

Abstract
Nine healthy subjects performed single rapid wrist movements from neutral to targets at 20 deg of flexion or extension in response to an auditory cue. Surface EMG was recorded from the wrist flexors and extensors together with wrist position. Movements in both directions were characterised by the usual triphasic pattern of EMG activity in agonist (AG1), antagonist (ANTAG) and again in agonist (AG2) muscles.
Single pulses of transcranial magnetic stimulation (TMS) were applied over the motor cortex at an intensity of 80 % of resting threshold at random times between 80 and 380 ms after the cue. We measured the peak-to-peak amplitude of the evoked motor potential (MEP) and the integrated EMG (IEMG) activity that preceded the MEP. In a separate set of experiments H reflexes were elicited in the wrist flexors instead of MEPs.
MEP amplitudes in the agonist muscle increased by an average of 10 ± 8 ms (range −1 to 23 ms) prior to the onset of the AG1 burst and were associated with an increase of over sevenfold in the MEP:IEMG ratio, irrespective of movement direction. Agonist H reflex amplitudes were linearly related to, and increased at the same time as, changes in agonist IEMG.
The principal ANTAG burst was not preceded by an increase in the antagonist muscle MEP:IEMG ratio. No relationship was found between the amplitude of the antagonist H reflexes and the preceding antagonist IEMG.
Five subjects showed an increase in the MEP:IEMG ratio preceding and during the initial part of the AG2 burst.
Our method of analysis shows that changes in motor cortical excitability mediating the initiation of movement occur much closer to the onset of EMG activity (less than 23 ms) than the 80–100 ms lead time previously reported. The lack of such changes before the onset of the ANTAG burst suggests that this may be initiated by a different, perhaps subcortical, mechanism.
Rapid, self-terminated movements about a single joint are produced by a triphasic pattern of EMG activity. An initial burst of activity in the agonist (AG1) accelerates the limb; an antagonist (ANTAG) burst brakes the movement at the end-position; and a second agonist (AG2) burst reduces terminal oscillations (for review see Berardelli et al. 1996). Several lines of evidence suggest that this pattern is planned centrally. For example, the bursts are present in deafferented patients with large fibre sensory neuropathies (Hallett et al. 1975; Rothwell et al. 1982; Sanes & Jennings, 1984), and the ANTAG and AG2 bursts are absent when subjects are not required volitionally to halt the movement themselves (Marsden et al. 1983; Mustard & Lee, 1987). However, apart from the onset of the AG1 burst, which is mediated by the primary motor cortex, there is very little evidence about whether later bursts are produced by activity in cortical or subcortical structures.
Evidence of the cortical role in the AG1 burst is particularly strong. Studies in behaving primates have analysed the relationship of motor cortical activity to movement in self-initiated and reaction time tasks. In self-initiated movements, changes in neuronal firing occur up to 400 ms before EMG activation (Cheney & Fetz, 1980). Since a proportion of this activity is in corticomotoneuronal cells that have monosynaptic connections to spinal motoneurones innervating the agonist muscle, the implication is that motor cortex contributes directly to the onset of EMG activity. In reaction time tasks, motor cortical activity still precedes contraction of the agonist muscle, but in this case by only 100 ms or less (Evarts, 1966; Lamarre et al. 1981). However, no distinction between corticomotoneuronal and other pyramidal tract neurones was made in these studies, so it is not possible to conclude with absolute certainty that the activity was facilitating spinal motoneurones, or whether it was destined to change the excitability of spinal interneuronal networks. Nevertheless, it seems highly likely that at least part, if not all, of the EMG activity was caused by activity in corticospinal projections.
Compatible results have been reported using transcranial magnetic stimulation (TMS) in humans. Most studies have involved reaction time tasks with a magnetic stimulus given over the motor cortex at different times in the reaction period. The size of the evoked EMG response (MEP) or the probability of its occurrence increased about 80–100 ms before onset of EMG activity in the agonist muscle (Tomberg & Caramia, 1991; Pascual-Leone et al. 1992a,b; Tarkka et al. 1995; Hoshiyama et al. 1996; Chen et al. 1998). By analogy with the primate data, it was supposed that motor cortical excitability increased 100 ms before movement onset, so that a given transcranial magnetic stimulus evoked a larger descending corticospinal volley than in subjects at rest. This larger volley was responsible for the increased MEP. A potential flaw in this argument is that MEP size depends on the excitability of spinal motoneurones as well as the amplitude of the descending corticospinal volley. Since spinal excitability was not tested in these studies (for example by measuring spinal reflex excitability or the ongoing level of EMG activity in the muscle under test), it is always possible that some of the changes in the MEP were caused by segmental rather than cortical effects.
The present experiments were designed to extend these previous human studies in two ways. First, we wished to re-examine the time at which motor cortical excitability changes prior to movement using H reflex and EMG measures for comparison with the effects of TMS. Second, we wanted to extend the study to later components of the triphasic EMG pattern. In agreement with others, we suggest that the motor cortex is involved in the initiation of the AG1 burst, but that its lead time can be much shorter than previous estimates of 100 ms. In addition the data appear to show that the ANTAG (and AG2) burst may be mediated by a different, perhaps subcortical, system. The preliminary results of these studies have been published in abstract form (MacKinnon & Rothwell, 1998).
METHODS
Experimental protocol
Nine subjects (7 males, 2 females, age range 27–45 years) volunteered to participate in the study after giving informed consent. All subjects were right-handed. Experiments were approved by the local ethical committee in accordance with the standards set by the Declaration of Helsinki. Subjects were seated with their right elbow at an angle of approximately 90 deg and their forearm immobilised in a cuff in a semi-supinated position. The hand was encased in a cuff with the fingers extended and the centre of rotation of the wrist joint aligned to be coaxial with the axis of rotation of a shaft connected to a potentiometer.
A simple auditory reaction time protocol was used to probe the changes in excitability preceding and over the time course of the triphasic EMG pattern (Fig. 1). Subjects were instructed to make movements ‘as quickly and accurately as possible’ from a neutral wrist position to a target location at a joint angle of 20 deg. Sets of trials consisted of consecutive movements of either wrist flexion or extension. Visual feedback of joint position was displayed on an oscilloscope. Movements were initiated by an auditory ‘go’ tone presented randomly every 5–15 s. A minimum of 20 practice trials were completed before data collection commenced to ensure that movements were fast, accurate and accompanied by a triphasic EMG pattern.
Figure 1. Examples of the angular displacement, velocity and rectified EMG for flexor carpi radialis (FCR) and extensor carpi radialis longus (ECRL) during wrist flexion and extension movements in a single subject.
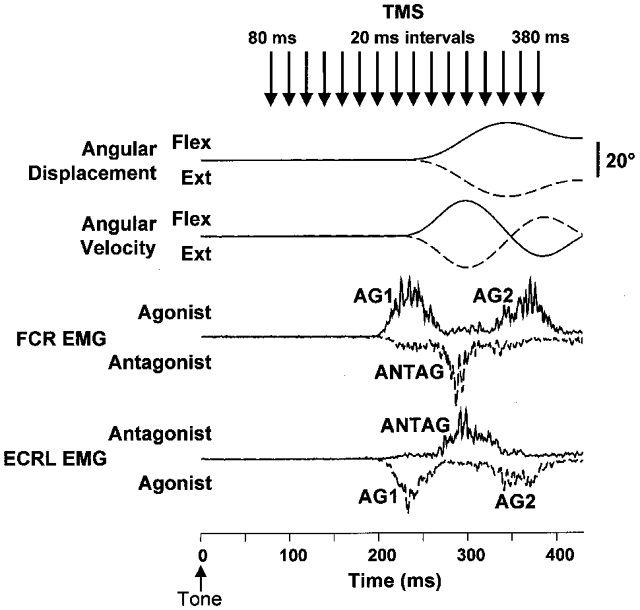
Each trace is the average of 15 trials. Continuous lines are for flexion trials and dashed lines are for extension trials. Note the characteristic triphasic pattern of EMG activity (AG1, ANTAG and AG2) associated with movements in both directions. The arrows at the top of the figure show the times that TMS was applied across trials. The auditory tone was presented at 0 ms.
