

Abstract
A mitochondrial complex comprising the voltage-dependent anion channel (outer membrane), the adenine nucleotide translocase (inner membrane) and cyclophilin-D (matrix) assembles at contact sites between the inner and outer membranes. Under pathological conditions associated with ischaemia and reperfusion the junctional complex ‘deforms’ into the permeability transition (PT) pore, which can open transiently, allowing free permeation of low Mr solutes across the inner membrane. This may be a critical step in the pathogenesis of lethal cell injury in ischaemia and reperfusion. Moreover, it is argued, the degree of pore opening may be an important determinant of the relative extent of apoptosis and necrosis under these conditions. In addition, mitochondria are the major sites of action of Bax and other apoptotic regulatory proteins of the Bcl-2 family. These proteins control a mitochondrial amplificatory loop in the apoptotic signalling pathway in which cytochrome c and other apoptogenic proteins of the mitochondrial intermembrane space are released into the cytosol. There are indications that the junctional complex, or components of it, may also mediate the action of Bax, but in a way that does not involve PT pore formation.
This article focuses on three mitochondrial proteins. The first of these, the voltage-dependent anion channel (VDAC), resides in the mitochondrial outer membrane, where it forms a large H2O-filled pore (2.5-3.0 nm diameter), allowing low Mr solutes to permeate freely and to gain access to the solute-specific transport systems of the inner membrane. The second, the adenine nucleotide translocase (ANT), is found in the inner membrane where it mediates ADP-ATP exchange, which is essential for the basic bioenergetic function of the organelle in exporting ATP to the cytosol. VDAC and ANT can interact strongly to form a complex. Since VDAC and ANT are the major integral membrane proteins of their respective membranes, mitochondria may contain many such junctional complexes at contact sites between the two membranes. The VDAC-ANT complex can recruit additional proteins depending on the function to be executed (Fig. 1). One of these is cyclophilin-D (CyP-D), the third protein protagonist of this article. CyP-D is located in the mitochondrial matrix, where it can facilitate protein conformational change.
Figure 1. VDAC-ANT complexes.
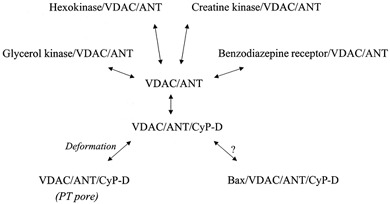
VDAC and ANT bind to each other and to a number of other proteins. These may, or may not, include Bax (see text). Under pathological conditions associated with ischaemia, the VDAC-ANT-CyP-D complex can deform into the PT pore, by which the inner membrane becomes permeabilized to low Mr solutes.
Current interest in the VDAC-ANT-CyP-D complex stems from its emerging involvement in cell death, in particular that caused by ischaemia and related conditions, i.e. hypoxia and hypoglycaemia (brain). Ischaemia impairs energy metabolism, culminating in cell death. With moderate periods of ischaemia the depressed cellular energy state is reversed on reperfusion, and the cells remain viable. However, with prolonged ischaemia, reperfused cells also become dysfunctional, and may even die, a phenomenon termed reperfusion injury. The mechanisms underlying these forms of cell death are of major importance. Most obviously, they define the severity of strokes and heart attacks. They also influence the outcome of clinical procedures in which reperfusion injury is encountered, e.g. cardiac bypass surgery (elective cardiac arrest) and organ transplantation. It is generally agreed that major metabolic factors in the pathogenesis of these forms of injury are the loss of ATP and adenine nucleotides, as they are degraded to nucleosides and bases, increases in intracellular Ca2+, and oxidative stress (reviewed in Crompton, 1999). These changes provoke both necrotic and apoptotic cell death. Characteristically, the core of ischaemic tissue becomes necrotic, while initially surviving cells in the surrounding regions that have been less severely compromised die subsequently by apoptosis (Veinot et al. 1997; Anversa et al. 1998). This duality is also seen when the critical metabolic factors are manipulated experimentally. Thus, neurons exposed to massive Ca2+ overload become necrotic, but with ‘moderate’ Ca2+ overload they undergo delayed death by apoptosis (Ankarcrona et al. 1995). Similarly, the form of cell death caused by oxidative stress changes from necrotic to apoptotic as the intensity of the insult lessens (Leist et al. 1997).
The VDAC-ANT-CyP-D complex has been implicated in both forms of cell death. In seeking to understand the role of the complex in cell death, however, it is important that we distinguish between the physiological process of programmed cell death, which specifically selects apoptosis, and the ‘accidental’ apoptosis that accompanies necrosis during ischaemic injury. Here, it is argued that ‘accidental’ apoptosis and necrosis may well share a common feature, namely ‘deformation’ of the VDAC-ANT-CyP-D complex into the permeability transition (PT) pore. In contrast, any participation of the complex in the apoptotic pathway during programmed cell death, when necrosis is excluded, must also exclude PT pore formation as a mechanism, and utilize a subtler mode of involvement.
VDAC-ANT-CyP-D complexes at intermembrane contact sites
Contact sites between the inner and outer mitochondrial membranes were first observed in electron micrographs over 30 years ago (Hackenbrock, 1968). Subsequent work detected an enrichment of VDAC and ANT at these sites, suggesting that an association of these two proteins could provide the basis for contact site formation (Brdiczka, 1991). When isolated by conventional protein purification procedures, VDAC-ANT complexes retain numerous other proteins (see for example Marzo et al. 1998), indicating that the contact sites can participate in various aspects of mitochondrial function. Much of this relates to the poor diffusibility of cytosolic ADP; this means that there is a need to bring ATP-consuming reactions close to the sites of ATP production and to make use of a more diffusible energy currency. Consequently, several cytosolic kinases bind to the junctional complexes. For example, hexokinase co-purifies with the VDAC-ANT complex, and ImmunoGold studies show enrichment of hexokinase at contact sites in vivo (Kottke et al. 1988); this creates a microdomain in which mitochondrially generated ATP can be used efficiently in the first step of glycolysis (McCabe, 1994). The association of creatine phosphokinase (intermembrane space) with the junctional complexes facilitates the use of the more diffusible creatine-creatine phosphate couple as a means of energy transport in the cytosol (Yoshizaki et al. 1990). However, other functions of the junctional complexes are also evident. Binding of the mitochondrial benzodiazepine receptor to VDAC in the junctional complexes (McEnery et al. 1992) may regulate the transfer of extramitochondrial cholesterol to the inner membrane of steroidogenic cells for conversion to pregnenolone (Papadopoulos et al. 1999). Thus the junctional complexes have emerged as a multifunctional reaction centre, assembling and disassembling the appropriate reaction microdomains, depending on the physiological process to be executed.
