

Abstract
Membrane currents evoked by repeated noxious heat stimuli (43–47 °C) of 3 s duration were investigated in acutely dissociated dorsal root ganglion (DRG) neurones of adult rats. The heat stimuli generated by a fast solution exchanger had a rise time of 114 ± 6 ms and a fall time of 146 ± 13 ms.
When heat stimuli were applied to heat-sensitive small (≤ 32·5 μm) DRG neurones, an inward membrane current (Iheat) with a mean peak of 2430 ± 550 pA was observed (n = 19). This current started to activate and deactivate with no significant latency with respect to the heat stimulus. The peak of Iheat was reached with a rise time of 625 ± 115 ms. When the heat stimulus was switched off Iheat deactivated with a fall time of 263 ± 17 ms.
During constant heat stimulation Iheat decreased with time constants of 4–5 s (inactivation). At the end of a 3 s heat stimulus the peak current was reduced by 44 ± 5 % (n = 19).
Current-voltage curves revealed outward rectifying properties of Iheat and a reversal potential of −6·3 ± 2·2 mV (n = 6). Inactivation was observed at all membrane potentials investigated (−80 to 60 mV); however, inactivation was more pronounced for inward currents (37 ± 5 %) than for outward currents (23 ± 6 %, P < 0·05).
When neurones were investigated with repeated heat stimuli (3 to 5 times) of the same temperature, the peak current relative to the first Iheat declined by 48 ± 6 % at the 3rd stimulus (n = 19) and by 54 ± 18 % at the 5th stimulus (n = 4; tachyphylaxis).
In the absence of extracellular Ca2+ (buffered with 10 mm EGTA) inactivation (by 53 ± 6 %) and tachyphylaxis (by 42 ± 7 % across three stimuli) were still observed (n = 8). The same was true when intracellular Ca2+ was buffered by 10 mm BAPTA (inactivation by 49 ± 4 %, tachyphylaxis by 52 ± 7 % across three stimuli; n = 13). Thus, inactivation and tachyphylaxis were mainly independent of intra- and extracellular Ca2+.
These results indicate that inactivation and tachyphylaxis of heat-evoked inward currents can be observed in vitro, similar to adaptation and suppression of action potential discharges elicited by comparably fast heat stimuli in vivo. Whereas the voltage dependence of Iheat resembles that of capsaicin-induced membrane currents (ICaps), the independence of inactivation and tachyphylaxis of Iheat from calcium is in clear contrast to ICaps. A similar difference in calcium dependence of inactivation has been reported between heat-evoked and capsaicin-induced currents through the cloned capsaicin receptor channel VR1. Thus, the properties of Iheat and of VR1 largely account for the adaptation and suppression of heat-evoked nociceptor discharges.
In contrast to other sensory modalities, pain does not decrease when a noxious stimulus is applied at constant intensity (Greene & Hardy, 1962). From this lack of adaptation on the perceptive level it has traditionally been implied that primary nociceptive afferents also do not adapt upon constant stimulation. This is in contrast to the results of recordings from these afferents, which exhibit pronounced adaptation for physical as well as chemical stimuli (Meyer et al. 1994). Peripheral adaptation of nociceptive nerve endings is compensated by central summation (Mendell & Wall, 1965; Price et al. 1977); this slow summation process of small fibre input to the dorsal horn of the spinal cord is known as wind-up. Since wind-up is specific for wide dynamic range (WDR) neurones, heat responses of nociceptive-specific neurones showed adaptation whereas WDR neurones in the spinal cord did not (Coghill et al. 1993). Compensation of peripheral adaptation to yield a constant pain sensation may be the major function of wind-up.
Nociceptive primary afferents act as proportional and differential sensors (PD sensors), exhibiting pronounced (up to 90 %) albeit slow adaptation (τ ≈2·5 s) when stimulated with mechanical or heat stimuli (Meyer & Campbell, 1981; Handwerker et al. 1987; Schneider et al. 1995; Treede et al. 1995). Recovery from this adapted state takes 10 min or longer (LaMotte & Campbell, 1978; Treede et al. 1998) and affects the dynamic response more than the static response (Treede, 1995). This long-lasting reduction of nociceptor discharge is called ‘suppression’.
The adaptation and suppression of nociceptive afferent action potential discharges may occur at two stages of the neural encoding process: (1) transduction of physical stimuli into generator potentials and (2) transformation of generator potentials into trains of action potentials. Whereas adaptation in the transformation process is supported by slowing of conduction velocity (Thalhammer et al. 1994; Schmelz et al. 1995; Serra et al. 1999) and slow kinetics of sensory neurone-specific sodium channels (Waxman et al. 1999), there is no evidence for adaptation in the transduction process so far. The transduction process for noxious heat stimuli has been studied using dissociated neurones from dorsal root ganglia (DRG) as models of their own terminals (Cesare & McNaughton, 1996; Kirschstein et al. 1997, 1999; Nagy & Rang, 1999a,b; Vyklický et al. 1999). Brief heat stimuli (< 1 s) were found to elicit inward currents (Iheat) in DRG neurones which did not adapt (Cesare & McNaughton, 1996) and were reproducible with stimulus repetition at short intervals (Kirschstein et al. 1997, 1999; Guenther et al. 1999). Heat stimuli with slowly increasing temperatures revealed a threshold temperature of about 43°C to evoke Iheat in DRG neurones (Vyklický et al. 1999). The correlate of adaptation in the transduction process would be inactivation of Iheat upon constant stimulation; suppression would be visible as tachyphylaxis upon repeated stimulation.
The transduction of heat stimuli into membrane currents has been suggested to be mediated by the heat- and capsaicin-sensitive vanilloid receptor VR1 (Caterina et al. 1997; Kirschstein et al. 1999). This receptor when transfected into human embryonic kidney (HEK293) cells shows a threshold of ∼45°C for activation (Tominaga et al. 1998). In DRG neurones, Iheat is partly antagonized by the VR antagonists Ruthenium Red and capsazepine (Kirschstein et al. 1999; Nagy & Rang, 1999b). Whereas the transduction of moderately noxious heat stimuli in DRG neurones is likely to be mediated by the capsaicin receptor VR1, a second heat transduction mechanism with a higher threshold has been described in capsaicin-insensitive DRG neurones (Nagy & Rang, 1999a). This transduction mechanism may involve the vanilloid receptor-like protein 1 (VRL1) which is activated by higher temperatures with a threshold of ∼52°C but which is not sensitive to capsaicin or capsazepine in VRL1-expressing HEK293 cells (Caterina et al. 1999).
The aim of this study was to test whether inward currents elicited by moderately noxious heat stimuli show signs of inactivation and tachyphylaxis when long stimulus durations, as used previously in vivo, were applied. As inactivation and tachyphylaxis in capsaicin-induced currents are calcium dependent (Docherty et al. 1996; Liu & Simon, 1998), we further investigated the influence of extracellular and intracellular calcium on inactivation and tachyphylaxis of Iheat.
Preliminary accounts of this study have appeared in abstract form (Schwarz et al. 2000).
