct (Lh) illustrates the slow inactivation time course of the Cav1.m family (data reproduced with permission from Döring et al. 1996).
However, substitution of sequence stretches does not always result in a transfer of the inactivation properties; implantation of part of Cav2.1 segment IIIS6 and the adjacent extracellular flanking pore loop into α11.2 produced a chimera with apparently faster inactivation kinetics than the donor Cav2.1 channel (Tang et al. 1993). Moreover, incorporation of a part of domain I of a slowly inactivating α11.1-subunit into α11.2 resulted in a chimera with a more transient inactivation than the recipient Cav1.2 channel (Parent et al. 1995).
Most strikingly, transfer of segment IVS6 from a slow inactivating Cav1.1 to the more rapidly inactivating Cav2.1 channel resulted in a chimera with significantly faster inactivation properties than native Cav2.1 (Döring et al. 1996; chimera AL23 in Fig. 2B) whereas implantation of the Cav1.2 sequence into segments IIIS5, IIIS6 and the connecting P-loop of α11.2 almost completely abolished fast inactivation in the resulting chimera (chimera AL20; Hering et al. 1996). Switching domains between α1-subunits of rapidly inactivating Cav2.3 and a Cav1.2 produced numerous changes in activation and inactivation gating (Spaetgens & Zamponi, 1999) suggesting, however, that domains II and III contain key inactivation determinants of Cav2.3 channels. Hans et al. (1999b) reported that the more transient inactivation kinetics of an α12.2-subunit were successfully transferred by inserting a sequence stretch between IVS3 and the P-region into an α12.1-subunit. In the same paper the authors demonstrated that a single point mutation (E1740R) in the P-loop of domain IV significantly accelerated channel inactivation properties and shifted the steady-state inactivation curve suggesting that this amino acid (E1740) plays an essential role in inactivation gating (Fig. 4A). Substitution of part of segment IVS5 in an α11.2-subunit by the equivalent sequence of a fast inactivating Na+ channel significantly slowed the inactivation kinetics of the resulting construct compared to α11.2 suggesting a role of the IVS5 segment in inactivation gating (Motoike et al. 1999).
Figure 4. Molecular determinants of Cav2.1 channel inactivation.
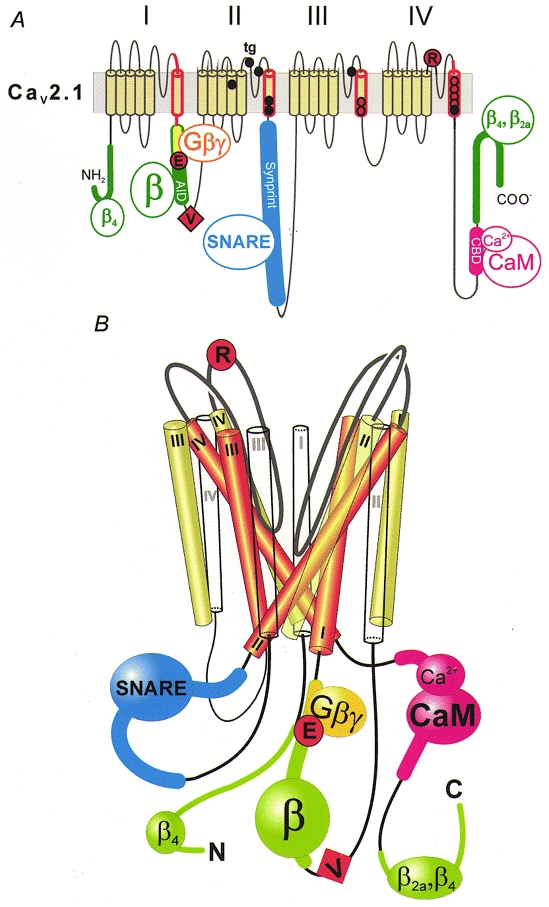
A, inactivation of Cav2.1 channels is affected by mutations in the α12.1-subunit and proteins interacting with intracellular loops. The scheme highlights crucial segments and the location of some key point mutations: segment IS6 and adjacent intra- and extracellular sequences (red; Zhang et al. 1994); mutations in the putative pore-lining region of segments IIIS6 and IVS6 (red circles; Hering et al. 1996, 1998; Sokolov et al. 2000); point mutations associated with FHM, shown as black circles (see Kraus et al. 1998, 2000, for functional studies); point mutations in the intracellular I-II loop (G-protein-interaction motif (yellow): QQIER→QQIEE; Herlitze et al. 1997) and the IVS5-IVS6 linker (R1740; Hans et al. 1999b), represented by large red circles; the additional valine of a Cav2.1 splice variant converting fast Q-type into slow P-type kinetics (V421; Bourinet et al. 1999), symbolized as a red square; the α-subunit interaction domain (AID) in the I-II loop and additional β-subunit interaction motifs on N- and C-terminal regions of α12.1, shown in green (Pragnell et al. 1994; Walker et al. 1998, 1999); SNARE protein interaction with the Synprint site on the domain II-III linker (Bezprozvanny et al. 1995; Rettig et al. 1996; Zhong et al. 1999), shown in blue; and the calmodulin (CaM)-binding domain (CBD; Lee et al. 1999) in the C-terminus, shown in pink. B, hypothetical scheme of the structural organization of Ca2+ channel segments S1 (white), S5 (green) and S6 (red) and the interaction with intracellular proteins. Analogous to the crystal structure of the KcsA channel (Doyle et al. 1998), the pore-forming S6 segments are arranged as an inverted ‘teepee’ (see also Hering et al. 1998). The loop interactions with intracellular proteins and the location of some crucial inactivation determinants are indicated.
