

Abstract
Cat ventricular myocytes loaded with [Ca2+]i- and pHi-sensitive probes were used to examine the subcellular mechanism(s) of the Ang II-induced positive inotropic effect. Ang II (1 μM) produced parallel increases in contraction and Ca2+ transient amplitudes and a slowly developing intracellular alkalisation. Maximal increases in contraction amplitude and Ca2+ transient amplitude were 163 ± 22 and 43 ± 8 %, respectively, and occurred between 5 and 7 min after Ang II administration, whereas pHi increase (0·06 ± 0·03 pH units) became significant only 15 min after the addition of Ang II. Furthermore, the inotropic effect of Ang II was preserved in the presence of Na+-H+ exchanger blockade. These results indicate that the positive inotropic effect of Ang II is independent of changes in pHi.
Similar increases in contractility produced by either elevating extracellular [Ca2+] or by Ang II application produced similar increases in peak systolic Ca2+ indicating that an increase in myofilament responsiveness to Ca2+ does not participate in the Ang II-induced positive inotropic effect.
Ang II significantly increased the L-type Ca2+ current, as assessed by using the perforated patch-clamp technique (peak current recorded at 0 mV: -1·88 ± 0·16 pA pF−1 in control vs. -3·03 ± 0·20 pA pF−1 after 6-8 min of administration of Ang II to the bath solution).
The positive inotropic effect of Ang II was not modified in the presence of either KB-R7943, a specific blocker of the Na+-Ca2+ exchanger, or ryanodine plus thapsigargin, used to block the sarcoplasmic reticulum function.
The above results allow us to conclude that in the cat ventricle the Ang II-induced positive inotropic effect is due to an increase in the intracellular Ca2+ transient, an enhancement of the L-type Ca2+ current being the dominant mechanism underlying this increase.
In the last few years several laboratories have explored the subcellular mechanisms of the positive inotropic effect of angiotensin II (Ang II). The outcome of this work turned out to be largely controversial. Although part of the controversy might arise from species differences (Ishihata & Endoh, 1995), opposite results were also reported in the same species. Experiments by Ikenouchi et al. (1994) in isolated rabbit myocytes indicated that the positive inotropic effect of Ang II was exclusively mediated by an increase in the myofilament responsiveness to Ca2+. In this study, no increase in the intracellular Ca2+ transients was detected. These results are in sharp contrast to those obtained later, showing that Ang II did increase the intracellular Ca2+ transient in rabbit ventricular myocytes (Skolnick et al. 1998). Results from Watanabe & Endoh (1998), also in rabbit heart, suggested that the positive inotropic effect of Ang II was due to an increase in both [Ca2+]i and myofilament responsiveness to Ca2+.
The subcellular mechanisms responsible for the described increase in either [Ca2+]i or the myofilament responsiveness to Ca2+ are also elusive. The L-type Ca2+ current (ICa) has been shown to be either increased (Kass & Blair, 1981; Allen et al. 1988; Kaibara et al. 1994), decreased (Habuchi et al. 1995) or unchanged (Ikenouchi et al. 1994; Ai et al. 1998). Moreover, whereas Kaibara et al. (1994) concluded that the enhancement in ICa was due to the increase in pHi, Ikenouchi et al. (1994) described in the same species an increase in pHi without significant changes in ICa. Using pharmacological tools, Talukder & Endoh (1997) concluded that the influx of Ca2+ through the L-type Ca2+ channels was one of the main mechanisms by which Ang II increases [Ca2+]i. However more recent experiments of the same group, working with the same species, suggested that the Na+-Ca2+ exchanger was the mechanism responsible for the Ang II-induced increase in [Ca2+]i (Fujita & Endoh, 1999).
Referring to the Ang II-induced increase in myofilament responsiveness to Ca2+, the pHi dependence of this mechanism (Ikenouchi et al. 1994) has been challenged by the finding that the positive inotropic effect of Ang II was also present in a physiological buffer (HCO3−/CO2) in which Ang II does not produce significant changes in pHi (Mattiazzi et al. 1997).
Considering the discrepant results summarised above, the present study was directed to examining in the cat ventricle the effect of Ang II on the fundamental events of excitation- contraction coupling, i.e. calcium transients, calcium currents, SR Ca2+ release and Na+-Ca2+ exchange, in an attempt to comprehensively answer the following questions: Is the positive inotropic effect of Ang II due to an increase in [Ca2+]i or to an increase in myofilament responsiveness to Ca2+? In any of these cases, what is (or are) the underlying mechanism(s) involved?
METHODS
Myocyte isolation
All experiments were performed in accordance with the guidelines for Animal Care of the Scientific Committee of the University of La Plata School of Medicine. Cats were anaesthetised by intra-peritoneal injection of sodium pentobarbitone (35 mg (kg body weight)−1). Myocytes were isolated according to the technique previously described (Aiello et al. 1998; Morgan et al. 1999) with some modifications. Briefly, the hearts were attached via the aorta to a cannula, excised and mounted in a Langendorff apparatus. They were then retrogradly perfused at 37°C at a constant perfusion pressure of 70-80 mmHg with Krebs-Henseleit (K-H) solution of the following composition (mm): 146.2 NaCl, 4.7 KCl, 1.35 CaCl2, 10.0 Hepes, 0.35 NaH2PO4, 1.05 MgSO4., 10.0 glucose (pH adjusted to 7.4 with NaOH). The solution was continuously bubbled with 100 % O2. After a stabilisation period of 4 min, the perfusion was switched to a nominally Ca2+-free K-H solution for 6 min. Hearts were then recirculated with collagenase (118 U ml−1), 0.1 mg ml−1 pronase and 1 % bovine serum albumin (BSA), in K-H solution containing 50 μm CaCl2. Perfusion continued until the hearts became flaccid (15-25 min). They were then removed from the perfusion apparatus by cutting at the atria-ventricular junction. The desegregated myocytes were separated from the undigested tissue and rinsed several times with a K-H solution containing 1 % BSA and 500 μm CaCl2. After each wash, myocytes were left for sedimentation for 10 min. Myocytes were kept in K-H solution at room temperature (20-22°C) until use. Only rod-shaped myocytes with clear and distinct striations and an obvious marked shortening and relaxation on stimulation were used. Experiments were performed at room temperature.
Indo-1 fluorescence and cell shortening measurements
The isolated myocytes were loaded at room temperature with the cell-permeant acetoxymethyl ester (AM) form of indo-1 (17 μm for 9 min) according to the bulk method described by Spurgeon et al. (1990) and left for de-esterification for 45 min. Cells were then placed on the stage of an inverted microscope (Nikon Diaphot 200) adapted for epifluorescence. Myocytes were continuously superfused with K-H solution (pH 7.4) at a constant flow of 1 ml min−1 and field stimulated via two platinum electrodes on either side of the bath (square waves, 2 ms duration and 20 % above threshold) at a constant frequency of 30 beats min−1. The excitation light was centred at 350 nm and the fluorescence emitted by the cell was recorded at 410 and 490 nm. Background fluorescence was subtracted from each signal before obtaining the 410 to 490 nm fluorescence ratio. The diastolic fluorescence ratio was measured as the mean value over a 100 ms period after the twitch was completed. Systolic fluorescence ratio was determined directly from the peak of the recorded ratio.
The stage of the microscope was illuminated with red light (640-750 nm) through its normal bright-field illumination optics to allow simultaneous measurements of fluorescence and shortening. Resting cell length and cell shortening were measured by a video-based motion detector (Crescent Electronics, Salt Lake City, UT, USA) and stored by software for an off-line analysis. Cell shortening was simultaneously monitored on a two-channel chart recorder (Gould RS 3200).
Indo-1-loaded myocytes were used to assess myofilament responsiveness to Ca2+ according to the previously described method (Shah et al. 1994). In brief, this method consists in plotting the instantaneous myocyte length versus simultaneously measured indo-1 fluorescence during single twitch contractions (phase-plane plot). In mechanically unloaded cells stimulated to contract from slack length, a dynamic equilibrium is achieved between cell relengthening and the decline in cytosolic Ca2+ during twitch relaxation. This implies that for interventions that increase contractility without changing myofilament responsiveness to Ca2+ (e.g. an increase in extracellular calcium, [Ca2+]o) the phase-plane plots describe a common trajectory during the relaxation phase. In contrast, perturbations that decrease myofilament responsiveness to Ca2+ (e.g. acidosis) shift the relaxation trajectory downward and rightward whereas interventions that increase myofilament responsiveness to Ca2+ (e.g. Ca2+-sensitizing drugs) have the opposite effect. The position of the trajectory of the relaxation phase reflects therefore the relative myofilament responsiveness to Ca2+.
pHi measurements
After enzymatic isolation, myocytes were loaded with the membrane-permeant acetoxymethyl ester form of the fluorescent H+-sensitive indicator SNARF-1 AM. Cell suspensions (2 ml) were exposed to a final concentration of 4 μm SNARF-1 AM and 0.6 % v/v DMSO. After 10 min, the myocytes were gently centrifuged for 2 min and resuspended in Hepes buffer and stored at room temperature until use. pHi and cell length were monitored on the stage of a modified inverted microscope, as previously described (Blank et al. 1992). After excitation at 530 ± 5 nm, the ratio of SNARF-1 AM emission at 590 ± 5 nm to that of 640 ± 5 nm was used as a measure of pHi according to an in vivo calibration, obtained from SNARF-1 AM-loaded myocytes exposed to solutions of varying pH values containing 140 mm KCl, 20 μm nigericin, 1 μm valinomycin and 1 μm carbonyl cyanide p-(trifluoromethoxy)-phenylhydrazone at room temperature.
