

Abstract
Although it has been known for over three decades that mitochondria are endowed with a complex array of Ca2+ transporters and that key enzymes of mitochondrial metabolism are regulated by Ca2+, the possibility that physiological stimuli that raise the [Ca2+] of the cytoplasm could trigger major mitochondrial Ca2+ uptake has long been considered unlikely, based on the low affinity of the mitochondrial transporters and the limited amplitude of the cytoplasmic [Ca2+] rises. The direct measurement of mitochondrial [Ca2+] with highly selective probes has led to a complete reversion of this view, by demonstrating that, after cell stimulation, the cytoplasmic Ca2+ signal is always paralleled by a much larger rise in [Ca2+] in the mitochondrial matrix. This observation has rejuvenated the study of mitochondrial Ca2+ transport and novel, unexpected results have altered long-standing dogmas in the field of calcium signalling. Here we focus on four main topics: (i) the current knowledge of the functional properties of the Ca2+ transporters and of the thermodynamic constraints under which they operate; (ii) the occurrence of mitochondrial Ca2+ uptake in living cells and the key role of local signalling routes between the mitochondria and the Ca2+ sources; (iii) the physiological consequences of Ca2+ transport for both mitochondrial function and the modulation of the cytoplasmic Ca2+ signal; and (iv) evidence that alterations of mitochondrial Ca2+ signalling may occur in pathophysiological conditions.
Setting the frame for mitochondrial Ca2+ transport: mechanism of energy conservation and non-equilibrium distribution
Energy conservation is achieved through charge separation at the inner mitochondrial membrane, whereby electrons derived from intermediary metabolism are transferred to oxygen through the respiratory chain in a process coupled to H+ pumping. Since the passive permeability to H+ is low, this process results in the establishment of a H+ electrochemical gradient (ΔμH). ΔμH is approximately 200 mV, and under physiological conditions most of the gradient is in the form of a membrane potential difference (ΔΨ) (Mitchell, 1966). This has major implications for Ca2+ transport and distribution, because for a divalent cation (z = 2 in the Nernst equation) the equilibrium condition (ΔμCa = 0) is given by:
Since the inner mitochondrial membrane possesses electrophoretic systems for Ca2+ transport, and since typical values of cytosolic free [Ca2+] ([Ca2+]c) oscillate between about 0.1 and 1 μm, for a membrane potential of -180 mV (negative inside) equilibrium matrix free [Ca2+] ([Ca2+]m) should be 0.1-1 M, which is 100000- to 1000000-fold higher than the measured range of [Ca2+]m. This displacement from equilibrium is due to the fact that Ca2+ distribution is modulated by kinetic rather than thermodynamic parameters (Azzone et al. 1977), in the sense that it represents a steady state where electrophoretic Ca2+ uptake is precisely matched by Ca2+ efflux via at least two separate pathways: (i) a Na+-independent Ca2+ efflux pathway, possibly a H+-Ca2+ exchanger, and (ii) a Na+-dependent Ca2+ efflux pathway, most probably a Na+-Ca2+ exchanger. An additional pathway for Ca2+ release may be represented by the permeability transition pore (PTP), a non-selective high-conductance channel, which is under intense scrutiny as both a potential target and an effector mechanism in cell death. The following paragraphs summarise the functional properties of these mitochondrial Ca2+ transport pathways (schematically represented in Fig. 1), whose molecular nature is still unsolved (see Bernardi, 1999, for a detailed review and original references).
Figure 1. Schematic representation of the mitochondrial Ca2+ transport pathways in energised mitochondria.
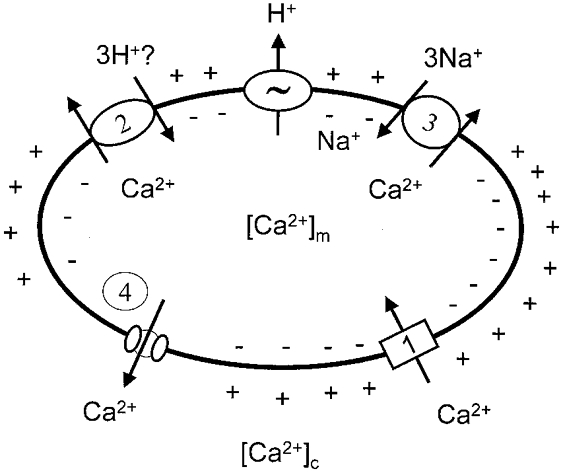
1, uptake pathways (uniporter, RaM). 2-4, efflux pathways: H+-Ca2+ (2) and Na+-Ca2+ (3) exchangers, and the permeability transition pore (4).
Pathways for Ca2+ uptake
Calcium uniporter
The Ca2+ uniporter is a mitochondrial channel that transports Ca2+ and Sr2+ but not Mg2+. In respiring mitochondria the kinetics of Ca2+ uptake become rapidly limited by the rate of H+ pumping as [Ca2+]o is raised above about 10 μm. This may lead to a serious underestimation of both the Vmax and the apparent Km for Ca2+. The true kinetic properties of the uniporter have been defined through the use of K+ diffusion potentials induced by valinomycin in respiratory-inhibited mitochondria. These studies established a Vmax in excess of 1400 nmol Ca2+ (mg protein)−1 min−1 and an apparent Km lower than 10 μm in sucrose-based media (Bragadin et al. 1979). The Ca2+ uniporter is regulated by a number of modulators (inhibitors and activators). Ruthenium compounds (typically ruthenium red, RR), represent a class of non-competitive inhibitors. A second class of inhibitors is divalent cations that are themselves transported by the uniporter (e.g. Sr2+, Mn2+, Ba2+ and lanthanides). Inhibition is generally competitive, but not all of the effects are exerted at the transport site(s) because the uniporter is regulated by metal ion binding sites that modulate the affinity for Ca2+. Mg2+, Mn2+ and Ca2+ itself can exemplify this class of effectors. In the millimolar range, Mg2+ changes the relationship between the rate of Ca2+ transport and [Ca2+]o from hyperbolic to sigmoidal, decreases the Vmax and increases the apparent Km for Ca2+ from 10 to about 50 μm (Bragadin et al. 1979). Mg2+ affects the uniporter by binding to a regulatory site(s) rather than to the transport site(s). Accordingly, Mg2+ is not transported by the uniporter, it does not affect the Ca2+ conductance (Heaton & Nicholls, 1976), and its effects can be mimicked by approximately 50-fold higher concentrations of monovalent cations like Li+ (Bragadin et al. 1979). Under specific conditions Mn2+ can stimulate rather than inhibit the kinetics of Ca2+ transport by counteracting the effects of Mg2+ through a mixed-type competition (Allshire et al. 1985). The uniporter is activated by external Ca2+ itself, and it undergoes deactivation upon removal of extramitochondrial Ca2+ (Kroner, 1986). Thus, at the low [Ca2+] values prevailing in the cytosol the activity of the uniporter could be extremely low, due also to inhibition by adenine nucleotides (ATP > ADP > AMP) (Litsky & Pfeiffer, 1997). Depolarisation after energy-dependent accumulation of Ca2+ is readily followed by Ca2+ release that is enhanced by RR (Igbavboa & Pfeiffer, 1988). Thus, under these conditions Ca2+ release is not occurring through the uniporter but rather via the voltage-dependent mitochondrial PTP, and it is accordingly inhibited by cyclosporin A (CsA). The uniporter is therefore not readily reversible, possibly because of the low [Ca2+]o at the onset of depolarisation, which deactivates the uniporter, and because a high membrane potential may be required to maintain it in a transport-competent conformation (Kapus et al. 1991).
