

Abstract
We hypothesized that enhanced voltage-gated Ca2+ channel current (VGCC) density in coronary smooth muscle cells of exercise-trained miniature Yucatan pigs is compensated by other cellular Ca2+ regulatory mechanisms to limit net myoplasmic free Ca2+ accumulation.
Whole-cell voltage clamp experiments demonstrated enhanced VGCC density in smooth muscle cells freshly dispersed from coronary arteries of exercise-trained vs. sedentary animals.
In separate experiments using fura-2 microfluorometry, we measured depolarization-induced (80 mm KCl) accumulation of myoplasmic free Ba2+ and free Ca2+. Both maximal rate and net accumulation of free Ba2+ in response to membrane depolarization were increased in smooth muscle cells isolated from exercise-trained pigs, consistent with an increased VGCC density. Depolarization also produced an enhanced maximal rate of free Ca2+ accumulation in cells of exercise-trained pigs; however, net accumulation of free Ca2+ was not significantly increased suggesting enhanced Ca2+ influx was compensated to limit net free Ca2+ accumulation.
Inhibition of sarco-endoplasmic reticulum Ca2+-transporting ATPase (SERCA; 10 μm cyclopiazonic acid) and/or sarcolemmal Na+-Ca2+ exchange (low extracellular Na+) suggested neither mechanism compensated the enhanced VGCC in cells of exercise-trained animals.
Local Ca2+-dependent inactivation of VGCC, assessed by buffering myoplasmic Ca2+ with EGTA in the pipette and using Ca2+ and Ba2+ as charge carriers, was not different between cells of sedentary and exercise-trained animals.
Our findings indicate that increased VGCC density is compensated by other cellular Ca2+ regulatory mechanisms to limit net myoplasmic free Ca2+ accumulation in smooth muscle cells of exercise-trained animals. Further, SERCA, Na+-Ca2+ exchange and local Ca2+-dependent inactivation of VGCC do not appear to function as compensatory mechanisms. Additional potential compensatory mechanisms include Ca2+ extrusion via plasma membrane Ca2+-ATPase, mitochondrial uptake, myoplasmic Ca2+-binding proteins and other sources of VGCC inactivation.
Exercise training-induced alterations in vasoreactivity of the coronary vasculature have been well documented (Bove & Dewey, 1985; Laughlin, 1985; Laughlin et al. 1989; DiCarlo et al. 1989; Rogers et al. 1991; Oltman et al. 1992; Bowles et al. 1995; Mombouli et al. 1996) and are generally associated with enhanced dilatation and reduced constriction in response to vasoactive agonists (Laughlin & McAllister, 1992; Parker et al. 1994). In vivo, the coronary circulation of exercise-trained animals exhibits enhanced adenosine-induced vasodilatation (Laughlin, 1985; Laughlin et al. 1989; DiCarlo et al. 1989) and attenuated α-adrenergic vasoconstriction (Bove & Dewey, 1985). Similarly, isolated coronary rings demonstrate increased relaxation to adenosine (Oltman et al. 1992) and diminished vasoconstriction to noradrenaline (norepinephrine) (Oltman et al. 1992) and endothelin (Bowles et al. 1995). Furthermore, simultaneous measurements of developed tension and myoplasmic free Ca2+ in arterial rings isolated from sedentary and exercise-trained pigs indicate that attenuated endothelin-induced contractile responses in exercise-trained animals are associated with reduced myoplasmic free Ca2+ levels (Bowles et al. 1995). In apparent contrast to these vascular adaptations with exercise training, Bowles et al. (1998) recently reported increased VGCC density in coronary smooth muscle cells isolated from exercise-trained vs. sedentary animals. The present study was undertaken to determine whether this enhanced Ca2+ current density is compensated in smooth muscle cells from exercise-trained animals to limit net myoplasmic free Ca2+ accumulation.
We also evaluated potential cellular mechanisms of Ca2+ regulation that may compensate the enhanced VGCC density of smooth muscle cells from exercise-trained animals. Experiments in which SERCA and/or sarcolemmal Na+-Ca2+ exchange were inhibited suggest neither mechanism compensates enhanced VGCC in coronary smooth muscle cells of exercise-trained animals. We also provide evidence that inactivation of VGCC by myoplasmic Ca2+ in the immediate vicinity of voltage-gated Ca2+ channels (local Ca2+) is not different between sedentary and exercise-trained animals. Taken together, our findings suggest increased VGCC density in smooth muscle cells of exercise-trained animals is compensated by other cellular Ca2+ regulatory mechanisms to limit net free Ca2+ accumulation; SERCA, Na+-Ca2+ exchange and local Ca2+-dependent inactivation of voltage-gated Ca2+ channels do not appear to function as compensatory mechanisms.
METHODS
Exercise training procedures
All animal protocols were in accordance with the Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research and Training and approved by the University of Missouri Animal Care and Use Committee. Adult female Yucatan miniature pigs (Charles River, Wilmington, MA, USA) were randomly assigned to either a sedentary or exercise-trained group. Exercise-trained pigs underwent 16 weeks of a progressive treadmill exercise training programme used extensively by our laboratories and described previously (Laughlin, 1985; Laughlin et al. 1989; Stehno-Bittel et al. 1991; Oltman et al. 1992; Bowles et al. 1995, 1998). Sedentary pigs were restricted to their pens (2 m × 4 m) for the 16 week duration. Pigs were given positive reinforcement for exercise by being fed after each training bout.
Efficacy of training
Treadmill performance tests were administered before and after completion of the 16 week exercise-training programme or sedentary pen confinement as described previously (Laughlin et al. 1989; Bowles et al. 1995, 1998). Effectiveness of the exercise-training programme was determined by comparing running time to exhaustion on treadmill performance tests, heart weight to body weight ratio and skeletal muscle oxidative enzyme activity of the exercise-trained vs. sedentary animals.
Removal of tissue
Isolation of coronary arteries
Following completion of the 16 week exercise training protocol or sedentary confinement, the animals were anaesthetized using ketamine (35 mg kg−1, i.m.), xylazine (2 mg kg−1, i.m.) and thiopental sodium (10 mg kg−1, i.v.) followed by administration of heparin (1000 u kg−1, i.v.). The hearts were removed, placed in ice-cold Krebs bicarbonate buffer (0–4°C) and weighed. Hearts were maintained in iced Krebs buffer during isolation of coronary arteries. With the aid of a dissection microscope, segments of the right coronary (RCA) and left circumflex coronary (LCX) arteries were trimmed of fat and connective tissue.
Muscle samples
After removal of the heart, as described above, samples were taken from the long, medial and lateral heads of the triceps brachii muscle for determination of citrate synthase activity as described previously (Stehno-Bittel et al. 1991; Oltman et al. 1992; Bowles et al. 1995, 1998).
Smooth muscle cell dissociation
Segments of RCA and LCX coronary arteries were cut longitudinally and pinned lumen-side up in low-Ca2+ physiological buffer containing 294 U ml−1 collagenase, 5 U ml−1 elastase, 2 mg ml−1 bovine serum albumin, 1 mg ml−1 soybean trypsin inhibitor, and 0·4 mg ml−1 DNase I. Cells were enzymatically dissociated by incubation in a 37°C shaking water bath for 20 min; this first fraction of dissociated cells, primarily consisting of endothelial cells, was removed. An additional 50 min enzymatic dissociation in fresh enzyme solution generated primarily smooth muscle cells. Dissociated smooth muscle cells from the LCX were then incubated with the acetoxymethyl ester form of the fluorescent Ca2+ indicator fura-2 (fura-2 AM; 2·5 μm) at 37°C for 25 min. Following fura-2 AM incubation, smooth muscle cells were washed in modified Eagle's minimal essential storage medium for 20 min before experiments were initiated. Dissociated smooth muscle cells from the RCA were placed in low-Ca2+ physiological mediim for use in voltage clamp experiments.