A particularly pure form of the junctional complex can be isolated using CyP-D. When mitochondrial membrane extracts (prepared using CHAPS as the detergent) were applied to a glutathione-S-transferase (GST)-CyP-D fusion protein affinity matrix, a 1:1 VDAC-ANT complex was selectively and tightly retained (Crompton et al. 1998; see also Woodfield et al. 1998). But why should VDAC-ANT bind CyP-D? CyP-D is the mitochondrial isoform of a family of cyclophilins found throughout the cell. Like all cyclophilins, CyP-D catalyses the cis-trans-isomerization of accessible Xaa-Pro peptide bonds in proteins. It can, therefore, catalyse protein conformational change. In addition, cyclophilins (or, at least, CyP-A in E. coli) preferentially bind cis-Pro isomers (Konno et al. 1996). In theory, then, catalysis may combine with isoform selection to stabilize a particular conformation of CyP-bound proteins. In the case of CyP-D, we know that it is located in the mitochondrial matrix (Johnson et al. 1999) and would bind to ANT of the VDAC-ANT complex. Accordingly, the fact that CyP-D fusions ‘pull down’ equimolar amounts of VDAC and ANT from extracts containing far more ANT than VDAC probably means that CyP-D establishes the particular ANT conformation that can also bind to VDAC (Crompton et al. 1998). In other words, CyP-D may have a general role in directing the assembly of the junctional complexes.
The VDAC-ANT-CyP-D complex in ischaemic injury
Under pathological conditions in vitro the VDAC- ANT- CyP-D complex can ‘deform’ into the so-called permeability transition (PT) pore. The PT pore was first detected in isolated mitochondria. When isolated mitochondria accumulate excessive amounts of Ca2+ in the presence of inorganic phosphate (Pi) and peroxides (or other pro-oxidants), and in the absence of external adenine nucleotides, the PT pore opens in the inner membrane (Hunter & Haworth, 1979; Al Nasser & Crompton, 1986a,b). Basic PT pore effectors are listed in Table 1. The internal diameter of the open pore has been estimated to be 2-2.5 nm, large enough to allow free diffusion of metabolites and hydrated metal ions across the inner membrane (Crompton & Costi, 1988, 1990). The pore is blocked by the cyclophilin ligand cyclosporin A (CSA; Crompton et al. 1988). The use of a differential photolabelling procedure with a photoactive CSA derivative positively identified CyP-D as the CSA receptor of the pore (Andreeva et al. 1995; Tanveer et al. 1996), in line with earlier approaches (McGuinness et al. 1990; Halestrap & Davidson, 1990; Griffiths & Halestrap, 1991). Subsequently CyP-D was used as an affinity matrix to identify VDAC and ANT as the other PT pore components. When the purified VDAC- ANT- recombinant CyP-D fusion (above) was incorporated into phospholipid vesicles, entrapped fluorescein sulphonate was released on addition of Ca2+ and Pi, and the release was blocked by CSA, in line with the basic properties of the PT pore. VDAC-ANT complexes containing CyP-D and other attached proteins show similar properties (Beutner et al. 1996, 1998). Thus, the VDAC-ANT- CyP-D complex most probably provides the core components of the PT pore. Although other proteins may associate with the complex, possibly modifying PT pore activity, the pore itself seems to comprise these three proteins.
Table 1.
PT pore effectors
| Agent | Comments | Reference |
|---|---|---|
| Ca2+ | Activates | Hunter & Haworth (1979) |
| > 1 μM extramitochondrial Ca2+ (resting) required | Duchen et al. (1993) | |
| Non-saturated by 25 μM matrix-free Ca2+ | Al Nasser & Crompton (1986a) | |
| Pi | Activates | Hunter & Haworth (1979) |
| Oxidants | Activate, e.g. peroxides | Crompton et al. (1987) |
| e.g. hyperoxia | Crompton & Andreeva (1993) | |
| ATP | Inhibits | |
| > 1.5 mm ATP required for full inhibition | Duchen et al. (1993) | |
| ADP | Facilitates open-closed pore interconversion | Crompton & Costi (1990) |
| CSA | Inhibits | Crompton et al. (1988) |
| 30 nM free CSA needed for cull inhibition (isolated mitochondria) | McGuiness et al. (1990) |
Principal agents are listed that promote and inhibit PT pore opening and that are relevant to its involvement in cell death.
How Ca2+, Pi and oxidative stress bring about PT pore formation from junctional complexes is unclear, but it seems likely that ‘deformation’ of ANT into an open-pore state most probably provides the basic pore structure in the inner membrane. In particular, arsenite can substitute for peroxides as the PT pore activator, indicative of vic-dithiol oxidation (Petronilli et al. 1994), and the use of a phenylarsine affinity matrix has tentatively identified ANT as the constituent with the relevant vic-dithiols (Halestrap et al. 1997). It is generally assumed that agents which trigger PT pore opening do so by causing CyP-D to bind to ANT, e.g. oxidative stress (Halestrap et al. 1997) and Ca2+ (Halestrap & Davidson, 1990), and that CyP-D is only recruited by ANT under the influence of these factors. However, our own studies reveal that CyP-D binds very tightly to VDAC-ANT (at least, in membrane extracts) irrespective of the presence or absence of Ca2+, and in the presence of dithiothreitol to maintain reduced thiols (Crompton et al. 1998). From this, it seems more likely that the VDAC-ANT-CyP-D complex is a permanent feature of healthy cells, in line with the proposed role (above) of CyP-D in directing the assembly of the junctional complexes.
Twelve years ago, we put forward a hypothesis that the PT pore might be a key lesion in the pathogenesis of necrotic cell death during ischaemia and reperfusion (Crompton & Costi, 1988; Crompton et al. 1988). This was based on the fact that the pore is opened in a synergistic manner by Ca2+, Pi, oxidative stress and falling ATP concentration, corresponding to the critical changes that unfold during ischaemia-reperfusion (Fig. 2). By rendering the inner membrane freely permeable to protons, PT pore opening would uncouple oxidative phosphorylation, so that mitochondria would hydrolyse ATP rather than synthesize it. We proposed a vicious cycle in which PT pore opening would impair energy metabolism, causing further Ca2+ deregulation, further PT pore opening, and so on, leading to cell death (reviewed in Crompton, 1990a,b). We also found that CSA was a potent PT pore inhibitor (Crompton et al. 1988), and that CSA protected cardiomyocytes against anoxia-induced injury (Nazareth et al. 1991; Crompton et al. 1992; Crompton & Andreeva, 1993). CSA protection has been confirmed in the reperfused heart (Griffiths & Halestrap, 1993), in cardiomyocytes subjected to reoxygenation (Griffiths et al. 2000), and in hepatocytes following anoxia (Pastorino et al. 1993), reoxygenation (Shimizu et al. 1994) and treatment with peroxides (Niemenen et al. 1995).
Figure 2. A model for the involvement of the PT pore in necrotic and apoptotic cell death during ischaemia and reperfusion.
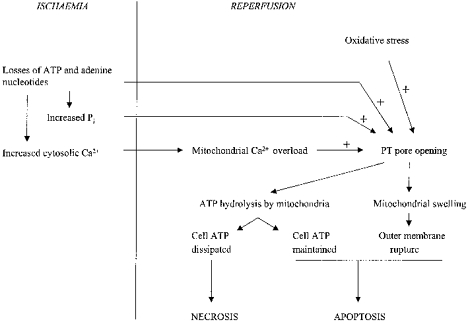
Ischaemia leads to ATP dissipation, with consequent rises in cell Ca2+ and Pi. On reperfusion, these three factors, together with oxidative stress, trigger PT pore opening. If widespread, PT pore opening overwhelms the ATP-generating capacity of the cell, and the cells become necrotic. If PT pore opening is more limited, enabling the cell to maintain sufficient ATP for viability, then outer membrane rupture may spark a caspase cascade, leading to apoptotis. The degree of PT pore opening may determine the balance between necrotic and apoptotic cell death.