METHODS
Acutely dissociated DRG neurones were obtained as previously described (Kirschstein et al. 1997, 1999). Briefly, adult Sprague-Dawley rats (110–310 g) of either sex were deeply anaesthetized with diethylether (Merck, Darmstadt, Germany) and rapidly decapitated. This method is in accordance with German national law on the principles for animal welfare and is approved by the representative for animal care and use of the University of Mainz. The spine was removed, chilled at 4°C in F12-Dulbecco’s modified Eagle’s medium (Sigma; adjusted to pH 7·4 by NaOH) containing 30 mm NaHCO3 (Merck), 100 i.u. ml−1 penicillin and 100 μg ml−1 streptomycin (Sigma). The F12 medium was equilibrated with 95 % O2–5 % CO2 throughout the whole preparation and dissociation procedure. Thoracic and lumbar DRGs were quickly dissected and freed from connective tissue. Neurones were dissociated at 37°C using collagenase CLS II (5–10 mg ml−1, 40–50 min; Biochrome, Berlin, Germany) and trypsin (0·2–1 mg ml−1, 10–12 min; Sigma) dissolved in the F12 medium. After trituration with fire polished Pasteur pipettes, neurones were plated on 35 mm diameter culture dishes, which also served as recording chambers, and stored at 37°C in a humidified 5 % CO2 atmosphere before being used for electrophysiological recordings 1–10 h after dissociation.
Electrophysiology
Whole-cell patch-clamp experiments were performed in extracellular solution (ES) containing (mm): 145 NaCl, 2·5 KCl, 10 Hepes, 10 glucose, 1·5 CaCl2 and 1·2 MgCl (pH adjusted to 7·4 at room temperature). Cell diameter, cross-sectional area, membrane capacity and resting membrane potential were measured for each investigated neurone. Only round or oval neurones up to a maximum diameter of 32·5 μm and without any processes were included in this study. The mean of the major and minor diameter was used to measure oval-shaped neurones. Nociceptive neurones are generally small (Petersen & LaMotte, 1991; Gold et al. 1996) and heat-sensitive neurones were previously found to have diameters between 20 and 32·5 μm (Kirschstein et al. 1997, 1999). Recordings were performed using an Axopatch 200A amplifier (Axon Instruments, Foster City, CA, USA; lowpass Bessel filter (4-pole) at 2 kHz) and pCLAMP software (version 6.0 and 8.0, Axon Instruments; sampling rate: 4 kHz). Neurones were clamped at −80 mV in voltage-clamp mode or held at a membrane potential of about −60 mV in current-clamp mode by applying current through the patch electrode. Excitability was tested by depolarizing current steps for each neurone. Cells without the capability to generate action potentials elicited by short (40 ms) depolarizing current pulses where excluded from further investigation. Patch pipettes were fabricated from borosilicate glass capillaries (Hilgenberg, Malsfeld, Germany) using a horizontal micropipette puller (P-87; Sutter, Novato, USA), fire polished and filled with a solution containing (mm): 160 KCl, 10 Hepes and 8·13 EGTA adjusted to pH 7·2 by KOH (RTip = 2–13 MΩ). In some experiments EGTA was replaced by 10 mm BAPTA and a mean period of 10 min was kept before applying heat stimuli to allow equilibration with intracellular solution (0 [Ca2+]i). In experiments with calcium-free ES (0 [Ca2+]o), 10 mm EGTA was added to the normal ES (pH adjusted to 7·4). Neurones were superperfused 30 s before and throughout the heat stimulations and interstimulus intervals with this buffered calcium-free solution. Current-voltage (I–V) curves of Iheat were obtained using fast depolarizing ramps (−80 to 60 mV in 200 ms every 310 ms). For these experiments patch pipettes were filled with a potassium-free solution containing (mm): 140 CsCl, 10 Hepes, 10 EGTA, 4 MgCl2 (pH 7·2). Before applying heat stimuli (10 s duration) the ramp protocol was repeated ∼100 times to inactivate voltage-dependent inward and outward currents (Liu et al. 1997).
Heat stimulation
A custom-made motor-driven solution changer with four parallel glass capillaries (outer diameter: 1·5 mm, inner diameter: 1·05 mm) was used to apply heated solutions. The flow of solution, driven by gravity, was adjusted to the same rate in each capillary (0·6–0·7 ml min−1, corresponding to a velocity of 12–13 μm ms−1). Every capillary had its own reservoir. Two of the capillaries perfused with heated solution were insulated with plastic tubes to minimize loss of temperature. A stepper motor, controlled by pCLAMP software, was used to switch between capillaries with superperfusing solutions of different temperature. ES in the two reservoirs connected to heated tubes was adjusted to pH 7·4 at 50°C. The solution in these tubes was heated to 65–80°C using a resistive device (HT 1.2; VETEC, Dummersdorf, Germany). Since there was a distance of about 14 cm between the heating device and the outlet, the solution cooled down to 43–47°C before reaching the bath. Each capillary was earthed and contained a miniature thermocouple (IT-1E; Physitemp, Clifton, USA) to measure the temperature of the solution close to the outlet (BAT-12 digital laboratory thermometer; Physitemp; 15 Hz lowpass filter). A further miniature thermocouple positioned in place of the neurone at a distance of about 150 μm from the outlets was used to measure the effective temperature of the applied stimulus off-line (see Fig. 1B). There was less than 1°C difference between the temperatures measured by the thermocouple inside and in front of the outlet in the steady state (see Results). The thermal response time of the setup was determined by rapidly immersing the thermocouple into heated ES (ΔT = 21 ± 1°C; n = 10). These measurements revealed a time constant of 95 ± 2 ms, indicating that the setup was too slow to register fast temperature steps exactly. Thus, open patch pipettes (RTip = 2–4 MΩ) in place of the neurone were used to record the time course of heat stimuli (see Fig. 1A, cf. Cesare & McNaughton, 1996).
Figure 1. Inward current elicited in a DRG neurone by a stepped temperature increase.