Myoplasmic free Ca2+ measurement
Fura-2-loaded cells were placed in a superfusion chamber and observed using an epifluorescence microscopy system (Nikon, Garden City, NY, USA). Excitation light from a 300 W xenon arc lamp, passed via a liquid light guide, was directed through alternating 340 and 380 nm bandpass filters. Fluorescence emission (510 nm) from user-specified regions of interest (selected smooth muscle cells) was synchronized with the appropriate excitation wavelength and reflected to an integrating CCD monochrome video camera (Cohu, San Diego, CA, USA) with a dichroic mirror. The microscope was equipped with a ×40 oil immersion objective with a numerical aperature of 1·3. Fluorescence images were acquired using InCa dual wavelength Ca2+ imaging software, version 2.1 (Intracellular Imaging, Cincinnati, OH, USA). This microfluorometry system provides individual traces of fura-2 fluorescence from multiple smooth muscle cells simultaneously. The fura-2 fluorescence ratio was collected for each cell throughout the experimental protocol. Background fluorescence was determined before the start of the experiment for on-line subtraction during data collection. After the subtraction of background fluorescence, images obtained at 340 and 380 nm were ratioed on a pixel-by-pixel basis. Final data for estimates of myoplasmic free Ca2+ are expressed as a fluorescence ratio (F340/F380) because of uncertainties in extrapolating in vitro calibrations to in situ measurements as described previously (Wagner-Mann et al. 1992). Cells were continually superfused (∼2·7 ml min−1) under gravity flow. All experiments were conducted at room temperature (22–25°C) and fluorescence data were sampled every 2 s.
For fura-2 microfluorometry studies, unless otherwise specified, cells were superfused with physiological saline solution (PSS) containing (mm): 2 CaCl2, 143 NaCl, 1 MgCl2, 5 KCl, 10 Hepes, and 10 glucose, pH 7·4. Cells were depolarized with PSS in which 80 mm KCl replaced equimolar amounts of NaCl. For low Na+ (5 mm Na+) protocols, Na+ was replaced with equimolar amounts of Li+. For Ba2+ protocols, Ca2+ was replaced with equimolar amounts of Ba2+ and low Na+ (5 mm Na+) was used to inhibit Ba2+ extrusion via Na+-Ca2+ exchange. Caffeine (5 mm), cyclopiazonic acid (CPA, 10 μm) and nifedipine (3 μm) additions were made directly to appropriate PSS and 80 mm KCl solutions as specified by the experimental protocol.
Whole-cell voltage clamp
In separate experiments, whole-cell Ca2+ and Ba2+ currents were determined using a standard whole-cell voltage clamp technique as described previously (Bowles et al. 1998). Cells were initially superfused with PSS during gigaseal formation. After whole-cell configuration, superfusate was switched to PSS with tetraethylammonium chloride (TEACl) substituted for NaCl and 2 mm Ca2+ or 10 mm Ba2+ as the charge carrier. Heat-polished glass pipettes (2–5 MΩ) were filled with a solution containing (mm): 120 CsCl, 10 TEACl, 1 MgCl2, 20 Hepes, 2 MgATP, 5 EGTA and 0·5 Tris.GTP. The pipette was connected to the 10 GΩ headstage of a Warner PC-501 patch clamp amplifier and advanced to the cell via a micromanipulator control. Junction potential was offset prior to contact between pipette and cell. After formation of a GΩ seal, pipette capacitance was cancelled and suction applied to achieve whole-cell configuration. Whole-cell currents were filtered through an eight-pole low-pass filter with a cut-off frequency of 400 Hz and digitized at 600 μs intervals. Data acquisition and analysis were accomplished using a Labmaster analog-to-digital converter and microcomputer equipped with AxoBASIC 1.0 software (Axon Instruments, Foster City, CA, USA). Current densities (pA pF−1) were obtained for each cell by normalization of whole-cell current to cell capacitance. Time to half-maximal (T½) decay of peak current for whole-cell configuration was determined using both 2 mm Ca2+ and 10 mm Ba2+ as external charge carriers. We used 10 mm Ba2+ to increase current magnitude in these cells. Importantly, Ba2+ current inactivation kinetics in smooth muscle cells have been shown previously to not be affected by current magnitude (Nilius et al. 1994). Capacity currents were measured for each cell during 10 ms pulses from a holding potential of −80 mV to a test potential of −70 mV. Capacity currents were filtered at a low-pass cut-off frequency of 8·4 kHz and digitized at 25 μs intervals. Leak subtraction was not performed. Cells were continuously perfused under gravity flow. All experiments were conducted at room temperature (22–25°C).
Statistical analysis
Treadmill test endurance time, heart weight to body weight ratio and citrate synthase activity were evaluated using Student's unpaired t test. Voltage-gated Ca2+ channel current-voltage (I–V) relationships were evaluated using analysis of variance and Student's unpaired t test for post hoc analyses. Analysis of variance and Student's unpaired t tests were used for comparison of T½ decay of peak current. Data for fura-2 experiments were analysed using one-way analysis of variance or split-plot repeated measures analysis of variance, as appropriate. Mean differences were ascertained using Fisher's least significant difference (LSD). Analyses for all experiments were performed on a per cell basis. For all analyses, a P value ≤ 0·05 was considered significant. Data are presented as means ± s.e.m., and values in parentheses reflect the number of animals and number of smooth muscle cells.
RESULTS
Training status
Effectiveness of the 16 week exercise training programme was demonstrated by significant (P < 0·05) increases in treadmill endurance time, skeletal muscle oxidative enzyme capacity and an increased heart weight to body weight ratio in exercise-trained animals. Treadmill endurance time increased in exercise-trained (21·6 ± 3·7 vs. 31·4 ± 4·2 min), but not sedentary animals (24·0 ± 3·8 vs. 24·9 ± 2·9 min), after completion of the 16 week exercise protocol or pen confinement, respectively. Citrate synthase activity was increased in the long heads (15·7 ± 5·3 vs. 11·1 ± 1·9 μmol min−1 g−1), in the medial heads (19·0 ± 5·3 vs. 15·7 ± 2·2 μmol min−1 g−1) and in the lateral heads (19·4 ± 6·7 vs. 14·2 ± 2·0 μmol min−1 g−1) of the triceps brachii muscle in exercise-trained vs. sedentary animals, respectively. Heart weight to body weight ratio also increased in exercise-trained vs. sedentary pigs (5·3 ± 0·2 vs. 4·8 ± 0·1 g kg−1).
Ca2+ channel current density
The effect of exercise training on whole-cell Ca2+ current was determined using 10 mm external Ba2+ as the charge carrier. I–V relationships for sedentary and exercise-trained animals are presented in Fig. 1. Current is plotted as peak inward current measured during a 260 ms step depolarization to the indicated membrane potential (Vm) from a holding potential of −80 mV. Current is normalized to cell membrane capacitance (pA pF−1). Cell capacitance (21 ± 1 vs. 22 ± 2 pF) was not different between smooth muscle cells isolated from sedentary (n = 4 animals, 8 cells) and exercise-trained (n = 4 animals, 11 cells) animals, respectively. These data confirm that exercise training increased peak VGCC density approximately twofold in smooth muscle cells isolated from conduit-sized coronary arteries as demonstrated previously (Bowles et al. 1998).
Figure 1. Current-voltage (I–V) relationships for whole-cell VGCCs from coronary smooth muscle of sedentary (sed) and exercise-trained (ex) pigs.