To determine Na+-H+ exchanger activity, intracellular acidosis was induced by the wash-out (10 min) of a transient (3 min) exposure of cells to 15 mm NH4Cl (first acid pulse). This procedure was repeated after either a 5 or a 20 min exposure to Ang II (second acid pulse). The rate of pHi recovery obtained by fitting a linear function to the initial recovery from acidosis was used to indicate changes in Na+-H+ exchanger activity (Vandenberg et al. 1994). The reproducibility of pHi recovery after two consecutive NH4Cl pulses was established in control experiments.
Perforated-patch recordings
Isolated cat ventricular myocytes were placed in a perfusion chamber and superfused with bath solution at a flow rate of 1.5 ml min−1. The perforated-patch configuration of the patch-clamp technique (Korn et al. 1991; Aiello et al. 1998) was used for voltage-clamp recordings with a patch-clamp amplifier (Axopatch 200A, Axon Instruments, Foster City, CA, USA). Patch pipettes were pulled with a PP-83 puller (Narishige, Tokyo, Japan) and fire-polished with a MF-83 Microforge (Narishige) to a final resistance of 1-3 MΩ when filled with pipette solution. The tip of the pipette was positioned above the cell, and its potential and capacitance were nullified. Whole-cell currents (filtered at 1 kHz) were digitally recorded directly to hard disk via an analog-to-digital converter (Digidata 1200, Axon Instruments) interfaced with an IBM clone computer running pCLAMP and Axotape software (Axon Instruments). Data analysis was performed with pCLAMP (Clampfit, Axon Instruments).
Voltage-clamp depolarising pulses (250 ms) were delivered at 0.2 Hz. A holding potential of -80 mV was used in all protocols to prevent slow inactivation and to minimise current rundown (McDonald et al. 1994). A 500 ms prepulse to -40 mV, used to inactivate sodium channels and potential T-type calcium channels, preceded the depolarizing test pulses to different potentials. Under the present recording conditions, no whole-cell currents were detected in the absence of Ca2+o (not shown). Nystatin produced good intracellular access after 15-20 min of seal formation. The calcium current amplitude was measured as peak inward current with reference to the current measured at the end of the test pulse. For each cell, capacitative current was recorded to determine the membrane capacitance and the currents were normalised for cell capacitance. The average cell capacitance was 148.7 ± 7.7 pF (n = 14).
In some experiments the perforated-patch configuration of the patch-clamp technique was used in combination with simultaneous measurements of cell length performed as described above. In these experimental groups the Ang II-induced increase in cell shortening was lower than in the other groups presented in this paper. The reason for this difference is not apparent to us. One possible cause might be the different experimental conditions used, i.e. perforated patch vs. intact myocytes.
The superfusion medium used to measure ICa had the following composition (mm): CsCl 5, NaCl 133, MgCl2 1, MgSO4 1.2, Hepes 10, tetraethylammonium chloride (TEA) 10, CaCl2 1.35 and glucose 10; pH was adjusted to 7.4 with NaOH. The internal (pipette) solution contained (mm): CsCl 140, MgCl2 1, NaCl 10, EGTA 1, Hepes 10 and nystatin 0.4 mg ml−1; pH was adjusted to 7.2 with NaOH. The superfusion medium used to measure action potentials and cell length had the following composition (mm): NaCl 138, MgCl2 1, MgSO4 1.2, Hepes 10, CaCl2 1.35 and glucose 10; pH was adjusted to 7.4 with NaOH. The pipette solution used in these experiments contained (mm): KCl 140, MgCl2 1, NaCl 10, EGTA 1, Hepes 10 and nystatin 0.4 mg ml−1; pH was adjusted to 7.2 with NaOH.
Materials
Collagenase type B was purchased from Worthington Biochemical Corp. (Lakewood, NJ, USA); pronase from Boerhinger Mannheim Corp. GmbH (Mannheim, Germany); BSA (essentially fatty acid free), thapsigargin and angiotensin II from Sigma Chemical Co. (St Louis, MO, USA); indo-1 AM and SNARF-1 AM from Molecular Probes Inc. (Eugene, OR, USA); and KB-R7943 (2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl]isothiourea methanesulphonate) from Kanebo (Osaka, Japan). All other chemicals were of the purest reagent grade available.
Statistics
All data are presented as means ±s.e.m. Comparisons within groups were assessed by Student’s paired or unpaired t test as appropriate. ANOVA was used when required as indicated in the text. A value of P < 0.05 was taken to indicate statistical significance.
RESULTS
Effect of Ang II on contraction and pHi
The effect of 1 μm Ang II on contraction and pHi was assessed in electrically stimulated cardiac myocytes. This concentration of Ang II was chosen based on the results of pilot experiments which indicated that the maximal positive inotropic effect of Ang II occurred in the concentration range of 0.5-1 μm, in agreement with previous reports (Koch-Weser, 1965; Ishihata & Endoh, 1993; Mattiazzi et al. 1997).
Figure 1A shows a representative example of the effect of Ang II on the unloaded myocyte contraction. Ang II induced a positive inotropic response, consisting in an initial increase in contraction that peaked between 5 and 7 min, followed by a slow decay that reached steady state after 20 min of incubation with the drug. The middle panel shows the individual twitch contractions in control and at 5 and 20 min after the addition of Ang II. The lower panel of Fig. 1A depicts a typical tracing of the effect of Ang II on pHi monitored continuously during 20 min. Note that 5 min after Ang II administration the positive inotropic effect had reached its maximum whereas pHi was not affected.
Figure 1. Effect of Ang II on contraction and pHi.
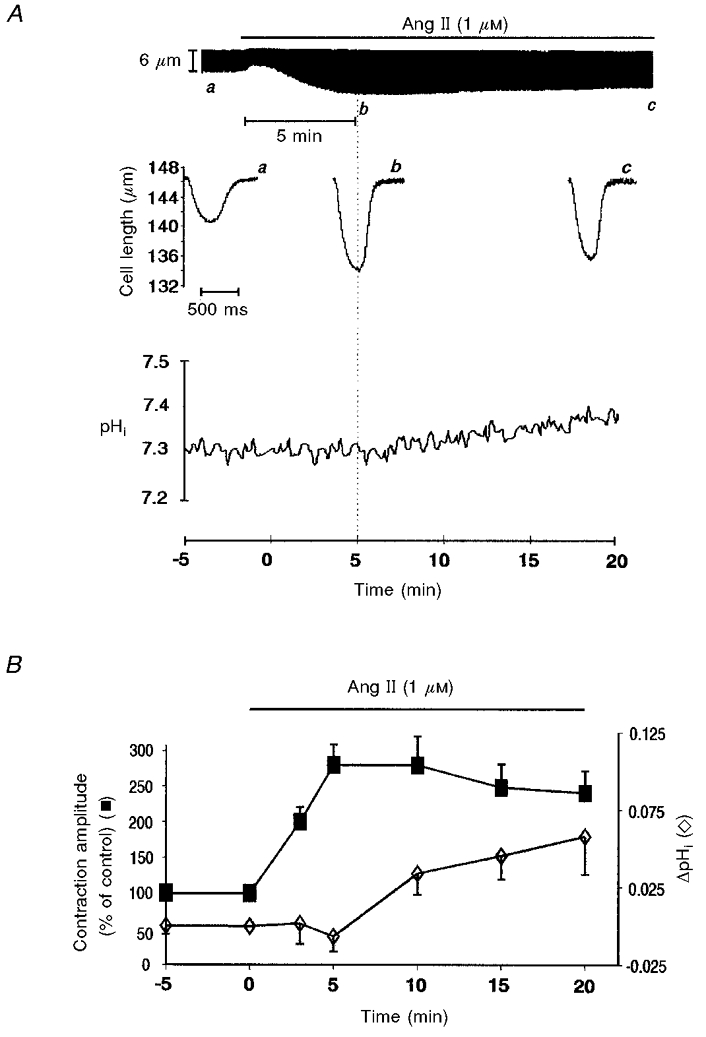
A, upper panel, a typical continuous recording of cell length showing the time course of the Ang II-induced positive inotropic effect. Middle panel, individual twitch contractions at the times indicated by the letters a–c on the continuous chart recording. Lower panel, effect of Ang II on pHi. Ang II produced a slow increase in pHi. B, the overall results of these experiments (n = 9) indicating the temporal dissociation between the positive inotropic and alkalising effect of Ang II. The Ang II-induced positive inotropic effect peaked before any significant change in pHi (ΔpHi) could be detected. The increase in pHi became significant when the positive inotropic effect of Ang II was already fading. Data points are means of five successive pHi and contraction amplitude measurements (sampling rate 0.5 Hz) from each experiment at every given time. Basal pHi value before Ang II administration was 7.31 ± 0.05.