Rapid uptake mode (RaM)
Ca2+ uptake is more efficient when energised mitochondria are exposed to trains of Ca2+ pulses of physiological concentration (about 400 nM) rather than to an identical steady [Ca2+]o for the same overall time. This mechanism can be reset in less than 0.75 s by a decrease in [Ca2+]o to between 100 and 200 nM. Like the Ca2+ uniporter, the RaM is inhibited by RR albeit at higher concentrations; unlike the uniporter, Mg2+ does not affect it. Gunter and co-workers favour the idea that it is mediated by a specific transport mechanism that might be responsible for mitochondrial Ca2+ uptake from [Ca2+]c transients in vivo (Sparagna et al. 1995). Up to now, however, no direct evidence for this type of mechanism in intact cells has been obtained.
Pathways for Ca2+ efflux
Na+-independent Ca2+ efflux
Respiring mitochondria maintain steady-state [Ca2+]o at a constant value of 0.25-1.0 μm. If RR is added to block the uniporter, a process of Ca2+ efflux ensues that is taken as evidence that RR-insensitive Ca2+ efflux (coupled to uniporter-mediated reuptake) was also occurring prior to the addition of RR via a specific pathway (Vasington et al. 1972). Many early studies of RR-insensitive Ca2+ efflux in mitochondria, however, have been complicated by the unrecognised contribution of the permeability transition. In the steady state, a small fraction of mitochondria may indeed undergo reversible depolarisations due to opening of the Ca2+-dependent PTP. Ca2+ released from this permeabilised fraction would be taken up by the polarised mitochondria, and addition of RR would result in what appears to be net Ca2+‘efflux’ (Riley & Pfeiffer, 1985). A reassessment of the Na+-independent pathway for Ca2+ efflux in rat liver mitochondria under conditions in which the occurrence of a permeability transition could be excluded established that: (i) it saturates at Ca2+ loads of 25 nmol (mg protein)−1; (ii) its Vmax is not influenced by the concentration of inorganic phosphate and does not exceed a rate of 1.2 nmol Ca2+ (mg protein)−1 min−1; and (iii) it extrudes Ca2+ against a gradient that is much higher than thermodynamically permissible for an electroneutral H+-Ca2+ exchanger (Wingrove & Gunter, 1986a). Thus, either Ca2+ efflux occurs via a nH+-Ca2+ exchanger with n > 2, or it has an active component. Accordingly, Ca2+ efflux is inhibited rather than stimulated by small depolarisations (Bernardi & Azzone, 1983).
Na+-dependent Ca2+ efflux
After the addition of RR, the rate of Ca2+ efflux can be stimulated by the addition of Na+ (Crompton et al. 1978). Ca2+ efflux is likely to occur via a Na+-Ca2+ exchanger that mediates physiological Ca2+ cycling. The Vmax ranges between 2.6 (liver) and 18 nmol Ca2+ (mg protein)−1 min−1 (heart). The dependence on Na+ is sigmoidal, with typical Km values of about 8-10 mm Na+. Ca2+ efflux is inhibited by Sr2+, Ba2+, Mg2+ or Mn2+, by RR above 5 nmol (mg protein)−1, by submicromolar concentrations of the membrane potential probe triphenylmethylphosphonium, and by a variety of compounds of pharmacological interest such as amiloride, trifluoperazine, diltiazem, verapamil, clonazepam, bepridil and CGP37157, a recently developed specific blocker (Cox & Matlib, 1993), and is stimulated by short-chain alcohols. It is modulated by matrix pH (optimum at pH 7.6) and by in vivo treatment with glucagon and β-adrenergic agonists. Ca2+ efflux is inhibited by antimycin A and protonophores, indicating that the transmembrane potential favours the exchange (Crompton et al. 1976). This is consistent with studies indicating that the likely stoichiometry is 3Na+:Ca2+ (Wingrove & Gunter, 1986b; Jung et al. 1995).
Permeability transition pore
The PTP is a high conductance, non-selective channel that exhibits a prominent dependence on matrix Ca2+ and is inhibited by CsA. Reversible PTP openings have been resolved both in individual isolated mitochondria (Huser et al. 1998) and in intact cells (Petronilli et al. 1999), but whether these are accompanied by Ca2+ release in situ remains unclear because evidence based on the effects of CsA alone may be ambiguous. On the one hand, PTP inhibition by CsA is transient in long time-frame experiments, and depends strictly on variables that cannot be controlled in vivo like matrix ADP and Mg2+ (which are required for inhibition) and Ca2+ (which counteracts it). On the other hand, CsA may also inhibit cyclophilin B-modulated Ca2+ transport in the endoplasmic reticulum (ER) (Bram & Crabtree, 1994), which complicates the interpretation of results at the cellular level. It should be noted that coupling of high Vmax uptake transport systems with slow and easily saturable release systems exposes mitochondria to the hazards of Ca2+ overload and that fast mitochondrial Ca2+ release demands charge compensation, which cannot be easily achieved through the impermeable inner membrane. In this respect the large size and lack of selectivity of the PTP confer the clear advantage of providing charge compensation within the channel itself, thus allowing maximal Ca2+ flux (i.e. at zero potential). This would guarantee fast Ca2+ release even for vanishingly small [Ca2+] gradients, and regulation would be achieved directly through modulation of the pore open time. Whether the PTP can function as a physiological mitochondrial Ca2+ release channel in vivo remains an intriguing possibility (Bernardi & Petronilli, 1996) that deserves further testing.
Mitochondrial Ca2+ transport in living cells
Based on the thermodynamic and kinetic properties of the transport pathways (see above) and the [Ca2+] measured in the cytoplasm of resting (∼0.1-0.2 μm) or stimulated (0.5-3 μm) cells, the general prediction was that, under physiological conditions, mitochondrial Ca2+ accumulation would be marginal (while significant uptake could possibly be achieved in pathological Ca2+ overload). Direct measurements of mitochondrial [Ca2+] with a highly specific probe, a targeted chimera of the Ca2+-sensitive photoprotein aequorin, proved the exact opposite (Rizzuto et al. 1992). In HeLa cells, stimulation with physiological stimuli acting on Gq-coupled receptors (and thus causing the release of Ca2+ from the ER) caused a large, rapid rise in [Ca2+] in the mitochondrial matrix ([Ca2+]m) (Fig. 2). Through the work of various laboratories, using widely different probes (recombinant aequorin or mitochondrially accumulated fluorescent probes), this observation has been extended to a large variety of cell types, including fibroblasts, endothelial and epithelial cells, cardiac and skeletal muscle cells, neurones and pancreatic β-cells, to name but a few (for a review, see Duchen, 1999). It is thus now accepted that in virtually every cell type, mitochondrial Ca2+ uptake invariably follows stimulation with an agonist causing a cytoplasmic [Ca2+] rise.
Figure 2. Agonist-dependent mitochondrial and cytoplasmic [Ca2+] increases in the presence and absence of the protonophore FCCP.