However, CSA protection cannot confidently be attributed to inhibition of CyP-D and the PT pore since cells contain numerous isoforms of CyP, any one of which may be the relevant target. Crucially, therefore, several laboratories have provided direct evidence for PT pore opening in ischaemia and related conditions. Lemasters and co-workers (Niemenen et al. 1995; Qian et al. 1997) loaded the cytosol of cells with the fluorescent probe calcein. The calcein remained cytosolic until the cells were subjected to hypoxia or oxidative stress, when it diffused into the mitochondria. Since the redistribution of calcein was blocked by CSA, PT pore opening was indicated. In a variant of this approach, Scorrano et al. (1999) loaded calcein into both the cytosolic and mitochondrial compartments, and then selectively quenched the cytosolic calcein fluorescence with cobalt ions. CSA-sensitive calcein loss from the mitochondria was observed under the influence of GD3 ganglioside, a further PT pore effector (below). Griffiths & Halestrap (1995) loaded perfused hearts with radiolabelled deoxyglucose, which becomes entrapped in the cytosol as deoxyglucose-6-phosphate. After tissue homogenization in the presence of EGTA (to close PT pores), the recovery of radiolabel in the mitochondrial fraction was used as an index of PT pore opening before homogenization. This technique detected PT pore opening following tissue ischaemia and reperfusion (Kerr et al. 1999). Duchen et al. (1998) imaged the inner membrane potential of heart mitochondria in situ with the fluorescent probe TMRME. TMRME accumulates in mitochondria and also photodecomposes with the production of reactive oxygen species. Individual mitochondria were seen to undergo occasional transient depolarizations, the occurrence of which was blocked both by an inhibitor of mitochondrial Ca2+ uptake and by CSA (Jacobsen & Duchen, 1998), consistent with transient opening of the PT pore under these conditions. Similar, transient PT pore opening was detected in isolated mitochondria subjected to oxidative stress (Huser et al. 1998). Taken as a whole, therefore, results from many laboratories over the last 12 years coalesce into a consistent pattern. Ischaemia and reperfusion can induce PT pore opening preceding cell death, and CSA is frequently beneficial in limiting the extent of lethal cell injury. Thus PT pore opening may well be an important factor in the progression to irreversible cell injury during ischaemia and reperfusion.
Mitochondria in apoptosis
Mitochondria are, of course, the well-known ‘powerhouses’ of the cell, producing ATP to drive the various physiological functions and to maintain life itself. But they are also intimately involved in cell demise. The first sign that mitochondria play an active role in apoptosis came in 1994 with the observation that induction of nuclear apoptotic changes (chromatin condensation, nuclear fragmentation) in extracts of Xenopus eggs required the presence of mitochondria (Newmeyer et al. 1994). The stage was thus set for a period of rapid advance, leading to radically new insights into mitochondrial function. Apoptosis is executed by caspases, proteolytic enzymes that are expressed constitutively as inactive proenzymes, and that are activated by cleavage of the N-terminal prodomain. Caspases can be grouped structurally and functionally into two classes. Class I caspases contain long prodomains and are mutually cleaved after aggregation into complexes, a process mediated by adaptor proteins. These caspases then cleave and activate class II caspases, which have short prodomains. For example, apoptosis in response to occupation of the tumour necrosis factor (TNF) superfamily of receptors leads to assembly of upstream procaspases and adaptor proteins at the plasma membrane receptor, forming a so-called DISC (death-inducing signalling complex). Here the upstream (class I) caspases are processed to their active forms, leading in turn to the activation of the downstream (class II) caspases. In some cells, receptor occupancy leads to rapid induction of the caspase cascade, and these cells have been termed type I cells (Scaffidi et al. 1999). But in many other types of cell, gross activation of upstream and downstream caspases is delayed. In these cells (type II cells), occupation of death receptors causes only a limited direct activation of upstream caspases at the DISC, and a mitochondrial amplificatory loop is required to spark the caspase cascade (Fig. 3).
Figure 3. The mitochondrial amplificatory loop.
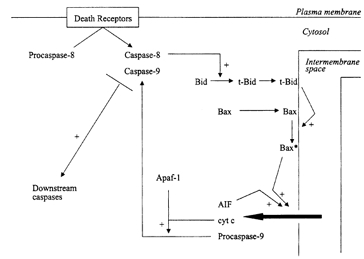
Mitochondria can be recruited into the apoptotic signalling pathway by Bid and Bax, which translocate from the cytosol to the mitochondrial outer membrane under apoptotic stimuli. Bid translocates as a truncated form (t-Bid) after cleavage by caspase-8. Bax is processed on the outer membrane to an active form (Bax*), a process possibly facilitated by t-Bid. Bax induces the release of apoptosis-inducing factor (AIF), procaspase-9, cytochrome c (cyt c) and other intermembrane space proteins to the cytosol. Cytochrome c binds to the adaptor protein Apaf-1, which is then able to bind procaspase-9 and -8, which are thus cleaved to their active forms.
Bid, Bax and the release of mitochondrial apoptogenic proteins
Mitochondria amplify the apoptotic signal by releasing apoptogenic proteins from the intermembrane space. The first to be recognized was cytochrome c (Liu et al. 1996). Released cytochrome c binds to the cytosolic (adaptor) protein Apaf-1, which is then able to recruit upstream caspases into a complex, where they are cleaved into their active forms (Zou et al. 1997). Of these, procaspase-9 is recruited most strongly. A proportion, at least, of procaspase-9 is also released from the mitochondrial intermembrane space (Krajewski et al. 1999), along with apoptosis-inducing factor (AIF), a flavoprotein that induces nuclear apoptotic changes (Susin et al. 1999). Current indications are that these apoptogenic proteins are released non-selectively together with other proteins of the intermembrane space, e.g. adenylate kinase and sulphite oxidase (100 kDa) (Pastorino et al. 1999; Kluck et al. 1999). The general loss of intermembrane space proteins suggests that large holes or breaks must appear in the outer membrane. The release of intermembrane space proteins is triggered by proapoptotic proteins of the Bcl-2 family, notably Bid, Bax and Bak. These proteins migrate to mitochondria under apoptotic stimuli. Bid is retained in the cytosol of healthy cells as an inactive precurser; this is cleaved by caspase-8 (Fig. 3) to produce a 15 kDa C-terminal fragment which binds avidly to mitochondria (Luo et al. 1998; Li et al. 1998). Bax is distributed diffusely as a monomer in the cytoplasm of unstimulated cells (Hsu & Youle, 1997; Wolter et al. 1997; Gross et al. 1998). The mechanisms leading to its translocation and binding to mitochondria are unclear; however, certain features are evident. It seems that Bax binds to mitochondria as both a peripheral and an integral membrane protein. The N-terminal region of Bax is inaccessible in the cytosolic and the peripherally associated states, but becomes reactive to trypsin and antibodies after membrane insertion (Goping et al. 1998; Nechushtan et al. 1999). The C-terminal region contains a hydrophobic stretch of about 20 amino acids, deletion of which impairs binding to mitochondria (Wolter et al. 1997) and to bilayers in general (Montessuit et al. 1999). From this it seems that integration of Bax into the mitochondrial outer membrane entails a profound conformational change such that the N-terminus becomes exposed on the membrane surface, while the C-terminus facilitates membrane insertion. The proapoptotic Bak may undergo similar changes. Thus, N-terminal-specific antibodies were unreactive with mitochondrially (peripherally?) associated Bak, but became reactive after induction of apoptosis (Griffiths et al. 1999). The conformational changes in Bax (and possibly also in Bak?) leading to membrane insertion may involve oligomerization. Gross et al. (1998) found that peripherally bound Bax was not cross-linkable (homobifunctional reagent), whereas it became so (forming dimers) on membrane insertion following a death signal. Moreover, cells transfected with chimeric FK506 binding protein (FKBP)-Bax became apoptotic when dimerization was enforced with the bivalent FKBP ligand FK1012.