Chimeras between Ca2+ channels with different inactivation properties represent valuable tools for investigating the structure-activity relationship of inactivation in Ca2+ channels. However, attempts to construct Ca2+ channel gain-of-function chimeras with respect to inactivation have been less successful than the transfer of drug sensitivity between different channel classes (see Striessnig et al. 1998 for review). Voltage-dependent inactivation in Ca2+ channels apparently involves structural elements in more than one domain (Fig. 1B), and in some cases the transfer of key amino acids from all four domains may be required. Most studies on chimeric channels highlight, nevertheless, the role of pore-forming S6 and S5 segments and adjacent extracellular and intracellular sequence stretches, particularly the S5-S6 linker regions.
Multiple inactivation determinants in pore-forming segments: key determinants at the inner vestibule of the pore
More detailed knowledge regarding the role of single amino acids in the pore-forming S6 segments comes from studies on the molecular mechanism of Ca2+ channel block by drugs such as phenylalkylamines (PAAs), 1,4-dihydropyridines (DHPs) and diltiazem (DIL) (Hockerman et al. 1997a; Striessnig et al. 1998). Hockerman et al. (1995, 1997b) systematically substituted the amino acids in Cav1.2 segments IIIS6 and IVS6 by alanine to analyse the potential role of pore-forming segments IIIS6 and IVS6 in Ca2+ channel inhibition by PAAs and DHPs. The S6 point mutants displayed not only changes in drug sensitivity but also manifold changes in inactivation gating compared to wild-type Cav1.2 channels (Fig. 3B and C). Furthermore, replacement of two glutamates in the P-loops in domains II and IV known to form part of the selectivity filter (E709Q, E1419Q; Hockerman et al. 1997a) caused significant changes in steady-state inactivation (Fig. 3C).
Figure 3. Multiple inactivation determinants in pore-forming segments IIIS6 and IVS6.
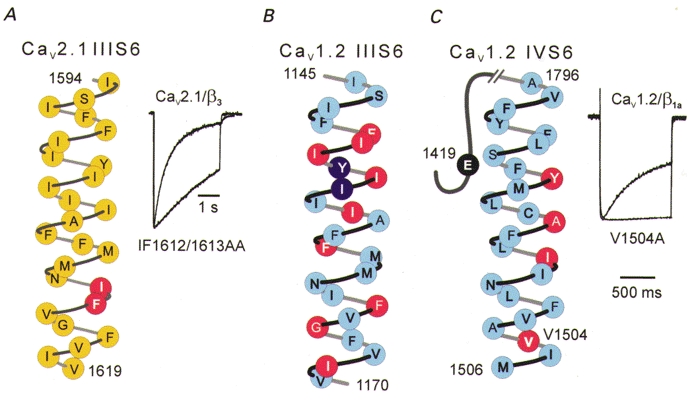
A, inactivation determinants in segment IIIS6 of a Cav2.1 channel (Sokolov et al. 2000). Substitution of two amino acids located close to the inner channel mouth (indicated in red) substantially slowed inactivation (current traces from Sokolov et al. 2000). B, inactivation determinants in segment IIIS6 of Cav1.2. Alanine substitutions for the indicated amino acids shifted the inactivation curves either to more depolarized voltages (> 5 mV, red) or in the hyperpolarizing direction (> 5 mV, purple) (unpublished data supplied by Drs G. Hockerman and T. Scheuer). C, inactivation determinants in Cav1.2 segment IVS6. Substitution of a single valine by alanine (V1504A) almost completely abolished fast inactivation (Ba2+ current traces reproduced with permission from Berjukow et al. 1999). The inactivation curve of the triple Cav1.2 mutant lacking the high-affinity PAA determinants in segment IVS6 (YAI; Hockerman et al. 1995) was shifted to the right. Substitution of glutamate in position E1419Q (black) induced a 6 mV shift of the inactivation curve to more positive potentials (Hockerman et al. 1997a).
We found, by mutating amino acids located in Cav1.2 and Cav2.1 segments IIIS6 and IVS6, that Ca2+ channel inactivation is particularly sensitive to amino acid substitution in the region of the inner channel mouth. One mutation in L-type segment IVS6 (V1504A; Berjukow et al. 1999) and a double mutation in segment IIIS6 of Cav2.1 (IF1612/1613AA; Sokolov et al. 2000) dramatically slowed the time course of fast inactivation (Fig. 3). Amino acid substitutions in segment IVS6 of Cav2.1 channels have substantial effects on the current decay (I1804Y, F1805M, S1808A, M1811I; Hering et al. 1996, 1998). The molecular mechanism by which amino acid substitutions in pore-forming segments affect channel inactivation remains to be elucidated.
Intracellular loops: interaction with β-subunits, G-proteins and syntaxin
Ca2+ channel inactivation is sensitive to structural changes in intracellular domain linkers. This has been demonstrated by Adams & Tanabe (1997) for Cav1.2/Cav1.1 chimeras consisting of α11.1 sequence in linkers between domains I and II and III and IV. A dramatic example of how structural changes in the loop between domains I and II can modulate Ca2+ channel inactivation gating was reported by Bourinet et al. (1999). By comparing the sequence of the I-II loop and the inactivation properties of two Cav2.1 splice variants (α1A-a and α1A-b), these authors discovered that a single valine insertion (V421) into the I-II loop of the α1A-a splice almost completely removed the fast inactivation thereby converting the kinetic phenotype of Q-type channels into that of slowly inactivating P-type Ca2+ channels (Fig. 4A). Cens et al. (1999) demonstrated that overexpression of the I-II loop of α12.1 accelerates current kinetics suggesting that the I-II loop may play the role of a ball peptide similar to the ball and chain mechanism in K+ channels. Cens et al. (1999) also hypothesize that Ca2+- and voltage-dependent inactivation share common molecular determinants.