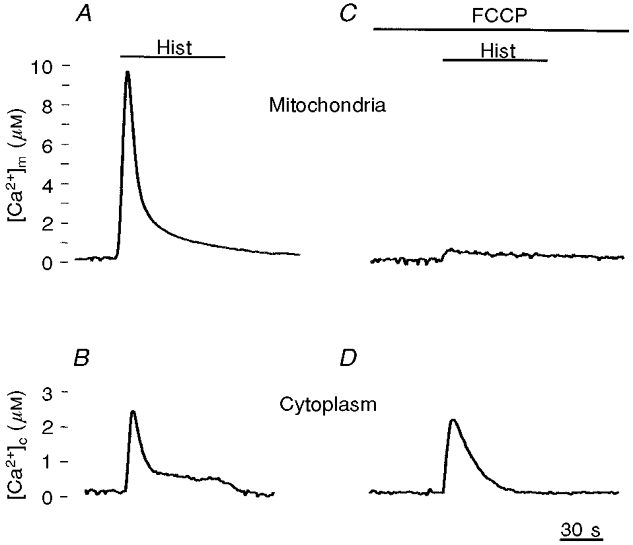
The traces show calibrated [Ca2+] values obtained in HeLa cells transiently expressing an aequorin chimera localised in the mitochondria (A and C) or in the cytoplasm (B and D). Detection and calibration of the luminescence signals were carried out as previously described (Brini et al. 1995). Where indicated, HeLa cells were challenged with histamine (100 μm; Hist), an agonist coupled to the generation of inositol 1,4,5-trisphosphate and thus the release of Ca2+ from intracellular stores. In C and D, the cells were treated with 5 μm FCCP (carbonyl cyanide p-(trifluoro-methoxy)-phenylhydrazone), which was added 1 min before the agonist and maintained throughout the stimulation.
The accumulation of Ca2+ occurs via the expected route for Ca2+ uptake, i.e. the Ca2+ uniporter, as highlighted by two simple observations: (i) collapse of the proton-motive force with a protonophore virtually abolishes mitochondrial Ca2+ uptake (Fig. 2); and (ii) injection of RR fully inhibits the uptake of Ca2+. As to the amplitude of the [Ca2+]m peak, values > 10 μm (and see below) are reached in < 5 s, thus revealing a striking discrepancy between a rapid rate of mitochondrial Ca2+ uptake in intact cells and the biochemical properties characterised in vitro (Rizzuto et al. 1993).
An easy way of rationalising conflicting observations is to invoke the existence of cellular microscopic environments where the concentration of the molecules participating in a given reaction is different from that measurable on a macroscopic cellular scale, the so called ‘bulk phase’. Though in many cases this may be an elegant way of concealing ignorance, ample theoretical and experimental evidence has accumulated demonstrating not only the existence but also the physiological importance of ‘microdomains’. This is particularly true for Ca2+, for which probes and measuring systems are far more accurate than for any other cellular parameter.
We followed the hypothesis that mitochondrial Ca2+ accumulation in living cells depended on the existence of a microheterogeneity in cellular Ca2+ concentration and in particular on localised hot spots of high Ca2+ concentration in the vicinity of the organelles. At the time this initial unexpected observation was made, alternative possibilities were considered, such as: (i) the existence within the cytoplasm of ‘factors’ that modify the kinetic parameters of the Ca2+ uniporter; (ii) underestimation by the classical probes of the real changes in bulk cytoplasmic Ca2+; and (iii) artefacts of the technique, at the time in its infancy.
Perhaps the most convincing evidence that only the microdomain hypothesis could explain the experimental findings was obtained in digitonin-permeabilised cells. Under these conditions the rate and extent of mitochondrial Ca2+ accumulation were almost perfectly reproduced by perfusing the cells with InsP3, the physiological agonist that mobilises Ca2+ from the ER (Rizzuto et al. 1993). In contrast, perfusion of buffered Ca2+ medium, with an up to 3- to 4-fold higher [Ca2+] than that measured in the bulk cytoplasm, resulted in sluggish mitochondrial Ca2+ uptake. Only when the buffered [Ca2+] was increased to 5-10 μm (10- to 20-fold greater than the level measured in the bulk cytosol) did the rate of mitochondrial Ca2+ accumulation approach that observed in intact cells (Rizzuto et al. 1993). In other words, if the functional interactions between the ER and mitochondria are kept intact (as in permeabilised cells), even in the absence of any soluble cytoplasmic factor the localised release of Ca2+ through the physiologically relevant channels results in efficient mitochondrial Ca2+ accumulation.
This initial observation was then reproduced in different cell types and using different probes to measure mitochondrial and cytoplasmic [Ca2+]. The vast majority of authors now agree that in living cells mitochondrial Ca2+ accumulation results from the close apposition of the organelles to either ER/SR Ca2+ release channels or to plasma membrane Ca2+ channels. An obvious prerequisite for such a hypothesis is the existence of morphologically identifiable close appositions between mitochondria and Ca2+ channels. A retrospective analysis of standard electron micrographs of various tissues reveals that indeed close apposition of mitochondria to ER cisternae occurs frequently. In some cell types, for example Purkinje neurones, the proximity of mitochondria to InsP3 receptor-rich ER cisternae has been reported repeatedly (Satoh et al. 1990). Similarly, in cardiac myocytes the distance between mitochondria and SR terminal cisternae, which contain the ryanodine receptors, has been reported to be as small as 5 nm (Ramesh et al. 1998). The possibility that these close appositions between the ER and mitochondria are an artefact of fixation is unlikely: (i) close interactions of ER and mitochondria can be also observed in rapidly frozen samples, where the cellular architecture is known to be highly preserved (Pezzati et al. 1997); and (ii) in intact cells, apposition of mitochondria to the ER (at a distance of less than 100 nm) is a common finding and involves a significant portion of the mitochondrial surface (Rizzuto et al. 1998) (Fig. 3).
Figure 3. 3-D imaging of mitochondrial and ER structure in living HeLa cells.

The two organelles were labelled with appropriately targeted chimeras of green fluorescent protein variants with different spectral properties. The images acquired at each focal plane were computationally deblurred and 3-D reconstructed. The mitochondrial and ER images are displayed in red and green, respectively, while the white areas are those in which overlap between the two images was detected. Reproduced with permission from Rizzuto et al. 1998.
A corollary of the concepts outlined above is that only a subfraction of the mitochondrial network should experience the Ca2+ hot spots. In fact, not only are the channels non-homogeneously distributed along the ER-SR and the plasma membrane, but also the morphological data indicate that the physical proximity of mitochondria to other membranes involves only part of their surface. Evidence for the heterogeneity of Ca2+ uptake within mitochondria upon physiological stimulation is supported by a number of indirect observations. For example, similar cytoplasmic Ca2+ increases result in different average accumulations of Ca2+ in the mitochondria depending on the source of Ca2+. In HeLa cells, for example, Ca2+ release from the stores is more efficient at inducing mitochondrial Ca2+ accumulation than Ca2+ influx through the plasma membrane (Rizzuto et al. 1993), while the opposite is true for rat pituitary GH3 cells (Pizzo et al. 1997). Furthermore, the apparent increases in [Ca2+], measured with targeted recombinant aequorin, are reduced upon repetitive activation of the same Ca2+ channels (e.g. in HeLa cells and in adrenal medullary cells) despite identical elevations in cytoplasmic [Ca2+] (Rizzuto et al. 1994). It has been argued that, since aequorin is irreversibly discharged upon binding of Ca2+ and emission of the photon, a selective consumption of the photoprotein occurs only in the domains of the highly interconnected mitochondrial network (Rizzuto et al. 1998) and/or in subpopulations of mitochondria exposed to high Ca2+ microdomains, where no probe remains available for a subsequent stimulation. We think that final proof for such heterogeneity still awaits conclusive evidence from the direct measurement of the [Ca2+] in single organelles, which is beyond the reach of currently available indicators like rhod-2 and similar dyes.