Figure 1B shows the overall results of these experiments indicating the time course of the effect of Ang II on contraction amplitude and pHi. Ang II induced a maximal increase in contraction amplitude of 163 ± 22 % before an increase in pHi became significant. The increase in pHi became statistically significant only after 15 min of administration of the drug reaching a maximal value of 0.06 ± 0.03 pH units after 20 min. At this time the positive inotropic effect of Ang II diminished from its peak value to 129 ± 14 % of control.
The increase in pHi produced by Ang II in Hepes buffer is due to the activation of the Na+-H+exchanger (Matsui et al. 1995; Camilión de Hurtado et al. 1998; Gunasegaram et al. 1999). To further explore the possible dependence of the Ang II-induced positive inotropic effect on the intracellular alkalisation two additional protocols were performed. (1) We examined, using the NH4Cl pulse procedure (see Methods), the activity of the Na+-H+ exchanger after 5 min of Ang II application, when the positive contractile response had reached its maximum level, and after 20 min, when the contractile response to Ang II had reached steady state. Figure 2A shows representative pHi recordings of two consecutive NH4Cl pulses that were superimposed for comparison. The profile of the second pulse, obtained after a 5 min incubation period with Ang II, was similar to that of the first (control) pulse, indicating that after 5 min, Ang II had still not produced activation of the Na+-H+ exchanger. Furthermore, there was no significant difference in the rate of pHi recovery from acidosis between the first (control) pulse (ΔpHi/Δt: 0.082 ± 0.02 pH units min−1) and the second (Ang II 5 min) pulse (0.084 ± 0.03 pH units min−1). However, when the cells were pretreated with Ang II for 20 min prior to the second pulse, pHi recovery from acidosis was accelerated (Fig. 2B) such that the rate of pHi recovery from acidosis was significantly increased from 0.075 ± 0.02 pH units min−1 in the first (control) pulse to 0.11 ± 0.01 pH units min−1 in the second (Ang II 20 min) pulse. (2) The effect of Ang II on contraction amplitude was examined in the presence and in the absence of the Na+-H+ exchanger blocker HOE 642 (HOE) (10 and 50 μm). Preliminary experiments showed that 10 μm HOE produced a significant inhibition of the rate of pHi recovery from an acid load (results not shown). This indicates that the concentrations of HOE used were enough to inhibit the activity of the Na+-H+ exchanger. HOE did not produce significant changes in basal contractility values. Administration of HOE up to 50 μm had no effect when it was applied after the Ang II-induced positive inotropic effect had reached steady state (n = 9) (Fig. 3A). Similarly, preincubation of the cells with 10 μm of the blocker did not prevent the positive inotropic effect of Ang II (n = 5) (Fig. 3B). These findings in isolated myocytes are in complete agreement with a previously published report from our laboratory in multicellular preparations showing that in feline myocardium the positive inotropic and alkalinising effect of Ang II are not linked phenomena (Mattiazzi et al. 1997).
Figure 2. Effect of Ang II on the pHi recovery from intracellular acidosis.
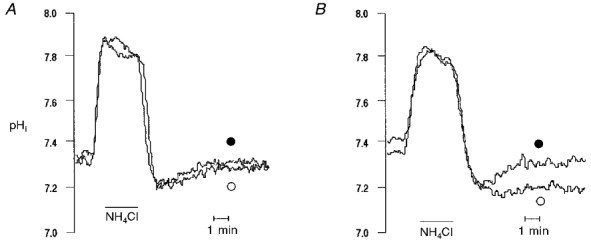
Representative pHi recordings obtained during two consecutive 15 mm NH4Cl pulses. ○, first pulse; •, second pulse performed after either 5 min (A) or 20 min (B) of exposure of cells to Ang II. The 5 min incubation period with Ang II failed to affect pHi recovery (n = 4) whereas a 20 min exposure to Ang II significantly accelerated recovery from acidosis (n = 5).
Figure 3. Failure of the Na+-H+ inhibitor HOE 642 to block the Ang II-induced positive inotropic effect.
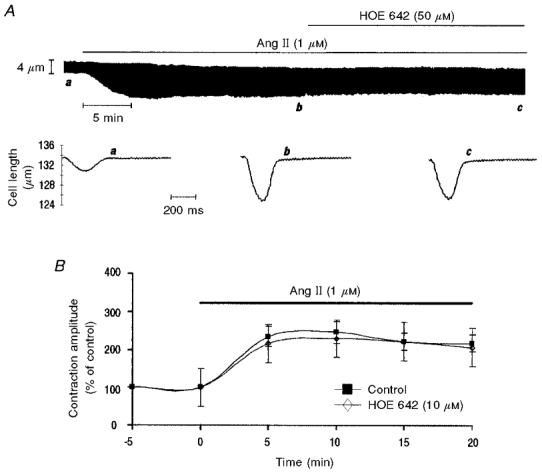
A, representative example of the effects of HOE added in the continued presence of Ang II. The sustained positive inotropic effect of Ang II was not affected upon addition of 50 μm HOE. B, overall results of the effect of Ang II alone and Ang II in the continued presence of HOE (10 μm) on contraction amplitude, expressed as a percentage of the control value. No significant difference in the positive inotropic effect elicited by Ang II was observed between control and HOE-treated cells.
Since the Ang II-induced positive inotropic effect cannot be attributed to an increase in myofilament responsiveness to Ca2+ due to intracellular alkalisation, alternative mechanisms that may be responsible for the Ang II-induced positive inotropic effect were examined, namely an increase in the intracellular Ca2+ transient and/or a pHi-independent increase in the myofilament responsiveness to Ca2+.
Effect of Ang II on myocyte contraction and intracellular calcium transient
Using indo-1-loaded cardiac myocytes we investigated the effect of 1 μm Ang II on contraction and the intracellular Ca2+ transient (CaiT). A representative example of the Ang II-induced positive inotropic effect and the associated CaiT are depicted in Fig. 4A. The initial increase in contraction amplitude elicited by Ang II followed by the slow decay was closely associated with a similar pattern of rise and slow decay in the CaiT amplitude and peak systolic [Ca2+]i. Neither diastolic cell length nor diastolic [Ca2+]i levels were significantly affected by Ang II. In some cells, the initial increase in contraction amplitude was preceded by a transient negative inotropic effect that was associated with a decrease in the CaiT. Figure 4B shows the overall results of these experiments (n = 5) indicating the time course of the effect of Ang II on contraction and CaiT amplitude. Similar results were obtained in four other cells using a lower Ang II concentration (0.5 μm) (data not shown). Considering the similar time course of the effect of Ang II on contraction and CaiT amplitude it seems reasonable to suggest that the Ang II-induced increase in [Ca2+]i constitutes the dominant mechanism for the positive inotropic effect observed.
Figure 4. Effect of Ang II on indo-1 transient and contraction amplitudes.
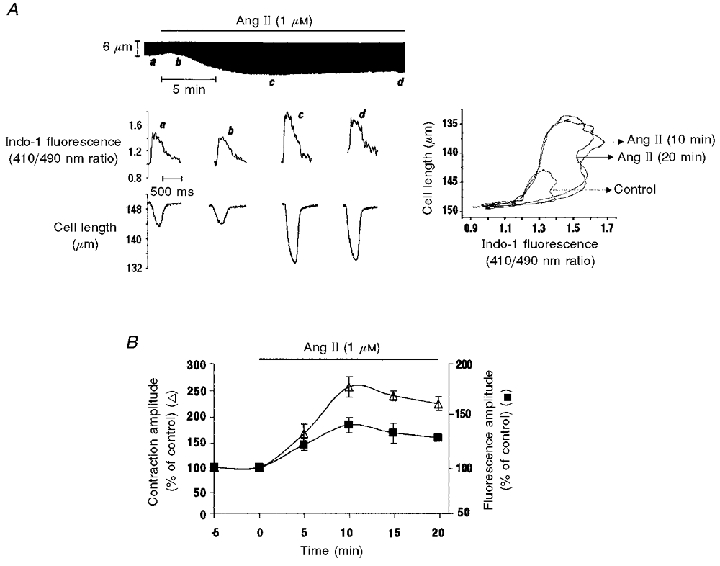
A, tracings of indo-1 fluorescence and cell length (lower panels) obtained at the times indicated by the letters in the continuous length recording (upper panel). The control condition (a) is followed by a transient decrease in contraction amplitude and in indo-1 fluorescence transient amplitude (b) and subsequently by a positive inotropic response that peaked and then decreased towards a steady state value. Trace c depicts the peak increase in contraction amplitude and trace d, the steady state contraction reached after 20 min of Ang II administration. These effects are associated with similar changes in indo-1 transient amplitude. On the right are the diagrams of indo-1 fluorescence vs. cell length (phase-plane plots) for contractions a, c and d, used to assess the myofilament responsiveness to Ca2+. The common trajectory during myocyte relaxation between the control and Ang II loops reflects the lack of effect of Ang II on myofilament responsiveness to Ca2+. B, the overall results of the time course of the effect of Ang II on contraction and indo-1 transient amplitude expressed as a percentage of the control value. Data are means ±s.e.m. of 5 cells. Both the Ang II-induced increase in contraction amplitude and the indo-1 transient increase and decay in parallel.