The picture that emerges is that Bax and, possibly, Bak are initially recruited by mitochondria in an inactive form, and become conformationally changed to their active forms on the membrane surface concomitant with membrane insertion. But how are these changes brought about? Intriguingly, there is evidence that the changes are facilitated by Bid (Desagher et al. 1999). In particular, mutations in Bid that impaired its capacity to bind to Bax also depressed the conformational change in Bax (N-terminal immunoreactivity), and Bid was unable to release cytochrome c from mitochondria prepared from Bax-deficient cell lines. Conversely, Bid-deficient mice were resistant to fas-mediated apoptosis (Yin et al. 1999). In addition, there are indications that Bax associates with anti-apoptotic Bcl-2 family proteins in unstimulated cells. Thus, coexpression of the fusion proteins Bax-green fluorescent protein and Bcl-2-blue fluorescent protein led to tight interactions between the two in mitochondria as detected by fluorescence resonance energy transfer (Mahajan et al. 1998). Bax and Bcl-XL coimmunoprecipitate from extracts of untreated cells, but this is greatly reduced under apoptotic stimuli (Griffiths et al. 1999). It may be, then, that Bid displaces Bax (and possibly also Bak?) from resident Bcl-2-Bcl-XL complexes. The ‘catalytic’ requirement for Bid may explain why extremely low amounts of it, relative to Bax, need to be added to isolated mitochondria to elicit cytochrome c release (Zou et al. 1997). One can speculate that the requirement for both Bid and Bax provides a ‘dual switch’ mechanism for release of intermembrane space proteins, thereby minimizing the risk of ‘unauthorized’ apoptosis.
Involvement of junctional complexes?
The translocation of Bax and truncated Bid to mitochondria under apoptotic stimuli raises the question of whether they target specific ‘receptor’ proteins on the mitochondrial surface. To date no ‘receptor’ as such has been positively identified. Nevertheless, there are gathering indications that the junctional complexes, or components of these complexes, participate in the action of Bax on mitochondria in some capacity.
Narita et al. (1998) provided various lines of evidence that Bax and Bcl-XL can bind to VDAC. Recombinant Bax coimmunoprecipitated with VDAC from mitochondrial extracts. Overexpressed Bax and VDAC could be cross-linked by cell-permeant reagents. A Bcl-XL affinity matrix bound both VDAC and ANT (Shimizu et al. 1999). The same group also analysed these interactions in liposomes. Sucrose permeation in VDAC proteoliposomes was largely blocked by recombinant Bcl-XL. Conversely, the low rate of sucrose permeation into VDAC proteoliposomes at low pH (pH 5.2) was enhanced by recombinant Bax. Thus Bax and Bcl-XL not only bind to VDAC, but also can modify its permeability properties. Indeed, Bax and Bak were able to induce the movement of cytochrome c (12 kDa) across VDAC proteoliposomes, although a larger protein (GST-green fluorescent protein) was retained. Further evidence comes from work with yeast. It is possible to mimic certain stages of mitochondrial involvement in apoptosis by transfection of Bax into yeast cells. This results in the loss of cytochrome c to the cytosol, and the cells die. Bax is non-toxic in petite cells (lacking mitochondria). Narita et al. (1998) found that Bax was unable to release cytochrome c from mitchondria isolated from VDAC-deficient yeast mutants, and that Bax action was restored after transfection with the VDAC1 isoform.
Other lines of enquiry point to a role for ANT. Bauer et al. (1999) screened an expression library for dominant, apoptosis-inducing genes. This led to the identification of the ANT1 isoform as a proapoptotic protein. Overexpression of ANT1 in a range of cell types produced extensive apoptosis, whereas overexpression of the ANT2 isoform was inocuous. The proapoptotic property of ANT1 was clearly independent of its transport activity (ADP-ATP exchange), since transport-deficient mutants were equally proapoptotic. Further, intriguing indications for an apoptotic role of ANT come from studies of virus-infected cells. Infected cells undergo apoptosis as an anti-viral defence mechanism, but the viruses fight back by producing proteins that suppress apoptosis of the host cells. Goldmacher et al. (1999) identified one such protein, vMIA, produced by human cytomegalovirus, by screening an expression library of the viral DNA. vMIA blocked fas-mediated apoptosis at a stage between Bid cleavage and cytochrome c release. vMIA in transfected cells was located to mitochondria (ImmunoGold studies) and coimmunoprecipitated ANT from cell extracts. Thus, vMIA may suppress the mitochondrial amplificatory loop by binding to ANT1. Marzo et al. (1998) examined the submitochondrial distribution of Bax (ImmunoGold studies) under the influence of the ANT ligand atractylate (which triggers cytochrome c release and apoptosis in a number of cell types). Atractylate was reported to bring about a translocation of mitochondrial Bax from outer to inner membrane sites. Taken as a whole, these data suggest that Bax may need to interact with the VDAC-ANT complex at some stage in its action on mitochondria. In line with this, Bax was observed to locate preferentially to intermembrane contact sites in mitochondria (De Jong et al. 1994).
There is gathering evidence that mitochondria may also contribute to apoptosis under the sphingomyelin signalling pathway and, again, junctional complexes have been implicated. Ceramide is a central signalling molecule of the pathway, released from sphingomyelin by a family of sphingomyelinases. Susin et al. (1997) showed that induction of nuclear apoptosis in a cell-free system could be brought about by inclusion of mitochondria and cytosol from ceramide-treated cells. Since nuclear apoptotic changes in this system can be triggered by the release of AIF from the mitochondrial intermembrane space, this suggests that a ceramide metabolite may provoke AIF release. The metabolite was identified by De Maria et al. (1997), who detected a transient accumulation of GD3 ganglioside 15 min after fas activation, and showed that overexpression of GD3 synthase brought about substantial apoptosis. Subsequently, GD3 ganglioside, but not C2 ceramide, was shown to induce release of cytochrome c from isolated mitochondria and to do so synergistically with Bax (Kristal & Brown, 1999; Pastorino et al. 1999; Scorrano et al. 1999). Both ceramide-induced apoptosis in cell-free systems (Susin et al. 1997) and GD3 ganglioside-induced loss of cytochrome c from isolated mitochondria are sensitive to CSA (Kristal & Brown, 1999), consistent with the involvement of CyP-D.