There are several lines of evidence suggesting that Ca2+ channel inactivation can be modulated by proteins interacting with intracellular loops. Pragnell et al. (1994) identified a consensus sequence for the binding of the Ca2+ channel β-subunits in the intracellular loop between domains I and II (Fig. 4). Additional determinants for α1-β-subunit interaction known to affect inactivation (see above) have been detected in the N- and C-terminus of Cav2.1 (Walker et al. 1998, 1999).
Functional studies confirmed that β-subunit interaction is a key determinant in Ca2+ channel inactivation with each β-subunit inducing individual gating effects. β2a-subunit interaction commonly causes the slowest current decay followed by β4-, β1a- and the β3-subunit (Sather et al. 1993; Stea et al. 1994; De Waard & Campbell, 1995; Sokolov et al. 1999). These effects occur irrespective of whether the current is carried by Ba2+ or Ca2+ ions (Cens et al. 1999). Some of the regulatory properties of the β2a-subunit are associated with the palmitoylation of two cysteines at its N-terminus (Chien et al. 1996; Qin et al. 1999). Recent data of Freise et al. (2000) indicate a role for the γ-subunit in inactivation of Cav1.1. A shift of the steady-state inactivation curve of Cav2.1 by coexpression of the γ-subunit subtypes γ-2 and γ-4 and an acceleration of the activation and inactivation time course of Cav3.1 induced by coexpression of the γ-5-subunit was reported by Klugbauer et al. (2000).
A motif in the intracellular I-II loop (QXXER; see Dolphin, 1998, for review) plays an essential role in G-protein interactions with the α12.1-, α12.2- and α12.3-subunits (De Waard et al. 1997). Single amino acid substitutions in this motif have pronounced effects on inactivation gating: substitution of an arginine in the G-protein-interaction motif of α12.1 by glutamate (QQIER→QQIEE; see Fig. 4A) reduced channel inactivation, the reverse mutation in α11.2 (QQLEE→QQLER) accelerated the Ba2+ current decay (Herlitze et al. 1997).
Further support for the hypothesis that protein interactions with intracellular loops play an essential role in Ca2+ channel inactivation comes from Bezprozvanny et al. (1995) who reported that coexpression of syntaxin with Cav2.2 or Cav2.1 channels in Xenopus oocytes enhances voltage-dependent inactivation (see Catterall, 1999, for review). The evidence shows that SNARE protein interaction with a synaptic protein interaction site (Synprint; Fig. 4) on the domain II-III linker of Cav2.1 and Cav2.2 channels represents a further mechanism by which Ca2+ channel inactivation can be determined by structural changes in intracellular domain linkers (Bezprozvanny et al. 1995; Zhong et al. 1999; Degtiar et al. 2000).
The C-terminus: Cav1.2 splice variants and Ca2+-dependent inactivation
The first evidence for a crucial role of the C-terminus in Ca2+ channel inactivation was reported by Soldatov et al. (1997) who identified a number of Cav1.2 channel splice variants. Coexpression of six Cav1.2 splice variants with auxiliary α2/δ- and β1-subunits in Xenopus oocytes resulted in currents with significantly different inactivation kinetics. Two of the isoforms (α1C,72 and α1C,86) contained C-terminal insertion sequences due to alternative splicing of exons 40–43. Corresponding Ba2+ currents displayed significantly faster inactivation kinetics. Another splice variant with alterations at the inner channel mouth in segment IS6 also displayed changes in inactivation kinetics (Soldatov et al. 1997, 1998).
Ca2+-dependent inactivation was first suggested to be caused by Ca2+ binding to a specific motif (EF-hand) at the C-terminus of Cav1.2 (De Leon et al. 1995). This sequence is analogous to the EF-hand motif known from Ca2+-binding proteins such as calmodulin (CaM) and parvalbumin and, except in Cav1.3, to a certain degree conserved in the C-terminus of all known Ca2+ channel α1-subunits. It is called EF-hand because the Ca2+-binding site represents a helix-loop-helix motif where the two helices in the original structure of parvalbumin were labelled as E and F. Ca2+-dependent inactivation of Cav1.2 can be eliminated if the EF-hand motif in α11.2 is replaced by corresponding residues of α12.3 (De Leon et al. 1995). However, the conformational changes during Ca2+-dependent inactivation are more complex. Bernatchez et al. (1998) revealed that a residue that is conserved within the EF-hand motif of Cav1 (E1537) contributes to voltage-dependent inactivation and hypothesized that this part of the intracellular C-terminus may play a role as an inactivation ball in voltage-dependent inactivation. Moreover, several groups have recently demonstrated that the ubiquitous Ca2+-binding protein CaM is the main sensor for Ca2+. CaM binds to the C-terminus of different Ca2+ channel classes thereby mediating Ca2+-dependent inactivation (Qin et al. 1999; Zuhlke et al. 1999; Peterson et al. 1999; Lee et al. 1999). It appears that the EF-hand motif and the consensus CaM-binding motif (including the crucial amino acids isoleucine and glutamine, so called ‘IQ’-motif; Zuhlke et al. 1999) that are located close to each other near the inner channel mouth at the C-terminus of Cav1.2 have a complementary role in Ca2+-dependent inactivation. Peterson et al. (2000) suggest that the EF-hand may support the transduction of the conformational changes induced by the Ca2+-CaM-binding step onto the α11.2. The CaM-binding domain on Cav2.1 (CBD; Lee et al. 1999) is illustrated in Fig. 4A.