Transcranial magnetic stimulation (TMS)
TMS was delivered using a figure-of-eight shaped coil (90 mm outer diameter; Magstim 200, Magstim Company Ltd, Whitland, Dyefield, UK) placed on the surface of the scalp over the optimal site to evoke MEPs in the right flexor carpi radialis (FCR) muscle. The coil was oriented to generate induced currents in a posterior-to-anterior direction. Resting threshold was determined by finding the scalp site that evoked optimal responses (maximal amplitude and minimum latency) in FCR using a suprathreshold stimulus, and then gradually decreasing the stimulus intensity until responses of 50–100 μV were evoked in three of five stimuli. Stimulus intensity was adjusted to 80 % of resting threshold intensity. Similar stimulus intensities have been used previously to probe changes in corticospinal excitability associated with reaching, grasping and precision lift (Lemon et al. 1995). Single stimuli were delivered randomly between 80 and 380 ms after the onset of the auditory tone (Fig. 1). Control trials were also included in which the tone was presented but TMS was not applied. Four sets of 75 movements were performed in the direction of wrist flexion and extension, resulting in 15 trials at each of 16 time intervals after the auditory tone and 60 control trials. In addition, three subjects participated in experiments in which TMS was delivered at suprathreshold intensities (110 % of resting threshold). This experiment was conducted to examine whether the timing of changes in the amplitude of MEPs was affected by stimulus intensity.
Recording and data analysis
Bipolar EMG activity was recorded from FCR and extensor carpi radialis longus (ECRL), differentially amplified (gain 500-1k), filtered (high-pass time constant, 3 ms; low-pass 3 dB cut-off, 1 kHz), and recorded, along with the joint position (high-pass time constant, 5 s; low-pass 3 dB cut-off, 100 Hz), using a 12 bit analog-to-digital converter (CED 1401, Cambridge Electronic Design, Cambridge, UK) at a sampling rate of 2000 Hz. Data were collected from 50 ms prior to the onset of the tone to 500 ms after the tone. Wrist angular velocity was determined by finite element differentiation of the angular displacement signal. Movement onset was defined as the time point when the velocity exceeded two standard deviations from baseline and was confirmed by visual inspection. Profiles of the EMG activity over the time course of the movement (Fig. 1) were determined by baseline correcting, full wave rectifying, and averaging the raw EMG signal with respect to the onset of movement.
The amplitude of the MEPs was quantified by calculating the peak-to-peak voltage of the evoked responses. EMG activity preceding the onset of the MEPs was quantified by calculating the area under the curve (IEMG) over an 8 ms time interval (5–13 ms after TMS) between the end of the TMS stimulus artifact and the onset of the MEP. The influence of the level of background EMG activity at the time of stimulation on the magnitude of the MEPs was assessed by calculating the MEP:IEMG ratio.
The timing of the MEPs was referenced to movement onset, and the responses were sorted into 10 ms bins and their magnitudes were averaged within each bin. Inter-subject means at each time bin were determined after normalisation of MEP, IEMG or ratio data to the maximum mean value for each subject. Differences in the amplitude of the MEPs, IEMG, or MEP:IEMG ratios between time bins were assessed using an analysis of variance (ANOVA) with repeated measures. Baseline IEMG was calculated over the time interval from 13 to 5 ms prior to the onset of the tone. Post hoc tests between responses at each time bin and baseline responses were conducted using Student's unpaired t tests with Bonferroni correction for multiple comparisons. Time delays between MEP and IEMG onset were calculated based on the time differences between data within the first MEP and IEMG time bins that were significantly different from baseline for each subject and movement direction (e.g. subject 1, FCR for flexion movements: first significant MEP time bin = −50 ms (data from −48 to −43 ms); first significant IEMG time bin = −30 ms (data from −27 to −21 ms); lead time = 21 ms, maximum range = 27 ms).
H reflex stimulation
An additional set of experiments was conducted with four subjects during which H reflexes were elicited over the time course of wrist flexion and extension movements using a similar protocol to the experiments described above. H reflexes were evoked by electrically stimulating the median nerve just proximal to the elbow crease. Stimuli were administered using a 500 μs pulse delivered through a pair of surface electrodes placed 2 cm apart with the cathode located proximal to the anode. Stimulus intensity was subsequently adjusted to evoke a clear H wave with an amplitude near 50 % of maximal H reflex amplitude. H waves were quantified by measuring the peak-to-peak amplitude of the responses. Due to differences in the control H waves across sets of movements, responses were subsequently expressed as a percentage of the average peak-to-peak H wave measured from control trials within a set of movements (100 × Hpeak-to-peak/Hcontrol). Responses were subsequently time referenced to the onset of initial velocity, sorted into 10 ms bins, averaged within bins, and normalised to the maximum mean response within subjects.
RESULTS
Triphasic EMG pattern
Movements to either flexion or extension were accompanied by a characteristic triphasic pattern of EMG activity. An example of the pattern of EMG activity for wrist flexion movements in a single subject is shown in Fig. 1. The mean onset time of the AG1 burst following the auditory ‘go’ tone was 216 ± 23 ms (mean ± 1 s.d.) and mean AG1 burst duration was 81 ± 9 ms. The electromechanical delay between the onset of the AG1 burst and movement onset averaged 36.5 ± 5.0 ms. Activity in the antagonist muscle consisted of an initial low level contraction that accompanied the AG1 burst (observed in 7 of 9 subjects), followed by a distinct increase in motor unit activity which we defined as the principal ANTAG burst. The initial antagonist activity began an average of 12 ± 6 ms after the onset of the AG1 burst, whereas the main ANTAG muscle burst began an average of 73 ± 13 ms after the onset of the AG1 burst and had a duration of 71 ± 11 ms. The AG2 burst commenced 118 ± 17 ms after the AG1 burst and had a duration of 97 ± 24 ms. A repeated measures ANOVA showed that reaction times were not significantly affected by TMS given at the intervals and intensities used in the present experiments (F = 0.342, d.f. = 16, P = 0.991).
Time-varying changes in MEPs and EMG
Figure 2 shows examples of rectified MEPs evoked by subthreshold TMS in FCR at four time intervals preceding the onset of wrist flexion movements (plots on the left) or the onset of deceleration for wrist extension movements (plots on the right) in an individual subject. Stimulation rarely elicited MEPs prior to 70 ms before movement onset. When the target muscle acted as the initial agonist (e.g. the flexors during wrist flexion movements), marked increases in the MEPs evoked by TMS were first observed in the time bin 51–60 ms prior to the onset of movement for both the wrist flexion and extension tasks. These early increases in the MEP amplitude were elicited in the absence of significant changes in background EMG activity. Maximal MEP amplitudes were typically evoked 0–20 ms before movement onset. In contrast, when the same muscle acted as the antagonist, changes in EMG activity were not preceded by increases in the amplitude of the MEPs. For example, note the small amplitude of the antagonist MEP evoked with stimulation 60 ms prior to the onset of deceleration despite an increase in background EMG activity.
Figure 2. Examples of MEPs evoked at four time intervals when FCR functioned as the initial agonist and antagonist.