It has been suggested that PT pore opening, leading to matrix expansion and physical rupture of the outer membrane, is the root mechanism for the release of intermembrane space proteins in apoptosis (see Marzo et al. 1998 and references therein). The various indications (above) that Bcl-2 family proteins may interact with the junctional complexes in mitochondria are clearly in line with this. In addition, the PT pore inhibitor, and ANT ligand, bongkrekate blocks apoptosis in several systems (e.g. dexamethasone-induced apoptosis in thymocytes; Zamzami et al. 1996), which is also consistent with this hypothesis. However, it should be noted that bongkrekate was also reported to inhibit cytochrome c-independent apoptosis induced by p53 (Li et al. 1999), so that interpretation of bongkrekate protection may not be straightforward. Indeed, it seems unlikely that PT pore opening would be used as a physiological (as opposed to pathological, below) mechanism of outer membrane rupture. PT pore opening would dissipate cellular ATP (above), yet ATP appears to be maintained in apoptosing cells until the release of cytochrome c is complete (e.g. Bossy-Wetzel et al. 1998). When the intracellular ATP concentration of apoptosing cells (fas ligand, staurosporine) was manipulated by changing the nutrient supply, only cells with initially maintained ATP levels developed nuclear apoptotic characteristics (Leist et al. 1997). Furthermore, Bax can release cytochrome c from isolated mitochondria in the absence of Ca2+, the basic trigger for PT pore opening (Eskes et al. 1998), and does so without detectable changes in matrix volume (Kluck et al. 1999). Nevertheless, there are numerous indications (above) that the VDAC-ANT-CyP-D complex, or components of it, participates in apoptotic signalling in some capacity, and it is interesting to speculate what this might be. One mechanism for outer membrane lysis would be to introduce asymmetry between the two leaflets, thus producing kinks, leading to transient breaks, in the bilayer. For example, lysophospholipids, containing a relatively large head group and a single narrow tail are an effective means of inducing transient breaks in phospholipid bilayers, so phospholipase recruitment/ activation at the outer membrane seems worth exploring. A redistribution of phospholipids between leaflets might also introduce sufficient membrane asymmetry to produce lysis and, in this context, it is interesting to note that the junctional complexes have been implicated in the transfer of cholesterol and phospholipids between the mitochondrial membranes (Ardail et al. 1991; Papadopoulos et al. 1999).
The VDAC-ANT-CyP-D complex in ‘accidental’ apoptosis associated with ischaemia
As outlined above, it is difficult to envisage PT pore opening as a physiological mechanism for outer membrane permeabilization, since it would cause ATP dissipation, and ATP loss leads to necrosis. It seems unlikely that a physiological signalling pathway (apoptosis) would utilize a mechanism (PT pore opening) that could easily produce the undesired outcome (necrosis). However, ‘accidental’ apoptosis in ischaemia presents a different picture, since ischaemia (hypoxia) and reperfusion (reoxygenation) produce both necrotic and apoptotic cell death (see Introduction). Thus, whereas it seems improbable that PT pore opening provides a physiological step in the apoptotic pathway during programmed cell death (what might be termed, in this context, ‘authorized’ apoptosis), since PT pore-activated cells could so easily slide into necrosis, it may well be a critical lesion in ‘accidental’ apoptosis in disease, when apoptotic and necrotic mechanisms coexist (Crompton, 2000).
Can PT pore opening rupture the outer membrane? When isolated mitochondria were incubated with supraphysiological levels of Ca2+ in the presence of Pi, cytochrome c was lost from the mitochondria concomitant with inner membrane depolarization, and both events were blocked by CSA, indicative of PT pore opening (Eskes et al. 1998; Pastorino et al. 1999). In cell-free systems, a range of agents that induce PT pore opening (atractylate, peroxides, diamide), produced mitochondrial matrix swelling, release of cytochrome c and nuclear apoptotic changes (Zamzami et al. 1996; Marchetti et al. 1996). From this, it seems that PT pore opening can lyse the outer membrane in vitro and, by extension, that any PT pore opening in vivo during ischaemia-reperfusion might result in ‘accidental’ loss of cytochrome c and other mitochondrial apoptogenic proteins to the cytosol. Intriguingly, Bax may contribute to this. Thus, it has been reported that Bax translocation to mitochondria can also be triggered by hypoxia; evidently hypoxia overrides the constraints that normally retain Bax in the cytosol (Saikumar et al. 1998). Moreover, in isolated mitochondria at least, Ca2+-induced PT pore opening and loss of cytochrome c are promoted by Bax (Jurgensmeier et al. 1998; Pastorino et al. 1999); this may be a further reflection of the interaction of Bax with the VDAC-ANT-CyP-D complex (above). To conclude, Bax and PT pore effectors (Ca2+, oxidative stress) may operate synergistically in ischaemia- reperfusion to elicit the release of apoptogenic proteins from the intermembrane space. In general agreement with this, ischaemia-induced release of intermembrane space proteins has been recorded (e.g. Krajewski et al. 1999).
But why should not all PT pore-activated cells become necrotic? After all, PT pore opening would be expected to lead to ATP dissipation and rapid loss of viability. While this may be generally true, one can envisage situations in which a limited degree of PT pore opening could be tolerated. For example, PT pore opening on reperfusion would only be immediately lethal (necrosis) if it led to the ATP-generating capacity of the cell being overwhelmed. A limited degree of PT pore opening, causing only a minor impairment in the efficiency of cellular energy transduction, and allowing maintenance of the cellular energy state, is quite consistent with cell viability. However, if PT pore opening, however limited, led to mitochondrial outer membrane rupture, then this might be sufficient to spark a caspase cascade, leading to delayed death by apoptosis. This model (Fig. 2) would explain why the form of cell death induced by basic triggers of PT pore opening (Ca2+ overload, oxidative stress) changes from necrotic to apoptotic as the severity of insult decreases, and why cells injured insufficiently by ischaemia-reperfusion to become necrotic can, nevertheless, die subsequently by apoptosis (see Introduction). According to this model, then, the form of cell death undergone by any ischaemic-reperfused cell would be determined by the degree of PT pore opening in that cell.
By limited PT pore opening, I mean limited to a few mitochondria within any cell. During oxidative stress, such highly localized effects could arise from correspondingly localized increases in cytosolic Ca2+. There is mounting evidence that mitochondria respond heterogeneously to increases in cytosolic Ca2+, depending on their proximity to the Ca2+ release channels in the endo(sarco)plasmic reticulum (Rizzuto et al. 1998; Duchen et al. 1998; Duchen, 1999). Mitochondria closely apposed to these channels experience high localized Ca2+ concentrations and rapidly accumulate high levels of Ca2+ on Ca2+ release from intracellular stores. Such mitochondria would be more prone to PT pore opening under conditions of oxidative stress. Consistent with this, astrocytes subjected to oxidative stress (photodecomposition of tetramethylrhodamine methylester; Jacobsen & Duchen, 1998) demonstrate highly localized PT pore opening. To conclude, therefore, cellular Ca2+ overload and oxidative stress during ischaemia and reperfusion may trigger PT pore opening and outer membrane rupture in endo(sarco)plasmic reticulum-juxtaposed mitochondria, thus initiating a caspase cascade. With increased intensity of insult, PT pore opening may be more extensive, overwhelming the capacity of the cell to maintain ATP levels, so that cells become necrotic (Fig. 2). But with lesser insults, PT pore opening in a small region of the cell may be sufficient to engage the apoptotic pathway.