A key question concerns the Ca2+ levels that are reached within the matrix when the organelles are exposed in situ to the high [Ca2+] microdomains. Targeted recombinant aequorin should be instrumental in their determination, given its unsurpassed selectivity of localisation. Furthermore, thanks to the use of mutated aequorin isoforms (Kendall et al. 1992) and of different coenzymes (Barrero et al. 1997), aequorin allows us to explore a very ample range of Ca2+ concentrations, from 0.1 μm up to 1-2 mm. Our initial estimates of the average peak levels of matrix Ca2+ concentrations in HeLa cells stimulated by histamine (4-5 μm; Rizzuto et al. 1993) were probably underestimated by a factor of 2-3, and a recalculation of the algorithm used to transform the crude luminescence signal into Ca2+ levels (Brini et al. 1995) rather suggests values of about 10-12 μm. A much more important cause of underestimation of the peak levels is due, however, to the heterogeneity of the responding mitochondrial population (see above). Due to the intrinsic calibration properties of aequorin, this underestimation can be larger than one order of magnitude. Using a low-affinity aequorin, Montero et al. (2000) have shown that in adrenal medullary cells the peak [Ca2+]m of the highly responding mitochondria can be as high as 500-700 μm. The peak levels of Ca2+ calculated with rhod-2 are much lower than those revealed by aequorin, and hardly different from those of the cytoplasm, i.e. 1-2 μm (Babcock et al. 1997). It is difficult to explain this discrepancy, particularly for estimates obtained in the same cell type. In our opinion, the intrinsic physicochemical characteristics of the different probes and the heterogeneity of the response within the organelle population may be at the basis of these very different results. In particular, the average aequorin signal is intrinsically biased towards the Ca2+ levels reached in the subpopulation of highly responding cells. For example, if 50 % of mitochondria respond (as has been suggested for the adrenal cells) with a Ca2+ rise of 500 μm, while the rest of the population reaches 1-2 μm, the average signal calculated with a low Ca2+ affinity aequorin would give an apparent mean peak of around 200 μm. The average signal of fluorescent dyes of the BAPTA family, such as rhod-2, in contrast, is biased towards the lower responding population. Assuming a situation such as that outlined above, a mitochondria-trapped dye with a Kd for Ca2+ of 1 μm would produce a signal that, on average, would be calibrated as an apparent increase of about 3 μm. The absolute levels of [Ca2+] reached within the mitochondrial matrix are of major relevance in coping with the activation of metabolic reactions occurring within the matrix (see below). If the high values indicated by aequorin are indeed correct, it can be predicted that most of the Ca2+-dependent enzymes located in the organelle matrix (that have a Kd for Ca2+ of around 1-10 μm) would not only be maximally activated, but also would remain so for prolonged periods of time. Increases within the 1-2 μm range, as suggested by the rhod-2 data, would lead to only partial activation and only for a short time (see below).
The mechanism and role of Ca2+ efflux from mitochondria in intact cells have been less extensively investigated. The lack of effect of CsA on Ca2+ efflux and the reduction in the rate of [Ca2+]m decrease upon treatment with the specific inhibitor CGP37157 (Brini et al. 1999; Montero et al. 2000; Colegrove et al. 2000; T. Pozzan & R. Rizzuto, unpublished data) suggest that under physiological conditions the Na+-Ca2+ exchange mechanism may be the predominant efflux pathway.
The physiological role of mitochondrial Ca2+ uptake
Mitochondrial metabolism
A first, obvious possibility stems from the fine biochemical work of Denton, McCormack, Hansford and co-workers, who, in the 1970s, demonstrated that the main function of mitochondria, oxidative phosphorylation, could be a Ca2+-regulated process, because three dehydrogenases of the Krebs cycle (pyruvate-, isocitrate and oxoglutarate dehydrogenase) are activated, with different mechanisms, by an increase in the [Ca2+] to which they are exposed (McCormack et al. 1990; Hansford, 1994). In the latter two cases, Ca2+ acts as an allosteric regulator, while in the case of pyruvate dehydrogenase a Ca2+-dependent phosphatase converts the enzyme complex into the active form. As a consequence, a [Ca2+] rise to micromolar levels in the mitochondrial matrix could increase the availability of reducing equivalents to the respiratory chain. This appears to actually occur in living cells, where the agonist-dependent Ca2+ rise is paralleled by a rise in the reduced form of NADH levels (Rizzuto et al. 1994).
When the phenomenon was investigated at the single cell level, a more sophisticated scenario emerged that shed further light not only on mitochondrial function but also on the significance of the spatio-temporal complexity of intracellular Ca2+ signalling. Indeed, much is known about the mechanisms that allow a variety of agonists to cause the periodic spiking of cytoplasmic Ca2+ concentration (the phenomenon commonly referred to as ‘Ca2+ oscillations’), but little is known about how these oscillations are perceived and decoded by a living cell. Pralong and co-workers showed that agonist-dependent Ca2+ spiking causes oscillatory increases in NADH levels (Pralong et al. 1994). Hajnoczky and co-workers went further and demonstrated that cytoplasmic Ca2+ oscillations, by causing parallel rises in the mitochondrial matrix, induce a rise in NADH levels that lasts longer that the Ca2+ rise itself (e.g. for a kinetic delay imposed by the rephosphorylation of pyruvate dehydrogenase) (Hajnoczky et al. 1995). As a consequence, a rapid pace of cytoplasmic, and hence, mitochondrial Ca2+ spiking causes a nearly sustained rise in NADH levels. In other words, cytoplasmic Ca2+ oscillations, through the effect on mitochondrial Ca2+ uptake, are decoded inside the mitochondria into a long-term activation of mitochondrial metabolism. Ca2+ oscillations proved to be the most efficient route for achieving such an effect, because they allow a repetitive, long-term activation of the dehydrogenases. Conversely, given that mitochondrial Ca2+ uptake is mediated by the transient generation of Ca2+ microdomains in the proximity of mitochondria and then rapidly slows down, a sustained rise in the cytoplasm causes a short-lived rise in mitochondrial [Ca2+] and hence in NADH levels (Hajnoczky et al. 1995).
The increased availability of NADH in the stimulated cell (i.e. a cell where Ca2+ has triggered energy-consuming processes such as secretion, motion or simply Ca2+ re-extrusion) will in turn allow the respiratory chain to match the increased energy demand with an increased rate of respiration, which rebuilds the proton-motive force used for ATP phosphorylation. If this is the case, the net effect should be that of increasing mitochondrial and eventually cytoplasmic ATP levels. To test this hypothesis, a mitochondrially targeted ATP probe based on the luminescent protein luciferase was constructed (Jouaville et al. 1999). The starting point in the development of this new technique was the observation that in a ‘cytoplasmic’ environment (which includes competing anions and proteins) the affinity of luciferase for ATP is markedly lower than that measured in saline solution (Allue et al. 1996). Thus, the in situ light emission of the photoprotein can be used to report variations of ATP levels in the physiological range (1-10 mm). Using this novel probe a set of interesting observations was made. The first is that agonists causing an intracellular Ca2+ signal increase mitochondrial, and subsequently cytosolic, ATP levels (Jouaville et al. 1999; Fig. 4). This is demonstrably due to an increased activity of the mitochondrial ATP synthase, because it is abolished by oligomycin (Fig. 4), and it strictly depends on the Ca2+ rise. Indeed, when the latter is dampened (e.g. by depleting Ca2+ from the ER or by chelating Ca2+ in the cytoplasm), the ATP rise is reduced proportionally (Jouaville et al. 1999). Finally, this ‘Ca2+ effect’ appears to constitute a long-lasting activation mechanism. In HeLa cells maintained in glycolytic conditions stimulation with Ca2+ does not cause a rise in ATP levels, but enhances the rise in ATP levels caused by the subsequent readdition of oxidisable substrates tens of minutes after the removal of the agonist (and hence well after the return of mitochondrial [Ca2+] to basal values) (Jouaville et al. 1999). As to the mechanisms allowing this long-term activation, they could include prolonged activation of pyruvate dehydrogenase activity (e.g. for a delay in the phosphorylation step), a direct effect on the respiratory chain, possibly dependent on a Ca2+-dependent increase in matrix volume (Robb-Gaspers et al. 1998), or other as yet unidentified mitochondrial targets.