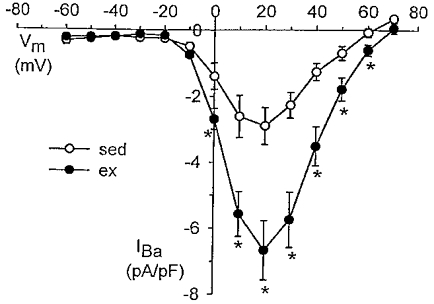
Currents were obtained using 10 mm Ba2+ as external charge carrier. Current is plotted as peak inward current measured during a 260 ms step depolarization to the membrane potential (Vm) indicated from a holding potential of −80 mV. Current is normalized to cell membrane capacitance (pA pF−1). Data are means ± s.e.m.; *P < 0·05, ex (n = 4 pigs, 11 cells) vs. sed (n = 4, 8).
Fura-2 measurements of Ba2+ influx
We further evaluated alterations in Ba2+ influx at the plasma membrane of single smooth muscle cells using the Ca2+ indicator, fura-2. Ba2+ produces a shift in the fura-2 excitation wavelength spectrum with increasing concentrations similar to that observed with Ca2+, although fura-2 has a higher affinity for Ca2+vs. Ba2+ (Schilling et al. 1989; Kwan & Putney, 1990). Ba2+ influx at the plasma membrane of smooth muscle cells is mediated mostly through Ca2+ channels (Benham & Tsien, 1987) and the Na+-Ca2+ exchanger (Condrescu et al. 1997). However, Ba2+ is not sequestered by intracellular organelles (Schilling et al. 1989; Kwan & Putney, 1990; Condrescu et al. 1997) or transported by ATP-dependent Ca2+ pumps (Schilling et al. 1989). Therefore, these unique characteristics of Ba2+ allow evaluation of unidirectional influx at the plasma membrane under conditions in which corresponding Ca2+ fluxes would be difficult to interpret due to multiple pathways for Ca2+ removal and sequestration.
Our protocol for assessment of myoplasmic free Ba2+ accumulation in response to membrane depolarization with 80 mm KCl and representative recordings from single cells isolated from both sedentary and exercise-trained animals are shown in Fig. 2A. Briefly, cells were initially exposed to 80 mm KCl followed by 5 mm caffeine both in the presence of 2 mm extracellular Ca2+ for the durations indicated by the horizontal line. Although the SR does not sequester Ba2+, we applied caffeine in this protocol to maintain consistency with our other fura-2 protocols described below. We then evaluated free Ba2+ accumulation in the presence of 80 mm KCl and low extracellular Na+ (5 mm), a method which has been employed previously to measure unidirectional divalent cation influx in smooth muscle cells (Liu et al. 1994). We assessed both the maximal rate and net free Ba2+ accumulation (area under the curve; AUC) throughout membrane depolarization (7 min) in sedentary and exercise-trained animals. AUC was obtained by subtracting the baseline fura-2 ratio for each cell (average of 10 data points prior to 80 mm KCl exposure) from each subsequent fura-2 data point throughout the 80 mm KCl exposure (between 11 and 18 min after the start of the experiment). These differences for each data point were then added together to obtain AUC.
Figure 2. Free Ba2+ accumulation during membrane depolarization (80 mm KCl) using fura-2 microfluorometry.
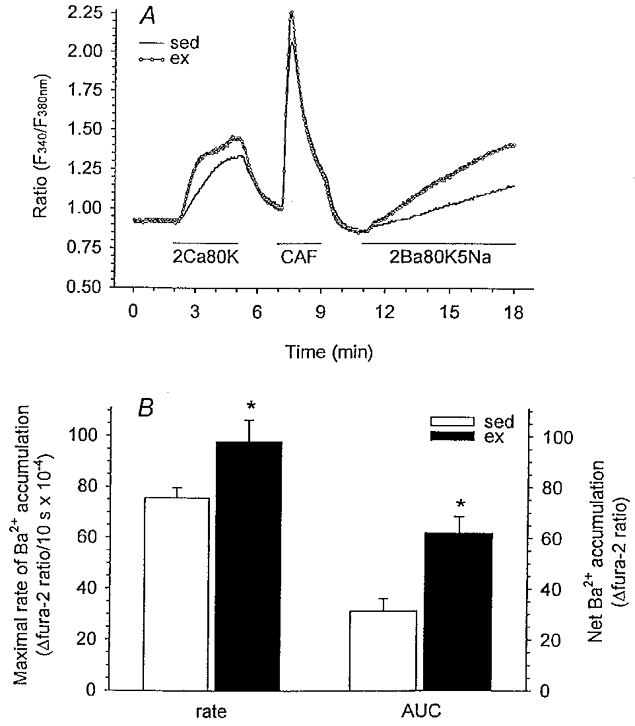
A, experimental protocol and representative recordings from single cells of both sedentary and exercise-trained pigs showing change in F340/F380 fluorescence ratio. Cells were superfused with PSS unless otherwise specified. Cells were exposed to 80 mm KCl (80K) and caffeine (CAF; 5 mm) in the presence of 2 mm extracellular Ca2+ (2Ca) for the durations indicated by the horizontal lines. Cells were then exposed to 80 mm KCl in the presence of 2 mm Ba2+ (2Ba; equimolar substitution for Ca2+) and low Na+ (5Na; 5 mm) for 7 min. B, maximal rate (10 s slope) and net (area under the curve, AUC) free Ba2+ accumulation for cells from sed (n = 6, 71) and ex (n = 5, 61) animals. Data are means ± s.e.m.; *P < 0·05, ex vs. sed.
Our data demonstrate that exercise training produced a 30 % increase in the maximal rate of free Ba2+ accumulation and a 98 % increase in net free Ba2+ accumulation (AUC) compared with sedentary pigs (Fig. 2B). Furthermore, the rate of free Ba2+ accumulation throughout membrane depolarization (slope of the time period 11–18 min, Fig. 2B) was significantly greater in cells isolated from exercise-trained vs. sedentary animals. These findings support our whole-cell Ca2+ current data indicating that membrane depolarization-induced divalent cation influx through voltage-gated Ca2+ channels is enhanced in exercise-trained pigs. In additional experiments we evaluated the effects of the Ca2+ channel blocker nifedipine (3 μm) on Ba2+ influx as illustrated in Fig. 3. Addition of nifedipine during membrane depolarization completely blocked Ba2+ influx as indicated by a levelling off of free Ba2+ accumulation (Fig. 3). Slope values representing Ba2+ influx for the time period 16–18 min were 8·4 ± 7·2 and −0·8 ± 2·2 in the absence and presence of nifedipine, respectively. These slope values indicate that in our experiments, Ba2+ influx occurred exclusively through dihydropyridine-sensitive Ca2+ channels.
Figure 3. Free Ba2+ accumulation during membrane depolarization (80 mm KCl) in the presence of the Ca2+ channel blocker, nifedipine.
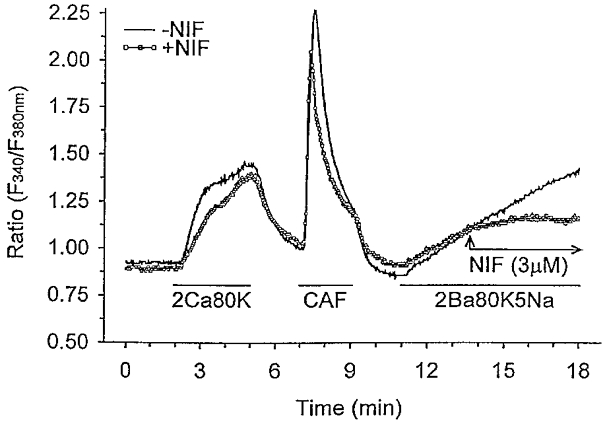
Control (-NIF; continuous line) experimental protocol is the same as presented in Fig. 2A. For experiments in the presence of nifedipine (+NIF; ○), a similar protocol was used with the addition of nifedipine (3 μm) between 13·5 and 18 min after the start of the experiment, as indicated by the arrows. Evaluation of the slope between 16 and 18 min indicates that free Ba2+ accumulation was abolished in the presence of nifedipine.