Determinants of slow inactivation in Ca2+ channels
Evidence for slow inactivation in Ca2+ channels comes from the slow phase in channel recovery kinetics after a sustained membrane depolarization. Interestingly, use-dependent Ca2+ channel blockers such as PAAs, DIL and mibefradil induce a slow component in recovery from inactivation suggesting that these drugs promote a conformation resembling the slow inactivated state (Hering et al. 1997; Aczél et al. 1998; Berjukow et al. 1999). A role for syntaxin as a physiological modulator of the slow inactivation of Cav2.2 was recently reported by Degtiar et al. (2000).
We have analysed the kinetics of the slow inactivation of α12.1 expressed together with β1a-, β2a-, β3- or β4-subunits. Our data clearly demonstrate that Cav2.1 channels proceed to the slow inactivated state from the fast inactivated state as well as directly from the open state. A reduction of fast voltage-dependent inactivation by coexpression of α12.1 with β2a increased channel state transitions into slow inactivation: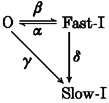
with α, β, γ and δ representing the rate constants for transitions between the open (O), fast inactivated (Fast-I) and slow inactivated (Slow-I) states. Thus, under certain circumstances open channels are even more willing to enter the slow inactivated channel conformation than channels in the fast inactivated state (i.e. γ > δ) (Sokolov et al. 2000). Tissue-specific expression patterns of different β-subunits or changes in the subunit assembly during development appear, therefore, as significant determinants of fast and slow inactivation in Cav2.1 channels. Further functional studies are required to identify the molecular determinants of slow inactivation in Ca2+ channels and to estimate the relative impacts of slow inactivation, fast inactivation of open channels and closed-state-dependent inactivation (Patil et al. 1998) on Ca2+ entry during trains of action potential-like stimuli under physiological conditions.
Inactivation determinants affect drug binding in the pore region
Some of the residues involved in inactivation modulate Ca2+ channel block by PAAs, DIL and DHPs. This led us to propose a ‘teepee’-dissociation model where mutations in the inner pore regions modulate inactivation gating by affecting the positions of S6 segments. This in turn modulates intracellular access, trapping or binding of PAAs and DIL in the channel pore (Hering et al. 1998; Berjukow et al. 1999). Indirect evidence for an inactivation mechanism involving conformational changes in the putative bundle-crossing region of S6 segments comes from recent experiments by Sokolov et al. (1999) demonstrating that the PAA sensitivity of a triple Cav2.1 mutant (α1A-PAA) is substantially influenced by a mutation in the I-II linker and by β-subunit interaction. The putative α-subunit interaction domain (AID) in the I-II linker and the mutation Arg387Glu are located far away from the putative drug-binding domain in the channel pore of the mutant α12.1-subunit. In particular, the β2a interaction slowed channel inactivation and correspondingly reduced the channel block by (–)gallopamil in a similar manner and to a comparable extent as previously reported for mutations close to the inner channel mouth in segment IIIS6 (Hering et al. 1997, 1998). Therefore, structural changes in intracellular linkers connected to S6 segments and S6 mutations might control the PAA sensitivity by similar mechanisms. Like PAAs and DIL, DHPs appear to bind in the pore region of Cav1 (key binding determinants are located on pore-forming segments IIIS5, IIIS6, IVS6 and connected P-loops; see Striessnig et al. 1998). Substitution of distinct inactivation determinants on pore-forming segment IVS6 destabilizes a DHP-induced inactivated channel conformation suggesting a synergism between intrinsic channel inactivation and DHP-induced inactivation (Berjukow et al. 2000).
Conclusions and outlook
The molecular determinants of inactivation are more widely spread over the Ca2+ channel protein than those in Na+ and K+ channels (Fig. 4). β interaction with different domains on the α1-subunit, point mutations in S6 segments (particularly the putative bundle-crossing region near the inner channel mouth; Fig. 4A), protein interactions with intracellular domain linkers, structural changes in the C-terminus induced by Ca2+-CaM interaction, alternative splicing or point mutations in the EF-hand motif all have pronounced affects on inactivation gating of Ca2+ channels.
Several hypotheses can be put forward for how these structural changes in different parts of the α1-subunits affect inactivation. The first hypothesis is that distortions of the putative ‘teepee’ structure of pore-forming S6 segments play a key role in inactivation gating. This assumption is in line with studies analysing the interaction of ‘inactivation-deficient’ Ca2+ channel mutants with PAAs, DIL and DHPs (Hering et al. 1998; Sokolov et al. 1999; Berjukow et al. 2000). Hence, structural changes in pore-forming S5 and S6 segments, domain linkers, P-loops and the C-terminus may all affect the orientation of drug-binding determinants in the channel pore thereby modulating drug sensitivity (Fig. 4). An alternative view could be that domain linkers (particular the I-II linker) and the C-terminus in Cav1.2 and Cav2.1 may form part of separate inactivation gates occluding the inner channel mouth (Bernatchez et al. 1998; Cens et al. 1999; Peterson et al. 2000; Stotz et al. 2000). Finally, the most complicated case where a receptor structure for an inactivation gate at the inner channel mouth is determined by the orientation of S6 segments also cannot be excluded. Nevertheless, at present it is more attractive to think that structural changes in the intracellular loops and C-terminus of Ca2+ channels affect the orientation of pore-forming segments rather than modulating a variety of distinct inactivation gates (Fig. 4).