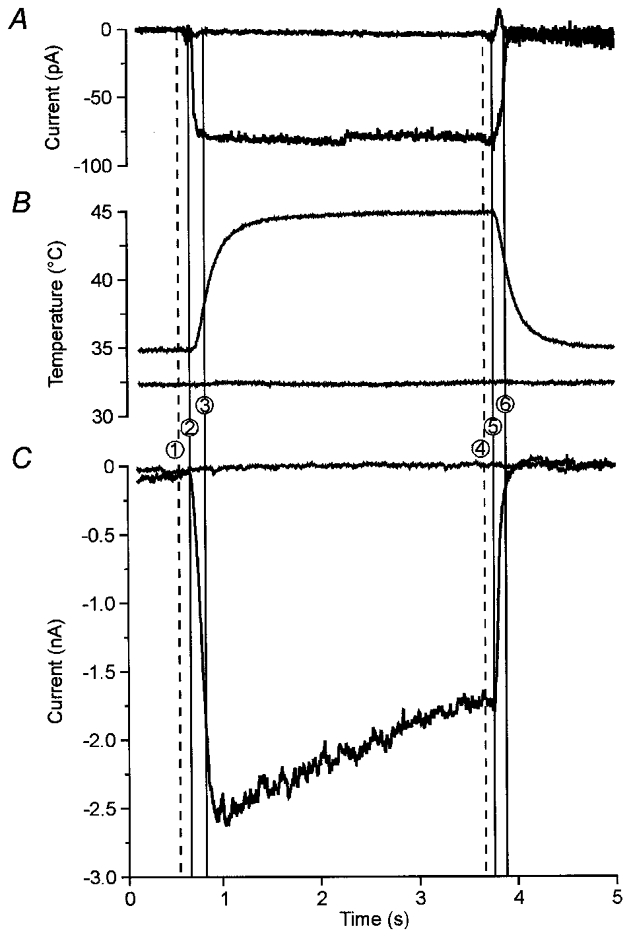
Stepped heat stimuli from 35 to 45 °C were applied by moving the boundary between two parallel flows (0·6 ml min−1 equivalent to 12 μm ms−1) of superperfusing medium at different temperatures across an open pipette (A), a fast thermocouple (B) and a small DRG neurone (C). A, the time course of the temperature change was obtained using the tip of an open pipette, which was positioned in place of the neurone. Switching the superperfusion from 35 to 45 °C induced a small inward current. The first vertical line (1) marks when the control pulse was sent to the solution changer, the second line (2) is set to the mean time (n = 26) of the first measurable effect of heat-induced open-pipette currents (onset latency) and the third line to the mean time when the plateau was reached (rise time; (3)). The three lines at the end of the stimulus indicate a second control pulse (4), mean time to the first visible recovery effect (offset latency;(5)) and mean time to return to baseline (fall time; (6)). These measurements revealed a change of temperature within about 140 ms at the neurone with a delay of about 100 ms after the control pulse. Stepping between solutions of the same temperature (30 °C) elicited only minor artefacts (upper line). B, effective temperature of the heat stimulus measured with a miniature thermocouple in place of the neurone. The thermocouple is fast enough to indicate the onset latency but not the time course of the temperature stimulus. The effective temperature in this case was 44·8 °C determined in the last second of the heat stimulus. Changing between solutions of the same temperature of 32·5 °C revealed no effect (lower line). C, in a DRG neurone (size: 27·5 μm, resting membrane potential: −43 mV) the heat stimulus elicited an inward current (Iheat), which reached a peak after a rise time of 236 ms. Afterwards the current decreased in spite of the constant stimulus temperature (inactivation). After termination of the heat stimulus Iheat immediately began to deactivate and recovered to baseline within 250 ms. Changing between solutions of the same temperature (33 °C) induced no visible current (upper line).
Protocol
After disruption of the cell membrane the neurones were tested for excitability by depolarizing current steps. Mechanically induced artefacts from the solution changer were measured by switching between two solutions of the same temperature between 29 and 35°C. These measurements revealed a mean outward current of 61 ± 161 pA (mean ± s.d.; n = 77). Inward currents exceeding 400 pA (mean + 3s.d. of the mechanically induced current) were considered specific heat responses, whereas inward currents <400 pA were considered non-specific effects. Of 77 neurones tested with one or more heat stimuli of 3 s, 49 were heat sensitive according to this definition (64 %). This proportion of heat-sensitive small, DRG neurones is in agreement with previous reports (Cesare & McNaughton, 1996; Kirschstein et al. 1997, 1999; Vyklický et al. 1999). In the present study only neurones were included which were tested at least three times with repeated noxious heat stimuli (3 s; 43–47°C) in voltage-clamp mode. A maximum variation of 1°C in the application tube was tolerated between the repeated heat stimuli. Variations of the holding current (Ihold) of maximally ± 400 pA before the first and the third stimulus were accepted. Nineteen of the heat-sensitive neurones investigated with normal extra- and intracellular solutions fulfilled these criteria. Additionally 8 heat-sensitive neurones were examined with 0 [Ca2+]o, 13 heat-sensitive neurones with 0 [Ca2+]i and 6 heat-sensitive neurones with voltage ramps using potassium-free intracellular solution; these are not included in the general statistics of heat sensitivity. In every dish only one neurone was tested with heated solutions.
Data analysis
Off-line measurements and statistical analyses were done using pCLAMP 8.0 (Axon Instruments) and EXCEL 97 (Microsoft). For illustration all recordings were filtered with a Gaussian filter (100 Hz), subsampled to 0·4 kHz and again filtered with a notch filter (centre frequency: 50 Hz, width 10 Hz). These filters were necessary only for the open-pipette recordings, but to be consistent were applied to all recordings. Onset and offset latency was measured from the control pulse for the stepper motor of the solution changer to the first visible effect. The rise time was determined from the first visible effect to the peak current. The fall time was determined as time needed from the last heat-induced effect to return to baseline. The decrease of current from the peak to the end of the heat stimulus is designated as inactivation, which is supposed to be a correlate of adaptation of action potential discharges reported in vivo (Meyer & Campbell, 1981; Treede et al. 1995). The time course of this decrease was linear in a semi-logarithmic plot and thus was characterized by a single time constant τ. The decrease of the peak current in response to repeated heat stimuli was designated as tachyphylaxis which is equivalent to suppression of action potential discharges to repetitive heat stimuli reported in vivo (LaMotte & Campbell, 1978; Adriaensen et al. 1984). I–V curves were obtained by subtracting the last background ramp before heat from the ramps during maximum Iheat and 2·5 s after the peak (Liu et al. 1997). Data are presented as means ± standard error of the mean (s.e.m.) if not otherwise indicated. Inactivation, tachyphylaxis and the effects of extracellular and intracellular calcium-free solutions were analysed using a 2-way ANOVA with Newman-Keuls post hoc test for ordered means (Statistica 4.5, StatSoft). Other effects were statistically analysed by Student’s t test for paired or unpaired data. Error probabilities with P ≤0·05 were considered statistically significant.
RESULTS
The time course of the heat stimulus was recorded with an open pipette in place of the neurone (n = 26). When the solution changer stepped between solutions from a baseline temperature of 32–35°C to a temperature of 45–46°C (ΔT = 10–13°C), a small inward current was elicited (see Fig. 1A). This current started with a delay of 110 ± 2 ms after the control pulse (onset latency) and reached a plateau of −93 ± 6 pA after a rise time of 114 ± 6 ms. When the heat stimulus was switched off, the current returned to the baseline within a fall time of 146 ± 13 ms (again delayed by 90 ± 4 ms with respect to the control pulse). Stepping between two solutions of the same temperature (n = 26) induced only small mechanical artefacts of −8 ± 3 pA (Fig. 1A). These measurements indicate that our system generates stepped temperature stimuli with a rise time similar to a feedback-controlled laser (Meyer et al. 1976). The effective temperature of the stimulus was measured with a thermocouple in place of the neurone. The step to a heated solution induced an asymptotic approximation to a plateau temperature visible in the time course recorded by the thermocouple (Fig. 1B). There was a difference of 0·2 ± 0·3°C (mean ± s.d., n = 27) between the temperature measured by the thermocouple inside the glass capillary and the plateau temperature measured by the thermocouple in front of its outlet. This plateau temperature indicates the effective temperature at the neurone, but the time course was too slow due to the response time of the temperature measurement setup (see Methods). There was no effect measurable with the thermocouple when stepped between two solutions of the same non-noxious temperature (Fig. 1B). Due to these results we used open-pipette currents as indicators of the time course of stepped heat stimuli, and readings from thermocouples inside the superfusion capillaries as indicators of the effective temperature.