Ang II and myofilament response to Ca2+
To assess whether a pHi-independent increase in the myofilament responsiveness to calcium also contributes to the Ang II-induced positive inotropic effect, two different approaches were used. (1) Phase plane diagrams (loops) of the instantaneous cell length versus the simultaneous indo-1 fluorescence (see Methods) at control and after 10 and 20 min of Ang II application (peak and steady state of the Ang II positive inotropic effect, respectively) were compared as illustrated in Fig. 4A, right panel. The relaxation phase of the loops obtained from control twitches, and after 10 and 20 min of Ang II application have a common trajectory indicating that the response of the myofilaments to Ca2+ was not changed after Ang II administration. Similar phase-plane diagrams were observed in five indo-1-loaded cells exposed to Ang II (1 μm). (2) The second approach consisted in comparing the increase in peak systolic Ca2+ induced by elevating [Ca2+]o with the increase in peak systolic Ca2+ evoked by Ang II (1 μm). Special care was taken to ensure a similar increase in contraction amplitude induced by either high [Ca2+]o or Ang II. Figure 5 shows a representative example obtained following this protocol. For a similar increase in contraction amplitude induced by either high [Ca2+]o or Ang II there was also a similar increase in peak systolic Ca2+. The mean effects (n = 8) of either elevating [Ca2+]o (ranging from 3 to 5 mm) or Ang II (1 μm) administration on contraction and CaiT amplitude and kinetics are provided in Fig. 6A and in Table 1. Figure 6B compares the relationship between the increase in peak systolic indo-1 fluorescence and peak shortening obtained after 10 and 20 min of Ang II and after increasing [Ca2+]o. The slopes of the lines fitted to the three different set of points were not significantly different (analysis of covariance). Taken together, these results are consistent with a lack of effect of Ang II on myofilament responsiveness to Ca2+ all along the time course of the positive inotropic effect of Ang II.
Figure 5. Lack of effect of Ang II on myofilament responsiveness to Ca2+.
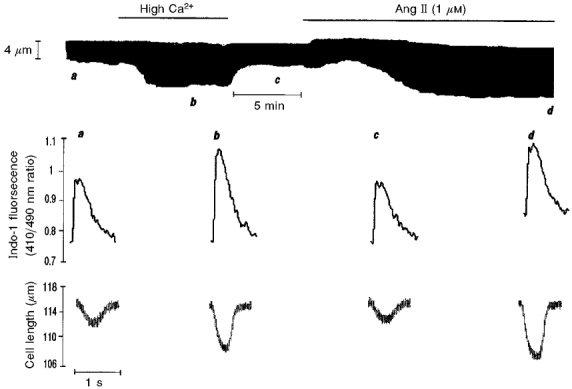
Typical continuous recording of cell length showing similar increases in cell shortening induced by either elevating [Ca2+]o or by addition of Ang II. Below are the actual tracings of the individual twitch contractions and the associated indo-1 fluorescence transients at the times indicated by letters a–d on the continuous chart recording.
Figure 6. Failure of Ang II to modulate myofilament responsiveness to Ca2+.
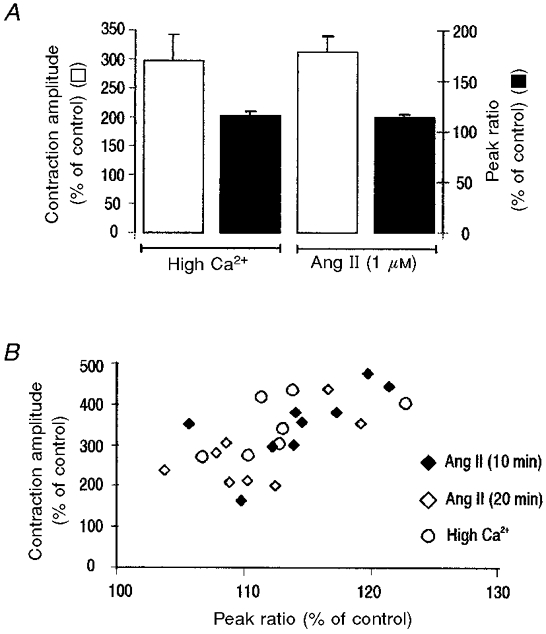
A, overall data of the effect of high [Ca2+]o and Ang II on contraction amplitude and peak indo-1 fluorescence (n = 8). Both interventions produced similar increases in contraction amplitude and peak fluorescence. B, relationship between the increase in peak systolic indo-1 fluorescence ratio and peak shortening after 10 min (peak) and 20 min (steady state) of the application of Ang II. The results are compared with the relationship obtained by elevating [Ca2+]o. All points follow a similar relationship.
Table 1.
Effect of high extracellular Ca2+ and Ang II on contraction and Ca2+ transient parameters of single cat myocytes
| Control | High Ca2+ | Control | Ang (1μM) | |
|---|---|---|---|---|
| Lo | 138 ± 6 | 138 ± 6 | 138 ± 6 | 138 ± 6 |
| TA (% of Lo) | 1.9 ± 0.4 | 4.7 ± 0.6* | 1.8 ± 0.3 | 4.2 ± 0.5* |
| t1/2 contraction (ms) | 134 ± 7 | 102 ± 11* | 135 ± 11 | 124 ± 12 |
| Indo-1 ratio (410/490) | ||||
| Diastolic | 1.01 ± 0.1 | 1.05 ± 0.1 | 0.97 ± 0.1 | 1.05 ± 0.1 |
| Systolic | 1.21 ± 0.1 | 1.37 ± 0.1* | 1.16 ± 0.1 | 1.32 ± 0.1* |
| Amplitude | 0.20 ± 0.03 | 0.32 ± 0.04* | 0.19 ± 0.02 | 0.27 ± 0.04* |
| t1/2 transient (ms) | 257 ± 12 | 210 ± 15* | 272 ± 15 | 253 ± 14 |
Lo, resting cell length; TA, twitch amplitue; t1/2 contraction, half-relaxation time of contraction; t1/2 transient, 50% relaxation of indo-1 fluorescence transient. Values are means ± S.E.M.; n = 8 for all values.
Significant vs. control value (P < 0.05).
Since Ang II enhances myocardial contractility by an increase in the CaiT without a significant modification of myofilament responsiveness to Ca2+, we next explored the possible mechanisms by which this increase in the CaiT may occur.
Does Ca2+ entry increase due to a prolongation of action potential duration?
Cardiac action potential duration (APD) lengthening after Ang II application has been previously reported (Kass & Blair, 1981; Morita et al. 1995). Prolongation of APD could favour Ca2+ influx through L-type Ca2+ channels and/or the reverse mode of the Na+-Ca2+ exchanger. In both cases, the prolongation of the APD would contribute to the increase in Ca2+ influx and therefore to the positive inotropic effect of Ang II. Conversely, prolongation of the APD could be the consequence of an increase in ICa. If this were the case, the prolongation of APD per se might not contribute to the increase in calcium influx. These possibilities were evaluated in the following current-clamp and voltage-clamp experiments performed in combination with simultaneous measurements of cell length. Figure 7 shows representative traces of action potentials (APs) recorded under current-clamp mode with the perforated-patch configuration of the patch-clamp technique. The corresponding contraction twitches are superimposed. Ang II induced a marked prolongation of APD. This Ang II-induced APD lengthening was associated with an increase of cell shortening within the first 10 min of Ang II treatment. At the peak of the Ang II response, average percentage increase of APD after 30 % (APD30), 50 % (APD50) and 90 % (APD90) of repolarisation time was 36.7 ± 8.1, 27.9 ± 6.4 and 21.7 ± 5.5 %, respectively (n = 8, P < 0.05 by repeated-measures ANOVA for paired values). The Ang II effect was significantly more important within the voltage range of the AP plateau than at more repolarised potentials, probably indicating that Ang II is predominantly affecting the currents that underlie plateau potentials, i.e. L-type Ca2+ currents (ICa).