References
- Al Nasser I, Crompton M. The reversible Ca-induced permeabilization of rat liver mitochondria. Biochemical Journal. 1986a;239:19–29. doi: 10.1042/bj2390019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Nasser I, Crompton M. The entrapment of the Ca indicator arsenazo III in the matrix space of rat liver mitochondria by permeabilization and resealing. Biochemical Journal. 1986b;239:31–40. doi: 10.1042/bj2390031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva L, Tanveer A, Crompton M. Evidence for the involvement of a mitochondrial cyclosporin A binding protein in the Ca-activated inner membrane pore of heart mitochondria. European Journal of Biochemistry. 1995;230:1125–1132. doi: 10.1111/j.1432-1033.1995.tb20664.x. [DOI] [PubMed] [Google Scholar]
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- Anversa P, Cheng W, Liu Y, Leri A, Redaelli G, Kajstura J. Apoptosis and myocardial infarction. Basic Research in Cardiolology. 1998;93(suppl 3):8–12. doi: 10.1007/s003950050195. [DOI] [PubMed] [Google Scholar]
- Ardail D, Lerme F, Louisot P. Involvement of contact sites in the phosphatidyl serine import into mitochondria. Journal of Biological Chemistry. 1991;266:7973–7981. [PubMed] [Google Scholar]
- Bauer MKA, Schubert A, Rocks O, Grimm S. Adenine nucleotide translocase1, a component of the permeability transition pore, can dominantly induce apoptosis. Journal of Cell Biology. 1999;147:1493–1499. doi: 10.1083/jcb.147.7.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner G, Ruck A, Riede B, Brdiczka D. Complexes betweem porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Biochimica et Biophysica Acta. 1998;1368:7–18. doi: 10.1016/s0005-2736(97)00175-2. [DOI] [PubMed] [Google Scholar]
- Beutner G, Ruck A, Riede B, Welte W, Brdiczka D. Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Letters. 1996;396:189–195. doi: 10.1016/0014-5793(96)01092-7. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Newmeyer DD, Green DR. Mitochondrial cytochrome c release in apoptosis occurs downstream of DEVD-specific caspase activation and independently of mitochondria depolarization. EMBO Journal. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brdiczka D. Contact sites between mitochondrial envelope membranes. Biochimica et Biophysica Acta. 1991;1071:291–312. doi: 10.1016/0304-4157(91)90018-r. [DOI] [PubMed] [Google Scholar]
- Crompton M. The role of Ca in the function and dysfunction of heart mitochondria. In: Langer GA, editor. Calcium and the Heart. New York: Raven Press; 1990a. pp. 167–195. [Google Scholar]
- Crompton M. The role of mitochondria in intracellular Ca regulation. In: Bronner F, editor. Intracellular Calcium Regulation. New York: Wiley-Liss; 1990b. pp. 181–211. [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochemical Journal. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Crompton M. Bax, Bid and the permeabilization of the mitochondrial outer membrane in apoptosis. Current Opinion in Cell Biology. 2000;12:414–441. doi: 10.1016/s0955-0674(00)00110-1. [DOI] [PubMed] [Google Scholar]
- Crompton M, Andreeva L. On the involvement of a mitochondrial pore in reperfusion injury. Basic Research in Cardiology. 1993;88:513–523. doi: 10.1007/BF00795416. [DOI] [PubMed] [Google Scholar]
- Crompton M, Costi A. Kinetic evidence for a heart mitochondrial pore activated by Ca, inorganic phosphate and oxidative stress. A potential mechanism for mitochondrial dysfunction during cellular Ca overload. European Journal of Biochemistry. 1988;178:489–501. doi: 10.1111/j.1432-1033.1988.tb14475.x. [DOI] [PubMed] [Google Scholar]
- Crompton M, Costi A. A heart mitochondrial Ca induced pore of possible relevance to reperfusion injury. Biochemical Journal. 1990;266:33–39. doi: 10.1042/bj2660033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Costi A, Hayat L. Evidence for the presence of a reversible Ca-dependent pore activated by oxidative stress in heart mitochondria. Biochemical Journal. 1987;245:914–918. doi: 10.1042/bj2450915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin A of a Ca dependent pore activated by inorganic phosphate and oxidative stress. Biochemical Journal. 1988;255:357–360. [PMC free article] [PubMed] [Google Scholar]
- Crompton M, McGuinness O, Nazareth W. The involvement of cyclosporin A binding proteins in regulating and uncoupling mitochondrial energy transduction. Biochimica et Biophysica Acta. 1992;1101:214–217. [PubMed] [Google Scholar]
- Crompton M, Virji S, Ward JM. Cyclophilin-D binds strongly to complexes of the voltage dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. European Journal of Biochemistry. 1998;258:729–735. doi: 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- De Jong D, Prins FA, Mason DY, Reed JC, Van Ommen GB, Kluin PM. Subcellular localization of the bcl-2 protein in malignant and normal lymphoid cells. Cancer Research. 1994;54:256–260. [PubMed] [Google Scholar]
- De Maria R, Lenti L, Malisan F, D’Agostino F, Tomassino B, Rippo MR, Testi R. Requirement for GD3 ganglioside in CD95 and ceramide induced apoptosis. Science. 1997;272:1652–1655. doi: 10.1126/science.277.5332.1652. [DOI] [PubMed] [Google Scholar]
- Desagher S, Osend-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou JC. Bid induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. Journal of Cell Biology. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. Journal of Physiology. 1999;516:1–17. doi: 10.1111/j.1469-7793.1999.001aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Leyssens A, Crompton M. Transient mitochondrial depolarisations reflect focal sarcoplasmic reticular calcium release in single rat cardiomyocytes. Journal of Cell Biology. 1998;142:975–988. doi: 10.1083/jcb.142.4.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, McGuinness O, Brown LA, Crompton M. On the involvement of a cyclosporin-A sensitive mitochondrial pore in myocardial reperfusion injury. Cardiovascular Research. 1993;27:1790–1794. doi: 10.1093/cvr/27.10.1790. [DOI] [PubMed] [Google Scholar]
- Eskes R, Antonsson B, Osen-Sand A, Montessuit S, Richter C, Sadoul R, Mazzei G, Nichols A, Martinou JC. Bax induced cytochrome c release from mitochondria is independent of the permeability transition pore but highly dependent on Mg ions. Journal of Cell Biology. 1998;143:217–224. doi: 10.1083/jcb.143.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmacher VS, Bartle LM, Skaletskaya R, Dionne CA, Kedersha NL, Vater CA, Han J, Lutz RJ, Watanabe S, McFarland ED, Kief ED, Mocarski ES, Chittenden T. A cytomegalovirus-encoded mitochondria- localized inhibitor of apoptosis structurally unrelated to bcl-2. Proceedings of the National Academy of Sciences of the USA. 1999;96:12536–12541. doi: 10.1073/pnas.96.22.12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goping IS, Gross A, Lavoie N, Nguyen M, Jemmerson R, Roth K, Korsmeyer SJ, Shore GC. Regulated targetting of Bax to mitochondria. Journal of Cell Biology. 1998;143:207–215. doi: 10.1083/jcb.143.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths EJ, Halestrap AP. Further evidence that cyclosporin A protects mitochondria from calcium overload by inhibition of a peptidylprolyl cis-trans-isomerase. Biochemical Journal. 1991;274:611–614. doi: 10.1042/bj2740611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths EJ, Halestrap AP. Protection by cyclosporin A of ischaemia/reperfusion induced damage in isolated hearts. Journal of Molecular and Cellular Cardiology. 1993;25:1461–1469. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- Griffiths EJ, Halestrap AP. Mitochondrial non specific pores remain closed during cardiac ischaemia but open upon reperfusion. Biochemical Journal. 1995;307:93–98. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths EJ, Ocampo CJ, Savage JS, Stern MD, Silverman HS. Protective effects of low and high doses of cyclosporin A against reoxygenation injury in isolated rat cardiomyocytes are associated with differential effects on mitochondrial calcium levels. Cell Calcium. 2000;27:87–95. doi: 10.1054/ceca.1999.0094. [DOI] [PubMed] [Google Scholar]