In DRG neurones stepping the temperature from a baseline temperature of 31–35°C to 43–47°C (ΔT = 10–13°C) elicited an inward membrane current (Iheat) after an onset latency of 116 ± 2 ms. This current was delayed 6 ms in comparison with open-pipette measurements but the difference did not reach significance. The peak of the heat-evoked current was −2430 ± 550 pA (n = 19). The mean rise time was 625 ± 115 ms and thus significantly longer (P < 0·001) than that of the stimulus. This result indicates that the peak of Iheat is reached about 500 ms after the peak temperature. During sustained heat stimulation the normalized current decreased by 27 ± 4 % after 2 s and by 44 ± 5 % after 3 s (inactivation of Iheat, Fig. 3A). After switching off the heat stimulus the current began to deactivate after an offset latency of 97 ± 4 ms (with respect to the control pulse) and thus was again slightly delayed compared with the stimulus offset (7 ms, n.s.). The current reached the baseline after a fall time of 263 ± 17 ms (n = 19; Fig. 1C). In comparison with the heat stimulus the fall time was also significantly longer (P < 0·001). In contrast to the observable inactivation of Iheat, the open-pipette current elicited by a temperature change did not show any signs of inactivation and was negligibly small (open-pipette current was less than 4 % of mean peak membrane current, Fig. 1A).
Figure 3. Inactivation of heat-evoked inward currents is independent of intra- and extracellular calcium.
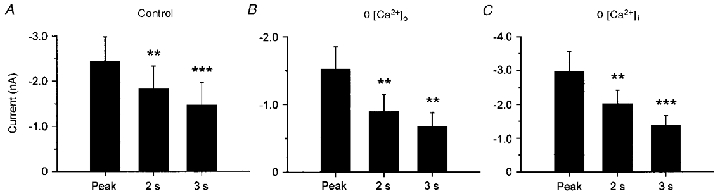
During sustained stimulation (3 s) heat-evoked inward currents progressively decreased (inactivation). The first bar in each graph represents the mean heat-evoked peak current, the second and third bar represent the mean current 2 and 3 s after stimulus onset. A, inactivation of Iheat in neurones investigated with normal intra- and extracellular solutions (n = 19). B, inactivation of Iheat in calcium-free extracellular solution (buffered by 10 mm EGTA, n = 8). C, inactivation in neurones where intracellular Ca2+ was buffered with 10 mm BAPTA in the patch electrode (n = 13). Data are presented as means ± s.e.m. (***P ≤ 0·001, **P ≤ 0·01 versus peak, Newman-Keuls post hoc test). Buffering intra- or extracellular calcium did not significantly affect inactivation.
The voltage dependence of inactivation was investigated with continuous voltage ramps. Iheat had outward rectifying properties and a reversal potential close to 0 mV (Vrev = −6·3 ± 2·2 mV, n = 6; Fig. 2A, squares). The time to peak was delayed under these experimental conditions (2·0 ± 0·4 s, P < 0·001, Student’s unpaired t test). Therefore, the degree of inactivation was determined 2·5 s after the peak of Iheat elicited by a heat stimulus of 10 s duration. The properties of Iheat were conserved in the inactivated state (Vrev = −7·0 ± 2·4 mV, n.s.; Fig. 2A, circles) but inactivation was more pronounced at membrane potentials negative to Vrev. Inactivation of heat-evoked inward currents was 37 ± 5 % in the negative-voltage range (−80 to −20 mV), that of heat-evoked outward currents in the positive-voltage range (10 to 60 mV) was 23 ± 6 % (P < 0·05, Student’s paired t test; Fig. 2B).
Figure 2. I–V relationship of heat-evoked currents before and during inactivation.

I–V curves were obtained using a repeated ramps protocol (−80 to 60 mV in 200 ms every 310 ms) and constant heat stimuli of 10 s duration were applied after inactivation of voltage-dependent inward and outward currents by the voltage ramps. A, I–V curves were determined at the peak of Iheat (□) and 2·5 s afterwards (○). Points are joined by polynominal curve fitting. B, plot of percentage reduction in Iheat 2·5 s after the peak current. The reduction in Iheat is divided by the respective peak current at each membrane potential. Data are presented as means ± s.e.m. (n = 6).
The contribution of Ca2+ to inactivation was investigated with calcium-free extracellular solution (0 [Ca2+]o), applied 30 s before and throughout the whole repeated stimulus protocol. The heat-evoked peak current of −1530 ± 400 pA (rise time 351 ± 71 ms, n = 8) was reduced by 53 ± 6 % within the 3 s of heat stimulation (Fig. 3B). This indicates that an influx of calcium from the extracellular space is not necessary for inactivation. However, calcium may also increase intracellularly through a release from intracellular stores. When intracellular calcium was buffered with 10 mm BAPTA in the patch electrode (0 [Ca2+]i) the mean heat-evoked peak current was −2960 ± 600 pA (n = 13, rise time 603 ± 103 ms). This peak current was reduced by 49 ± 4 % within 3 s (Fig. 3C). Statistical analysis with 2-way ANOVA revealed that the peak membrane currents and the degree of inactivation with 0 [Ca2+]o or 0 [Ca2+]i were not significantly different from control conditions (treatment: F(2,37) = 0·96; interaction: F(2,74) = 1·47; n.s.), but inactivation during the stimuli was significant (F(2,74) = 33·66; P < 0·001). When normalized to the peak amplitude, the percentage reduction in Iheat within the 3 s stimuli was also independent of extra- or intracellular Ca2+ (one-way ANOVA: F(2,37) = 0·73, n.s.).
To analyse the effect of repeated application of the same heat stimuli on the peak of Iheat, some neurones were investigated with five consecutive stimuli (mean interstimulus interval: 31 ± 6 s; n = 4). The peak current decreased substantially across the five heat stimuli (tachyphylaxis of Iheat). When normalized to the peak current elicited by the first heat stimulus, the 2nd peak Iheat was reduced by 11 ± 20 %, the 3rd by 45 ± 13 %, the 4th by 50 ± 18 % and the 5th by 54 ± 18 %. The major degree of tachyphylaxis was reached after three repeated heat applications. Therefore, the first three stimuli are enough to characterize this phenomenon of Iheat (Fig. 4).
Figure 4. Repetitive application of heat stimuli induces tachyphylaxis of heat-evoked inward currents.
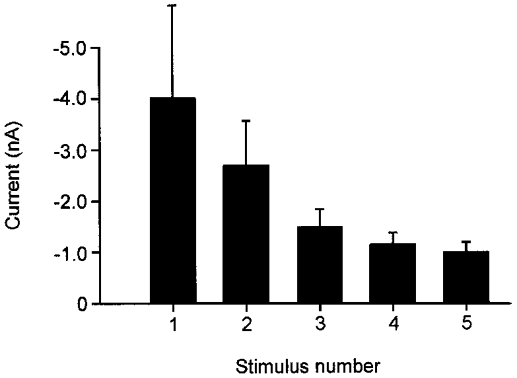
When neurones were tested repetitively with five heat stimuli the peak current of Iheat progressively decreased (tachyphylaxis). Most of the tachyphylaxis occurred across the first three stimuli. Each bar represents the mean peak current (n = 4).