Figure 4. Agonist-dependent mitochondrial [Ca2+] (A and C) and [ATP] (B and D) increases in the presence and absence of the ATP synthase inhibitor oligomycin.
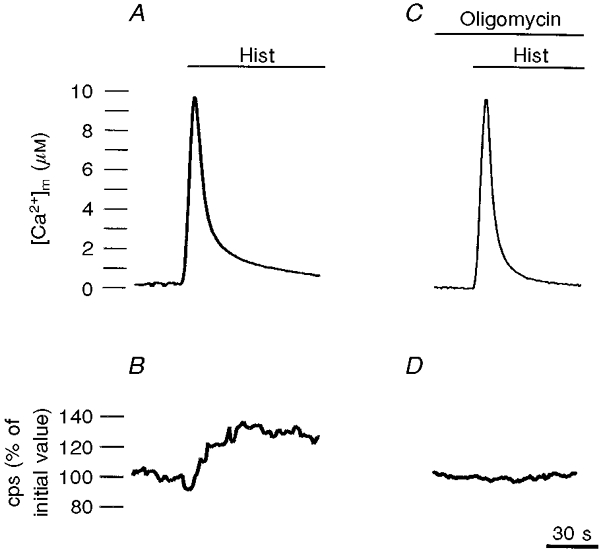
The calibrated [Ca2+] values were obtained as in Fig. 2, while ATP levels were inferred from light emission of a luciferase chimera targeted to the mitochondria (Jouaville et al. 1999). cps, counts per second. Where indicated, the cells were treated with 100 μm histamine. In C and D, the cells were treated with 2 μm oligomycin, which was added before the agonist and maintained throughout the stimulation.
In keeping with the complex spatio-temporal patterns in Ca2+ signalling, modulation of ATP production could also occur with a defined, functionally significant micro-heterogeneity. This concept is supported by emerging evidence. Landolfi and co-workers showed that ATP microdomains may exist in the proximity of ER Ca2+ pumps, based on the effect on ER Ca2+ refilling (Landolfi et al. 1998). Kennedy and co-workers showed that the agonist-induced ATP rises are more sustained in the subplasmalemmal space and in mitochondria than in the bulk cytosol of insulin-secreting cells, as indicated by luciferase chimeras specifically targeted to these compartments (Kennedy et al. 1999). Finally, we would like to mention that the role of mitochondrial Ca2+ in modulating ATP levels may also play a direct signalling role in specialised cell systems such as the pancreatic β-cell, where control is exerted directly on insulin secretion. We refer the reader to the work of Wollheim's group for a detailed account of these important observations (Maechler & Wollheim, 2000, this issue).
Mitochondria and the determination of the spatio-temporal pattern of Ca2+ signalling
The obvious relevance of mitochondrial Ca2+ uptake for the control of organelle function, as well as the general consensus that only a small fraction of Ca2+ released from the ER (or entering from the plasma membrane channels) actually ends up in the mitochondria, has led to an underestimation of the role of mitochondria in controlling the cytosolic Ca2+ rise. In other words, mitochondrial Ca2+ uptake was considered relevant for conveying an activatory signal to these organelles, but not significant in the overall balance of cellular Ca2+ homeostasis.
In the past few years, converging evidence that this is not the case has emerged from the work of various groups and there is now an established consensus for the concept that mitochondria participate in shaping the spatio-temporal complexity of calcium signalling in at least three different ways.
(i) Mitochondria, clustered in a defined portion of a polarised cell, could act as an ‘immobile buffer’ preventing, or delaying, the spread of the Ca2+ signal to the rest of the cell. For example, in the highly polarised pancreatic acinar cells, in some cases the Ca2+ signal is restricted to the apical pole, thus inducing the release of enzyme-containing granules, while in other cases it also spreads to the rest of the cell, thus reaching the nucleus and causing long-term effects dependent on the activation of gene transcription. Petersen and co-workers have recently demonstrated that a layer of mitochondria localised at the base of the granule-rich area plays a key role in restricting the effect of weaker agonists to the secretory pole. If the ability of mitochondria to accumulate Ca2+ is abolished, the signal conveyed by the same agonists is converted into a global signal extending to the basolateral portion of the cell (Tinel et al. 1999).
(ii) Cross-talk between the intracellular stores and the mitochondria could ‘bypass’ the bulk cytosol and communication could be restricted to regions of close contact with the ER. Indeed, it can be envisaged that mitochondrial Ca2+ uptake participates in dissipating the microdomains of high [Ca2+] at the mouth of the ER channels, which, in turn, exert a feedback control on the channel. The first demonstration of this possibility came from the work of Lechleiter and co-workers in Xenopus oocytes. They analysed the propagation pattern of cytosolic Ca2+ waves and demonstrated that mitochondrial energisation enhances Ca2+ release through the InsP3-gated channel by increasing Ca2+ uptake into mitochondria and thereby dissipating the Ca2+ microdomain at the mouth of the channel (Jouaville et al. 1995). More recently, similar evidence was obtained in mammalian cells. In primary cultures of hepatocytes, Hajnoczky and co-workers demonstrated that mitochondrial Ca2+ uptake suppressed the positive effects of Ca2+ on the InsP3 receptor, thus reducing Ca2+ release at submaximal doses of agonists (Csordas et al. 1999; Hajnoczky et al. 1999). In astrocytes, Boitier and co-workers showed that mitochondrial Ca2+ uptake reduces the diffusion of Ca2+ waves, thus exerting a negative feedback control on the explosive recruitment of InsP3 receptors (Boiter et al. 1999). In BHK-1 fibroblasts, Landolfi and co-workers showed that local Ca2+ buffering by mitochondria (as well as ATP production) exerts the opposite effect on the InsP3 receptor, as inhibition of mitochondrial Ca2+ uptake and/or of ATP production reduces the rate of Ca2+ release (Landolfi et al. 1998). These results are not surprising, nor contradictory, given the known bell-shaped Ca2+ response curve of the InsP3 receptor, with significant differences among the various isoforms (e.g. type 2 lacks the inhibition at high [Ca2+]; Ramos-Franco et al. 1998). Rather, they highlight how mitochondria can finely tune the microenvironment of the ER Ca2+ release sites, which in turn is the real determinant of the amplitude and spatio-temporal pattern of the Ca2+ increase.
(iii) Transient openings of the PTP could induce organellar Ca2+-induced Ca2+ release, and the ensuing depolarisation and/or Ca2+ rise could rapidly travel inside the cell via the mitochondrial network providing further complexity to the influence of these organelles in shaping the global Ca2+ signal of a cell (Ichas et al. 1997).