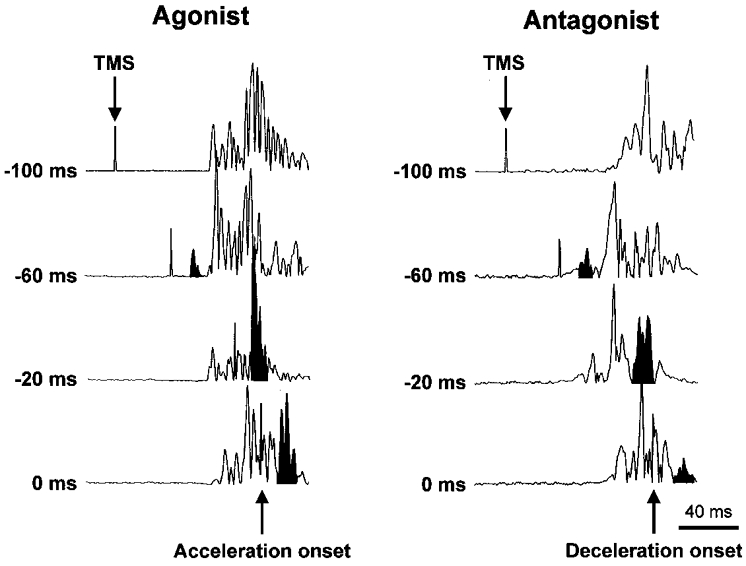
The times to the left of each plot indicate the timing of TMS expressed relative to the onset of acceleration for the agonist plots and relative to the onset of deceleration for the antagonist plots. Note the marked increase in the agonist MEPs 60 ms before movement onset in the absence of background EMG. In contrast, when the same muscle functioned as the antagonist, small MEPs were evoked despite increases in background EMG.
An example of the time-varying changes in the MEPs and IEMG of FCR for a single subject is shown in Fig. 3. A marked increase in the amplitude of the agonist MEPs preceded the AG1 burst by two time bins (range of 9–29 ms). Differences in the rate of rise of the agonist MEP amplitude and IEMG were most evident when the data were expressed as the ratio MEP:IEMG. In this subject, there was an increase of over sixteenfold in the MEP:IEMG ratio that peaked 41–50 ms prior to the onset of movement and returned to baseline levels near movement onset. Similarly, agonist MEPs and the MEP:IEMG ratio increased prior to the onset of the AG2. As discussed below, increases in MEP amplitude preceding the AG2 burst were not observed in all subjects. In contrast, antagonist muscle activity was not preceded by increases in the amplitude of antagonist MEPs. As a result, MEP:IEMG ratios did not change significantly from baseline levels. Differences between the AG1 and ANTAG ratios were observed despite similar peak IEMG values (1.72 ± 0.75 and 2.28 ± 1.21 mV ms, respectively) and rates of increase in IEMG (0.06 and 0.08 mV ms ms−1, respectively). There was a small increase in the ratio prior to the onset of the initial low amplitude antagonist activity, but this change was not significantly different from baseline.
Figure 3. Time-varying changes in the amplitude of the MEPs, IEMG and MEP:IEMG ratios in FCR during wrist flexion or extension movements in a single subject.
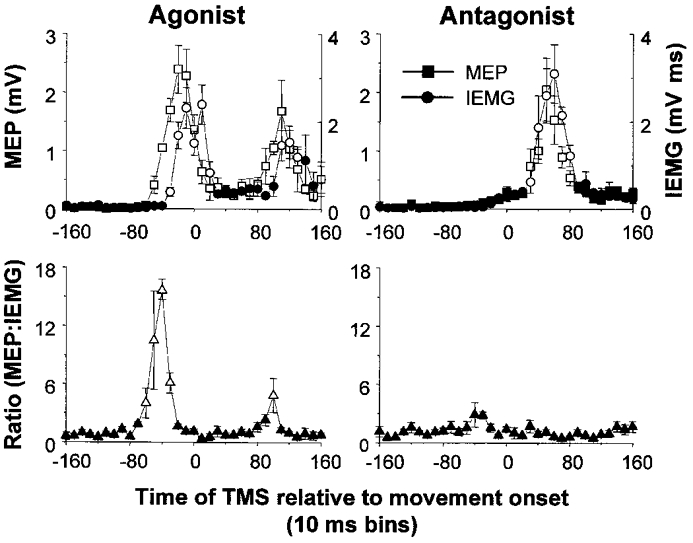
Plots on the left show profiles when FCR functioned as the agonist (wrist flexion) and plots on the right show profiles when FCR functioned as the antagonist (wrist extension). Responses have been sorted into 10 ms time bins. In this subject, MEPs increased prior to the onset of the AG1 and AG2 bursts. The phase advance resulted in a marked increase in the MEP:IEMG ratio prior to both agonist bursts. In contrast, MEPs and IEMGs increased within the same time bins when the same muscle functioned as the antagonist, resulting in no significant change in the MEP:IEMG ratio from baseline levels. Open symbols denote values that were significantly different from baseline (P < 0.05). Error bars are one standard error of the mean.
Similar trends in the time-varying profiles of the MEPs and MEP:IEMG ratios were observed in all subjects. Plots of the average normalised MEPs and IEMG across all subjects for both FCR and ECRL are shown in Fig. 4. Increases in the amplitude of the MEPs relative to background were observed in time bins ranging from 50 to 70 ms prior to the onset of movement, but did not reach significance across subjects until 50 ms (F = 8.129, d.f. = 37, P < 0.02) before movement onset. Increases in the MEP amplitude preceded the onset of the AG1 burst by two or three time bins (mean lead time = 21 ± 8 ms, range 10–34 ms). Similar lead times were observed regardless of whether FCR (Fig. 4A) or ECRL (Fig. 4B) was the initial agonist. When the differences in the timing of the MEPs (average onset of 16 ms) and IEMG (5–13 ms) relative to the stimulus were accounted for, the MEP increases led the AG1 onset by an average of 10 ± 8 ms (range −1 to 23 ms).
Figure 4. Time-varying changes in the amplitude of the MEPs, IEMG and MEP:IEMG ratios in FCR and ECRL when the same muscle functioned as an agonist and antagonist averaged across nine subjects.
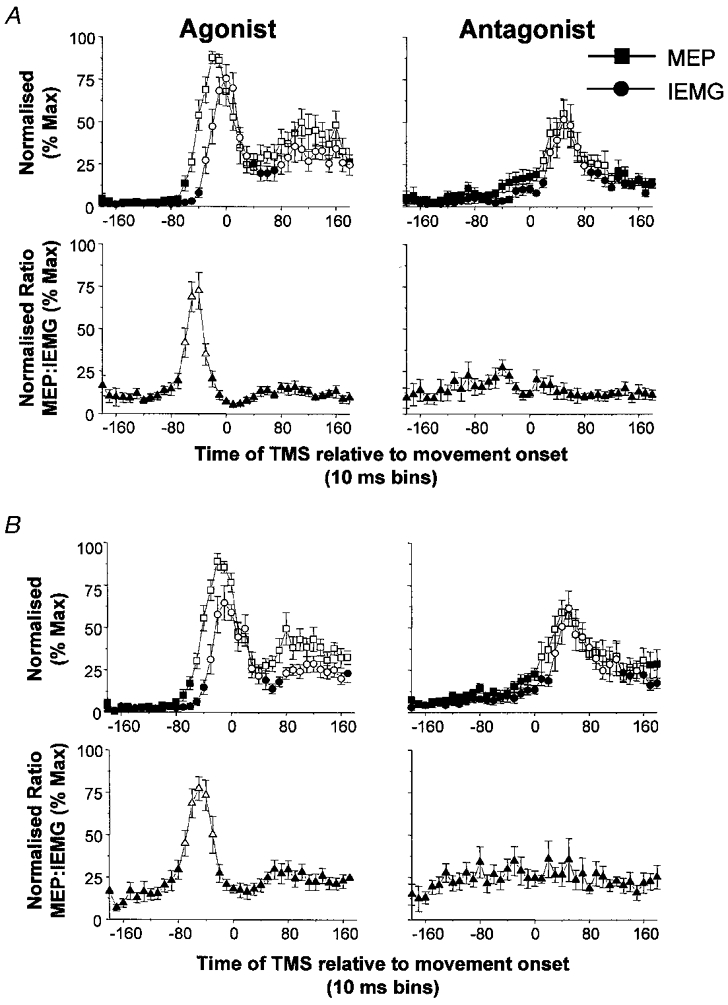
A, data for FCR; B, data for ECRL. Squares, MEP; circles, IEMG; triangles, MEP:IEMG ratio. Open symbols denote values that were significantly different from baseline (P < 0.05). Values have been normalised to the maximum average response within subjects. Increases in agonist MEPs preceded the AG1 burst by two or three time bins (range = 10–34 ms). The early increase in the agonist MEPs resulted in marked increases in the MEP:IEMG ratio prior to, and during the rising phase of, the AG1 burst. In contrast, changes in antagonist MEPs were accompanied by proportionate changes in pre-existing IEMG, resulting in no significant changes in the ratio from baseline levels. Error bars are one standard error of the mean.