Fura-2 measurements of free Ca2+ accumulation
Subsequent fura-2 studies evaluating changes in myoplasmic free Ca2+ allowed us to assess whole-cell regulation of intracellular free Ca2+. Our protocol for evaluation of free Ca2+ accumulation in response to membrane depolarization with 80 mm KCl and representative recordings from single cells isolated from both sedentary and exercise-trained animals are shown in Fig. 4A. Briefly, cells were exposed to 80 mm KCl and caffeine in the presence of 2 mm extracellular Ca2+ for the durations indicated by the horizontal lines. The use of caffeine depletes the SR adequately to potentiate SERCA activity and allows the examination of the role of Ca2+ uptake by SERCA in compensating the enhanced depolarization-induced Ca2+ influx observed in cells of exercise-trained animals. We evaluated both the maximal rate and net free Ca2+ accumulation (AUC) in response to high KCl-induced membrane depolarization following SR depletion in sedentary and exercise-trained animals. This protocol was the general format used for all subsequent experiments described in this paper. Myoplasmic free Ca2+ accumulation in response to high KCl-induced membrane depolarization is dependent on Ca2+ influx, Ca2+ extrusion at the plasmalemma, and Ca2+ sequestration by intracellular organelles. Our findings from these experiments demonstrated that the maximal rate of free Ca2+ accumulation in response to membrane depolarization was 22 % greater in smooth muscle cells of exercise-trained vs. sedentary animals (Fig. 4B). However, in contrast to our findings with Ba2+, net myoplasmic free Ca2+ accumulation (AUC) throughout the depolarization period was not significantly different between exercise-trained and sedentary animals (Fig. 4B). These data suggest that the enhanced maximal rate of free Ca2+ accumulation is compensated by additional Ca2+ regulatory mechanisms insensitive to Ba2+, thus limiting net myoplasmic free Ca2+ accumulation in smooth muscle cells of exercise-trained animals.
Figure 4. Free Ca2+ accumulation during membrane depolarization (80 mm KCl) using fura-2 microfluorometry.
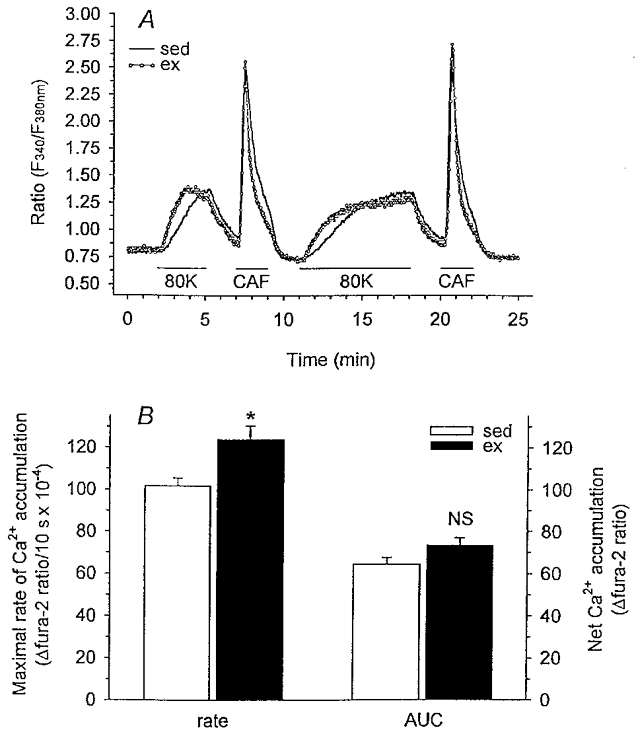
A, experimental protocol and representative recordings from single cells isolated from sedentary and exercise-trained animals showing change in F340/F380 ratio. Cells were superfused with PSS unless otherwise specified. Cells were exposed to 80 mm KCl and caffeine (5 mm) in the presence of 2 mm extracellular Ca2+ for the durations indicated by the horizontal lines. B, maximal rate (10 s slope) and net (AUC) free Ca2+ accumulation for cells from sed (n = 7, 84) and ex (n = 6, 76) animals. Data are means ± s.e.m.; *P < 0·05, ex vs. sed.
To investigate potential mechanisms of Ca2+ removal and/or sequestration that may compensate the enhanced Ca2+ influx in smooth muscle cells isolated from exercise-trained animals, we systematically evaluated the contribution of SERCA and/or the Na+-Ca2+ exchanger to Ca2+ removal from the myoplasm during membrane depolarization. The protocols for these experiments and representative recordings from single cells of sedentary animals are illustrated in Fig. 5A. These experimental protocols are similar to that presented in Fig. 4A with the addition of CPA and/or low Na+ for the 11 min period shown by the horizontal line in Fig. 5A.
Figure 5. Evaluation of free Ca2+ accumulation in the presence of SERCA and/or Na+-Ca2+ exchange inhibition during membrane depolarization (80 mm KCl).
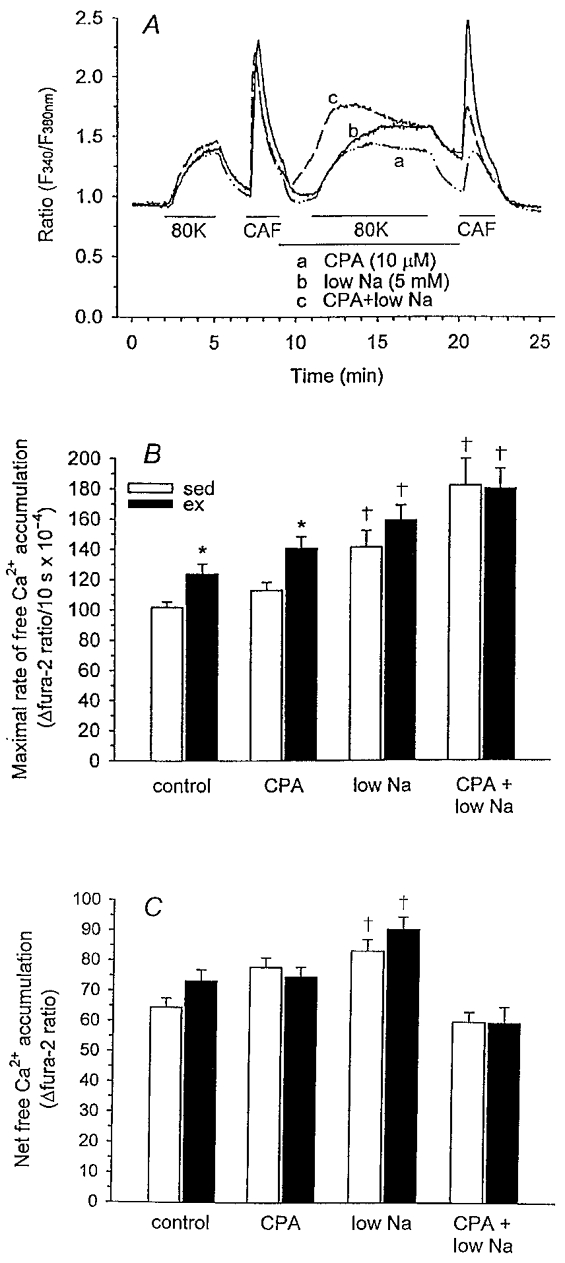
A, experimental protocols are similar to that used in Fig. 4A with the addition of CPA and/or low Na+ for the 11 min period shown by the horizontal line. Representative recordings from single cells of sedentary animals are presented for each protocol showing the change in F340/F380 fluorescence ratio. B, maximal rate (10 s slope). C, net free Ca2+ accumulation (AUC) for cells from sedentary and exercise-trained animals for all protocols. n values for control protocol as presented in Fig. 4 legend. CPA protocol: sed, n = 6, 66; ex, n = 6, 62. NCX protocol: sed, n = 5, 56; ex, n = 5, 45. Data are means ± s.e.m.: *P < 0·05, vs. respective sed; †P < 0·05, vs. control protocol.