To investigate the influence of extra- or intracellular calcium on tachyphylaxis, calcium was buffered extra- or intracellularly (see Methods) and three repetitive heat stimuli were applied under these different conditions. In neurones investigated under control conditions the peak Iheat decreased by 48 ± 6 % from the 1st to the 3rd stimulus (Fig. 5A; n = 19; mean interstimulus interval: 33 ± 3 s). When extracellular Ca2+ was buffered with 10 mm EGTA the mean peak current decreased by 42 ± 7 % across three stimuli (Fig. 5B; n = 8; mean interstimulus interval: 35 ± 1 s). Buffering intracellular Ca2+ with 10 mm BAPTA led to a decrease by 52 ± 7 % across three stimuli (Fig. 5C; n = 13; mean interstimulus interval: 34 ± 2 s). Thus buffering intracellular or extracellular calcium did not change the degree of tachyphylaxis compared with normal conditions (treatment: F(2,37) = 0·94, interaction: F(4,74) = 0·94; n.s.), whereas tachyphylaxis across the three repeated applications within each treatment was significant (F(2,74) = 16·18, P < 0·001).
Figure 5. Tachyphylaxis of heat-evoked inward currents is independent of intra- and extracellular calcium.
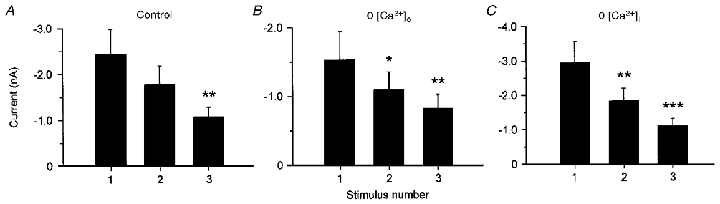
Each graph shows the mean heat-evoked peak currents of three consecutive heat stimuli. A, tachyphylaxis of Iheat under control conditions (n = 19). B, tachyphylaxis investigated when extracellular Ca2+ was buffered with. 10 mm EGTA (n = 8). C, tachyphylaxis in neurones where intracellular Ca2+ was buffered with 10 mm BAPTA (n = 13). Data are presented as means ± s.e.m. (***P ≤ 0·001, **P ≤ 0·01, *P ≤ 0·05 versus peak of 1st stimulus, Newman-Keuls post hoc test). Currents decreased significantly across the repeated heat stimulation, but buffering intra- or extracellular Ca2+ did not change the degree of tachyphylaxis.
The effect of repeated stimulation on inactivation of Iheat during the 3 s heat stimuli was evaluated as well. As already demonstrated, the inactivation of Iheat within the 1st heat stimulus was 44 ± 5 % of the peak current. Within the 2nd application the normalized current inactivated by 37 ± 6 % and by 44 ± 6 % during the 3rd application of the heat stimulus (Fig. 6). There was no significant difference of the normalized inactivation between the three heat stimuli (F(2,74) = 1·45; n.s.) and between the different treatments (F(2,37) = 0·48; interaction: F(4,74) = 2·17; n.s; data not shown).
Figure 6. Inactivation does not change within repeated heat stimuli.
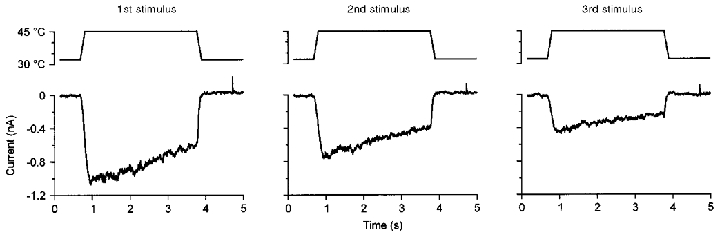
Currents induced by three repeated heat stimuli of 45 °C in a heat-sensitive neurone (size: 30 μm, resting membrane potential −52 mV, interstimulus intervals 35 and 36 s). Across the three heat responses the peak current decreased (tachyphylaxis) but the degree of relative inactivation of each current was almost identical over the three stimuli (40, 45 and 52 %).
Semi-logarithmic graphical representation of Iheat was linear throughout the 3 s measurement time suggesting a monoexponential decay in this time interval. Since a few neurones exhibited very little inactivation, the distribution of time constants (τ) was skewed and thus values were log transformed to yield a normal distribution. Again neither the repeated stimulation (F(2,74) = 0·46) nor the treatment (F(2,37) = 1·21, interaction F(4,74) = 1·39) affected the time constants. The time constant for inactivation was 4·4 s within the first stimulus (log τ = 0·64 ± 0·07), 4·9 s within the second stimulus (log τ = 0·69 ± 0·05) and 4·7 s within the third stimulus (log τ = 0·67 ± 0·07) (n = 40, each).
DISCUSSION
This study has shown that inward currents evoked by moderate heat stimuli (43–47°C) in nociceptive DRG neurones activate and deactivate rapidly. The currents inactivate with a time constant of 4–5 s during a constant heat stimulus, and they show pronounced tachyphylaxis at an interstimulus interval of 30 s. Neither omission of extracellular calcium, nor buffering of intracellular calcium, affected the inactivation or tachyphylaxis of heat-evoked currents.
Inactivation and tachyphylaxis of heat-evoked and capsaicin-induced inward currents
The voltage dependence of heat-evoked currents (Iheat) with a reversal potential near 0 mV indicates that heat-induced currents are carried through non-selective cation channels. This finding, as well as the outward rectification of Iheat are consistent with results previously described in cultured DRG neurones for whole-cell and single-channel currents (Cesare & McNaughton, 1996; Kirschstein et al. 1999; Nagy & Rang, 1999b) and in excised patches of VR1 transfected HEK293 cells (Tominaga et al. 1998). Inactivation of Iheat was observed for all membrane potentials tested in the present study. The outward rectification increased during inactivation as also reported for capsaicin-induced inward currents (ICaps) (Piper et al. 1999). Thus for both Iheat and ICaps, inactivation appears to be more pronounced for inward currents than for outward currents.
The finding that inactivation and tachyphylaxis of Iheat were independent of the extra- and intracellular calcium concentrations is in contrast to inactivation and tachyphylaxis of ICaps. Both inactivation and tachyphylaxis of ICaps depend on a rise in intracellular calcium and activation of calcium-dependent phosphatases such as calcineurin (Cholewinski et al. 1993; Docherty et al. 1996; Liu & Simon, 1996). If ICaps in DRG neurones is a current carried through the VR1 ion channel, channel closing by dephosphorylation of the VR1 protein may underly inactivation and tachyphylaxis. Since the capsaicin receptor VR1 is also a heat-sensitive ion channel (Caterina et al. 1997), inactivation of Iheat should be similar to that of ICaps and, therefore, depend on activation of calcium-dependent phosphatases. This suggestion is supported by the demonstration that sensitization of Iheat by bradykinin involves a phosphorylation step by PKCɛ (Cesare et al. 1999a). The molecular mechanisms of sensitization on the one hand and inactivation and/or tachyphylaxis on the other may therefore simply be complementary aspects of the same process, i.e. regulation of the heat sensitivity of an ion channel by phosphorylation (Cesare et al. 1999b). This prediction, however, was not supported by our study.