Previous studies have consistently reported MEP lead times relative to EMG onset in the range of 80–100 ms and not less than 23 ms as reported above. Several of those studies examined the timing of changes in corticospinal excitability by expressing the probability of evoking an MEP relative to EMG onset (Starr et al. 1988;Pascual-Leone et al. 1992a,b). Accordingly, we plotted the probability of evoking an MEP relative to movement onset, but unlike previous studies, we also plotted the probability of measuring IEMG activity that was greater than 2 s.d. above baseline levels (Fig. 5). The probability of evoking an MEP increased 80–100 ms prior to movement onset, but in keeping with the peak-to-peak MEP and IEMG data, these changes preceded increases in IEMG probability by no more than three time bins (average lead time = 3 ± 22 ms). In four subjects, changes in MEP probability occurred within the same time bin, or after, the changes in IEMG probability.
Figure 5. Time course of the changes in the probability of evoking an MEP or recording IEMG levels of greater than two standard deviations from baseline when the muscle functioned as an agonist (A) and antagonist (B).
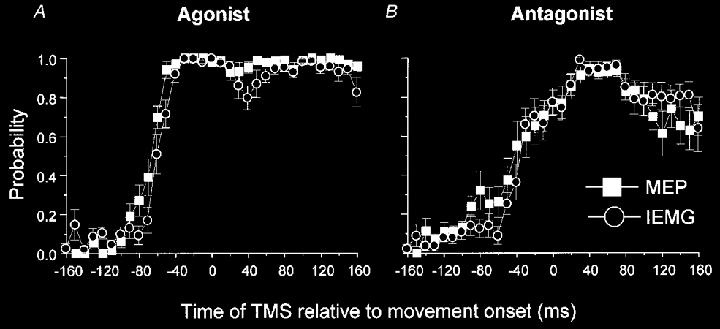
Data from FCR and ECRL have been pooled and averaged across subjects. When the muscle functioned as the agonist, the probability of evoking an MEP increased prior to changes in IEMG probability, but the lead time was less than 28 ms. Similarly, when the muscle functioned as the antagonist the probability of evoking an MEP increased before the changes in IEMG probability associated with the initial antagonist burst, but not prior to the principal ANTAG burst. Error bars are one standard error of the mean.
The AG1 burst was always preceded by a significant increase in the MEP:IEMG ratio, irrespective of whether the initial agonist was a wrist flexor or extensor (wrist flexion: F = 6.86, d.f. = 37, P < 0.001; wrist extension: F = 11.71, d.f. = 37, P < 0.001) (Fig. 4). This ratio increased an average of 62 ± 6 ms prior to movement onset, peaked at over seven times baseline values, and had an average duration of 50 ± 13 ms. These large ratios were primarily derived from MEPs that were elicited in the absence of significant increases in background EMG activity.
We further explored the relationship between the MEP and IEMG by conducting linear regressions between the two variables. If the time bins from −60 to −20 ms were excluded from the regression analysis, a significant linear relationship existed between the amplitude of the agonist MEPs and agonist IEMG (r = 0.92, P < 0.0001) (Fig. 6A). The relationship between the agonist MEPs and IEMG over the −60 to −20 ms time bins was best fitted by a second-order polynomial equation (y = 16.5 + 2.8x − 0.03x2; r = 0.94, P < 0.0001). These data further illustrate that over the time period of 60 to 20 ms prior to movement onset the MEP amplitudes increased disproportionately with respect to the magnitude of the preceding EMG activity.
Figure 6. Relationships between the amplitude of the MEPs and IEMG when muscles functioned as the agonist (A) and antagonist (B).
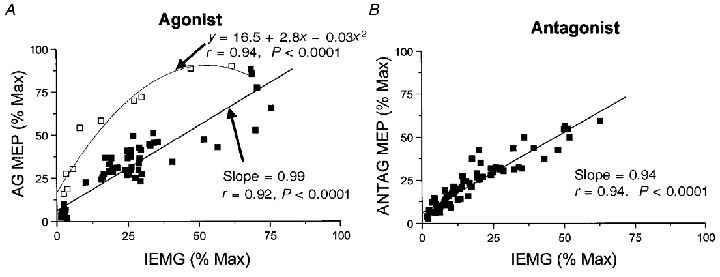
A, a linear regression was performed and a line of best fit drawn through agonist muscle data points from −180 to −70 ms and −10 to 180 ms (▪). Data points correspond to normalised responses averaged across subjects for both FCR and ECRL. The fit of the line to these data was highly significant (r = 0.92, P < 0.0001). Data points from −60 to −20 ms (□) were best fitted by a second-order polynomial function (r = 0.94, P < 0.0001). B, a linear regression was conducted and a line of best fit drawn through all antagonist muscle data points (-180 to 180 ms). A linear relationship (r = 0.94, P < 0.0001) between the antagonist MEPs and IEMG was maintained throughout the movement.
The initial phase of antagonist EMG activity was preceded by an increase in the amplitude of the MEPs and a corresponding increase in the MEP:IEMG ratio in four subjects. This early increase in the MEPs was observed only in FCR for wrist extension movements (Fig. 4A). Across subjects, the early increase in the ratio preceding the initial antagonist burst did not reach significance. The initial antagonist activity was not preceded by an increase in MEP amplitude during wrist flexion movements in all subjects, and changes in MEP amplitude were accompanied by changes in IEMG within the same time bin.
In marked contrast to the AG1 burst, the principal ANTAG burst was not preceded by a disproportionate increase in the amplitude of the MEPs. Changes in the amplitude of the MEPs occurred within the same time bin as changes in IEMG. For this reason, no significant change in the MEP:IEMG ratio was observed prior to, or over the time course of, the ANTAG burst (wrist flexion: F = 1.129, d.f. = 37, P = 0.306; wrist extension: F = 1.383, d.f. = 37, P = 0.100). Linear regression analysis between the antagonist MEPs and IEMG showed a significant linear relationship (r = 0.94, P < 0.0001) between the two variables throughout the time course of the movement (Fig. 6B). These data suggest that the observed changes in amplitude of the antagonist MEPs were closely related to the changes in pre-existing EMG activity.
Time-varying changes in agonist MEPs and IEMG associated with the AG2 burst differed across subjects. Five subjects showed an increase in MEP amplitude preceding the AG2 burst (see Fig. 3). This was reflected in a transient increase in the MEP:IEMG ratio over one or more time bins. In the remaining four subjects, no changes in the ratio were observed in association with the AG2 burst. When averaged across subjects, no significant changes in the MEP:IEMG ratio associated with the AG2 burst were observed.
Suprathreshold stimulation
Three subjects participated in experiments using the same protocol described above, but with TMS at 110 % of resting threshold. Suprathreshold stimulation resulted in time-varying changes in MEP amplitude that paralleled the time course observed with subthreshold stimulation. The amplitudes of the changes in MEPs and MEP:IEMG ratios were markedly reduced with suprathreshold stimulation and did not reach significance at the P < 0.05 level. Nonetheless, increases in MEP amplitude above baseline were observed in time bins 40 to 70 ms prior to movement onset (average = 53 ± 15 ms) and overlapped with the time range observed for increases in agonist MEP amplitude with subthreshold stimulation. Similar to the results obtained with subthreshold stimulation, changes in antagonist MEP amplitude occurred at the same time as the changes in antagonist IEMG. Therefore, suprathreshold stimulation had no effect on the observed timing of the changes in the MEPs.