Inhibition of SR Ca2+ sequestration
CPA, a reversible inhibitor of SERCA, was used to evaluate the contribution of SR Ca2+ sequestration in compensating the enhanced Ca2+ influx in exercise-trained animals. Findings from these experiments are presented in Fig. 5B and C in comparison with the control protocol (in the absence of CPA). CPA had little effect on either maximal rate or net free Ca2+ accumulation in smooth muscle cells from both sedentary and exercise-trained animals. The maximal rate of free Ca2+ accumulation remained significantly enhanced in smooth muscle cells from exercise-trained animals (Fig. 5B) and net free Ca2+ accumulation (AUC) was not different between sedentary and exercise-trained pigs (Fig. 5C); these results were similar to those observed in the absence of CPA (control protocol; Fig. 5B and C). Our finding that SERCA inhibition had a negligible influence on net free Ca2+ accumulation in either exercise-trained or sedentary smooth muscle cells suggests that the SR does not function to compensate the enhanced Ca2+ influx demonstrated in smooth muscle cells from exercise-trained animals. Our findings also indicate that caffeine-releasable SR Ca2+ stores in cells of sedentary (6 animals, 66 cells) vs. exercise-trained (6 animals, 62 cells) animals were not different after CPA application (Δfura-2 ratio, 0·37 ± 0·02 vs. 0·36 ± 0·02, respectively), suggesting similar inhibition of SERCA activity in both groups of pigs. Additionally, caffeine-releasable SR Ca2+ stores in the absence of SERCA inhibition in sedentary (n = 7 animals, 84 cells) and exercise-trained (n = 6 animals, 76 cells) groups (Δfura-2 ratio, 1·18 ± 0·06 vs. 1·19 ± 0·07, respectively) were not different, suggesting similar SR Ca2+ store capacity. These data indicate that enhanced SR Ca2+ uptake does not compensate increased VGCC in coronary smooth muscle cells isolated from exercise-trained animals to limit net free Ca2+ accumulation.
Inhibition of Ca2+ extrusion via Na+-Ca2+ exchange
We also evaluated the potential contribution of Na+-Ca2+ exchange activity to the extrusion of Ca2+ from the cell. During depolarization with 80 mm KCl, the membrane potential of smooth muscle cells typically increases to approximately −15 mV (Ito et al. 1980) causing reversal of the Na+-Ca2+ exchanger and, therefore, Ca2+ influx at the exchanger (Sturek et al. 1992). During steady-state depolarization, low extracellular Na+ would have little further influence on the reversal of the exchanger. However, during the initial development of membrane depolarization following exposure to 80 mm KCl, the Na+-Ca2+ exchanger may contribute to alterations in Ca2+ handling of coronary smooth muscle cells of sedentary vs. exercise-trained animals. Therefore, this low Na+ protocol was used to evaluate a potential role for the Na+-Ca2+ exchanger as a compensatory mechanism for enhanced Ca2+ influx during the early stages of depolarization in smooth muscle cells of exercise-trained pigs. Our data from these experiments are presented in Fig. 5B and C and indicate that decreased extracellular Na+ produced significant but similar increases in maximal rate and net free Ca2+ accumulation (AUC) of cells from both sedentary and exercise-trained animals. These increases in maximal rate and net free Ca2+ accumulation may result from impaired Ca2+ extrusion or enhanced Ca2+ entry at the exchanger. However, similar increases in sedentary and exercise-trained groups indicate that Na+-Ca2+ exchange activity does not compensate the enhanced Ca2+ influx demonstrated in cells from exercise-trained animals to limit net free Ca2+ accumulation. Furthermore, our data also demonstrate that inhibition of Na+-Ca2+ exchange abolished the significant difference observed in the maximal rate of free Ca2+ accumulation between cells of sedentary and exercise-trained animals in the control protocol. Conversely, if Na+-Ca2+ exchange activity compensated the enhanced VGCC observed in smooth muscle cells of exercise-trained pigs, inhibition of Na+-Ca2+ exchange would potentiate the difference between cells of sedentary and exercise-trained animals.
Simultaneous inhibition of Ca2+ removal via SR and Na+-Ca2+ exchange
Previous studies have demonstrated that inhibition of one cellular mechanism of Ca2+ removal may prompt an additional regulatory mechanism to compensate to some extent for its absence (Bers & Bridge, 1989). For this reason, we simultaneously inhibited SERCA and the sarcolemmal Na+-Ca2+ exchanger to unequivocally evaluate the role of either pathway of Ca2+ removal as a potential compensatory mechanism for increased Ca2+ current density in cells of exercise-trained animals. Our data from these experiments are presented in Fig. 5B and C and indicate that simultaneous inhibition of both the SERCA and Na+-Ca2+ exchanger produced significant but similar increases in maximal rate of free Ca2+ accumulation in cells from both sedentary and exercise-trained animals. Furthermore, net free Ca2+ accumulation (AUC) was similar between cells of sedentary and exercise-trained animals following inhibition of both mechanisms of Ca2+ removal. These findings further suggest that SERCA and Na+-Ca2+ exchange do not compensate the enhanced Ca2+ influx demonstrated in cells from exercise-trained animals. Interestingly, simultaneous inhibition of SERCA and Na+-Ca2+ exchange produced a gradual increase in myoplasmic free Ca2+ prior to membrane depolarization (between 9 and 11 min after the start of the experiment, Fig. 5A, representative trace c).The subsequently diminished depolarization-induced net free Ca2+ accumulation (AUC) compared with control data illustrated in Fig. 5C suggests that these cells may possess a regulatory mechanism to limit maximal myoplasmic free Ca2+ concentrations and, perhaps, thereby protect the cell from Ca2+ concentrations which may compromise cell viability.
Ca2+-dependent inactivation of voltage-gated Ca2+ channels
We also evaluated inactivation of voltage-gated Ca2+ channel current for both Ca2+ and Ba2+ in the presence of EGTA (5 mm). EGTA generally buffers Ca2+ or Ba2+ at a distance of approximately 100 nm from voltage-gated Ca2+ channels (Gutnick et al. 1989); thus, local Ca2+ gradients near the channel that are not buffered by EGTA can influence inactivation of the channel. In contrast, more ‘distant’ Ca2+-dependent inactivation of voltage-gated Ca2+ channels is greatly attenuated in the presence of EGTA. Furthermore, Ba2+ does not directly inactivate voltage-gated Ca2+ channels (Ohya et al. 1988); therefore, using Ba2+ as charge carrier in the presence of high EGTA results in little to no local Ca2+-dependent inactivation of voltage-gated Ca2+ channels.
The voltage clamp template for these experiments is illustrated in Fig. 6A. The holding potential was −80 mV and the cells were depolarized to the test potential that elicited peak current for Ca2+ or Ba2+; generally +10 mV for both charge carriers. Representative current traces from a single cell for both Ca2+ and Ba2+ are presented in Fig. 6A. The time for the peak current to decay to half-maximal amplitude (T½ decay) was used to assess Ca2+ current inactivation. As illustrated in Fig. 6B, T½ decay of peak current was significantly shorter in the presence of Ca2+vs. Ba2+, suggesting that local Ca2+ gradients did play a role in Ca2+ channel inactivation. However, T½ decay of peak current was not significantly different between cells of sedentary and exercise-trained animals in the presence of either Ca2+ or Ba2+ as charge carrier. These findings indicate that local Ca2+-dependent inactivation of the voltage-gated Ca2+ channel did not compensate for the enhanced VGCC observed in exercise-trained animals. Furthermore, our findings that T½ decay of peak current was not significantly different between cells of sedentary and exercise-trained animals in the presence of Ba2+ as charge carrier suggest that the voltage-dependent inactivation of voltage-gated Ca2+ channels in cells of sedentary and exercise-trained pigs was not different.