- Griffiths GJ, Dubrez L, Morgan CP, Jones NA, Whitehouse J, Dive C, Hickman JA. Cell damage induced conformational changes of the proapoptotic Bak in vivo precede the onset of apoptosis. Journal of Cell Biology. 1999;144:903–914. doi: 10.1083/jcb.144.5.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A, Jockel J, Wei MC, Korsmeyer SJ. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO Journal. 1998;17:3878–3885. doi: 10.1093/emboj/17.14.3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenbrock CR. Chemical and physical fixation of isolated mitochondria in low energy and high energy states. Proceedings of the National Academy of Sciences of the USA. 1968;61:598–605. doi: 10.1073/pnas.61.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Davidson AM. Inhibition of Ca induced mitochondrial swelling by cyclosporin A is probably caused by the inhibitor binding to matrix peptidylprolyl cis-trans- isomerase and preventing its interacting with the adenine nucleotide translocase. Biochemical Journal. 1990;268:153–160. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Woodfield KY, Connern CP. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transtion by affecting nucleotide binding to the adenine nucleotide translocase. Journal of Biological Chemistry. 1997;272:3346–3354. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- Hsu YT, Youle RJ. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. Journal of Biological Chemistry. 1998;272:10777–10787. doi: 10.1074/jbc.273.17.10777. [DOI] [PubMed] [Google Scholar]
- Hunter PR, Haworth RA. The Ca induced membrane transition in mitochondria. Archives of Biochemistry and Biophysics. 1979;195:468–477. doi: 10.1016/0003-9861(79)90373-4. [DOI] [PubMed] [Google Scholar]
- Huser J, Rechenmacher CE, Blatter LA. Imaging the permeability pore transition in isolated mitochondria. Biophysical Journal. 1998;74:2129–2137. doi: 10.1016/S0006-3495(98)77920-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson D, Duchen MR. Fluorescence imaging of the mitochondrial permeability transition in rat cortical astrocytes in culture. Journal of Physiology. 1998;506.P:75. [Google Scholar]
- Johnson N, Khan A, Virji S, Ward JM, Crompton M. Import and processing of heart mitochondrial cyclophilin-D. European Journal of Biochemistry. 1999;263:353–359. doi: 10.1046/j.1432-1327.1999.00490.x. [DOI] [PubMed] [Google Scholar]
- Jurgensmeier JM, Xie S, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proceedings of the National Academy of Sciences of the USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr PM, Suleiman MS, Halestrap AP. Reversal of permeability transition during recovery of hearts from ischaemia and its enhancement by pyruvate. American Journal of Physiology. 1999;276:H496–502. doi: 10.1152/ajpheart.1999.276.2.H496. [DOI] [PubMed] [Google Scholar]
- Kluck RM, Degli Esposti M, Perkins G, Renken C, Kuwana T, Bossy-Wetzel E, Goldberg M, Allen T, Barker MJ, Green DR, Newmeyer DD. The proapoptotic proteins Bid and Bax cause a limited permeabilization of the mitochondrial outer membrane that is enhanced by cytosol. Journal of Biological Chemistry. 1999;47:809–822. doi: 10.1083/jcb.147.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno M, Ito M, Hayano T. The substrate binding site in E. coli cyclophilin A preferably recognizes a cis-proline isomer or a highly distorted form of the trans isomer. Journal of Molecular Biology. 1996;256:897–908. doi: 10.1006/jmbi.1996.0136. [DOI] [PubMed] [Google Scholar]
- Kottke M, Adam M, Reisinger I. Mitochondrial boundary membrane sites in brain: points of hexokinase and creatine kinase location, and control of Ca transport. Biochimica et Biophysica Acta. 1988;935:87–102. doi: 10.1016/0005-2728(88)90111-9. [DOI] [PubMed] [Google Scholar]
- Krajewski S, Krajewska M, Ellerby LM, Welsh K, Xie Z, Deveraux QL. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischaemia. Proceedings of the National Academy of Sciences of the USA. 1999;96:5752–5759. doi: 10.1073/pnas.96.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristal BS, Brown AM. Apoptogenic ganglioside GD3 directly induces the mitochondrial permeability transition. Journal of Biological Chemistry. 1999;274:23169–23175. doi: 10.1074/jbc.274.33.23169. [DOI] [PubMed] [Google Scholar]
- Leist M, Single B, Castaldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate concentration: a switch in the decision between apoptosis and necrosis. Journal of Experimental Medicine. 1997;185:1481–1486. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of Bid by caspase-8 mediates mitochondrial damage in the fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Li P-F, Dietz R, Von Harsdorf R. p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome c independent apoptosis blocked by bcl-2. EMBO Journal. 1999;18:6027–6036. doi: 10.1093/emboj/18.21.6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of the apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Luo X, Budihardjo I, Zou N, Slaughter C, Wang X. Bid, a bcl-2 interacting protein, mediates cytochrome c release from mitochondria in response to activation cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- McCabe ER. The microcompartmentation of energy metabolism at the outer mitochondrial membrane. Journal of Bioenergetics and Biomembranes. 1994;26:317–321. doi: 10.1007/BF00763103. [DOI] [PubMed] [Google Scholar]
- McEnery MW, Snowman AM, Trifiletti RR, Snyder SH. Isolation of the mitochondrial benzodiazepine receptor: association with the voltage dependent anion channel and the adenine nucleotide carrier. Proceedings of the National Academy of Sciences of the USA. 1992;89:3170–3174. doi: 10.1073/pnas.89.8.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuinness OM, Yafei N, Costi A, Crompton M. The presence of two classes of high affinity cyclosporin A binding sites in mitochondria. Evidence that the minor component is involved in the opening of an inner membrane Ca-dependent pore. European Journal of Biochemistry. 1990;194:671–679. doi: 10.1111/j.1432-1033.1990.tb15667.x. [DOI] [PubMed] [Google Scholar]
- Mahajan NP, Linder K, Berry G, Gordon GW, Heim R, Herman B. Bax and bcl-2 interactions in mitochondria probed with green fluorescent protein and fluorescence resonance energy transfer. Nature Biotechnology. 1998;16:547–553. doi: 10.1038/nbt0698-547. [DOI] [PubMed] [Google Scholar]