In conclusion, it appears that a number of crucial questions remain. What is the structural basis of the β-subunit modulation of Ca2+ channel inactivation? What is the molecular mechanism underlying the numerous changes in inactivation produced by point mutations in pore-forming segments? Are structural changes in the pore region and β-subunit modulation interdependent? How do the different inactivation mechanisms in Ca2+ channels finally modulate the interaction of Ca2+ channel α1-subunits with blockers such as PAAs, DIL and DHPs? Have the fast and slow voltage-dependent inactivation and the Ca2+-dependent inactivation common structural determinants? We anticipate that detailed functional studies of the different inactivation mechanisms (Fast-I, Slow-I, Ca-I; Fig. 1A) on Ca2+ channels expressed in recombinant systems combined with new structural information will provide the answers.
Acknowledgments
The authors thank Dr A. Hughes for comments on the manuscript. This work was supported by FWF grants P12649-MED, P12828-MED, a grant from the Else Kröner-Fresenius-Stiftung and a grant from the Austrian National Bank (to S.H.).
References
- Aczél S, Kurka B, Hering S. Mechanism of voltage- and use-dependent block of class A Ca2+ channels by mibefradil. British Journal of Pharmacology. 1998;125:447–454. doi: 10.1038/sj.bjp.0702092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams B, Tanabe T. Structural regions of the cardiac Ca channel α subunit involved in Ca-dependent inactivation. Journal of General Physiology. 1997;110:379–389. doi: 10.1085/jgp.110.4.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. Journal of General Physiology. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berjukow S, Gapp F, Aczél S, Sinnegger MJ, Mitterdorfer J, Glossmann H, Hering S. Sequence differences between α1C and α1S Ca2+ channel subunits reveal structural determinants of a guarded and modulated benzothiazepine receptor. Journal of Biological Chemistry. 1999;274:6154–6160. doi: 10.1074/jbc.274.10.6154. [DOI] [PubMed] [Google Scholar]
- Berjukow S, Marksteiner R, Gapp F, Sinnegger MJ, Hering S. Molecular mechanism of calcium channel block by isradipine: Role of a drug-induced inactivated channel conformation. Journal of Biological Chemistry. 2000;275:22114–22120. doi: 10.1074/jbc.M908836199. [DOI] [PubMed] [Google Scholar]
- Bernatchez G, Talwar D, Parent L. Mutations in the EF-hand motif impair the inactivation of barium currents of the cardiac α1C channel. Biophysical Journal. 1998;75:1727–1739. doi: 10.1016/S0006-3495(98)77614-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Elementary and global aspects of calcium signalling. The Journal of Physiology. 1997;499:291–306. doi: 10.1113/jphysiol.1997.sp021927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Scheller RH, Tsien RW. Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature. 1995;378:623–626. doi: 10.1038/378623a0. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Sutton K, Slaymaker S, Mathews E, Monteil A, Zamponi GW, Nargeot J, Snutch TP. Splicing of α1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nature Neuroscience. 1999;5:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- Brehm P, Eckert R. Calcium entry leads to inactivation of calcium channel in Paramecium. Science. 1978;202:1203–1206. doi: 10.1126/science.103199. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Interactions of presynaptic Ca2+ channels and snare proteins in neurotransmitter release. Annals of the New York Academy of Sciences. 1999;868:144–159. doi: 10.1111/j.1749-6632.1999.tb11284.x. [DOI] [PubMed] [Google Scholar]
- Cens T, Restituito S, Galas S, Charnet P. Voltage and calcium use the same molecular determinants to inactivate calcium channels. Journal of Biological Chemistry. 1999;274:5483–5490. doi: 10.1074/jbc.274.9.5483. [DOI] [PubMed] [Google Scholar]
- Chien AJ, Carr KM, Shirokov RE, Rios E, Hosey MM. Identification of palmitoylation sites within the L-type calcium channel β2a subunit and effects on channel function. Journal of Biological Chemistry. 1996;271:26465–26468. doi: 10.1074/jbc.271.43.26465. [DOI] [PubMed] [Google Scholar]
- Degtiar VE, Scheller RH, Tsien RW. Syntaxin modulation of slow inactivation of N-type calcium channels. Journal of Neuroscience. 2000;20:4355–4367. doi: 10.1523/JNEUROSCI.20-12-04355.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leon M, Wang Y, Jones L, Perez-Reyes E, Wei X, Soong TW, Snutch TP, Yue DT. Essential Ca2+-binding motif for Ca2+-sensitive inactivation of L-type Ca2+ channels. Science. 1995;270:1502–1506. doi: 10.1126/science.270.5241.1502. [DOI] [PubMed] [Google Scholar]