Time-varying changes in the H reflex
H reflexes were elicited in FCR over the time course of wrist flexion and extension movements in four subjects. Changes in the amplitudes of the H and M waves and IEMG prior to, and during, these movements are shown in Fig. 7. The amplitude of the agonist H reflex began to increase no more than one time bin prior to the AG1 burst. Furthermore, changes in agonist H reflexes paralleled corresponding changes in the IEMG throughout the movement. Stimulus intensity remained constant as shown by the stability of the M wave amplitude.
Figure 7. Relationships between the amplitude of the H wave and IEMG when FCR functioned as an agonist and antagonist.
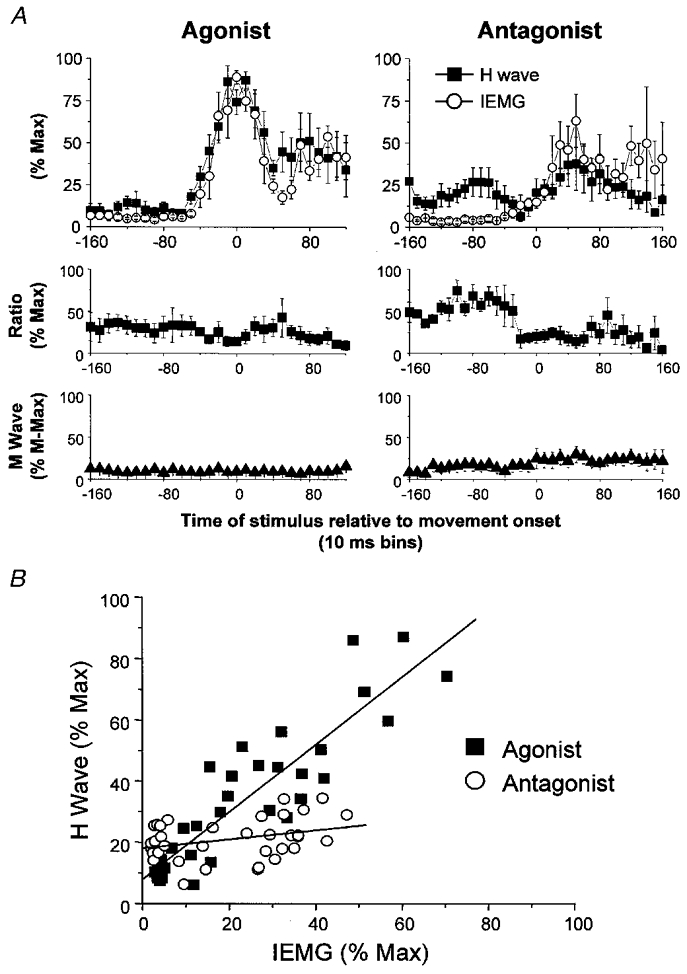
A, time-varying changes in the amplitude of the H wave, IEMG, H wave:IEMG ratio and M wave, when FCR functioned as an agonist (plots on the left) and an antagonist (plots on the right), averaged across four subjects. Increases in agonist H waves preceded the AG1 burst by no more than one time bin. The initial antagonist burst was preceded by a small facilitation of the H wave whereas the principal ANTAG burst was not preceded by an increase in the H wave. The H wave:IEMG ratio did not change when FCR functioned as an agonist, but decreased at EMG onset when FCR functioned as the antagonist. M wave amplitudes remained the same throughout the movement irrespective of whether FCR was the agonist or antagonist. Error bars are one standard error of the mean. B, a linear regression was performed between the amplitude of the H wave and IEMG when FCR functioned as the agonist (▪) and antagonist (○) and a line of best fit was drawn through all data points (-180 to 180 ms). The linear relationship was significant for the agonist data (r = 0.90, P < 0.0001), but not for the antagonist data (r = 0.33, P > 0.05).
When FCR functioned as the antagonist there was a slight facilitation of the H reflex amplitude prior to the onset of the AG1 burst above that observed when the same muscle acted as the agonist. This facilitation could not be accounted for by differences in background EMG or joint position. There was a marked suppression of the H wave amplitude beginning near the onset of the AG1 burst and during the period of agonist H reflex facilitation. A recovery of the H wave amplitude to slightly greater than baseline levels occurred in parallel with the rising phase of the principal ANTAG burst. An upward drift in the M wave was observed in one of the four subjects and is reflected in the slight increase in the grand average M wave during wrist extension movements. On average, the peak amplitude of the agonist H reflex was 2.6 times larger than the antagonist peak reflex, whereas the peak agonist IEMG was only 1.7 times larger than the antagonist peak IEMG. Linear regression analysis showed that the amplitude of the agonist FCR H waves was tightly coupled to the magnitude of the accompanying IEMG (y = 7.9 + 1.1x, r = 0.90, P < 0.0001) (Fig. 7B). In contrast, amplitudes of the antagonist H waves and IEMG were unrelated (r = 0.33, P > 0.05).
DISCUSSION
In the present study we measured the amplitude or the probability of evoking an MEP at different times before and during the triphasic EMG pattern that accompanied rapid extension or flexion movements at the wrist. The results show that the amplitude of the MEP relative to the background level of EMG increases prior to the onset of AG1, but that there is no such increase before the principal ANTAG burst. We conclude that the mechanisms responsible for the initiation of the AG1 and principal ANTAG burst are different. In addition, the lead time of the MEP changes relative to the EMG was much shorter in the present study than in previous reports (0–23 ms versus 80–100 ms). This suggests that under conditions when reactions have to be as fast as possible, there is only minimal delay between activation of cortical and spinal motoneurones. There is no need for a slow build-up of spinal excitability as implied by previous results in monkey and human experiments.
Changes in corticospinal excitability preceding AG1
In agreement with previous reports (Rossini et al. 1988; Starr et al. 1988; Tomberg & Caramia, 1991; Pascual-Leone et al. 1992a,b;Tarkka et al. 1995; Hoshiyama et al. 1996; Chen et al. 1998; Davey et al. 1998), we found that the amplitude of MEPs evoked by a constant test stimulus increased prior to the onset of the AG1 burst. In five of the nine subjects, the increase was limited to MEPs in the agonist, suggesting selective facilitation of the projections to the agonist muscle. In the other four subjects, there was also a small increase in the size of MEPs recorded simultaneously from the antagonist muscle (see below).
In contrast with the previous reports we found that the time interval between increases in MEP amplitude and AG1 was only 0–23 ms rather than 80–100 ms. We think that this difference is due to the way that we measured EMG timings. Unlike other workers, we never tried to estimate the onset of AG1 directly in every trial. Instead we quantified in each individual trial the level of EMG just before MEP onset. This removed the need to make an arbitrary decision about the onset of EMG activity. We could therefore produce much more accurate information about occasions when the MEP appeared to increase in size without concomitant changes in background EMG. Such activity may have been ignored by other methods which concentrated on the latency of the main AG1 burst.
Our technique relied on making measurements of mean MEP amplitude over several trials. When we tried to improve the sensitivity of the method, by plotting the probability of evoking a response relative to the time of movement onset (Fig. 5), we obtained very similar results. Increases in MEP probability occurred about 80–100 ms before movement. Although this was 20–40 ms earlier than when we had measured changes in peak-to-peak MEP amplitude, the probability approach also changed the onset time of EMG activity by the same amount. In keeping with the amplitude data, increases in MEP probability led changes in the probability of measuring increased IEMG by no more than 28 ms.
Despite the relatively small difference between the onset of facilitation in the MEP and EMG, there was a very clear change in the way the MEP was recruited by transcranial stimulation at the time of onset of the AG1 burst. This was shown by expressing MEP amplitude relative to IEMG as the MEP:IEMG ratio. At rest and during most of the triphasic EMG activity, the amplitude of the MEP was directly proportional to the level of ongoing EMG. A similar, approximately linear relationship has been described during tonic contractions for responses in biceps and tibialis anterior (Kischka et al. 1993). However, just before, and overlapping with the onset of the AG1 burst, there was a sharp increase in the ratio of MEP:IEMG that lasted for 40–50 ms.