Figure 7. Representative traces of action potentials (current clamp) and cell shortening recorded simultaneously, before and after 5 and 10 min of application of Ang II to the bath solution.
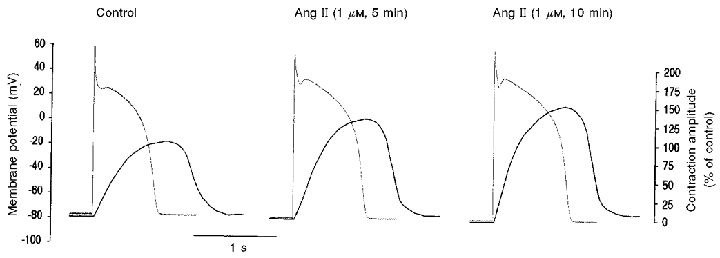
The contraction amplitude was related to that of the control peak shortening which was considered as 100 %. Ang II induced an increase in cell shortening and a lengthening of APD.
In order to evaluate whether the Ang II-induced positive inotropic effect was due to an enhancement of ICa and/or of the reverse mode of the Na+-Ca2+ exchange secondary to APD lengthening, a voltage protocol where voltage and time were controlled was used. Figure 8A shows representative traces of the effect of Ang II on cell shortening at two different simulated APs, a short AP (plateau of 200 ms of duration; Fig. 8A, upper traces) and a long AP (plateau of 400 ms of duration; Fig. 8A, lower traces). Under both these conditions, exposure of the myocyte to Ang II induced a similar increase in cell shortening to that measured under current-clamp configuration. Figure 8B depicts overall results of cell shortening provoked by the current-clamp and voltage-clamp protocols of Figs 7 and 8A. No significant differences were observed in the Ang II-induced positive inotropic effect between contractions provoked by current-clamp stimulus and those produced by protocols in which time duration changes were controlled. These results indicate that Ang II-induced APD lengthening per se does not contribute to the Ang II-induced positive inotropic effect.
Figure 8. Effects of Ang II on contractions evoked by simulated voltage-clamped action potentials.
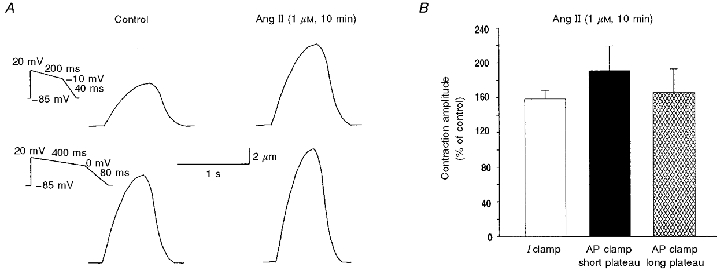
A, representative traces of cell shortening evoked by the voltage protocols shown on the left (voltage clamp), before and after 10 min of exposure of the myocyte to Ang II. B, mean percentage increase of peak shortening in relation to control for contractions evoked by current-clamp stimulus (Fig. 7, n = 8) and by voltage-clamp protocols simulating short and long action potentials (A, n = 6 and 5, respectively). No significant changes were observed among these groups.
Does Ca2+ entry increase due to a direct effect of Ang II on ICa?
The following step was directed to the study of the effects of Ang II on ICa. Since results reported previously using the whole-cell configuration of patch-clamp technique are highly controversial (Allen et al. 1988; Kaibara et al. 1994; Ikenouchi et al. 1994; Ai et al. 1998), we used, as stated above, a more physiological approach, the perforated-patch technique. Figure 9A shows the time course of the effect of Ang II on the peak ICa evoked at 0 mV. The representative traces corresponding to the points indicated in the figure are shown below. Application of Ang II to the bath induced an increase in ICa. This increase in the current started after 2 min, reached a maximum plateau value (75 % above control) after 7-11 min, began to decrease after this period of time, and finally reached a new steady-state value (50 % above control) after 16-20 min of exposure of the myocyte to Ang II. Six other myocytes exhibited this behaviour, the mean increase above control ICa being 77.7 ± 6.3 % at the peak effect of Ang II (8-15 min) and 62.1 ± 8.7 % after 20 min of exposure of the myocytes to the peptide (P < 0.05; n = 7). Figure 9B depicts the mean current density-voltage relations at control and after 7-8 min of Ang II treatment. A significant increase in current density was observed in the range of voltage between -15 and +45 mV after application of Ang II to the bath solution.
Figure 9. Ang II increases ICa.
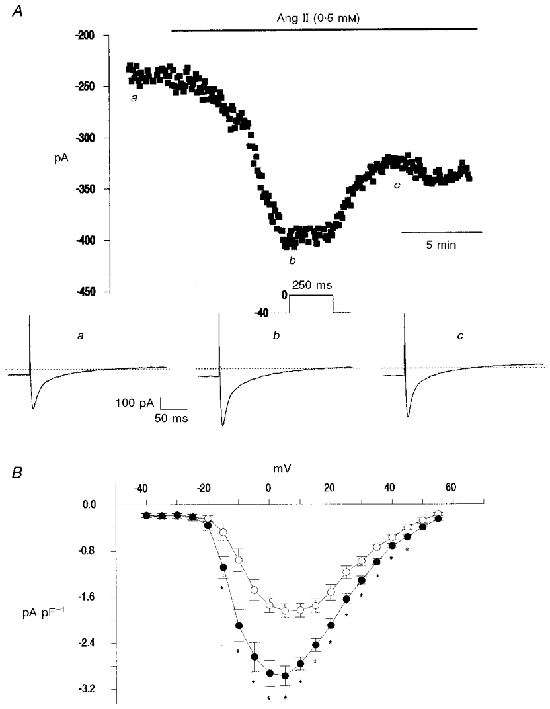
A, time course of the effects of Ang II on peak ICa current evoked by a step to 0 mV in a single myocyte. Representative traces of ICa corresponding to the points indicated in the upper panel are shown below. B, current density-voltage relations for mean data of peak current density collected from six myocytes in the absence and the presence of Ang II. A significant increase in ICa was observed after application of Ang II to the bath solution.
The increase in ICa evoked by Ang II and the close temporal association between this effect and the increase in the CaiT would indicate that the increase in ICa is a main determinant of the Ang II-induced increase in the CaiT and myocardial contractility. However, other mechanisms that could also contribute to this effect should be considered.
Role of the Na+-Ca2+ exchanger in the Ang II-induced positive inotropic effect
Enhanced Ca2+ entry via the reverse mode of the Na+-Ca2+ exchanger may also play a role in the Ang II-induced positive inotropic effect. To test this possibility, we studied the effect of Ang II in the presence of 2.5 μm of KB-R7943 (KB), a specific blocker of the reverse mode of the Na+-Ca2+ exchanger (Iwamoto et al. 1996). This concentration of KB blocked the increase in [Ca2+]i after activation of the reverse mode of the Na+-Ca2+ exchanger (Fig. 10A, inset) produced by reduction of external Na+ from 143 to 70 mm (NaCl replaced with choline chloride). A similar method of challenging the cell with a Na+ gradient sufficient to make the Na+-Ca2+ exchanger work in the reverse mode was previously used by Ladilov et al. (1999). Although KB produced a significant decrease of cell shortening (45 ± 10 %), the percentage increase in contraction amplitude produced by Ang II in the presence of KB was not significantly different from that produced in the absence of the Na+-Ca2+ blocker (n = 5) (Fig. 10A, chart recording and Fig. 10C). In the presence of KB Ang II increased the CaiT amplitude by 27 ± 3 %. This increment was not significantly different from that produced by Ang II in the absence of the Na+-Ca2+ exchanger blocker. These results are in agreement with results obtained in additional experiments performed in cat papillary muscles contracting isometrically. In these preparations KB concentrations up to 5 μm produced only a modest decrease in basal contractility (7 ± 1.9 %, n = 12). This concentration of KB failed to significantly affect the positive inotropic effect of 0.5 and 1 μm Ang II (results not shown).
Figure 10. Failure of KB and Ry to prevent the Ang II-induced positive inotropic effect.