- De Waard M, Campbell KP. Subunit regulation of the neuronal α1A Ca2+ channel expressed in Xenopus oocytes. The Journal of Physiology. 1995;485:619–634. doi: 10.1113/jphysiol.1995.sp020757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Waard M, Liu H, Walker D, Scott VE, Gurnett CA, Campbell KP. Direct binding of G-protein βγ complex to voltage-dependent calcium channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. Mechanisms of modulation of voltage-dependent calcium channels by G proteins. The Journal of Physiology. 1998;506:3–11. doi: 10.1111/j.1469-7793.1998.003bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC, Wyatt CN, Richards J, Beattie RE, Craig P, Lee J-H, Cribbs LL, Volsen SG, Perez-Reyes E. The effect of α2-δ and other accessory subunits on expression and properties of the calcium channel α1G. The Journal of Physiology. 1999;519:35–45. doi: 10.1111/j.1469-7793.1999.0035o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döring F, Degtiar VE, Grabner M, Striessnig J, Hering S, Glossman H. Transfer of L-type calcium channel IVS6 segment increases phenylalkylamine sensitivity of α1A. Journal of Biological Chemistry. 1996;271:11745–11749. doi: 10.1074/jbc.271.20.11745. [DOI] [PubMed] [Google Scholar]
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Ellinor PT, Yang J, Sather WA, Zhang JF, Tsien RW. Ca2+ channel selectivity at a single locus for high-affinity Ca2+ interactions. Neuron. 1995;15:1121–1132. doi: 10.1016/0896-6273(95)90100-0. [DOI] [PubMed] [Google Scholar]
- Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW, Catterall WA. Nomenclature of voltage-gated calcium channels. Neuron. 2000;25:533–535. doi: 10.1016/s0896-6273(00)81057-0. [DOI] [PubMed] [Google Scholar]
- Fletcher CF, Lutz CM, O'Sullivan TN, Shaughnessy JD, Jr, Hawkes R, Frankel WN, Copeland NG, Jenkins NA. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87:607–617. doi: 10.1016/s0092-8674(00)81381-1. [DOI] [PubMed] [Google Scholar]
- Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. [DOI] [PubMed] [Google Scholar]
- Freise D, Held B, Wissenbach U, Pfeifer A, Trost C, Himmerkus N, Schweig U, Freichel M, Biel M, Hofmann F, Hoth M, Flockerzi V. Absence of γ subunit of the skeletal muscle dihydropyridine receptor increases L-type Ca2+ currents and alters channel inactivation properties. Journal of Biological Chemistry. 2000;275:14476–14481. doi: 10.1074/jbc.275.19.14476. [DOI] [PubMed] [Google Scholar]
- Ganitkevich V Ya, Shuba MF, Smirnov SV. Calcium-dependent inactivation of potential-dependent calcium inward current in an isolated guinea-pig smooth muscle cell. The Journal of Physiology. 1987;392:431–449. doi: 10.1113/jphysiol.1987.sp016789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Sekido Y, Maximov A, Saad M, Forgacs E, Latif F, Wei MH, Lerman M, Lee JH, Perez-Reyes E, Bezprozvanny I, Minna JD. Functional properties of a new voltage-dependent calcium channel α2δ auxiliary subunit gene (CACNA2D2) Journal of Biological Chemistry. 2000;275:12237–12242. doi: 10.1074/jbc.275.16.12237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabner M, Wang Z, Hering S, Striessnig J, Glossmann H. Transfer of 1,4-dihydropyridine sensitivity from L-type to class A (BI) calcium channels. Neuron. 1996;16:207–218. doi: 10.1016/s0896-6273(00)80037-9. [DOI] [PubMed] [Google Scholar]
- Hans M, Luvisetto S, Williams ME, Spagnolo M, Urrutia A, Tottene A, Brust PF, Johnson EC, Harpold MM, Stauderman KA, Pietrobon D. Functional consequences of mutations in the human α1A calcium channel subunit linked to familial hemiplegic migraine. Journal of Neuroscience. 1999a;19:1610–1619. doi: 10.1523/JNEUROSCI.19-05-01610.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hans M, Urrutia A, Deal C, Brust PF, Stauderman K, Ellis SB, Harpold MM, Johnson EC, Williams ME. Structural elements in domain IV that influence biophysical and pharmacological properties of human α1A-containing high-voltage-activated calcium channels. Biophysical Journal. 1999b;76:1384–1400. doi: 10.1016/S0006-3495(99)77300-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering S, Aczél S, Grabner M, Döring F, Berjukow S, Mitterdorfer J, Sinnegger MJ, Striessnig J, Degtiar VE, Wang Z, Glossmann H. Transfer of high sensitivity for benzothiazepines from L-type to class A (BI) calcium channels. Journal of Biological Chemistry. 1996;271:24471–24475. doi: 10.1074/jbc.271.40.24471. [DOI] [PubMed] [Google Scholar]
- Hering S, Aczél S, Kraus RL, Berjukow S, Striessnig J, Timin EN. Molecular mechanism of use-dependent calcium channel block by phenylalkylamines: role of inactivation. Proceedings of the National Academy of Sciences of the USA. 1997;94:13323–13328. doi: 10.1073/pnas.94.24.13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering S, Berjukow S, Aczél S, Timin EN. Calcium channel block and inactivation: common molecular determinants. Trends in Pharmacological Sciences. 1998;19:439–443. doi: 10.1016/s0165-6147(98)01258-9. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Hockerman GH, Scheuer T, Catterall WA. Molecular determinants of inactivation and G protein modulation in the intracellular loop connecting domains I and II of the calcium channel α1A subunit. Proceedings of the National Academy of Sciences of the USA. 1997;94:1512–1516. doi: 10.1073/pnas.94.4.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. Sunderland, MA, USA: Sinauer; 1992. [Google Scholar]
- Hockerman GH, Johnson BD, Abbott MR, Scheuer T, Catterall WA. Molecular determinants of high affinity phenylalkylamine block of L-type calcium channels in transmembrane segment IIIS6 and the pore region of the α1 subunit. Journal of Biological Chemistry. 1997a;272:18759–18765. doi: 10.1074/jbc.272.30.18759. [DOI] [PubMed] [Google Scholar]