- Marchetti P, Castedo M, Susin SA, Zamzami N, Hirsch T, Macho A, Haeffner A, Hirsch F, Geuskens M, Kroemer G. Mitochondrial permeability transition is a central coordinating event of apoptosis. Journal of Experimental Medicine. 1996;184:1155–1166. doi: 10.1084/jem.184.3.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HLE, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G. Bax and the adenine nucleotide translocase cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2035. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- Marzo I, Brenner C, Zamzami N, Susin SA, Beutner G, Brdiczka D, Remy R, Xie Z, Reed JC, Kroemer G. The permeability transition pore complex: a target for apoptosis regulation by caspases and bcl-2 related proteins. Journal of Experimental Medicine. 1998;187:1261–1271. doi: 10.1084/jem.187.8.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montessuit S, Mazzei G, Magnenat E, Antonsson B. Expression and purification of full length human Bax. Protein Expression and Purification. 1999;15:202–206. doi: 10.1006/prep.1998.1010. [DOI] [PubMed] [Google Scholar]
- Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H, Tsujimoto Y. Bax interacts with the permeability transition pore to induce the permeability transition and cytochrome c release in isolated mitochondria. Proceedings of the National Academy of Sciences of the USA. 1998;95:14681–14686. doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazareth W, Yafei N, Crompton M. Inhibition of anoxia-induced injury in heart myocytes by cyclosporin A. Journal of Molecular and Cellular Cardiology. 1991;23:1351–1354. doi: 10.1016/0022-2828(91)90181-k. [DOI] [PubMed] [Google Scholar]
- Nechushtan A, Smith CL, Hsu YT, Youle RJ. Conformation of the Bax C-terminal region regulates subcellular location and cell death. EMBO Journal. 1999;18:2330–2341. doi: 10.1093/emboj/18.9.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newmeyer DD, Farschon DM, Reed JC. Cell free apoptosis in Xenopus egg extracts: inhibition by bcl-2 and requirement for an organelle fraction enriched in mitochondria. Cell. 1994;79:353–364. doi: 10.1016/0092-8674(94)90203-8. [DOI] [PubMed] [Google Scholar]
- Niemenen A-L, Saylor AK, Tesfai SA, Herman B, Lemasters JJ. Contribution of the mitochondrial permeability transition to lethal injury after exposure of hepatocytes to t-butylhydroperoxide. Biochemical Journal. 1995;307:99–106. doi: 10.1042/bj3070099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos V, Dharmarajan AM, Li H, Culty M, Lemay M, Sridaran R. Mitochondrial peripheral-type benzodiazepine receptor expression. Correlation with gonadotropin-releasing hormone agonist induced apoptosis in corpus luteum. Biochemical Pharmacology. 1999;58:1389–1393. doi: 10.1016/s0006-2952(99)00215-4. [DOI] [PubMed] [Google Scholar]
- Pastorino JG, Snyder JW, Serroni A, Hoek JL, Farber JL. Cyclosporin and carnitine prevent the anoxic death of cultured hepatocytes by inhibiting the mitochondrial permeability transition. Journal of Biological Chemistry. 1993;268:13791–13798. [PubMed] [Google Scholar]
- Pastorino JG, Tafani M, Rothman RJ, Marcineviciute A, Hoek FB, Farber JL. Functional consequences of the sustained or transient activation by Bax of the mitochondrial permeability transition pore. Journal of Biological Chemistry. 1999;274:31734–31739. doi: 10.1074/jbc.274.44.31734. [DOI] [PubMed] [Google Scholar]
- Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonte S, Bernardi P. The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation-reduction state of vicinal thiols. Journal of Biological Chemistry. 1994;269:16638–16642. [PubMed] [Google Scholar]
- Qian T, Niemenen A-L, Herman B, Lemasters JJ. Mitochondrial permeability transition in pH-dependent reperfusion injury to hepatocytes. American Journal of Physiology. 1997;273:C1783–1797. doi: 10.1152/ajpcell.1997.273.6.C1783. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Saikumar P, Dong Z, Patel Y, Hall K, Hopfer U, Weinberg J, Venkatachalam MA. Role of hypoxia-induced Bax translocation and cytochrome c release in reoxygenation injury. Oncogene. 1998;17:3401–3415. doi: 10.1038/sj.onc.1202590. [DOI] [PubMed] [Google Scholar]
- Scaffidi C, Schmitz I, Zha J, Korsmeyer SJ, Krammer PH, Peter ME. Differential modulation of apoptosis sensitivity in CD95 type 1 and type 2 cells. Journal of Biological Chemistry. 1999;274:22532–22538. doi: 10.1074/jbc.274.32.22532. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Petronilli B, Di Lisa F, Bernardi P. Commitment to apoptosis by GD3 ganglioside depends on opening of the permeability transition pore. Journal of Biological Chemistry. 1999;274:22581–22585. doi: 10.1074/jbc.274.32.22581. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Kamiike W, Hatanaka N, Miyata T. Beneficial effects of cyclosporin on reoxygenation induced injury in hypoxic rat liver. Transplantation. 1994;57:1526–1537. [PubMed] [Google Scholar]
- Shimizu S, Narita N, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlet DR, Abersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterisation of mitochondrial apoptosis inducing factor. Nature. 1999;397:441–450. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Susin SA, Zamzami N, Larochette N, Marzo I, Brenner C, Hirsch T, Petit PX, Geuskens M, Kroemer G. A cytofluorimetric assay of nuclear apoptosis induced in a cell free system: application to ceramide induced apoptosis. Experimental Cell Research. 1997;236:397–403. doi: 10.1006/excr.1997.3733. [DOI] [PubMed] [Google Scholar]
- Tanveer A, Virji S, Andreeva L, Totty NF, Hsuan JJ, Ward JM, Crompton M. Involvement of cyclophilin-D in the activation of a mitochondrial pore by Ca and oxidative stress. European Journal of Biochemistry. 1996;238:166–172. doi: 10.1111/j.1432-1033.1996.0166q.x. [DOI] [PubMed] [Google Scholar]
- Veinot JP, Gattinger PA, Fliss H. Early apoptosis in human myocardial infarcts. Human Pathology. 1997;28:485–495. doi: 10.1016/s0046-8177(97)90039-3. [DOI] [PubMed] [Google Scholar]
- Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi X-G, Youle RJ. Movement of Bax from mitochondria to cytosol during apoptosis. Journal of Cell Biology. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfield K, Ruck A, Brdiczka D, Halestrap AP. Direct demonstration of specific interactions between cyclophilin-D and the adenine nucleotide translocase confirm their role in the mitochondrial permeability transition. Biochemical Journal. 1998;336:287–290. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Roth KA, Korsmeyer SJ. Bid-deficient mice are resistant to fas-induced hepatocellular apoptosis. Nature. 1999;400:886–891. doi: 10.1038/23730. [DOI] [PubMed] [Google Scholar]
- Yoshizaki K, Watari H, Radda GK. Role of phosphocreatine in energy transport in skeletal muscle of bullfrog studied by 31P-NMR. Biochimica et Biophysica Acta. 1990;1051:44–50. doi: 10.1016/0167-4889(90)90186-h. [DOI] [PubMed] [Google Scholar]
- Zamzami N, Susin SA, Marchetti P, Hirsch T, Gomez-Monterey R, Castedo MJ, Kroemer G. Mitochondrial control of nuclear apoptosis. Journal of Experimental Medicine. 1996;183:1533–1542. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homogous to C. elegans CED4, participates in cytochrome c dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]