There are two possible explanations of this effect. One relies on changes in excitability of spinal motoneurones, the other on changes in supraspinal structures. A spinal effect could occur in the following way. IEMG measures activity only in discharging motoneurones whilst the response of the motoneuronal pool to a given descending input also depends on the excitability of the subliminal fringe of motoneurones that are not firing but which can be recruited to fire by additional descending input. If the size or excitability of this fringe were increased just before the onset of the AG1 burst, then responses to TMS would also be facilitated. We included the H reflex experiments to test this possibility. Although there is some recent evidence that not all motor units recruited in an H reflex are the same as those recruited in an MEP, the results from spatial facilitation experiments involving H reflexes and MEPs show there must be at least some overlap between the respective motoneurone pools (Morita et al. 1999). Given that this is the case, then we might have expected to see an effect on the H reflex equivalent to that on the MEP, but this did not happen. The H wave:IEMG ratio did not change in the period around the start of the AG1 burst (Fig. 7A), suggesting that the relative size of the subliminal fringe remained constant. If this were the case then the only way the MEP:IEMG ratio could have changed is for the magnetic stimulus to have recruited a larger input to the spinal motoneurone pool. Since there is no presynaptic control of the corticomotoneuronal synapse, and since the majority of the MEP at low intensities of stimulation is thought to be due to activity in monosynaptic corticospinal connections (Day et al. 1989), this suggests that the increased response to magnetic stimulation must have occurred because of an effect at the cortical level.
Experiments in monkeys and humans have shown that the size of the corticospinal volley evoked by TMS is sensitive to the level of cortical activity at the time the stimulus is given (Baker et al. 1994; Di Lazzaro et al. 1998). Thus we conclude that just prior to the onset of detectable EMG in the periphery, there is a subthreshold increase in the excitability of corticomotoneurones or local cortical interneurones, and that this accounts for the increase in the MEP:IEMG ratio. An important point is that the present data do not suggest that there is any great delay over and above the corticospinal conduction time between the build-up of cortical and spinal excitability. Spinal motoneurones are activated within milliseconds of the motor cortical output systems accessed by TMS, at least in the rapid reaction time movements studied here. Lead times between motor cortical firing and spinal motoneurone activation may be greater for movement tasks in which the timing of the imperative stimulus to initiate movement is known, such as self-paced movements. Chen et al. (1998) have reported an approximately 20 ms increase in the lead time of changes in corticospinal excitability preceding self-paced movements compared to simple reaction movements. However, for the methodological reasons cited above, these lead time differences may be artefactual.
The very short interval between increases in cortical and spinal excitability raises the possibility that non-corticospinal pathways might mediate some of the early effects. Valls-Solé et al. (1999) recently proposed that, under certain conditions, ballistic movements might be triggered by output from subcortical, perhaps reticular structures. Given the overlap between the timing of increases in MEP and EMG, this opens the possibility that such structures might participate in the onset of movement in the present experiments.
Initiation of antagonist muscle activity
As pointed out in the Introduction, there is good evidence that the antagonist burst can be initiated by central mechanisms, even though afferent feedback may be involved in modulating the magnitude (Sanes & Jennings, 1984) and timing of the bursts (Ghez & Martin, 1982). Activity in antagonist muscles during rapid single-joint movements can typically be divided into two phases: an early phase characterised by low amplitude activity beginning 20–30 ms after the onset of the AG1 burst and a late phase consisting of a rapid rise in motor unit activity beginning near the end of the AG1 burst (Gottlieb et al. 1989; Cooke & Brown, 1990; Wild & Corcos, 1997). Because the timing of the two bursts is differentially affected by changes in movement distance (Gottlieb et al. 1989) or lesions of the cerebellum (Wild & Corcos, 1997) the two phases are usually thought to be under separate control.
In four of the nine subjects, the early phase of antagonist EMG activity was preceded by an increase in the amplitude of the antagonist MEPs and the MEP:IEMG ratio. This only happened when FCR was used as the antagonist during wrist extension movements, and was never seen in ECRL during flexion movements. Indeed, the early part of the ANTAG burst was much larger in these subjects in the FCR than the ECRL, and began at the same time as the AG1 burst. In monkeys, some corticospinal neurones project to both wrist flexors and extensors (Cheney et al. 1985), and may be active when the wrist is stabilised by co-contraction in muscles acting around the joint. The co-contraction during the early part of the ANTAG burst could reflect activity in this system. Early increases in the antagonist MEP amplitude were unlikely to be due to changes in segmental excitability since spinal reflexes, as tested with the H reflex technique, were suppressed over this time period. Thus we suggest that increased MEPs sometimes seen around the onset of the initial low amplitude activity in the antagonist muscle are the result of increased excitability of neurones within the primary motor cortex.
MEP behaviour at the time of the principal antagonist burst was quite different. The principal ANTAG burst was never preceded by an increase in the excitability of the corticospinal pathway. The tight linear relationship between the amplitude of the antagonist MEPs and IEMG throughout the movement (Fig. 6B) shows that the changes in the amplitude of antagonist MEPs could be fully explained by changes in preceding EMG activity. Therefore, we were unable to find evidence for a period of subthreshold increase in cortical excitability prior to the principal ANTAG burst.
The absence of an increase in the MEP:IEMG ratio prior to the onset of the principal ANTAG burst does not rule out a contribution from primary motor cortical neurones to the facilitation of antagonist motoneurones. One possibility is that the excitability of antagonist corticomotoneurones was sufficiently raised in association with the early antagonist muscle activity such that there was no time lag between changes in cortical and spinal excitability. However, this seems unlikely in view of the fact that (1) there was very little early antagonist activity in some subjects, particularly in the ECRL, and (2) there was a clear increase in the MEP:IEMG ratio at about the time of onset of AG2 in five of the subjects, even though the level of background activity was much higher than that preceding the principal antagonist burst. An alternative is that there was no or minimal change in excitability of motor cortical projections to the antagonist and that the change in EMG activity was produced by non-cortical mechanisms. In other words, even though the AG1 might be produced by cortical activity, the ANTAG might be mainly generated subcortically.
Such a proposal is consistent with the limited data available from animal studies. These have shown that even if a motor cortical unit fires in association with a muscle that contracts during AG1, it may not fire at all if the same muscle is active during the ANTAG burst (Hore & Flament, 1988; Kalaska et al. 1989; Crammond & Kalaska, 1996; Sergio & Kalaska, 1997). Even if cortical activity is seen in association with the ANTAG burst, it often occurs later than for the AG1 burst. Increases in firing appear to begin near the onset of deceleration and therefore after the onset of the principal ANTAG burst (Lamarre et al. 1983; Flament & Hore, 1988; Hore & Flament, 1988; Sergio & Kalaska, 1998). These results suggest that antagonist motor cortical neurones are responding to afferent feedback from the movement, rather than driving the onset of the ANTAG burst. However, it should be noted that no study has specifically tested the temporal relationship between the onset of firing of corticomotoneurones projecting to the antagonist motoneurone pool and the onset of the ANTAG burst.
Hore and colleagues have proposed that the onset time of the ANTAG burst is calculated by the cerebellum in order to avoid delays that would occur if its onset were reliant on afferent feedback from the movement. Thus, cooling of the deep cerebellar nuclei, and lesions of the cerebellum in monkeys and humans delay the onset of the ANTAG burst and cause hypermetria (Flament & Hore, 1986; Hore et al. 1991). Under these conditions, the ANTAG activity appears to become driven by afferent input from the periphery. This delays its onset and, because of time delays in the system, produces terminal oscillations at the movement endpoint. If the cerebral cortex is not involved in the initiation of the ANTAG burst, it may be that cerebellar output drives the ANTAG burst via direct connections to subcortical structures.