- Hockerman GH, Johnson BD, Scheuer T, Catterall WA. Molecular determinants of high affinity phenylalkylamine block of L-type calcium channels. Journal of Biological Chemistry. 1995;270:22119–22122. doi: 10.1074/jbc.270.38.22119. [DOI] [PubMed] [Google Scholar]
- Hockerman GH, Peterson BZ, Johnson BD, Catterall WA. Molecular determinants of drug binding and action on L-type calcium channels. Annual Review of Pharmacology and Toxicology. 1997b;37:361–396. doi: 10.1146/annurev.pharmtox.37.1.361. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Lacinova L, Klugbauer N. Voltage-dependent calcium channels: from structure to function. Reviews of Physiology Biochemistry and Pharmacology. 1999;139:33–87. doi: 10.1007/BFb0033648. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Jen J. Calcium channelopathies in the central nervous system. Current Opinion in Neurobiology. 1999;9:274–280. doi: 10.1016/s0959-4388(99)80040-3. [DOI] [PubMed] [Google Scholar]
- Jun K, Piedras-Renteria ES, Smith SM, Wheeler DB, Lee SB, Lee TG, Chin H, Adams ME, Scheller RH, Tsien RW, Shin HS. Ablation of P/Q-type Ca2+ channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the α1A-subunit. Proceedings of the National Academy of Sciences of the USA. 1999;96:15245–15250. doi: 10.1073/pnas.96.26.15245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugbauer N, Dai S, Specht V, Lacinova L, Marais E, Bohn G, Hofmann F. A family of γ-like calcium channel subunits. FEBS Letters. 2000;470:189–197. doi: 10.1016/s0014-5793(00)01306-5. [DOI] [PubMed] [Google Scholar]
- Kraus RL, Sinnegger MJ, Glossmann H, Hering S, Striessnig J. Familial hemiplegic migraine mutations change α1A Ca2+ channel kinetics. Journal of Biological Chemistry. 1998;273:5586–5590. doi: 10.1074/jbc.273.10.5586. [DOI] [PubMed] [Google Scholar]
- Kraus RL, Sinnegger MJ, Koschak A, Glossmann H, Stenirri S, Carrera P, Striessnig J. Three new familial hemiplegic migraine mutants affect P/Q-type Ca2+ channel kinetics. Journal of Biological Chemistry. 2000;275:9239–9243. doi: 10.1074/jbc.275.13.9239. [DOI] [PubMed] [Google Scholar]
- Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- Lee KS, Marban E, Tsien RW. Inactivation of calcium channels in mammalian heart cells: joint dependence on membrane potential and intracellular calcium. The Journal of Physiology. 1985;364:395–411. doi: 10.1113/jphysiol.1985.sp015752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoike HK, Bodi I, Nakayama H, Schwartz A, Varadi G. A region in IVS5 of the human cardiac L-type calcium channel is required for the use-dependent block by phenylalkylamines and benzothiazepines. Journal of Biological Chemistry. 1999;274:9409–9420. doi: 10.1074/jbc.274.14.9409. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- Parent L, Gopalakrishnan M, Lacerda AE, Wie X, Perez-Reyes E. Voltage-dependent inactivation in a cardiac-skeletal chimeric calcium channel. FEBS Letters. 1995;360:144–150. doi: 10.1016/0014-5793(95)00090-v. [DOI] [PubMed] [Google Scholar]
- Patil PG, Brody DL, Yue DT. Preferential closed-state inactivation of neuronal calcium channels. Neuron. 1998;20:1027–1038. doi: 10.1016/s0896-6273(00)80483-3. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E, Lee JH, Cribbs L-L. Molecular characterization of two members of the T-type calcium channel family. Annals of the New York Academy of Sciences. 1999;868:131–143. doi: 10.1111/j.1749-6632.1999.tb11283.x. [DOI] [PubMed] [Google Scholar]
- Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- Peterson BZ, Lee JS, Mulle JG, Wang Y, De Leon M, Yue DT. Critical determinants of Ca2+-dependent inactivation within an EF-hand motif of L-type Ca2+ channels. Biophysical Journal. 2000;78:1906–1920. doi: 10.1016/S0006-3495(00)76739-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomp JJ, Vergouwe MN, Van den Maagdenberg AM, Ferrari MD, Frants RR, Molenaar PC. Abnormal transmitter release at neuromuscular junctions of mice carrying the tottering α1A Ca2+ channel mutation. Brain. 2000;123:463–471. doi: 10.1093/brain/123.3.463. [DOI] [PubMed] [Google Scholar]
- Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP, Campbell KP. Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature. 1994;368:67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- Qin N, Olcese R, Bransby M, Lin T, Birnbaumer L. Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proceedings of the National Academy of Sciences of the USA. 1999;96:2435–2438. doi: 10.1073/pnas.96.5.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig J, Sheng ZH, Kim DK, Hodson CD, Snutch TP, Catterall WA. Isoform-specific interaction of the α1A subunits of brain Ca2+ channels with the presynaptic proteins syntaxin and SNAP-25. Proceedings of the National Academy of Sciences of the USA. 1996;93:7363–7368. doi: 10.1073/pnas.93.14.7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sather WA, Tanabe T, Zhang JF, Mori Y, Adams ME, Tsien RW. Distinctive biophysical and pharmacological properties of class A (BI) calcium channel α1 subunits. Neuron. 1993;11:291–303. doi: 10.1016/0896-6273(93)90185-t. [DOI] [PubMed] [Google Scholar]