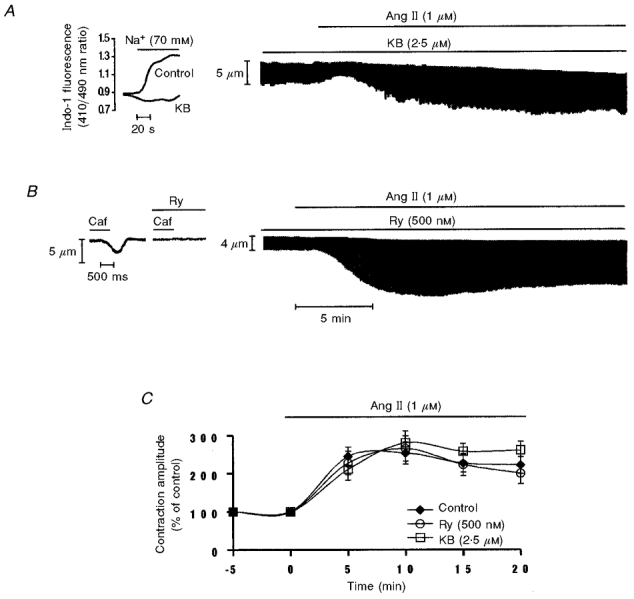
A, representative example of the effect of Ang II in the continued presence of KB (2.5 μm) on myocyte contraction. The continuous chart recording shows a typical biphasic contractile response to Ang II in a myocyte pretreated and continuously perfused with the Na+-Ca2+ blocker, KB. The inset to the left of the figure shows that KB was able to prevent the increase in Ca2+ entry by activation of the reverse mode of the Na+-Ca2+exchanger produced by external Na+ reduction. B, typical continuous chart recording of contraction amplitude in response to Ang II in the continued presence of Ry (500 nM). Ang II exerts a typical positive inotropic response even in the presence of Ry. The inset to the left shows a representative example of the effect of caffeine (15 mm) applied in the absence and presence of Ry (500 nM). Rapid application of caffeine induced a phasic contraction that was not reproducible after cells were treated with Ry. Similar results were obtained in 5 other cells. C, overall data of the time course of the contractile effects of Ang II alone, Ang II in the continued presence of KB or Ang II in the continued presence of Ry. Data are means ±s.e.m.; n = 5 cells per group.
Role of the SR in the Ang II-induced positive inotropic effect
SR Ca2+ release plays a key role in the excitation-contraction coupling mechanism of mammalian heart (Fabiato, 1983, 1985). We therefore investigated the role of the SR as a possible target for the Ang II-induced positive inotropic effect. For this purpose, the SR was functionally inhibited by pretreatment of the cells with ryanodine (Ry; 500 nM). Caffeine-induced contractions provoked in the presence and in the absence of Ry were used to assess the ability of Ry (500 nM) to inhibit SR function. The tracings in Fig. 10B (inset) show that application of 15 mm caffeine induced a phasic contraction that was not reproducible when caffeine was applied after the incubation with Ry, indicating the failure of the SR to accumulate and release Ca2+ in the presence of Ry. The chart recording in Fig. 10B depicts a representative example of the effect of Ang II in the continued presence of Ry. Although Ry produced a significant decrease in cell shortening (53 ± 7 %) associated with a prolongation of time to peak shortening (37 ± 7 %) and in half-relaxation time (61 ± 12 %), the percentage increase in contraction amplitude produced by Ang II in the presence of Ry was not significantly different from that produced by Ang II alone. The Ang II-induced positive inotropic effect observed in the presence of Ry was associated with a 35 ± 9 % increase in the CaiT amplitude that was not significantly different from that produced by Ang II alone. The unexpected finding that the SR seems not to be necessary for the Ang II-induced positive inotropism led us to examine the role of the SR in the presence of a more typical inotropic intervention, like elevating [Ca2+]o. These experiments demonstrated that an increase in cell shortening similar to that evoked by Ang II, produced by increasing [Ca2+]o, was not blunted in the presence of 500 nM Ry (4 mm[Ca2+]o: 191 ± 13 %vs. 4 mm[Ca2+]o+ 500 nM Ry: 187 ± 19 %; n = 6). Figure 10C shows the overall results of the effect of 1 μm Ang II on contractile amplitude in the absence and in the presence of Ry. Similar results were obtained in three other cells using 500 nM thapsigargin (Thaps) plus 1 μm Ry. These results indicate that the presence of a functional SR is not necessary for Ang II to be able to develop a typical positive inotropic response.
DISCUSSION
Physiological peptides such as endothelin-1 and Ang II have been recently recognised as potential modulators of cardiac contractility, producing positive inotropic effects in several species including the human. Plasma levels of Ang II overlap the range of the threshold concentrations for the positive inotropic effect of the peptide (Koch-Weser, 1965; Catt et al. 1970). The physiological importance of Ang II as a modulator of myocardial contractility is strengthened further, in light of experimental evidence showing that Ang II levels in cardiac tissue may reach fairly high concentrations (over 100 times plasma levels; Dell’Italia et al. 1997).
The subcellular mechanisms responsible for the positive inotropic effect of Ang II are still a matter of major controversy. Different results obtained in several mammalian species may be due at least in part to species-dependent variations which appear as a common feature among agents that increase phosphoinositide turnover (Endoh et al. 1991).
The cat is a mammalian species in which Ang II exerts a pronounced positive inotropic effect (Koch-Weser, 1965; Mattiazzi et al. 1997). The underlying mechanisms of this effect have not been previously investigated and constitute the aim of the present study. For this purpose we selected from pilot experiments the concentrations of Ang II that evoked maximal inotropic effects. The findings are the following: (1) the main mechanism involved in the overall shape of Ang II-induced positive inotropic effect is a pHi-independent increase in the CaiT; (2) no significant changes in myofilament responsiveness to Ca2+ were detected; and (3) the increase in the CaiT was mainly determined by an increase in ICa.
Increases in the CaiT vs. increases in myofilament responsiveness to Ca2+; role of pHi
The present experiments have shown that similar contractility increases produced by either Ang II administration or by increasing [Ca2+]o were associated with similar increases in the CaiT (Figs 5 and 6). These results, together with those obtained with the phase-plane analysis of cell shortening vs.[Ca2+]i (Fig. 4, right panel) measured either at the peak of the Ang II-induced positive inotropic response or when the positive inotropic effect of Ang II reached steady state, indicate that in the cat ventricle Ang II does not produce changes in myofilament responsiveness to Ca2+ and that at least in this species the overall shape of the Ang II-induced positive inotropic effect is entirely determined by changes in the CaiT.
The temporal dissociation between the increase in pHi and in contractility produced by Ang II, plus the fact that the Ang II-induced positive inotropic effect was preserved in the presence of HOE, indicate that Ang II-induced intracellular alkalosis does not modulate myofilament responsiveness to Ca2+. Considering the results of Fabiato & Fabiato (1978), this is not an unexpected finding, since using skinned cardiac fibres they demonstrated that an increase in pHi of 0.4 units was needed to produce only a modest increase in tension (approximately 30 % at pCa 6.0). Furthermore, a pHi shift towards alkalosis did not produce a significant change in maximal activated force, as acidosis did (Fabiato & Fabiato, 1978). Therefore, if these results can be extrapolated to feline myocardium and to a more intact preparation like the isolated myocytes, the pHi increase of 0.06 pH units produced by Ang II in these preparations may not be sufficient to evoke a detectable increase in myofilament responsiveness to Ca2+. Previous experiments by Ikenouchi et al. (1994) in rabbit heart failed to detect a significant increase in the CaiT after Ang II administration. It was concluded from these results that the increase in contractility produced by Ang II (more than 100 %) was entirely due to a pHi-dependent increase in myofilament responsiveness to Ca2+. However, the Ang II-induced increase in pHi reported in these experiments (approximately 0.2 pH units), although higher than the one detected in the cat ventricle (Mattiazzi et al. 1997 and the present results), would again, according to Fabiato & Fabiato’s results, not be sufficient to completely account for the increase in contractility observed. Indeed, more recent experiments demonstrated that Ang II also induced an increase in the CaiT in rabbit heart (Skolnick et al. 1998). Other experimental evidence indicates that the Ang II-induced activation of the Na+-H+ exchanger cannot be taken as a synonym of intracellular alkalosis (Boron et al. 1989; Ganz et al. 1989; Mattiazzi et al. 1997; Camilión de Hurtado et al. 1998). In this scenario, working in the more physiological bicarbonate buffer, Ang II has been shown to activate both an alkalinising mechanism, the Na+-H+ exchanger (Mattiazzi et al. 1997; Camilión de Hurtado et al. 1998) and an acidifying mechanism, the Na+-independent Cl−/HCO3− exchanger (Camilión de Hurtado et al. 1998), the net result being a lack of change in pHi. However, under these conditions, the positive inotropic effect of Ang II was still present (Mattiazzi et al. 1997). Therefore the conclusion that the increase in pHi plays a predominant role in determining the increase in contractility induced by Ang II is not straightforward and should be supported by increases in pHi of a magnitude sufficient to explain a cause-and-effect relationship. Our findings indicate that the positive inotropic effect of Ang II is not related to a pHi change in isolated myocytes from the cat ventricle. This conclusion is in agreement with previous results in the same species in multicellular preparations (Mattiazzi et al. 1997).