Figure 6. Evaluation of local Ca2+-dependent inactivation of voltage-gated Ca2+ channels during membrane depolarization.
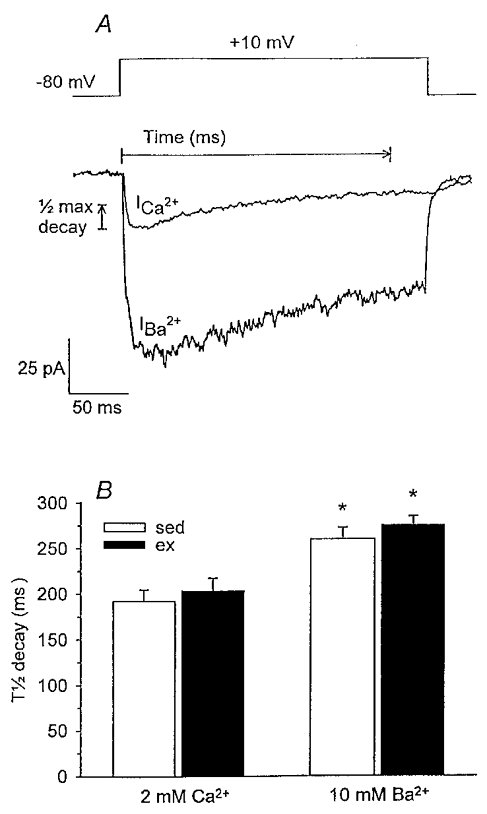
A, voltage clamp template and representative current traces from a single cell using both Ca2+ and Ba2+ as charge carriers were elicited by 260 ms step depolarizations to +10 mV from a holding potential of −80 mV. Measures for calculation of T½ decay of peak Ca2+ current are illustrated. B, T½ decay of peak current was significantly shorter in the presence of Ca2+vs. Ba2+ for cells of both sed (n = 8, 36) and ex (n = 8, 29) animals. However, T½ decay was not significantly different between cells of sed and ex animals in the presence of Ca2+ or Ba2+. Data are means ± s.e.m.; *P < 0·05 vs. respective Ca2+ current.
DISCUSSION
In the present study, we document the novel finding that increased Ca2+ influx through voltage-gated Ca2+ channels observed in smooth muscle cells from exercise-trained animals is compensated such that net free Ca2+ accumulation in sedentary vs. exercise-trained animals was not significantly different. Inhibition of SERCA and/or sarcolemmal Na+-Ca2+ exchange did not support a role for either mechanism in compensating the enhanced VGCC in exercise-trained animals. Also, Ca2+-dependent inactivation of VGCC by local Ca2+ gradients was not different between sedentary and exercise-trained animals and therefore did not appear to compensate the initial increase in Ca2+ influx. The present study also confirmed previous findings using whole-cell voltage clamp experiments that exercise training does indeed increase peak VGCC density in coronary smooth muscle cells.
Previous studies have shown that chronic exercise training attenuates contractile responses of coronary arterial rings to vasoactive agents (Oltman et al. 1992; Bowles et al. 1995), which are associated with reduced myoplasmic free Ca2+ levels (Bowles et al. 1995). Paradoxically, in the same experiments, the rate of endothelin-stimulated divalent cation influx was significantly greater in arterial rings from exercise-trained vs. sedentary animals (Bowles et al. 1995). More recent findings of enhanced VGCC density in coronary smooth muscle cells from exercise-trained animals (Bowles et al. 1998) prompted our hypothesis that enhanced Ca2+ influx via voltage-gated Ca2+ channels is compensated to limit net free Ca2+ accumulation in smooth muscle cells of exercise-trained animals.
Evaluation of Ba2+ accumulation in the presence of low extracellular Na+ has been used previously as a measure of unidirectional divalent cation influx in smooth muscle cells (Liu et al. 1994). Ba2+ is transported via voltage-gated Ca2+ channels (Benham & Tsien, 1987) and the Na+-Ca2+ exchanger (Condrescu et al. 1997) but is a poor substrate for ATP-dependent Ca2+ pumps (Schilling et al. 1989). Furthermore, our results in the presence of nifedipine indicate that accumulation of intracellular Ba2+ during high KCl-induced membrane depolarization was exclusively dependent on influx through dihydropyridine-sensitive L-type Ca2+ channels. In contrast, Ca2+ can be removed from the myoplasm by a variety of cellular mechanisms and thus accumulation of myoplasmic free Ca2+ is dependent upon the balance of influx and efflux/sequestration pathways. Therefore, increased net free cation accumulation (AUC) in the presence of Ba2+, but not Ca2+, suggests compensation for exercise training-enhanced Ca2+ influx via the increased activity of additional Ca2+ uptake and/or extrusion mechanisms. Our experiments also demonstrated a faster rate of rise in free Ca2+vs. Ba2+ in response to membrane depolarization (Fig. 4vs. 2). Although Ba2+ current is typically larger than Ca2+ current through VGCC, the faster rate of rise in free Ca2+vs. Ba2+ appears to reflect the increased binding affinity of fura-2 for Ca2+ as documented previously (Schilling et al. 1989; Kwan & Putney, 1990).
We conducted experiments to evaluate the relative role of potential cellular mechanisms of Ca2+ extrusion and/or sequestration that may function to compensate enhanced VGCC density and thus limit net free Ca2+ accumulation in smooth muscle cells from exercise-trained animals. While maximal rate of free Ca2+ accumulation remained significantly elevated in cells from exercise-trained vs. sedentary animals, SERCA inhibition had a negligible influence on maximal rate and net free Ca2+ accumulation in cells from both groups of animals (Fig. 5B and C). These data suggest that SR Ca2+ uptake made minimal contribution to Ca2+ removal during membrane depolarization in sedentary and exercise-trained animals, and provide support for another Ca2+ regulatory mechanism as the primary mechanism for the compensation of enhanced VGCC density.
In the presence of 80 mm KCl, the plasma membrane is depolarized to values more positive than the reversal potential for the Na+-Ca2+ exchanger and therefore the exchanger contributes to Ca2+ influx rather than efflux (Sturek et al. 1992). However, we were concerned that during the early stages of depolarization when changes in membrane potential are more dynamic and have not yet attained reversal potential or steady state, Na+-Ca2+ exchange may contribute to Ca2+ removal from the intracellular space. Therefore, we applied low extracellular Na+ superfusate for 2 min prior to high KCl-induced membrane depolarization. Our data indicate that inhibition of Na+-Ca2+ exchange activity produced significant, but similar, increases in maximal rate and net free Ca2+ accumulation in cells from both sedentary and exercise-trained animals. These findings indicate that Na+-Ca2+ exchange activity alters Ca2+ influx in smooth muscle cells from sedentary and exercise-trained animals to a similar extent and thus provides evidence against Na+-Ca2+ exchange activity as the compensating mechanism for increased VGCC density in cells of exercise-trained pigs. The increase in maximal rate and net free Ca2+ accumulation (AUC) in the presence of Na+-Ca2+ exchange inhibition was probably the result of enhanced Ca2+ influx via the exchanger during 80 mm KCl-induced membrane depolarization. Preliminary experiments indicate a further reduction of extracellular Na+ to 5 mm in the presence of 80 mm KCl-induced depolarization has no further influence on the membrane potential of smooth muscle cells (M. Sturek, unpublished observations). These findings suggest that the increases in maximal rate and net free Ca2+ accumulation in the presence of low Na+ are the result of Ca2+ influx via Na+-Ca2+ exchange and are not a response to greater membrane depolarization. Subsequent experiments in which SERCA and Na+-Ca2+ exchange were inhibited simultaneously unequivocally demonstrate that neither of these Ca2+ regulatory mechanisms compensates the increased VGCC density to limit net free Ca2+ accumulation in cells of exercise-trained pigs.