- Sokolov S, Weiß RG, Kurka B, Gapp F, Hering S. Inactivation determinant in the I-II loop of the Ca2+ channel α1-subunit and β-subunit interaction affect sensitivity for the phenylalkylamine (–)gallopamil. The Journal of Physiology. 1999;519:315–322. doi: 10.1111/j.1469-7793.1999.0315m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov S, Weiß RG, Timin EN, Hering S. Modulation of slow inactivation in class A Ca2+ channels by β-subunits. The Journal of Physiology. 2000;527:445–454. doi: 10.1111/j.1469-7793.2000.t01-1-00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldatov NM, Oz M, O'Brien KA, Abernethy DR, Morad M. Molecular determinants of L-type Ca2+ channel inactivation. Segment exchange analysis of the carboxyl-terminal cytoplasmic motif encoded by exons 40–42 of the human α1C subunit gene. Journal of Biological Chemistry. 1998;273:957–963. doi: 10.1074/jbc.273.2.957. [DOI] [PubMed] [Google Scholar]
- Soldatov NM, Zuhlke RD, Bouron A, Reuter H. Molecular structures involved in L-type calcium channel inactivation. Role of the carboxyl-terminal region encoded by exons 40–42 in α1C subunit in the kinetics and Ca2+ dependence of inactivation. Journal of Biological Chemistry. 1997;272:3560–3566. doi: 10.1074/jbc.272.6.3560. [DOI] [PubMed] [Google Scholar]
- Spaetgens RL, Zamponi GW. Multiple structural domains contribute to voltage-dependent inactivation of rat brain α(1E) calcium channels. Journal of Biological Chemistry. 1999;274:22428–22436. doi: 10.1074/jbc.274.32.22428. [DOI] [PubMed] [Google Scholar]
- Stea A, Tomlinson WJ, Soong TW, Bourinet E, Dubel SJ, Vincent SR, Snutch TP. Localization and functional properties of a rat brain α1A calcium channel reflect similarities to neuronal Q- and P-type channels. Proceedings of the National Academy of Sciences of the USA. 1994;91:10576–10580. doi: 10.1073/pnas.91.22.10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stotz SC, Hamid J, Spaetgens RL, Jarvis SE, Zamponi GW. Fast inactivation of voltage-dependent calcium channels. A hinged-lid mechanism? Journal of Biological Chemistry. 2000;275:24575–24582. doi: 10.1074/jbc.M000399200. [DOI] [PubMed] [Google Scholar]
- Striessnig J, Grabner M, Mitterdorfer J, Hering S, Sinnegger MJ, Glossmann H. Structural basis of drug binding to L Ca2+ channels. Trends in Pharmacological Sciences. 1998;19:108–115. doi: 10.1016/s0165-6147(98)01171-7. [DOI] [PubMed] [Google Scholar]
- Tang S, Yatani A, Bahinski A, Mori Y, Schwartz A. Molecular localization of regions in the L-type calcium channel critical for dihydropyridine action. Neuron. 1993;11:1013–1021. doi: 10.1016/0896-6273(93)90215-d. [DOI] [PubMed] [Google Scholar]
- Tareilus E, Schoch J, Breer H. Ca2+-dependent inactivation of P-type calcium channels in nerve terminals. Journal of Neurochemistry. 1994;62:2283–2291. doi: 10.1046/j.1471-4159.1994.62062283.x. [DOI] [PubMed] [Google Scholar]
- Wakamori M, Yamazaki K, Matsunodaira H, Teramoto T, Tanaka I, Niidome T, Sawada K, Nishizawa Y, Sekiguchi N, Mori E, Mori Y, Imoto K. Single tottering mutations responsible for the neuropathic phenotype of the P-type calcium channel. Journal of Biological Chemistry. 1998;273:34857–34867. doi: 10.1074/jbc.273.52.34857. [DOI] [PubMed] [Google Scholar]
- Walker D, Bichet D, Campbell KP, De Waard M. A β4 isoform-specific interaction site in the carboxyl-terminal region of the voltage-dependent Ca2+ channel α1A subunit. Journal of Biological Chemistry. 1998;273:2361–2367. doi: 10.1074/jbc.273.4.2361. [DOI] [PubMed] [Google Scholar]
- Walker D, Bichet D, Geib S, Mori E, Cornet V, Snutch TP, Mori Y, De Waard M. A new β subtype-specific interaction in α1A subunit controls P/Q-type Ca2+ channel activation. Journal of Biological Chemistry. 1999;274:12383–12390. doi: 10.1074/jbc.274.18.12383. [DOI] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+ channel inactivation. Proceedings of the National Academy of Sciences of the USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams ME, Feldman DH, McCue AF, Brenner R, Velicelebi G, Ellis SB, Harpold MM. Structure and functional expression of α1, α2, and β subunits of a novel human neuronal calcium channel subtype. Neuron. 1992;8:71–84. doi: 10.1016/0896-6273(92)90109-q. [DOI] [PubMed] [Google Scholar]
- Wu LG, Westenbroek RE, Borst JGG, Catterall WA, Sakmann B. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. Journal of Neuroscience. 1999;19:726–736. doi: 10.1523/JNEUROSCI.19-02-00726.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JF, Ellinor PT, Aldrich RW, Tsien RW. Molecular determinants of voltage-dependent inactivation in calcium channels. Nature. 1994;372:97–100. doi: 10.1038/372097a0. [DOI] [PubMed] [Google Scholar]
- Zhong H, Yokoyama CT, Scheuer T, Catterall WA. Reciprocal regulation of P/Q-type Ca2+ channels by SNAP-25, syntaxin and synaptotagmin. Nature Neuroscience. 1999;2:939–941. doi: 10.1038/14721. [DOI] [PubMed] [Google Scholar]
- Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the α1A-voltage-dependent calcium channel. Nature Genetics. 1997;15:62–69. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]
- Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]