A recent study using a nominally calcium-free solution for repeated ramped heat stimuli reported that tachyphylaxis of Iheat was independent of extracellular calcium (Vyklický et al. 1999). In those experiments, calcium was present in the extracellular solution between stimuli; therefore the intracellular calcium concentration may have been increased by calcium released from intracellular stores and/or by activation of store-operated calcium channels. In fact, their original traces (Fig. 6 in Vyklický et al. 1999) display inward currents upon re-admission of extracellular calcium, similar to a typical current through store-operated calcium channels (for review see Holda et al. 1998). In our study, calcium-free extracellular solution was superfused throughout the repeated stimulation protocol, and in addition we used a rapid chelator intracellularly (BAPTA; Tsien, 1980). The tachyphylaxis under these conditions cannot be explained by a rise of intracellular calcium. Tachyphylaxis of inward currents evoked by zingerone or olvanil in nociceptive DRG neurones is also calcium independent, although both substances are assumed to act on vanilloid receptors, and their actions are blocked by the competitive vanilloid receptor antagonist capsazepine (Liu & Simon, 1998). Thus, there may be different vanilloid receptors or different modes of activation, and the actions of noxious heat may be more similar to actions of zingerone and olvanil than to those of capsaicin.
In HEK293 cells expressing the cloned VR1, inactivation and tachyphylaxis to heat were independent of calcium, whereas capsaicin-induced inactivation and tachyphylaxis were absent in calcium-free media (Tominaga et al. 1998). Thus the fact that inactivation of ICaps in DRG neurones was mostly calcium dependent, whereas that of Iheat was not, does not contradict the model that both may be carried through VR1. The potency of capsazepine to block currents through VR1 was smaller for activation by heat than by capsaicin (Caterina et al. 1997; Tominaga et al. 1998). This may explain the tenfold lower potency of capsazepine to antagonize Iheat compared with ICaps in DRG neurones (Kirschstein et al. 1999; Nagy & Rang, 1999b). The classification of two types of vanilloid receptors (R-type: resiniferatoxin-like, and C-type: capsaicin-like) had been based on different structure-activity relations for ligand binding and channel opening (Acs et al. 1996). Recently, however, it was reported that VR1 exhibits the properties of both R-type and C-type vanilloid receptors (Szallasi et al. 1999). Thus, whereas there is substantial evidence for the existence of multiple classes of vanilloid receptors and multiple heat-transduction pathways, the properties of VR1 have also proved to be quite complex and this polymodal sensor in the nociceptive transduction pathway explains many findings that had previously been attributed to separate pathways (Caterina & Julius, 1999).
Potential calcium-independent mechanisms of inactivation of heat-evoked currents
While a spontaneous dephosphorylation after short heat applications as used in our experiments is unlikely to be due to the high activation energy of phosphate bonds, it has been shown that heat shocks of a similar temperature (43–45°C) but much longer duration (10 min) caused a selective dephosphorylation of several proteins (Ohta et al. 1989; Henle et al. 1990). Extended exposure of cells, including DRG neurones (Amin et al. 1995), to elevated temperatures induces enhanced synthesis of heat-shock proteins which leads to a reduced sensitivity to subsequent stress. The heat-shock protein Hsp72 has been found to stimulate dephosphorylation of stress kinase JNK (Volloch et al. 1999). This process contributes to protecting cells from apoptosis. Furthermore, it is conceivable that the activity of phosphatases like phosphatase 1, which is tonically active in dorsal root ganglion neurones (Dolphin, 1992), is rapidly enhanced by an increase in temperature.
There are examples of other channel-linked receptors like the nicotinic acetylcholine receptor (see e.g. Fig. 7 in Mulle et al. 1992) and the NMDA receptor where desensitization is partly calcium independent. In the case of the NMDA receptor-channel complex a calcium-independent mechanism of inactivation is related to an allosteric modulation at the glycine binding site, which has to be occupied to open the channel (Mayer et al. 1989; Parsons et al. 1993). Binding of NMDA or glutamate at the agonist binding site reduces the affinity of the glycine site. As a consequence, glycine diffuses away, leading to a reduction in current through the NMDA receptor channel during constant stimulation with an agonist. Similar allosteric interactions are conceivable between the heat-sensing element and the capsaicin binding site of VR1.
Although subunit composition and locations of temperature sensor and capsaicin binding site of VR1 are not known at present, the predicted membrane topology of the heat-sensitive ion channel VR1 (Caterina et al. 1997) is comparable with that of voltage-gated channels (6-transmembrane region channels), and both classes of channels are gated by physical stimuli. Thus, one may speculate, whether the inactivation mechanisms of Iheat could be similar to those of voltage-gated sodium channels, in that temperature changes may directly cause reversible folding and de-folding of the channel protein (two processes: fast opening, slow inactivation) leading to transitions between three different states of the channel (closed, opened, inactivated).
Comparison with in vivo data from nociceptive afferents
The heat-evoked action potential discharges of C-fibre nociceptors adapt with a time constant of about 2·5 s (Treede, 1995) and those of type II A-fibre nociceptors with a time constant of about 2·8 s (Treede et al. 1998). These rates of adaptation are slow, but the discharge rate may decrease by up to 90 % during a heat stimulus of 30 s duration (Meyer & Campbell, 1981). The constant pain elicited by such a stimulus is explained by central summation and/or recruitment of type I A-fibre nociceptors, which exhibit a slow heat-transduction mechanism with a high threshold (Meyer & Campbell, 1981). The inactivation rate of Iheat in our data (time constant 4–5 s) was somewhat slower than that of adaptation of action potential discharges in vivo. In their recent review paper, McNaughton’s group show an example of inactivation of Iheat that also has a time constant of about 4·5 s during the first 3 s (Cesare et al. 1999b). Other studies on Iheat either used stimulus durations that were too short to determine inactivation or used ramp stimuli. While we cannot exclude that additional mechanisms in the transformation process of action potential generation contribute to nociceptor adaptation, inactivation of Iheat may fully account for nociceptor adaptation to heat stimuli in vivo. Interestingly, adaptation in corneal afferents of the cat is also independent of calcium, similar to the inactivation of Iheat (Belmonte et al. 1994).
Suppression of nociceptor discharges across repeated stimulation at interstimulus intervals of 30 s amounts to ∼60 % at the second and ∼75 % at the third stimulus in A-fibres in vivo (Treede et al. 1998). Heat-evoked discharge in C-fibres of monkeys in vivo was suppressed by ∼60 % across the first three stimuli whereas, similar to tachyphylaxis of Iheat in the present study, further stimuli displayed only weak suppression (LaMotte & Campbell, 1978). Pain ratings in human subjects decreased across repeated heat stimuli (LaMotte & Campbell, 1978; Adriaensen et al. 1984). Tachyphylaxis of Iheat (40–50 % across three stimuli) provides at least a partial basis for suppression of C-fibre discharge, which in turn was suggested to explain the characteristics of concomitant reduction of pain sensations in humans.
The processes of inactivation and tachyphylaxis may be two manifestations of a single mechanism. From in vivo experiments it is known that adaptation affects the dynamic response more than the static response (Treede, 1995): after repetitive heat stimulation the dynamic response disappears but the static response remains. Under the in vitro conditions in our experiments, however, the degree of inactivation remained nearly the same during repetitive heat stimulation. Thus inactivation and tachyphylaxis in vitro seem to be at least partly independent of each other.
In conclusion, inward currents elicited by moderately noxious heat stimuli in small DRG neurones inactivate during prolonged stimulation with time constants of 4–5 s and show signs of tachyphylaxis when activated repetitively. Neither inactivation nor tachyphylaxis depends on extra- or intracellular calcium. These phenomena provide at least a partial explanation for the adaptation and suppression of action potential discharges of primary nociceptive afferents that are observed in response to heat stimuli applied in vivo.