Ca2+ removal from smooth muscle myoplasm occurs primarily via Na+-Ca2+ exchange, plasma membrane Ca2+-ATPase (PMCA) and SERCA (Nazer & van Breemen, 1998), while the role of mitochondria in Ca2+ regulation remains controversial (Drummond & Tuft, 1999; Ganitkevich, 1999). Previous studies have documented that following stimulated Ca2+ influx, PMCA-mediated extrusion accounts for approximately 50 % of intracellular Ca2+ removal (Furukawa et al. 1988; Nazer & van Breemen, 1998) with SERCA and Na+-Ca2+ exchange equally accounting for the remaining 50 %, in the presence of experimentally induced collapse of the mitochondrial potential gradient (Nazer & van Breemen, 1998). In the present study, Ca2+ removal during 80 mm KCl-induced membrane depolarization would be primarily dependent on PMCA, SERCA and, potentially, mitochondrial uptake since the membrane potential would be more positive than the reversal potential for Na+-Ca2+ exchange (Ito et al. 1980). Therefore, in the light of our findings that Na+-Ca2+ exchange and SERCA contribute to Ca2+ removal to a similar extent in sedentary and exercise-trained pigs, enhanced PMCA activity or mitochondrial uptake potentially compensates the enhanced Ca2+ influx in exercise-trained animals.
Previous studies have suggested that increased spontaneous release of Ca2+ from the SR towards the plasma membrane in smooth muscle cells isolated from exercise-trained miniature pigs may contribute to enhanced subsarcolemmal Ca2+ gradients (Stehno-Bittel et al. 1991; Stehno-Bittel & Sturek, 1992). We hypothesized that enhanced Ca2+ concentrations in the vicinity of voltage-gated Ca2+ channels (increased local Ca2+) of exercise-trained animals may function to rapidly inactivate VGCC and thereby attenuate net free Ca2+ accumulation. To test this hypothesis, we evaluated Ca2+ current inactivation in the presence of high EGTA to eliminate more distant Ca2+-dependent inactivation. Distant Ca2+-dependent inactivation of voltage-gated Ca2+ channels may involve activation of an intracellular mediator, such as a Ca2+-dependent phosphatase, which might dephosphorylate the Ca2+ channel, rendering it inactive (Chad, 1988). Furthermore, because Ba2+ does not directly inactivate voltage-gated Ca2+ channels (Ohya et al. 1988), the more rapid T½ decay in the presence of Ca2+vs. Ba2+ indicates that local Ca2+-dependent inactivation of voltage-gated Ca2+ channels does exist in these cells. However, our findings suggest that local Ca2+-dependent inactivation of the voltage-gated Ca2+ channel is similar in sedentary and exercise-trained animals.
Our data indicate that the enhanced rate of free Ca2+ accumulation in smooth muscle cells from exercise-trained animals may be compensated by increased Ca2+ removal ultimately producing increased movement of Ca2+ (both influx and efflux) across the plasma membrane. The resultant increase in Ca2+ movement across the plasma membrane without any increase in net free Ca2+ accumulation (AUC) may increase the Ca2+ concentration in a restricted subdomain of the plasma membrane as originally proposed by Rasmussen et al. (1989). This change in subsarcolemmal Ca2+ could potentially regulate the activities of numerous membrane-associated Ca2+-sensitive proteins and thus, in turn, strongly influence cellular Ca2+ efflux and influx mechanisms. For example, increased subsarcolemmal Ca2+ concentrations in smooth muscle cells from our exercise-trained pigs might stimulate Ca2+-dependent K+ channel current and subsequently hyperpolarize the cell, thereby limiting subsequent Ca2+ influx through voltage-gated Ca2+ channels (Guia et al. 1999). Increased subsarcolemmal Ca2+ concentrations could also directly activate mechanisms of Ca2+ extrusion (i.e. PMCA) or potentially provide a direct inhibition at L-type Ca2+ channels to limit Ca2+ influx. Additional studies have endorsed the presence of a subsarcolemmal Ca2+ regulatory process in smooth muscle cells, dissociated from global Ca2+ regulation, which may alter cellular contractile states (Stehno-Bittel et al. 1991; Stehno-Bittel & Sturek, 1992; Sturek et al. 1992; Ganitkevich & Isenberg, 1996; Guia et al. 1999). The presence of an increased Ca2+ concentration in a restricted subdomain of the plasma membrane might provide a mechanistic link between the cellular adaptation of increased Ca2+ influx and the general functional adaptation of enhanced vasodilatation and reduced vasoconstriction in the coronary circulation of exercise-trained animals.
In conclusion, we report the novel finding that the increased Ca2+ influx observed in smooth muscle cells from exercise-trained animals is compensated by other cellular Ca2+ regulatory mechanisms, and thus net myoplasmic free Ca2+ accumulation in cells of sedentary vs. exercise-trained animals is not significantly different. We also verify previous findings that exercise training produces adaptations in coronary smooth muscle cells that increase VGCC density. Our evaluations of cellular mechanisms that may function to compensate enhanced VGCC density in exercise-trained pigs indicate that neither Na+-Ca2+ exchange nor SERCA plays a compensatory role in Ca2+ removal. Furthermore, local Ca2+-dependent inactivation of voltage-gated Ca2+ channels does not appear to compensate enhanced VGCC. Our findings suggest that increased Ca2+ extrusion via PMCA, Ca2+ sequestration by mitochondria, increased Ca2+-binding protein number/affinity, or inactivation of voltage-gated Ca2+ channels by means other than local Ca2+-dependent inactivation, are potential compensatory mechanisms for the enhanced rate of free Ca2+ accumulation in smooth muscle cells from exercise-trained animals.
Acknowledgments
The computer programming expertise of Dr Nancy Dietz contributed significantly to data analyses for these studies and is gratefully acknowledged. The authors also appreciate the technical contributions of Pam Thorne, Tammy Strawn and Denise Holiman to this project. These studies were supported by research funds from the National Institutes of Health, Program Project PO1-HL52490. C.L.H. was supported by a predoctoral fellowship from the American Heart Association, Missouri Affiliate.