The concept that mitochondrial Ca2+ homeostasis plays a fundamental role in controlling the cytosolic Ca2+ response should by no means be limited to the prevailing route of the Ca2+ rise of non-excitable cells (Ca2+ release from the ER via the InsP3-gated channel). Work from numerous laboratories has now demonstrated that mitochondria also participate in modulating [Ca2+]c when the source of Ca2+ is the extracellular medium and the rise is mediated by the opening of plasma membrane channels. In neurones, mitochondria placed in the proximity of the plasma membrane channels determine the kinetic profile of the [Ca2+]c rise, acting probably as fixed buffers that first limit the peak amplitude, and then contribute to the long-lasting plateau phase of the [Ca2+]c rise by gradually releasing Ca2+ via the Na+-Ca2+ exchanger (Werth & Thayer, 1994; White & Reynolds, 1995; Colegrove et al. 2000). The role of mitochondria in regulating Ca2+ influx is not limited to neuronal channels. In T lymphocytes, Hoth and co-workers showed that Ca2+ uptake by a resident mitochondrial pool enhances the ubiquitous current activated by the release of ER Ca2+ (commonly referred to as ‘capacitative calcium entry’, CCE) by relieving the Ca2+ inhibition in the microdomain at the mouth of the channel (Hoth et al. 1997).
In conclusion, although the physiological effects and molecular mechanisms responsible for the functional interactions are different in the various cell systems, it can be concluded that mitochondria experience a two-sided, close relationship with the cellular Ca2+ pools that allows them to invariably undergo a major rise in matrix [Ca2+] when a Ca2+ signal is elicited in a cell. On the one hand, this allows the stimulation of mitochondrial metabolism to match the increased cell needs while on the other, it places a fixed buffer of high capacity in the proximity of Ca2+-sensitive release pathways, thus allowing different modes of regulation of Ca2+ release. How these interactions are formed and modified, and how they in turn affect the long-term cellular responses, are interesting biological questions that need to be addressed.
Pathophysiological alterations of mitochondrial Ca2+ signalling
As discussed, mitochondrial Ca2+ uptake depends on a large thermodynamic force for accumulation and a kinetic limitation that allows high rates of accumulation only transiently, i.e. when the organelles are exposed to high [Ca2+] microdomains generated at the mouth of the opening channels. Such an arrangement restricts the timing of Ca2+ uptake to the earliest phases of the cytosolic Ca2+ rise, and thus prevents the cell from dissipating the ΔΨ generated by the respiratory chain in futile Ca2+ cycling across the mitochondrial inner membrane throughout agonist stimulation. However, at least in principle, it could be negatively affected in a number of pathophysiological conditions that affect mitochondrial structure and function. We discuss here only a few examples where an alteration of mitochondrial Ca2+ handling could be shown and discuss the possible implication of these alterations in generating the cellular phenotype.
Mitochondrial diseases
Mitochondrial diseases are a heterogeneous group of disorders sharing a genetic defect involving mitochondrial proteins and the common morphological hallmark of altered mitochondrial number and/or shape. As to the molecular defect, numerous pathogenic mutations have been identified in mitochondrial DNA (mtDNA), the 16.5 kb circular DNA encoding 13 polypeptides that are either components of the respiratory chain complexes or of the mitochondrial ATPase. The defects range from large scale deletions, such as those found in Kearns-Sayre syndrome and progressive external ophtalmoplegia (PEO), to point mutations in transfer RNAs (tRNAs), such as those found in MELAS (myopathy, encephalopathy, lactic acidosis and stroke-like episodes) and in MERRF (myoclonic epilepsy with ragged red fibres), and to point mutations in structural genes, such as those found in Leber's optic atrophy or NARP (neuropathy, ataxia and retinitis pigmentosa) (Wallace, 1994; Schon et al. 1997). While the molecular identity of the mutations is in many cases known, the pathway(s) that leads to the functional cell defect is often obscure. Indeed, in many cases biochemical analysis of the various disorders, which may reveal comparable, partial deficiencies of mitochondrial function, does not clarify the cellular and clinical specificity of the various disorders. We investigated the possibility that genetic defects affecting the generation of the proton gradient across the mitochondrial membrane (and thus the driving force for Ca2+ accumulation) could impair mitochondrial Ca2+ homeostasis. We thus carried out experiments in transmitochondrial cell lines (‘cybrids’) harbouring in the same nuclear background either normal mtDNAs, mtDNAs harbouring the tRNAlys mutation of MERRF, or mtDNAs harbouring the ATPase6 mutation of NARP. We observed that the mitochondrial Ca2+ response to agonists was drastically reduced in MERRF cybrids, while the response in NARP cells was normal, the cytoplasmic response being normal in all cases. The impairment of the Ca2+ response caused a smaller increase in ATP levels upon agonist stimulation, as monitored by recombinant luciferase. Interestingly, an inhibitor of the mitochondrial efflux pathways caused a significant restoration of both the Ca2+ and the ATP response, thus implying that the defect in mitochondrial Ca2+ homeostasis is a relevant event in the cellular pathogenesis of this disorder and, potentially, a therapeutic target (Brini et al. 1999).
The oncoprotein Bcl-2
A second, interesting example is the effect of the oncoprotein Bcl-2 on subcellular Ca2+ homeostasis. While the role of Bcl-2 and related proteins in the control of apoptotic cell death is widely accepted, its mechanism of action is still unsolved. Bcl-2 can form ion channels in lipid bilayers, and it is widely distributed in intracellular membranes including the outer mitochondrial membrane. Among other possibilities, it has been proposed that Bcl-2 could act on Ca2+ signalling by affecting ion fluxes across the organelle membranes (Minn et al. 1997). We utilised targeted aequorins to specifically monitor [Ca2+] in the cytosol, in the mitochondrial matrix and in the lumen of intracellular Ca2+ stores (the ER and the Golgi apparatus) and revealed that Bcl-2 overexpression alters Ca2+ handling in all these compartments (Pinton et al. 2000). The key event was an increase in the Ca2+ leak from the intracellular stores, which consequently attained a lower state of filling. Thus, upon agonist stimulation the amount of Ca2+ released from the ER (and that accumulated in the mitochondria) was significantly reduced. Also, in this case the mitochondrial Ca2+ response was reduced, but this was caused by a global reduction of the cellular Ca2+ signal rather than an intrinsic limitation of the capacity of mitochondria to accumulate Ca2+. The relevance for cell survival of the alteration in Ca2+ signalling in the mitochondria (in which Ca2+ overload has been proposed to be a major apoptotic signal) and in the other cell compartments is the aim of studies currently underway in our laboratories.
Acknowledgments
We wish to thank Drs Marisa Brini, Luisa Filippin and Paolo Pinton for experimental work, helpful discussions and critical reading of the manuscript. Research in the authors’ laboratories was supported by grants from the Italian University and Health Ministries, the Italian Space Agency (AGI), the European Union, the Italian National Research Council (CNR), Telethon, the Italian Cancer Research Association (AIRC) and the Armenise-Harvard Foundation.