Acknowledgments
This study contains essential parts of the MD thesis of S. Schwarz which will be submitted to the Faculty of Medicine, Johannes Gutenberg University Mainz, Germany. The authors appreciate the technical assistance of G. Günther. This research was supported by the Deutsche Forschungsgemeinschaft Grant Tr236/11-1.
References
- Acs G, Lee J, Marquez VE, Blumberg PM. Distinct structure-activity relations for stimulation of 45Ca uptake and for high affinity binding in cultured rat dorsal root ganglion neurons and dorsal root ganglion membranes. Molecular Brain Research. 1996;35:173–182. doi: 10.1016/0169-328x(95)00204-6. [DOI] [PubMed] [Google Scholar]
- Adriaensen H, Gybels J, Handwerker HO, van Hees J. Suppression of C-fibre discharges upon repeated heat stimulation may explain characteristics of concomitant pain sensations. Brain Research. 1984;302:203–211. doi: 10.1016/0006-8993(84)90232-4. [DOI] [PubMed] [Google Scholar]
- Amin V, Cumming DVE, Coffin RS, Latchman DS. The degree of protection provided to neuronal cells by a pre-conditioning stress correlates with the amount of heat shock protein 70 it induces and not with the similarity of the subsequent stress. Neuroscience Letters. 1995;200:85–88. doi: 10.1016/0304-3940(95)12074-e. [DOI] [PubMed] [Google Scholar]
- Belmonte C, Gallar J, Lopez-Briones LG, Pozo MA. Polymodality in nociceptive neurons: experimental models of chemotransduction. In: Urban L, editor. NATO ASI Series, Cellular Mechanisms of Sensory Processing. H 79. Berlin: Springer; 1994. pp. 87–117. [Google Scholar]
- Caterina MJ, Julius D. Sense and specificity: a molecular identity for nociceptors. Current Opinion in Neurobiology. 1999;9:525–530. doi: 10.1016/S0959-4388(99)00009-4. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature. 1999;398:436–441. doi: 10.1038/18906. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, Mcnaughton PA. Specific involvement of PKC-ɛ in sensitization of the neuronal response to painful heat. Neuron. 1999a;23:617–624. doi: 10.1016/s0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- Cesare P, Mcnaughton P. A novel heat-activated current in nociceptive neurons and its sensitization by bradykinin. Proceedings of the National Academy of Sciences of the USA. 1996;93:15435–15439. doi: 10.1073/pnas.93.26.15435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare P, Moriondo A, Vellani V, Mcnaughton PA. Ion channels gated by heat. Proceedings of the National Academy of Sciences of the USA. 1999b;96:7658–7663. doi: 10.1073/pnas.96.14.7658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholewinski A, Burgess GM, Bevan S. The role of calcium in capsaicin-induced desensitization in rat cultured dorsal root ganglion neurons. Neuroscience. 1993;55:1015–1023. doi: 10.1016/0306-4522(93)90315-7. [DOI] [PubMed] [Google Scholar]
- Coghill RC, Mayer DJ, Price DD. Wide dynamic range but not nociceptive-specific neurons encode multidimensional features of prolonged repetitive heat pain. Journal of Neurophysiology. 1993;69:703–716. doi: 10.1152/jn.1993.69.3.703. [DOI] [PubMed] [Google Scholar]
- Docherty RJ, Yeats JC, Bevan S, Boddeke HWGM. Inhibition of calcineurin inhibits the desensitization of capsaicin evoked currents in cultured dorsal root ganglion neurones from adult rats. Pflügers Archiv. 1996;431:828–837. doi: 10.1007/s004240050074. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. The effect of phosphatase inhibitors and agents increasing cyclic-AMP-dependent phosphorylation on calcium channel currents in cultured rat dorsal root ganglion neurones: interaction with the effect of G protein activation. Pflügers Archiv. 1992;421:138–145. doi: 10.1007/BF00374820. [DOI] [PubMed] [Google Scholar]
- Gold MS, Dastmalchi S, Levine JD. Co-expression of nociceptor properties in dorsal root ganglion neurons from the adult rat in vitro. Neuroscience. 1996;71:265–275. doi: 10.1016/0306-4522(95)00433-5. [DOI] [PubMed] [Google Scholar]
- Greene LC, Hardy JD. Adaptation of thermal pain in the skin. Journal of Applied Physiology. 1962;17:693–696. doi: 10.1152/jappl.1962.17.4.693. [DOI] [PubMed] [Google Scholar]
- Guenther S, Reeh PW, Kress M. Rises in [Ca2+]i mediate capsaicin- and proton-induced heat sensitization of rat primary nociceptive neurons. European Journal of Neuroscience. 1999;11:3143–3150. doi: 10.1046/j.1460-9568.1999.00734.x. [DOI] [PubMed] [Google Scholar]
- Handwerker HO, Anton F, Reeh PW. Discharge patterns of afferent cutaneous nerve fibers from the rat’s tail during prolonged noxious mechanical stimulation. Experimental Brain Research. 1987;65:493–504. doi: 10.1007/BF00235972. [DOI] [PubMed] [Google Scholar]
- Henle KJ, Norris JS, Lumpkin CK. Heat-induced protein dephosphorylation in Chinese hamster ovary cells. Biomedica Biochimica Acta. 1990;49:35–44. [PubMed] [Google Scholar]
- Holda JR, Klishin A, Sedova M, Hüser J, Blatter LA. Capacitative calcium entry. News in Physiological Sciences. 1998;13:157–163. doi: 10.1152/physiologyonline.1998.13.4.157. [DOI] [PubMed] [Google Scholar]
- Kirschstein T, Büsselberg D, Treede R-D. Coexpression of heat-evoked and capsaicin-evoked inward currents in acutely dissociated rat dorsal root ganglion neurons. Neuroscience Letters. 1997;231:33–36. doi: 10.1016/s0304-3940(97)00533-8. [DOI] [PubMed] [Google Scholar]
- Kirschstein T, Greffrath W, Büsselberg D, Treede R-D. Inhibition of rapid heat responses in nociceptive primary sensory neurons of the rat by vanilloid receptor antagonists. Journal of Neurophysiology. 1999;82:2853–2860. doi: 10.1152/jn.1999.82.6.2853. [DOI] [PubMed] [Google Scholar]
- Lamotte RH, Campbell JN. Comparison of responses of warm and nociceptive C-fiber afferents in monkey with human judgments of thermal pain. Journal of Neurophysiology. 1978;41:509–528. doi: 10.1152/jn.1978.41.2.509. [DOI] [PubMed] [Google Scholar]
- Liu L, Lo Y-C, Chen I-J, Simon SA. The responses of rat trigeminal ganglion neurons to capsaicin and two nonpungent vanilloid receptor agonists, olvanil and glyceryl nonamide. Journal of Neuroscience. 1997;17:4101–4111. doi: 10.1523/JNEUROSCI.17-11-04101.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Simon SA. Capsaicin-induced currents with distinct desensitization and Ca2+ dependence in rat trigeminal ganglion cells. Journal of Neurophysiology. 1996;75:1503–1514. doi: 10.1152/jn.1996.75.4.1503. [DOI] [PubMed] [Google Scholar]