Changes in the H reflex
We could only evaluate the H reflex in FCR since it is difficult to obtain in the ECRL muscle. Nevertheless, we observed striking changes in the amplitude of the H reflexes depending upon whether the muscle functioned as the agonist or antagonist. During wrist flexion movements the amplitude of the FCR H reflexes was linearly related to the magnitude of the preceding IEMG. In contrast, H reflexes remained low in amplitude throughout the movement when FCR was the antagonist muscle. The antagonist H reflex also showed a particular pattern of temporal modulation. H reflex amplitudes were suppressed prior to and during the onset of the AG1 burst and then recovered to just above baseline levels during the rising phase of the ANTAG burst (Fig. 7). A similar suppression of H reflexes has previously been described prior to voluntary movements (Rüegg, 1989). The low amplitudes of the H reflexes and the absence of a linear relationship between the H reflex and IEMG suggests that presynaptic inhibition of the group I afferents from the spindles of the lengthening muscle is maintained throughout the movement.
Initiation of the AG2 burst
Time-varying profiles of the changes in corticospinal excitability preceding the AG2 burst were different across subjects. In five subjects the AG2 burst was preceded by a small increase in the amplitude of the MEPs and MEP:IEMG ratios. In the other subjects, there was no evidence of a change in MEP amplitude that could not be explained by preceding changes in IEMG. Mills & Kimiskidis (1996) have previously shown that the biceps AG2 burst is preceded by an increase in the MEP amplitude evoked by TMS. Paradoxically, they also showed a similar pattern of changes in MEPs evoked in the first dorsal interosseous muscle by transcranial magnetic but not electrical stimulation, in the absence of AG2 EMG activity. The authors interpreted these data to reflect phasic changes in motor cortical excitability that, for specific movements, is responsible to the generation of the AG2 burst. Under normal circumstances, the timing and magnitude of the AG2 burst are more variable within subjects than the AG1 and ANTAG bursts (Hallett & Marsden, 1979; Berardelli et al. 1996). It has been postulated that the function of the AG2 burst is to stabilise the limb at the endpoint of movement by dampening oscillations (Berardelli et al. 1996). Considering that the average onset timing of the AG2 burst relative to the onset of the AG1 was 118 ± 17 ms, there is unlikely to be sufficient time for a voluntary correction of the movement prior to the AG2 burst. However, there is time for reafferent feedback from the movement to modify the discharge of motor cortical neurones. We favour the hypothesis that the AG2 is generated by reafferent feedback to cerebello-thalamocortical pathways whereby changes in motor cortical firing are mediated by discrepancies between internal representations of the movement and the actual movement. Those subjects who made relatively constant errors across trials, necessitating corrective commands from the motor cortex, might be those that showed an early increase in the MEP amplitude preceding the AG2.
Conclusions
The command to generate the triphasic pattern of muscle activity is generally thought to be programmed centrally in advance of movement. The present results are consistent with the idea that the AG1 burst is mediated by descending volleys from corticospinal neurones in the primary motor cortex. However they suggest that the onset of cortical firing can be much closer to the onset of EMG activity than previously proposed. An unexpected feature was the lack of evidence for cortical involvement in the ANTAG burst. We propose that the cerebellum initiates the ANTAG burst through subcortical pathways. The mechanisms responsible for initiation of the AG2 burst are less clear.
Acknowledgments
We wish to thank Drs R. Hashimoto, C. Civardi and N. Modugno for valuable assistance with the data collection. We would also like to thank Mr P. Asselman and Mr R. Bedlington for technical assistance. Dr C. D. MacKinnon was supported by a Research Fellowship from the Heart and Stroke Foundation of Canada.
References
- Baker SNB, Olivier E, Lemon RN. Recording the pyramidal volley evoked by transcranial magnetic stimulation in a conscious monkey. Experimental Brain Research. 1994;99:529–533. doi: 10.1007/BF00228989. [DOI] [PubMed] [Google Scholar]
- Berardelli A, Hallett M, Rothwell JC, Agostino R, Manfredi M, Thompson PD, Marsden CD. Single-joint rapid arm movements in normal subjects and in patients with motor disorders. Brain. 1996;119:661–674. doi: 10.1093/brain/119.2.661. [DOI] [PubMed] [Google Scholar]
- Chen R, Yaseen Z, Cohen LG, Hallett M. Time course of corticospinal excitability in reaction time and self-paced movements. Annals of Neurology. 1998;44:317–325. doi: 10.1002/ana.410440306. [DOI] [PubMed] [Google Scholar]
- Cheney PD, Fetz EE. Functional classes of primate corticomotoneuronal cells and their relation to active force. Journal of Neurophysiology. 1980;44:773–791. doi: 10.1152/jn.1980.44.4.773. [DOI] [PubMed] [Google Scholar]
- Cheney PD, Fetz EE, Palmer SS. Patterns of facilitation and suppression of antagonist forelimb muscles from motor cortex sites in the awake monkey. Journal of Neurophysiology. 1985;53:805–820. doi: 10.1152/jn.1985.53.3.805. [DOI] [PubMed] [Google Scholar]
- Cooke JD, Brown SH. Movement-related phasic muscle activation. II. Generation and functional role of the triphasic pattern. Journal of Neurophysiology. 1990;63:465–472. doi: 10.1152/jn.1990.63.3.465. [DOI] [PubMed] [Google Scholar]
- Crammond DJ, Kalaska JF. Differential relation of discharge in primary motor cortex and premotor cortex to movements versus actively maintained postures during a reaching task. Experimental Brain Research. 1996;108:45–61. doi: 10.1007/BF00242903. [DOI] [PubMed] [Google Scholar]
- Davey NJ, Rawlinson SR, Maskill DW, Ellaway PH. Facilitation of a hand muscle response to stimulation of the motor cortex preceding a simple reaction task. Motor Control. 1998;2:241–250. doi: 10.1123/mcj.2.3.241. [DOI] [PubMed] [Google Scholar]
- Day BL, Dressler D, Maertens de noordhout A, Marsden CD, Nakashima K, Rothwell JC, Thompson PD. Electrical magnetic stimulation of human motor cortex: surface EMG and single motor unit responses. The Journal of Physiology. 1989;412:449–473. doi: 10.1113/jphysiol.1989.sp017626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di lazzaro V, Restuccia D, Oliviero A, Profice P, Ferrara L, Insola A, Mazzone P, Tonali P, Rothwell JC. Effects of voluntary contraction on descending volleys evoked by transcranial magnetic stimulation in conscious humans. The Journal of Physiology. 1998;508:625–633. doi: 10.1111/j.1469-7793.1998.625bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evarts EV. Pyramidal tract activity associated with a conditioned hand movement in the monkey. Journal of Neurophysiology. 1966;29:1011–1027. doi: 10.1152/jn.1966.29.6.1011. [DOI] [PubMed] [Google Scholar]
- Flament D, Hore J. Movement and electromyographic disorders associated with cerebellar dysmetria. Journal of Neurophysiology. 1986;35:1221–1233. doi: 10.1152/jn.1986.55.6.1221. [DOI] [PubMed] [Google Scholar]
- Flament D, Hore J. Relations of motor cortex neural discharge to kinematics of passive and active elbow movements in the monkey. Journal of Neurophysiology. 1988;60:1268–1284. doi: 10.1152/jn.1988.60.4.1268. [DOI] [PubMed] [Google Scholar]