References
- Allshire A, Bernardi P, Saris NE. Manganese stimulates calcium flux through the mitochondrial uniporter. Biochimica et Biophysica Acta. 1985;807:202–209. doi: 10.1016/0005-2728(85)90123-9. [DOI] [PubMed] [Google Scholar]
- Allue I, Gandelman O, Dementieva E, Ugarova N, Cobbold P. Evidence for rapid consumption of millimolar concentrations of cytoplasmic ATP during rigor-contracture of metabolically compromised single cardiomyocytes. Biochemical Journal. 1996;319:463–469. doi: 10.1042/bj3190463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzone GF, Pozzan T, Massari S, Bragadin M, Dell'Antone P. H+/site ratio and steady state distribution of divalent cations in mitochondria. FEBS Letters. 1977;78:21–24. doi: 10.1016/0014-5793(77)80264-0. [DOI] [PubMed] [Google Scholar]
- Babcock DF, Herrington J, Goodwin PC, Park YB, Hille B. Mitochondrial participation in the intracellular Ca2+ network. Journal of Cell Biology. 1997;136:833–844. doi: 10.1083/jcb.136.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrero MJ, Montero M, Alvarez J. Dynamics of [Ca2+] in the endoplasmic reticulum and cytoplasm of intact HeLa cells: a comparative study. Journal of Biological Chemistry. 1997;272:27694–27699. doi: 10.1074/jbc.272.44.27694. [DOI] [PubMed] [Google Scholar]
- Bernardi P. Mitochondrial transport of cations: Channels, exchangers and permeability transition. Physiological Reviews. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Azzone GF. Regulation of Ca2+ efflux in rat liver mitochondria. Role of membrane potential. European Journal of Biochemistry. 1983;134:377–383. doi: 10.1111/j.1432-1033.1983.tb07578.x. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Petronilli V. The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. Journal of Bioenergetics and Biomembranes. 1996;28:131–138. doi: 10.1007/BF02110643. [DOI] [PubMed] [Google Scholar]
- Boiter E, Rea R, Duchen MR. Mitochondria exert a negative feedback on the propagation of intracellular Ca2+ waves in rat cortical astrocytes. Journal of Cell Biology. 1999;17:795–808. doi: 10.1083/jcb.145.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragadin M, Pozzan T, Azzone GF. Kinetics of Ca2+ carrier in rat liver mitochondria. Biochemistry. 1979;18:5972–5978. doi: 10.1021/bi00593a033. [DOI] [PubMed] [Google Scholar]
- Bram RJ, Crabtree GR. Calcium signalling in T cells stimulated by a cyclophilin B-binding protein. Nature. 1994;371:355–358. doi: 10.1038/371355a0. [DOI] [PubMed] [Google Scholar]
- Brini M, Marsault R, Bastianutto C, Alvarez J, Pozzan T, Rizzuto R. Transfected aequorin in the measurement of cytosolic Ca2+ concentration ([Ca2+]c): a critical evaluation. Journal of Biological Chemistry. 1995;270:9896–9903. doi: 10.1074/jbc.270.17.9896. [DOI] [PubMed] [Google Scholar]
- Brini M, Pinton P, King MP, Davidson M, Schon EA, Rizzuto R. A calcium signaling defect in the pathogenesis of a mitochondrial DNA inherited oxidative phosphorylation deficiency. Nature Medicine. 1999;5:951–954. doi: 10.1038/11396. [DOI] [PubMed] [Google Scholar]
- Colegrove SL, Albrecht MA, Friel DD. Quantitative analysis of mitochondrial Ca2+ uptake and release pathways in sympathetic neurons – Reconstruction of the recovery after depolarization-evoked [Ca2+](i) elevations. Journal of General Physiology. 2000;115:371–388. doi: 10.1085/jgp.115.3.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DA, Matlib MA. A role for the mitochondrial Na+-Ca2+ exchanger in the regulation of oxidative phosphorylation in isolated heart mitochondria. Journal of Biological Chemistry. 1993;268:938–947. [PubMed] [Google Scholar]
- Crompton M, Capano M, Carafoli E. The sodium-induced efflux of calcium from heart mitochondria. A possible mechanism for the regulation of mitochondrial calcium. European Journal of Biochemistry. 1976;69:453–462. [Google Scholar]
- Crompton M, Moser R, Ludi H, Carafoli E. The interrelations between the transport of sodium and calcium in mitochondria of various mammalian tissues. European Journal of Biochemistry. 1978;82:25–31. doi: 10.1111/j.1432-1033.1978.tb11993.x. [DOI] [PubMed] [Google Scholar]
- Csordas G, Thomas AP, Hajnoczky G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO Journal. 1999;18:96–108. doi: 10.1093/emboj/18.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. The Journal of Physiology. 1999;516:1–17. doi: 10.1111/j.1469-7793.1999.001aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnoczky G, Hager R, Thomas AP. Mitochondria suppress local feedback activation of inositol 1,4,5-trisphosphate receptors by Ca2+ Journal of Biological Chemistry. 1999;274:14157–14162. doi: 10.1074/jbc.274.20.14157. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Hansford RG. Physiological role of mitochondrial Ca2+ transport. Journal of Bioenergetics and Biomembranes. 1994;26:495–508. doi: 10.1007/BF00762734. [DOI] [PubMed] [Google Scholar]
- Heaton GM, Nicholls DG. The calcium conductance of the inner membrane of rat liver mitochondria and the determination of the calcium electrochemical gradient. Biochemical Journal. 1976;156:635–646. doi: 10.1042/bj1560635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. Journal of Cell Biology. 1997;137:633–648. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huser J, Rechenmacher CE, Blatter LA. Imaging the permeability pore transition in single mitochondria. Biophysical Journal. 1998;74:2129–2137. doi: 10.1016/S0006-3495(98)77920-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89:1145–1153. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Igbavboa U, Pfeiffer DR. EGTA inhibits reverse uniport-dependent Ca2+ release from uncoupled mitochondria. Possible regulation of the Ca2+ uniporter by a Ca2+ binding site on the cytoplasmic side of the inner membrane. Journal of Biological Chemistry. 1988;263:1405–1412. [PubMed] [Google Scholar]
- Jouaville LS, Ichas F, Holmuhamedov EL, Camacho P, Lechleiter JD. Synchronization of calcium waves by mitochondrial substrates in Xenopus laevis oocytes. Nature. 1995;377:438–441. doi: 10.1038/377438a0. [DOI] [PubMed] [Google Scholar]
- Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proceedings of the National Academy of Sciences of the USA. 1999;96:13807–13812. doi: 10.1073/pnas.96.24.13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung DW, Baysal K, Brierley GP. The sodium-calcium antiport of heart mitochondria is not electroneutral. Journal of Biological Chemistry. 1995;270:672–678. doi: 10.1074/jbc.270.2.672. [DOI] [PubMed] [Google Scholar]
- Kapus A, Szaszi K, Kaldi K, Ligeti E, Fonyo A. Is the mitochondrial Ca2+ uniporter a voltage-modulated transport pathway? FEBS Letters. 1991;282:61–64. doi: 10.1016/0014-5793(91)80444-8. [DOI] [PubMed] [Google Scholar]
- Kendall JM, Sala-Newby G, Ghalaut V, Dormer RL, Campbell AK. Engineering the Ca2+-activated photoprotein aequorin with reduced affinity for calcium. Biochemical and Biophysical Research Communications. 1992;187:1091–1097. doi: 10.1016/0006-291x(92)91309-e. [DOI] [PubMed] [Google Scholar]
- Kennedy HJ, Pouli AE, Ainscow EA, Jouaville LS, Rizzuto R, Rutter GA. Glucose generates sub-plasma membrane ATP microdomains in single islet beta-cells. Potential role for strategically located mitochondria. Journal of Biological Chemistry. 1999;274:13281–13291. doi: 10.1074/jbc.274.19.13281. [DOI] [PubMed] [Google Scholar]