- Liu L, Simon SA. The influence of removing extracellular Ca2+ in the desensitization responses to capsaicin, zingerone and olvanil in rat trigeminal ganglion neurons. Brain Research. 1998;809:246–252. doi: 10.1016/s0006-8993(98)00853-1. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Vyklický L, Jr, Clements J. Regulation of NMDA receptor desensitization in mouse hippocampal neurons by glycine. Nature. 1989;338:425–427. doi: 10.1038/338425a0. [DOI] [PubMed] [Google Scholar]
- Mendell LM, Wall PD. Responses of single dorsal cord cells to peripheral cutaneous unmyelinated fibres. Nature. 1965;206:97–99. doi: 10.1038/206097a0. [DOI] [PubMed] [Google Scholar]
- Meyer RA, Campbell JN. Myelinated nociceptive afferents account for the hyperalgesia that follows a burn to the hand. Science. 1981;213:1527–1529. doi: 10.1126/science.7280675. [DOI] [PubMed] [Google Scholar]
- Meyer RA, Campbell JN, Raja SN. Peripheral neural mechanisms of nociception. In: Wall PD, Melzack R, editors. Textbook of Pain. Edinburgh: Churchill Livingstone; 1994. pp. 13–44. [Google Scholar]
- Meyer RA, Walker RE, Mountcastle VB. A laser stimulator for the study of cutaneous thermal and pain sensations. IEEE Transactions on Biomedical Engineering. 1976;BME-23:54–60. doi: 10.1109/tbme.1976.324616. [DOI] [PubMed] [Google Scholar]
- Mulle C, Choquet D, Korn H, Changeux J-P. Calcium influx through nicotinic receptor in rat central neurons: its relevance to cellular regulation. Neuron. 1992;8:135–143. doi: 10.1016/0896-6273(92)90115-t. [DOI] [PubMed] [Google Scholar]
- Nagy I, Rang H. Noxious heat activates all capsaicin-sensitive and also a sub-population of capsaicin-insensitive dorsal root ganglion neurons. Neuroscience. 1999a;88:995–997. doi: 10.1016/s0306-4522(98)00535-1. [DOI] [PubMed] [Google Scholar]
- Nagy I, Rang HP. Similarities and differences between the responses of rat sensory neurons to noxious heat and capsaicin. Journal of Neuroscience. 1999b;19:10647–10655. doi: 10.1523/JNEUROSCI.19-24-10647.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta Y, Nishida E, Sakai H, Miyamoto E. Dephosphorylation of cofilin accompanies heat shock-induced nuclear accumulation of cofilin. Journal of Biological Chemistry. 1989;264:16143–16148. [PubMed] [Google Scholar]
- Parsons CG, Zong X, Lux HD. Whole cell and single channel analysis of the kinetics of glycine-sensitive N-methyl-D-aspartate receptor desensitization. British Journal of Pharmacology. 1993;109:213–221. doi: 10.1111/j.1476-5381.1993.tb13556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen M, Lamotte RH. Relationships between capsaicin sensitivity of mammalian sensory neurons, cell size and type of voltage gated Ca-currents. Brain Research. 1991;561:20–26. doi: 10.1016/0006-8993(91)90744-g. [DOI] [PubMed] [Google Scholar]
- Piper AS, Yeats JC, Bevan S, Docherty RJ. A study of the voltage dependence of capsaicin-activated membrane currents in rat sensory neurones before and after acute desensitization. The Journal of Physiology. 1999;518:721–733. doi: 10.1111/j.1469-7793.1999.0721p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DD, Hu JW, Dubner R, Gracely RH. Peripheral suppression of first pain and central summation of second pain evoked by noxious heat pulses. Pain. 1977;3:57–68. doi: 10.1016/0304-3959(77)90035-5. [DOI] [PubMed] [Google Scholar]
- Schmelz M, Forster C, Schmidt R, Ringkamp M, Handwerker HO, Torebjörk HE. Delayed responses to electrical stimuli reflect C-fiber responsiveness in human microneurography. Experimental Brain Research. 1995;104:331–336. doi: 10.1007/BF00242018. [DOI] [PubMed] [Google Scholar]
- Schneider W, Slugg RM, Turnquist BP, Meyer RA, Campbell JN. An electromechanical stimulator system for neurophysiological and psychophysical studies of pain. Journal of Neuroscience Methods. 1995;60:61–68. doi: 10.1016/0165-0270(94)00220-b. [DOI] [PubMed] [Google Scholar]
- Schwarz S, Greffrath W, Büsselberg D, Treede R-D. Inactivation and desensitization of heat-evoked inward currents in nociceptive primary sensory neurons of rats. Pflügers Archiv. 2000;439(suppl. 6):R335. doi: 10.1111/j.1469-7793.2000.00539.x. (Abstract) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra J, Campero M, Ochoa J, Bostock H. Activity-dependent slowing of conduction differentiates functional subtypes of C fibres innervating human skin. The Journal of Physiology. 1999;515:799–811. doi: 10.1111/j.1469-7793.1999.799ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szallasi A, Blumberg PM, Annicelli LL, Krause JE, Cortright DN. The cloned rat vanilloid receptor VR1 mediates both R-type binding and C-type calcium response in dorsal root ganglion neurons. Molecular Pharmacology. 1999;56:581–587. doi: 10.1124/mol.56.3.581. [DOI] [PubMed] [Google Scholar]
- Thalhammer JG, Raymond SA, Popitzbergez FA, Strichartz GR. Modality-dependent modulation of conduction by impulse activity in functionally characterized single cutaneous afferents in the rat. Somatosensory and Motor Research. 1994;11:243–257. doi: 10.3109/08990229409051392. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Treede R-D. Peripheral acute pain mechanisms. Annals of Medicine. 1995;27:213–216. doi: 10.3109/07853899509031961. [DOI] [PubMed] [Google Scholar]
- Treede R-D, Meyer RA, Campbell JN. Myelinated mechanically insensitive afferents from monkey hairy skin: heat response properties. Journal of Neurophysiology. 1998;80:1082–1093. doi: 10.1152/jn.1998.80.3.1082. [DOI] [PubMed] [Google Scholar]
- Treede R-D, Meyer RA, Raja SN, Campbell JN. Evidence for two different heat transduction mechanisms in nociceptive primary afferents innervating monkey skin. The Journal of Physiology. 1995;483:747–758. doi: 10.1113/jphysiol.1995.sp020619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY. New calcium indicators and buffers with high selectivity against magnesium and protons: design, synthesis, and properties of prototype structures. Biochemistry. 1980;19:2396–2404. doi: 10.1021/bi00552a018. [DOI] [PubMed] [Google Scholar]
- Volloch V, Gabai VL, Rits S, Sherman MY. ATPase activity of the heat shock protein hsp72 is dispensable for its effects on dephosphorylation of stress kinase JNK and on heat-induced apoptosis. FEBS Letters. 1999;461:73–76. doi: 10.1016/s0014-5793(99)01428-3. [DOI] [PubMed] [Google Scholar]
- Vyklický L, Vlachová V, Vitásková Z, Dittert I, Kabát M, Orkand RK. Temperature coefficient of membrane currents induced by noxious heat in sensory neurones in the rat. The Journal of Physiology. 1999;517:181–192. doi: 10.1111/j.1469-7793.1999.0181z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Dib-Hajj S, Cummins TR, Black JA. Sodium channels and pain. Proceedings of the National Academy of Sciences of the USA. 1999;96:7635–7639. doi: 10.1073/pnas.96.14.7635. [DOI] [PMC free article] [PubMed] [Google Scholar]