Subcellular mechanisms of the Ang II-induced increase in the CaiT
There are different and not mutually exclusive mechanisms by which Ang II may increase the CaiT: (1) an increase in Ca2+ entry through L-type calcium channels or the reverse mode of the Na+-Ca2+ exchanger; (2) a decrease in Ca2+ efflux through the forward mode of the Na+-Ca2+ exchanger; and (3) an IP3-mediated Ca2+ release from intracellular calcium stores.
The results obtained in voltage-clamp experiments demonstrated that Ang II produced a significant increase in ICa with a time course similar to that of the positive inotropic effect (Fig. 9). The increase in ICa evoked by Ang II in cardiac myocytes has been detected in some but not all previous studies. In fact ICa has been found to be increased (Kass & Blair, 1981; Allen et al. 1988; Kaibara et al. 1994), unchanged (Ikenouchi et al. 1994; Ai et al. 1998) or diminished by Ang II (Habuchi et al. 1995). The reason for these discrepant results is not apparent to us. They may be explained at least in part by the different species used and/or the different techniques employed to measure ICa. The technique used in the present experiments allows the preservation of the intracellular milieu, which is essential to detect the effects of hormones mediated by second messengers, as is the case for Ang II. Recent experiments have shown, indeed, that the increase in ICa evoked by α-adrenoceptor stimulation could be detected only when either perforated patch-clamp or cell-attached single channel recording was used, two conditions that preserve the intracellular environment (Liu & Kennedy, 1998; Zhang et al. 1998).
The subcellular mechanisms that determine the increase in ICa were not assessed directly in the present study. However, this increase seems unlikely to be mediated by an Ang II-induced increase in pHi as suggested by Kaibara et al. (1994), since this increase occurred before a significant increment in pHi could be detected in parallel experiments.
The present experiments showed in addition that the Ang II-induced prolongation of the APD does not seem to contribute to the positive inotropic effect of the peptide: When the APD was not allowed to increase by using voltage-clamped simulated APs, the Ang II-induced positive inotropic effect was still detected. Thus, the prolongation of the APD produced by Ang II appears to be a consequence rather than a cause of the increase in ICa.
Recent experiments in the rabbit heart have suggested that the Na+-Ca2+ exchanger may represent the main mechanism by which the CaiT increases after Ang II administration (Fujita & Endoh, 1999). A possible contribution of the Na+-Ca2+ exchanger to the positive inotropic effect of Ang II has also been suggested by Barry et al. (1995). The rationale behind this suggestion was that the increase in intracellular Na+ produced by Ang II through the activation of the Na+-H+ exchanger would lead to a decrease in Na+ gradient which would favour a Ca2+ influx via the reverse mode of the exchanger. The increase in intracellular Na+ load produced by Na+-H+ exchanger activation could also produce a reduction of Ca2+ efflux via the forward mode of the Na+-Ca2+ exchanger and thus increase [Ca2+]i. Moreover, it has been shown in in vitro experiments that Ang II directly activates the Na+-Ca2+ exchanger mechanism (Ballard & Schaffer, 1996). In light of the present experiments it is hard to conceive that the Na+-Ca2+ exchanger plays a significant role in the Ang II-induced increase in the CaiT for the following reasons: (1) no significant changes in pHi were detected at the moment in which the positive inotropic effect of Ang II was maximum; (2) the presence of HOE did not modify the positive inotropic effect of Ang II; and (3) the positive inotropic effect and the increase in the CaiT produced by Ang II were both preserved in the presence of the specific blocker of the reverse mode of the Na+-Ca2+ exchanger, KB.
An increase in Ca2+ release by the SR evoked by IP3 also seems unlikely since the amplitude of the CaiT produced by Ang II was not significantly modified after functional blockade of the SR by Ry and Thaps. Thus the possibility that IP3 plays a significant role in the positive inotropic effect of Ang II would require that IP3 released Ca2+ from a Ca2+ store that was insensitive to caffeine, different from that blocked by Ry, like the one that has been described for rat atrial cells (Vigne et al. 1990) and in addition, that was insensitive to the SERCA2 inhibitor Thaps. We are not aware of such a reservoir in ventricular cells. The failure of Ry plus Thaps to affect the positive inotropic effect of Ang II shown in the present results is in line with the results of Kentish et al. (1990), which showed that Ca2+ release by IP3 does not play a significant role in the excitation-contraction coupling of cardiac muscle.
In summary the present results demonstrate for the first time that in feline myocardium the Ang II-induced positive inotropic effect is exclusively mediated by an increase in the CaiT which is mainly due to the activation of ICa. Two additional major findings of this study are that the increase in the CaiT and the activation of ICa occurred independently of changes in pHi.
Acknowledgments
This study was supported by a grant (PICT 05-00149-02423) from the Agencia Nacional de Promoción Científica y Tecnológica, Argentina. We are most grateful to Dr H. E. Cingolani for his helpful comments on the manuscript and to Dr Wolfgang Scholz from Hoechst AG for kindly supplying the drug HOE 642. The technical assistance of Mrs Mónica Rando is gratefully acknowledged. M.G.V.P., E.A.A. and A.R.M. are Established Investigators of The Consejo Nacional de Investigaciones y Técnicas (CONICET), Argentina.
References
- Ai T, Horie M, Obayashi K, Sasayama S. Accentuated antagonism by angiotensin II on guinea-pig cardiac L-type Ca-currents enhanced by β-adrenergic stimulation. Pflügers Archiv. 1998;436:168–174. doi: 10.1007/s004240050619. [DOI] [PubMed] [Google Scholar]
- Aiello A, Vila-Petroff MG, Mattiazzi A, Cingolani HE. Evidence for an electrogenic Na+-HCO3− symport in rat cardiac myocytes. The Journal of Physiology. 1998;512:137–148. doi: 10.1111/j.1469-7793.1998.137bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen IS, Cohen NM, Dhallan RS, Gaa ST, Lederer WJ, Rogers TB. Angiotensin II increases spontaneous contractile frequency and stimulates calcium current in cultured neonatal rat heart myocytes: insights into the underlying biochemical mechanisms. Circulation Research. 1988;62:524–534. doi: 10.1161/01.res.62.3.524. [DOI] [PubMed] [Google Scholar]
- Ballard C, Schaffer S. Stimulation of the Na+/Ca2+ exchanger by phenylephrine, angiotensin II and endothelin 1. Journal of Molecular and Cellular Cardiology. 1996;28:11–17. doi: 10.1006/jmcc.1996.0002. [DOI] [PubMed] [Google Scholar]
- Barry WH, Matsui H, Bridge JH, Spitzer KW. Excitation-contraction coupling in ventricular myocytes: effects of angiotensin II. Advances in Experimental and Medical Biology. 1995;382:31–39. doi: 10.1007/978-1-4615-1893-8_4. [DOI] [PubMed] [Google Scholar]
- Blank PS, Silverman HS, Chun OY, Hogue BA, Stern MD, Hansford RG, Lakatta EG, Capogrossi MC. Cytosolic pH measurements in single cardiac myocytes using carboxy-seminaphthorhodafluor-1. American Journal of Physiology. 1992;263:H276–284. doi: 10.1152/ajpheart.1992.263.1.H276. [DOI] [PubMed] [Google Scholar]
- Boron WF, Boyarsky G, Ganz M. Regulation of intracellular pH in renal mesangial cells. Annals of the New York Academy of Sciences. 1989;574:321–332. doi: 10.1111/j.1749-6632.1989.tb25168.x. [DOI] [PubMed] [Google Scholar]
- Camilión deHurtado MC, Alvarez BV, Pérez NG, Ennis IL, Cingolani HE. Angiotensin II activates Na+-independent Cl−-HCO3−exchange in ventricular myocardium. Circulation Research. 1998;82:473–481. doi: 10.1161/01.res.82.4.473. [DOI] [PubMed] [Google Scholar]
- Catt KJ, Cain MD, Coghlan JP, Zimmet PZ, Cran E, Best JB. Metabolism and blood levels of Angiotensin II in normal subjects, renal disease, and essential hypertension. Circulation Research. 1970;27:177–193. [PubMed] [Google Scholar]