- Kroner H. Ca2+ ions, an allosteric activator of calcium uptake in rat liver mitochondria. Archives of Biochemistry and Biophysics. 1986;251:525–535. doi: 10.1016/0003-9861(86)90360-7. [DOI] [PubMed] [Google Scholar]
- Landolfi B, Curci S, Debellis L, Pozzan T, Hofer A. Ca2+ homeostasis in the agonist-sensitive internal store: Functional interactions between mitochondria and the ER measured in situ in intact cells. Journal of Cell Biology. 1998;142:1235–1243. doi: 10.1083/jcb.142.5.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litsky ML, Pfeiffer DR. Regulation of the mitochondrial Ca2+ uniporter by external adenine nucleotides: the uniporter behaves like a gated channel which is regulated by nucleotides and divalent cations. Biochemistry. 1997;36:7071–7080. doi: 10.1021/bi970180y. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiological Reviews. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- Maechler P, Wollheim CB. Mitochondrial signals in glucose-stimulated insulin secretion in the beta cell. The Journal of Physiology. 2000;529:49–56. doi: 10.1111/j.1469-7793.2000.00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Velez P, Schendel SL, Liang H, Muchmore SW, Fesik SW, Fill M, Thompson CB. Bcl-x(L) forms an ion channel in synthetic lipid membranes. Nature. 1997;385:353–357. doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- Mitchell P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biological Reviews of the Cambridge Philosophical Society. 1966;41:445–502. doi: 10.1111/j.1469-185x.1966.tb01501.x. [DOI] [PubMed] [Google Scholar]
- Montero M, Alonso MT, Carnicero E, Cuchillo-Ibanez I, Albillos A, Garcia AG, Garcia-Sancho J, Alvarez J. Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nature Cell Biology. 2000;2:57–61. doi: 10.1038/35000001. [DOI] [PubMed] [Google Scholar]
- Petronilli V, Miotto G, Canton M, Colonna R, Bernardi P, Di Lisa F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes of mitochondrial calcein fluorescence. Biophysical Journal. 1999;76:725–734. doi: 10.1016/S0006-3495(99)77239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzati R, Bossi M, Podini P, Meldolesi J, Grohovaz F. High-resolution calcium mapping of the endoplasmic reticulum-Golgi-exocytic membrane system. Electron energy loss imaging analysis of quick frozen-freeze dried PC12 cells. Molecular Biology of the Cell. 1997;8:1501–1512. doi: 10.1091/mbc.8.8.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R. Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2 overexpressing cells. Journal of Cell Biology. 2000;148:857–862. doi: 10.1083/jcb.148.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzo P, Fasolato C, Pozzan T. Dynamic properties of an inositol 1,4,5-trisphosphate- and thapsigargin-insensitive calcium pool in mammalian cell lines. Journal of Cell Biology. 1997;138:355–366. doi: 10.1083/jcb.136.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pralong WF, Spat A, Wollheim CB. Dynamic pacing of cell metabolism by intracellular Ca2+ transients. Journal of Biological Chemistry. 1994;269:27310–27314. [PubMed] [Google Scholar]
- Ramesh V, Sharma VK, Sheu SS, Franzini-Armstrong C. Structural proximity of mitochondria to calcium release units in rat ventricular myocardium may suggest a role in Ca2+ sequestration. Annals of the New York Academy of Sciences. 1998;853:341–344. doi: 10.1111/j.1749-6632.1998.tb08295.x. [DOI] [PubMed] [Google Scholar]
- Ramos-Franco J, Fill M, Mignery GA. Isoform-specific function of single inositol 1,4,5-trisphosphate receptor channels. Biophysical Journal. 1998;75:834–839. doi: 10.1016/S0006-3495(98)77572-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley WW, Jr, Pfeiffer DR. Relationships between Ca2+ release, Ca2+ cycling, and Ca2+-mediated permeability changes in mitochondria. Journal of Biological Chemistry. 1985;260:12416–12425. [PubMed] [Google Scholar]
- Rizzuto R, Bastianutto C, Brini M, Murgia M, Pozzan T. Mitochondrial Ca2+ homeostasis in intact cells. Journal of Cell Biology. 1994;126:1183–1194. doi: 10.1083/jcb.126.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ concentration that are sensed by neighboring mitochondria. Science. 1993;262:744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Simpson AWM, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature. 1992;358:325–328. doi: 10.1038/358325a0. [DOI] [PubMed] [Google Scholar]
- Robb-Gaspers LD, Burnett P, Rutter GA, Denton RM, Rizzuto R, Thomas AP. Integrating cytosolic calcium signals in mitochondrial metabolic responses. EMBO Journal. 1998;17:4987–5000. doi: 10.1093/emboj/17.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Ross CA, Villa A, Supattapone S, Pozzan T, Snyder SH, Meldolesi J. The inositol 1,4,5,-trisphosphate receptor in cerebellar Purkinje cells: quantitative immunogold labeling reveals concentration in an ER subcompartment. Journal of Cell Biology. 1990;111:615–624. doi: 10.1083/jcb.111.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon EA, Bonilla E, DiMauro S. Mitochondrial DNA mutations and pathogenesis. Journal of Bioenergetics and Biomembranes. 1997;29:131–149. doi: 10.1023/a:1022685929755. [DOI] [PubMed] [Google Scholar]
- Sparagna GC, Gunter KK, Sheu SS, Gunter TE. Mitochondrial calcium uptake from physiological-type pulses of calcium. A description of the rapid uptake mode. Journal of Biological Chemistry. 1995;270:27510–27515. doi: 10.1074/jbc.270.46.27510. [DOI] [PubMed] [Google Scholar]
- Tinel H, Cancela JM, Mogami H, Gerasimenko JV, Gerasimenko OV, Tepikin AV, Petersen OH. Active mitochondria surrounding the pancreatic acinar granule region prevent spreading of inositol trisphosphate-evoked local cytosolic Ca2+ signals. EMBO Journal. 1999;18:4999–5008. doi: 10.1093/emboj/18.18.4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasington FD, Gazzotti P, Tiozzo R, Carafoli E. The effect of ruthenium red on Ca2+ transport and respiration in rat liver mitochondria. Biochimica et Biophysica Acta. 1972;256:43–54. doi: 10.1016/0005-2728(72)90161-2. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial DNA mutations in diseases of energy metabolism. Journal of Bioenergetics and Biomembranes. 1994;26:241–250. doi: 10.1007/BF00763096. [DOI] [PubMed] [Google Scholar]
- Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. Journal of Neuroscience. 1994;14:348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondria and Na+/Ca2+ exchange buffer glutamate-induced calcium loads in cultured cortical neurons. Journal of Neuroscience. 1995;15:1318–1328. doi: 10.1523/JNEUROSCI.15-02-01318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingrove DE, Gunter TE. Kinetics of mitochondrial calcium transport. I. Characteristics of the sodium-independent calcium efflux mechanism of liver mitochondria. Journal of Biological Chemistry. 1986a;261:15159–15165. [PubMed] [Google Scholar]
- Wingrove DE, Gunter TE. Kinetics of mitochondrial calcium transport. II. A kinetic description of the sodium-dependent calcium efflux mechanism of liver mitochondria and inhibition by ruthenium red and by tetraphenylphosphonium. Journal of Biological Chemistry. 1986b;261:15166–15171. [PubMed] [Google Scholar]