- Ghez C, Martin JH. The control of rapid limb movement in the cat. III. Agonist-antagonist coupling. Experimental Brain Research. 1982;45:115–125. doi: 10.1007/BF00235770. [DOI] [PubMed] [Google Scholar]
- Gottlieb GL, Corcos DM, Agarwal GC. Organizing principles for single joint movements: I. A speed-insensitive strategy. Journal of Neurophysiology. 1989;62:342–357. doi: 10.1152/jn.1989.62.2.342. [DOI] [PubMed] [Google Scholar]
- Hallett M, Marsden CD. Ballistic flexion of the human thumb. The Journal of Physiology. 1979;294:33–50. doi: 10.1113/jphysiol.1979.sp012913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett M, Shahani BT, Young RR. EMG analysis of stereotyped voluntary movements in man. Journal of Neurology Neurosurgery and Psychiatry. 1975;38:1154–1162. doi: 10.1136/jnnp.38.12.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hore J, Flament D. Changes in motor cortex neural discharge associated with the development of cerebellar limb ataxia. Journal of Neurophysiology. 1988;60:1285–1303. doi: 10.1152/jn.1988.60.4.1285. [DOI] [PubMed] [Google Scholar]
- Hore J, Wild B, Diener H-C. Cerebellar dysmetria at the elbow, wrist, and fingers. Journal of Neurophysiology. 1991;65:563–571. doi: 10.1152/jn.1991.65.3.563. [DOI] [PubMed] [Google Scholar]
- Hoshiyama M, Kitamura Y, Koyama S, Watanabe S, Shimojo M, Kakigi R. Reciprocal changes of motor evoked potentials preceding voluntary movement in humans. Muscle and Nerve. 1996;19:125–131. doi: 10.1002/(SICI)1097-4598(199602)19:2<125::AID-MUS1>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Kalaska JF, Cohen DA, Hyde ML, Prud'homme M. A comparison of movement direction-related versus load direction-related activity in primate motor cortex, using a two-dimensional reaching task. Journal of Neuroscience. 1989;9:2080–2102. doi: 10.1523/JNEUROSCI.09-06-02080.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kischka U, Fajfr R, Fellenberg T, Hess CW. Facilitation of motor evoked potentials from magnetic brain stimulation in man: a comparative study of different target muscles. Journal of Clinical Neurophysiology. 1993;10:505–512. doi: 10.1097/00004691-199310000-00008. [DOI] [PubMed] [Google Scholar]
- Lamarre Y, Busby L, Spidalieri G. Fast ballistic arm movements triggered by visual, auditory, and somesthetic stimuli in the monkey. I. Activity of precentral neurons. Journal of Neurophysiology. 1983;50:1343–1358. doi: 10.1152/jn.1983.50.6.1343. [DOI] [PubMed] [Google Scholar]
- Lamarre Y, Spidalieri G, Lund JP. Patterns of muscular and motor cortical activity during a simple arm movement in the monkey. Canadian The Journal of Physiology and Pharmacology. 1981;59:748–756. doi: 10.1139/y81-111. [DOI] [PubMed] [Google Scholar]
- Lemon RN, Johansson RS, Westling G. Corticospinal control during reach, grasp, and precision lift in man. Journal of Neuroscience. 1995;15:6145–6156. doi: 10.1523/JNEUROSCI.15-09-06145.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackinnon CD, Rothwell JC. Time-varying changes in corticospinal excitability accompanying the triphasic EMG pattern. The Journal of Physiology. 1998;506.P:114P. doi: 10.1111/j.1469-7793.2000.00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden CD, Obeso JA, Rothwell JC. Function of the antagonist muscle during fast limb movements in man. The Journal of Physiology. 1983;335:1–13. doi: 10.1113/jphysiol.1983.sp014514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills KR, Kimiskidis V. Motor cortex excitability during ballistic forearm and finger movements. Muscle and Nerve. 1996;19:468–473. doi: 10.1002/(SICI)1097-4598(199604)19:4<468::AID-MUS7>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Morita H, Baumgarten J, Petersen N, Christensen LO, Nielsen J. Recruitment of extensor-carpi-radialis motor units by transcranial magnetic stimulation and radial-nerve stimulation in human subjects. Experimental Brain Research. 1999;128:557–562. doi: 10.1007/s002210050881. [DOI] [PubMed] [Google Scholar]
- Mustard BE, Lee RG. Relationship between EMG patterns and kinematic properties for flexion movements at the human wrist. Experimental Brain Research. 1987;66:247–256. doi: 10.1007/BF00243302. [DOI] [PubMed] [Google Scholar]
- Pascual-Leone A, Brasil-Neto JP, Valls-Solé J, Cohen LG, Hallett M. Simple reaction time to focal transcranial magnetic stimulation. Comparison with reaction time to acoustic, visual and somatosensory stimuli. Brain. 1992a;115:109–122. doi: 10.1093/brain/115.1.109. [DOI] [PubMed] [Google Scholar]
- Pascual-Leone A, Valls-Solé J, Wassermann EM, Brasil-neto J, Cohen LG, Hallett M. Effects of focal transcranial magnetic stimulation on simple reaction time to acoustic, visual and somatosensory stimuli. Brain. 1992b;115:1045–1059. doi: 10.1093/brain/115.4.1045. [DOI] [PubMed] [Google Scholar]
- Rossini PM, Zarola F, Stalberg E, Caramia M. Premovement facilitation of motor-evoked potentials in man during transcranial magnetic stimulation of the central motor pathways. Brain Research. 1988;458:20–30. doi: 10.1016/0006-8993(88)90491-x. [DOI] [PubMed] [Google Scholar]
- Rothwell JC, Traub MM, Day BL, Obeso JA, Thomas PK, Marsden CD. Manual motor performance in a deafferented man. Brain. 1982;105:515–542. doi: 10.1093/brain/105.3.515. [DOI] [PubMed] [Google Scholar]
- Rüegg DG. Ia afferents of the antagonist are inhibited presynaptically before the onset of a ballistic muscle contraction in man. Experimental Brain Research. 1989;74:663–666. doi: 10.1007/BF00247372. [DOI] [PubMed] [Google Scholar]
- Sanes JN, Jennings VA. Centrally programmed patterns of muscle activity in voluntary motor behaviour of humans. Experimental Brain Research. 1984;54:23–32. doi: 10.1007/BF00235815. [DOI] [PubMed] [Google Scholar]
- Sergio LE, Kalaska JF. Systematic changes in directional tuning of motor cortex cell activity with hand location in the workspace during generation of static isometric forces in constant spatial directions. Journal of Neurophysiology. 1997;78:1170–1174. doi: 10.1152/jn.1997.78.2.1170. [DOI] [PubMed] [Google Scholar]
- Sergio LE, Kalaska JF. Changes in the temporal pattern of primary motor cortex activity in a directional isometric force versus limb movement task. Journal of Neurophysiology. 1998;80:1577–1583. doi: 10.1152/jn.1998.80.3.1577. [DOI] [PubMed] [Google Scholar]
- Starr A, Caramia M, Zarola F, Rossini PM. Enhancement of motor cortical excitability in humans by non-invasive electrical stimulation appears prior to voluntary movement. Electroencephalography and Clinical Neurophysiology. 1988;70:26–32. doi: 10.1016/0013-4694(88)90191-5. [DOI] [PubMed] [Google Scholar]
- Tarkka IM, Mckay WB, Sherwood AM, Dimitrijevic MR. Early and late motor evoked potentials reflect preset agonist-antagonist organization in lower limb muscles. Muscle and Nerve. 1995;18:276–282. doi: 10.1002/mus.880180303. [DOI] [PubMed] [Google Scholar]
- Tomberg C, Caramia MD. Prime mover muscle in finger lift or finger flexion reaction times: identification with transcranial magnetic stimulation. Electroencephalography and Clinical Neurophysiology. 1991;81:319–322. doi: 10.1016/0168-5597(91)90019-t. [DOI] [PubMed] [Google Scholar]
- Valls-Solé J, Rothwell JC, Goulart F, Cossu G, Muñoz E. Patterned ballistic movements triggered by a startle in healthy humans. The Journal of Physiology. 1999;516:931–938. doi: 10.1111/j.1469-7793.1999.0931u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild B, Corcos D. Cerebellar hypermetria: reduction in the early component of the antagonist electromyogram. Movement Disorders. 1997;12:604–607. doi: 10.1002/mds.870120420. [DOI] [PubMed] [Google Scholar]