- Dell’Italia LJ, Meng QC, Balcells E, Wei C, Palmer R, Hageman JD, Hankens GH, Oparil S. Compartmentalization of angiotensin II generation in the dog heart. Evidence for independent mechanisms in intravascular and interstitial spaces. Journal of Clinical Investigation. 1997;100:253–258. doi: 10.1172/JCI119529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh M, Hiramoto T, Ishihata A, Takanashi M, Inui J. Myocardial α1-adrenoceptors mediate positive inotropic effect and changes in phosphatidylinositol metabolism: species differences in receptor distribution and the intracellular coupling process in mammalian ventricular myocardium. Circulation Research. 1991;68:1179–1190. doi: 10.1161/01.res.68.5.1179. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. American Journal of Physiology. 1983;245:C1–14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. Journal of General Physiology. 1985;85:247–289. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Effects of pH on the myofilaments and the sarcoplasmic reticulum of skinned cells from cardiac and skeletal muscles. The Journal of Physiology. 1978;276:233–255. doi: 10.1113/jphysiol.1978.sp012231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita S, Endoh M. Influence of a Na+/H+ exchange inhibitor ethylisopropylamiloride, Na+/Ca2+ exchange inhibitor KB-R7943 and their combination on the increases in contractility and Ca2+ transient induced by angiotensin II in isolated adult rabbit ventricular myocytes. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1999;360:575–584. doi: 10.1007/s002109900123. [DOI] [PubMed] [Google Scholar]
- Ganz MB, Boyarsky G, Sterzel RB, Boron WF. Arginine vasopressin enhances pHi regulation in the presence of HCO3− by stimulating three acid-base transport systems. Nature. 1989;337:648–651. doi: 10.1038/337648a0. [DOI] [PubMed] [Google Scholar]
- Gunasegaram S, Haworth RS, Hearse DJ, Avkiran M. Regulation of sarcolemmal Na+/H+ exchanger activity by angiotensin II in adult rat ventricular myocytes: opposing actions via AT1versus AT2 receptors. Circulation Research. 1999;85:919–930. doi: 10.1161/01.res.85.10.919. [DOI] [PubMed] [Google Scholar]
- Habuchi Y, Lu LL, Morikawa J, Yoshimura M. Angiotensin II inhibition of L-type Ca2+ current in sinoatrial node cell of rabbits. American Journal of Physiology. 1995;268:H1053–1060. doi: 10.1152/ajpheart.1995.268.3.H1053. [DOI] [PubMed] [Google Scholar]
- Ikenouchi I, Barry WH, Bridge JHB, Weiberg EO, Apstein CS, Lorell H. Effects of angiotensin II on intracellular Ca2+ and pH in isolated rabbit hearts and myocytes loaded with the indicator indo-1. The Journal of Physiology. 1994;480:203–215. doi: 10.1113/jphysiol.1994.sp020353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihata A, Endoh M. Pharmacological characteristics of the positive inotropic effect of angiotensin II in the rabbit ventricular myocardium. British Journal of Pharmacology. 1993;108:999–1005. doi: 10.1111/j.1476-5381.1993.tb13497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihata A, Endoh M. Species-related differences in inotropic effects of angiotensin II in mammalian ventricular muscle: receptors, subtypes and phosphoinositide hydrolysis. British Journal of Pharmacology. 1995;114:447–453. doi: 10.1111/j.1476-5381.1995.tb13247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto T, Watano T, Shigekawa M. A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+exchange in cells expressing NCX1. Journal of Biological Chemistry. 1996;271:22391–22397. doi: 10.1074/jbc.271.37.22391. [DOI] [PubMed] [Google Scholar]
- Kaibara M, Matiarai S, Yano K, Kameyama M. Involvement of Na+/H+ antiporter in the regulation of L-type Ca2+ channel current by angiotensin II in rabbit ventricular myocytes. Circulation Research. 1994;75:1121–1125. doi: 10.1161/01.res.75.6.1121. [DOI] [PubMed] [Google Scholar]
- Kass RS, Blair ML. Effects of angiotensin II on membrane current in cardiac Purkinje fibers. Journal of Molecular and Cellular Cardiology. 1981;13:797–809. doi: 10.1016/0022-2828(81)90237-6. [DOI] [PubMed] [Google Scholar]
- Kentish JC, Barsotti RJ, Lea TJ, Mulligan IP, Patel JR, Ferenczi MA. Calcium release from cardiac sarcoplasmic reticulum induced by photorelease of calcium or Ins(1,4,5)P3. American Journal Physiology. 1990;258:H610–615. doi: 10.1152/ajpheart.1990.258.2.H610. [DOI] [PubMed] [Google Scholar]
- Koch-Weser J. Nature of the inotropic action of angiotensin II on ventricular myocardium. Circulation Research. 1965;16:230–237. doi: 10.1161/01.res.16.3.230. [DOI] [PubMed] [Google Scholar]
- Korn SJ, Marty A, Connor JA, Horn R. Perforated patch recording. Methods in Neurosciences. 1991;4:364–373. [Google Scholar]
- Ladilov Y, Haffner S, Balser-Schafer C, Maxeiner H, Piper HM. Cardioprotective effects of KB-R7943: a novel inhibitor of the reverse mode of NA+/Ca2+ exchanger. American Journal of Physiology. 1999;276:H1868–1876. doi: 10.1152/ajpheart.1999.276.6.H1868. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Kennedy RH. α1-Adrenergic activation of L-type Ca current in rat ventricular myocytes: perforated patch-clamp recordings. American Journal of Physiology. 1998;274:H2203–2207. doi: 10.1152/ajpheart.1998.274.6.H2203. [DOI] [PubMed] [Google Scholar]
- McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiological Reviews. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- Matsui H, Barry WH, Livsey C, Spitzer KW. Angiotensin II stimulates sodium-hydrogen exchange in adult rabbit ventricular myocytes. Cardiovascular Research. 1995;29:215–221. [PubMed] [Google Scholar]
- Mattiazzi A, Pérez NG, Vila-Petroff MG, Alvarez BV, Camilión de Hurtado MC, Cingolani HE. Dissociation between the positive and alkalinizing effects of angiotensin II in feline myocardium. American Journal of Physiology. 1997;272:H1131–1136. doi: 10.1152/ajpheart.1997.272.3.H1131. [DOI] [PubMed] [Google Scholar]
- Morgan PE, Aiello EA, Chiappe de Cingolani GE, Mattiazzi AR, Cingolani HE. Chronic administration of nifedipine induces upregulation of functional calcium channels in rat myocardium. Journal of Molecular and Cellular Cardiology. 1999;31:1873–1883. doi: 10.1006/jmcc.1999.1019. [DOI] [PubMed] [Google Scholar]
- Morita H, Kimura J, Endoh M. Angiotensin II activation of a chloride current in rabbit cardiac myocytes. The Journal of Physiology. 1995;483:119–130. doi: 10.1113/jphysiol.1995.sp020572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah AM, Spurgeon HA, Sollott SJ, Talo A, Lakatta EG. 8-Bromo-cGMP reduces the myofilament response to Ca2+ in intact cardiac myocytes. Circulation Research. 1994;74:970–978. doi: 10.1161/01.res.74.5.970. [DOI] [PubMed] [Google Scholar]
- Skolnick RL, Litwin SE, Barry WH, Spitzer KW. Effect of angiotensin II on pHi, [Ca2+]i, and contraction in rabbit ventricular myocytes from infarcted hearts. American Journal of Physiology. 1998;275:H1788–1797. doi: 10.1152/ajpheart.1998.275.5.H1788. [DOI] [PubMed] [Google Scholar]
- Spurgeon HA, Stern MD, Baartz G, Raffaeli S, Hansford RG, Talo A, Lakatta EG, Capogrossi MC. Simultaneous measurements of Ca2+, contraction, and potential in cardiac myocytes. American Journal of Physiology. 1990;258:H574–586. doi: 10.1152/ajpheart.1990.258.2.H574. [DOI] [PubMed] [Google Scholar]
- Talukder MAH, Endoh M. Pharmacological differentiation of synergistic contribution of L-type Ca2+ channels and Na+/H+ exchange to the positive inotropic effect of phenylephrine, endothelin-3 and angiotensin II in rabbit ventricular myocardium. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1997;355:87–96. doi: 10.1007/pl00004923. [DOI] [PubMed] [Google Scholar]
- Vandenberg JI, Metcalfe JC, Grace AA. Intracellular pH recovery during respiratory acidosis in perfused hearts. American Journal of Physiology. 1994;266:C489–497. doi: 10.1152/ajpcell.1994.266.2.C489. [DOI] [PubMed] [Google Scholar]
- Vigne P, Breittmayer JP, Marsault R, Frelin C. Endothelin mobilizes Ca2+ from a caffeine- and ryanodine- insensitive intracellular pool in rat atrial cells. Journal of Biological Chemestry. 1990;265:6782–6787. [PubMed] [Google Scholar]
- Watanabe A, Endoh M. Relationship between the increase in Ca2+ transient and contractile force induced by angiotensin II in aequorin-loaded rabbit ventricular myocardium. Cardiovascular Research. 1998;37:524–531. doi: 10.1016/s0008-6363(97)00287-3. [DOI] [PubMed] [Google Scholar]
- Zhang S, Hiraoka M, Hirano Y. Effects of α1-adrenergic stimulation on L-type Ca2+ current in rat ventricular myocytes. Journal of Molecular and Cellular Cardiology. 1998;30:1955–1965. doi: 10.1006/jmcc.1998.0758. [DOI] [PubMed] [Google Scholar]