References
- Benham CD, Tsien RW. Calcium-permeable channels in vascular smooth muscle: voltage-activated, receptor-operated, and leak channels. In: Mandel LJ, Eaton DC, editors. Cell Calcium and the Control of Membrane Transport. New York: Rockefeller University Press; 1987. pp. 49–78. [PubMed] [Google Scholar]
- Bers DM, Bridge JHB. Relaxation of rabbit ventricular muscle by Na-Ca exchange and sarcoplasmic reticulum calcium pump: ryanodine and voltage sensitivity. Circulation Research. 1989;65:334–342. doi: 10.1161/01.res.65.2.334. [DOI] [PubMed] [Google Scholar]
- Bove AA, Dewey JD. Proximal coronary vasomotor reactivity after exercise training in dogs. Circulation. 1985;71:620–625. doi: 10.1161/01.cir.71.3.620. [DOI] [PubMed] [Google Scholar]
- Bowles DK, Laughlin MH, Sturek M. Exercise training alters the Ca2+ and contractile responses of coronary arteries to endothelin. Journal of Applied Physiology. 1995;78:1079–1087. doi: 10.1152/jappl.1995.78.3.1079. [DOI] [PubMed] [Google Scholar]
- Bowles DK, Hu Q, Laughlin MH, Sturek M. Exercise training increases L-type calcium current density in coronary smooth muscle. American Journal of Physiology. 1998;275:H2159–2169. doi: 10.1152/ajpheart.1998.275.6.H2159. [DOI] [PubMed] [Google Scholar]
- Chad JE. Control of the generation and removal of calcium-mediated inactivation of the calcium current in Helix aspersa neurons. In: Grinnel AD, Armstrong D, Jackson MB, editors. Calcium and Ion Channel Modulation: A Tribute to Roger Eckert. New York: Plenum; 1988. pp. 197–214. [Google Scholar]
- Condrescu M, Chernaya G, Kalaria V, Reeves JP. Barium influx mediated by the cardiac sodium-calcium exchanger in transfected chinese hamster ovary cells. Journal of General Physiology. 1997;109:41–51. doi: 10.1085/jgp.109.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicarlo SE, Blair RW, Bishop VS, Stone HL. Daily exercise enhances coronary resistance vessel sensitivity to pharmacological activation. Journal of Applied Physiology. 1989;66:421–428. doi: 10.1152/jappl.1989.66.1.421. [DOI] [PubMed] [Google Scholar]
- Drummond RM, Tuft RA. Release of Ca2+ from the sarcoplasmic reticulum increases mitochondrial [Ca2+] in rat pulmonary artery smooth muscle cells. The Journal of Physiology. 1999;516:139–147. doi: 10.1111/j.1469-7793.1999.139aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa KI, Tawada Y, Shigekawa M. Regulation of the plasma membrane Ca2+ pump by cyclic nucleotides in cultured vascular smooth muscle cells. Journal of Biological Chemistry. 1988;263:8058–8065. [PubMed] [Google Scholar]
- Ganitkevich VY. Clearance of large Ca2+ loads in single smooth muscle cell: examination of the role of mitochondrial Ca2+ uptake and intracellular pH. Cell Calcium. 1999;25:29–42. doi: 10.1054/ceca.1998.0001. [DOI] [PubMed] [Google Scholar]
- Ganitkevich VY, Isenberg G. Dissociation of subsarcolemmal from global cytosolic [Ca2+] in myocytes from guinea-pig coronary artery. The Journal of Physiology. 1996;490:305–318. doi: 10.1113/jphysiol.1996.sp021145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guia A, Wan X, Courtemanche M, Leblanc N. Local Ca2+ entry through L-type Ca2+ channels activates Ca2+-dependent K+ channels in rabbit coronary myocytes. Circulation Research. 1999;84:1032–1042. doi: 10.1161/01.res.84.9.1032. [DOI] [PubMed] [Google Scholar]
- Gutnick MJ, Lux HD, Swandulla D, Zucker H. Voltage-dependent and calcium-dependent inactivation of calcium channel current in identified snail neurons. The Journal of Physiology. 1989;412:197–220. doi: 10.1113/jphysiol.1989.sp017611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Kitamura K, Kuriyama H. Nitroglycerine and catecholamine actions on smooth muscle cells of the canine coronary artery. The Journal of Physiology. 1980;309:171–183. doi: 10.1113/jphysiol.1980.sp013502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan CY, Putney JW. Uptake and intracellular sequestration of divalent cations in resting and methacholine-stimulated mouse lacrimal acinar cells. Journal of Biological Chemistry. 1990;265:678–684. [PubMed] [Google Scholar]
- Laughlin MH. Effects of exercise training on coronary transport capacity. Journal of Applied Physiology. 1985;58:468–476. doi: 10.1152/jappl.1985.58.2.468. [DOI] [PubMed] [Google Scholar]
- Laughlin MH, Mcallister RM. Exercise training-induced coronary vascular adaptation. Journal of Applied Physiology. 1992;73:2209–2225. doi: 10.1152/jappl.1992.73.6.2209. [DOI] [PubMed] [Google Scholar]
- Laughlin MH, Overholser KA, Bhatte MJ. Exercise training increases coronary transport reserve in miniature swine. Journal of Applied Physiology. 1989;67:1140–1149. doi: 10.1152/jappl.1989.67.3.1140. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jones AW, Sturek M. Increased barium influx and potassium current in stroke-prone spontaneously hypertensive rats. Hypertension. 1994;23:1091–1095. doi: 10.1161/01.hyp.23.6.1091. [DOI] [PubMed] [Google Scholar]
- Mombouli JV, Nakashima N, Hamra M, Vanhoutte PM. Endothelium-dependent relaxation and hyperpolarization evoked by bradykinin in canine coronary arteries: enhancement by exercise training. British Journal of Pharmacology. 1996;117:413–418. doi: 10.1111/j.1476-5381.1996.tb15206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazer MA, Van breemen C. A role for the sarcoplasmic reticulum in Ca2+ extrusion from rabbit inferior vena cava smooth muscle. American Journal of Physiology. 1998;274:H123–131. doi: 10.1152/ajpheart.1998.274.1.H123. [DOI] [PubMed] [Google Scholar]
- Nilius B, Kitamura K, Kuriyama I. Properties of inactivation of calcium channel currents in smooth muscle cells of rabbit portal vein. Pflügers Archiv. 1994;426:239–246. doi: 10.1007/BF00374777. [DOI] [PubMed] [Google Scholar]
- Ohya Y, Kitamura K, Kuriyama H. Regulation of calcium current by intracellular calcium in smooth muscle cells of rabbit portal vein. Circulation Research. 1988;62:375–383. doi: 10.1161/01.res.62.2.375. [DOI] [PubMed] [Google Scholar]
- Oltman CL, Parker JL, Adams HR, Laughlin MH. Effects of exercise training on vasomotor reactivity of porcine coronary arteries. American Journal of Physiology. 1992;263:H372–382. doi: 10.1152/ajpheart.1992.263.2.H372. [DOI] [PubMed] [Google Scholar]
- Parker JL, Oltman CL, Muller JM, Myers PR, Adams HR, Laughlin MH. Effects of exercise training on regulation of tone in coronary arteries and arterioles. Medicine and Science in Sports and Exercise. 1994;26:1252–1261. [PubMed] [Google Scholar]
- Rasmussen H, Barrett P, Zawalich W, Isales C, Stein P, Smallwood J, Mccarthy R, Bollag W. Cycling of Ca2+ across the plasma membrane as a mechanism for generating a Ca2+ signal for cell activation. Annals of the New York Academy of Sciences. 1989;568:73–80. doi: 10.1111/j.1749-6632.1989.tb12492.x. [DOI] [PubMed] [Google Scholar]
- Rogers PJ, Miller TD, Bauer BA, Brum JM, Bove AA, Vanhoutte PM. Exercise training and responsiveness of isolated coronary arteries. Journal of Applied Physiology. 1991;71:2346–2351. doi: 10.1152/jappl.1991.71.6.2346. [DOI] [PubMed] [Google Scholar]
- Schilling WP, Rajan L, Strobl-Jager E. Characterization of the bradykinin-stimulated calcium influx pathway of cultured vascular endothelial cells. Journal of Biological Chemistry. 1989;264:12838–12848. [PubMed] [Google Scholar]
- Stehno-Bittel L, Laughlin MH, Sturek M. Exercise training depletes sarcoplasmic reticulum calcium in coronary smooth muscle. Journal of Applied Physiology. 1991;71:1764–1773. doi: 10.1152/jappl.1991.71.5.1764. [DOI] [PubMed] [Google Scholar]
- Stehno-Bittel L, Sturek M. Spontaneous sarcoplasmic reticulum calcium release and extrusion from bovine, not porcine, coronary artery smooth muscle. The Journal of Physiology. 1992;451:49–78. doi: 10.1113/jphysiol.1992.sp019153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturek M, Kunda K, Hu Q. Sarcoplasmic reticulum buffering of myoplasmic calcium in bovine coronary artery smooth muscle. The Journal of Physiology. 1992;451:25–48. doi: 10.1113/jphysiol.1992.sp019152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner-Mann C, Hu Q, Sturek M. Multiple effects of ryanodine on intracellular free Ca2+ in smooth muscle cells from bovine and porcine coronary artery: modulation of sarcoplasmic reticulum function. British Journal of Pharmacology. 1992;105:903–911. doi: 10.1111/j.1476-5381.1992.tb09076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]