

Abstract
In many non-excitable cells Ca2+ influx is mainly controlled by the filling state of the intracellular Ca2+ stores. It has been suggested that this store-mediated or capacitative Ca2+ entry is brought about by a physical and reversible coupling of the endoplasmic reticulum with the plasma membrane. Here we provide evidence for an additional, non-capacitative Ca2+ entry mechanism in human platelets.
Changes in cytosolic Ca2+ and Sr2+ were measured in human platelets loaded with the fluorescent indicator fura-2.
Depletion of the internal Ca2+ stores with thapsigargin plus a low concentration of ionomycin stimulated store-mediated cation entry, as demonstrated upon Ca2+ or Sr2+ addition. Subsequent treatment with thrombin stimulated further divalent cation entry in a concentration-dependent manner.
Direct activation of protein kinase C (PKC) by phorbol-12-myristate-13-acetate or 1-oleoyl-2-acetyl-sn-glycerol also stimulated divalent cation entry, without evoking the release of Ca2+ from intracellular stores. Cation entry evoked by thrombin or activators of PKC was abolished by the PKC inhibitor Ro-31-8220.
Unlike store-mediated Ca2+ entry, jasplakinolide, which reorganises actin filaments into a tight cortical layer adjacent to the plasma membrane, did not inhibit divalent cation influx evoked by thrombin when applied after Ca2+ store depletion, or by activators of PKC. Thrombin also activated Ca2+ entry in platelets in which the release from intracellular stores and store-mediated Ca2+ entry were blocked by xestospongin C. These results indicate that the non-capacitative divalent cation entry pathway is regulated independently of store-mediated entry and does not require coupling of the endoplasmic reticulum and the plasma membrane.
These results support the existence of a mechanism for receptor-evoked Ca2+ entry in human platelets that is independent of Ca2+ store depletion. This Ca2+ entry mechanism may be activated by occupation of G-protein-coupled receptors, which activate PKC, or by direct activation of PKC, thus generating non-capacitative Ca2+ entry alongside that evoked following the release of Ca2+ from the intracellular stores.
The cytosolic Ca2+ concentration ([Ca2+]i) modulates a large number of cellular processes ranging from short term responses such as contraction and secretion to longer term modulation of cell growth (Monteith & Roufogalis, 1995). Stimulation of non-excitable cells by various agonists results in a rise in [Ca2+]i, which consists of two components: release of Ca2+ from the intracellular stores and the entry of Ca2+ across the plasma membrane (Rink & Sage, 1990; Parekh & Penner, 1997).
The mechanisms involved in the generation of Ca2+ signals in platelets have much in common with those operating in other non-excitable cells. Thus platelets are a good model for the study of Ca2+ signalling in normal human non-excitable cells. In these cells, store-mediated Ca2+ entry (SMCE), a mechanism regulated by the degree of filling of the intracellular Ca2+ stores (Putney, 1990; Sage, 1997), is a major pathway for Ca2+ influx (Sage et al. 1990). The mechanism of activation of SMCE is not fully understood and current hypotheses fall into two main categories: those suggesting a diffusible messenger that gates plasma membrane channels and those suggesting a conformational coupling between elements in the endoplasmic reticulum (possibly the InsP3 receptors) and the plasma membrane (Berridge, 1995). Recently, the physical coupling model has received support from studies which show that the activation of SMCE shares properties with those of the secretory mechanism (Patterson et al. 1999; Putney, 1999; Yao et al. 1999; Rosado et al. 2000). These studies suggest that SMCE is mediated by a physical and reversible coupling of the endoplasmic reticulum and the plasma membrane and that the actin cytoskeleton might play a key regulatory role in the activation and maintenance of SMCE (Rosado et al. 2000).
In platelets there is in addition evidence for the existence of a purinoceptor-activated receptor-operated channel (Sage & Rink, 1986; Mackenzie et al. 1996). Adenine nucleotides have been shown to be able to evoke Ca2+ entry in human platelets with a short latency via a P2x1 purinoceptor (Sage & Rink, 1986; Mackenzie et al. 1996). This purinoceptor-operated non-selective cation channel can be blocked by desensitisation of the P2 x1 receptors by addition of the selective P2 x1 receptor agonist α,β-methylene ATP (Mackenzie et al. 1996).
Here we provide evidence for a novel mechanism for receptor-evoked divalent cation entry in human platelets, which is independent of the filling state of the intracellular Ca2+ stores and different from the purinoceptor-activated receptor-operated cation entry. In this mechanism Ca2+ entry occurs through a divalent cation permeable channel that is activated via protein kinase C (PKC).
METHODS
Platelet preparation
Experiments were carried out on human blood platelets obtained from healthy drug-free volunteers who gave written informed consent, with local ethical committee approval and in accordance with the Declaration of Helsinki. Fura-2-loaded platelets were prepared as described previously (Rosado & Sage, 2000b). Briefly, blood was mixed with a one-sixth volume of acid/citrate dextrose anticoagulant containing (mm): 85 sodium citrate, 78 citric acid and 111 D-glucose. Platelet-rich plasma was then prepared by centrifugation for 5 min at 700 g and aspirin (100 μm) and apyrase (40 μg ml−1) were added. The platelet-rich plasma was incubated at 37°C with 2 μm fura-2 AM for 45 min. Cells were then collected by centrifugation at 350 g for 20 min and resuspended in Hepes-buffered saline (HBS) containing (mm): 145 NaCl, 10 Hepes, 10 D-glucose, 5 KCl, 1 MgSO4, pH 7.45, and supplemented with 0.1 % (w/v) bovine serum albumin (BSA) and 40 μg ml−1 apyrase.
Measurement of [Ca2+]i
Fluorescence was recorded from 1.5 ml aliquots of a magnetically stirred platelet suspension (108 cells ml−1) at 37°C using a Cairn Research Spectrophotometer (Cairn Research Ltd) with excitation wavelengths of 340 and 380 nm and emission at 500 nm. Changes in [Ca2+]i were monitored using the fura-2 340 nm/380 nm fluorescence ratio and calibrated in terms of [Ca2+]i according to the method of Grynkiewicz et al. (1985).
Determination of Sr2+ entry
In a number of experiments, Sr2+ was used to monitor divalent cation entry. This was done to avoid complications arising from stimulation of the platelet plasma membrane Ca2+-ATPase by PKC (Rink & Sage, 1987), since Sr2+ is transported with lower affinity than Ca2+ by this Ca2+-ATPase (Graf et al. 1982). Sr2+ entry was measured in Ca2+-free HBS containing EGTA (100 μm) to minimise the effects of contaminating Ca2+. Cytosolic Sr2+ was monitored using the fura-2 340 nm/380 nm fluorescence ratio. Store depletion-evoked Sr2+ entry was calculated using the integral of the rise in the 340 nm/380 nm fluorescence ratio for 2.5 min after addition of SrCl2. Thrombin-evoked Sr2+ influx was measured as the integral of the rise in the 340 nm/380 nm fluorescence ratio above basal levels for 1 min after addition of thrombin in the presence of external Sr2+. When platelets were preincubated with various compounds, Sr2+ entry was corrected by subtraction of the change in the 340 nm/380 nm fluorescence ratio (due to leakage of the indicator) that occurred when Sr2+ was added to vehicle-treated (non-depleted) controls.
Confocal microscopy
Samples of platelet suspension (200 μl) were transferred to 200 μl ice-cold 3 % (w/v) formaldehyde in PBS (mm: 137 NaCl, 2.7 KCl, 5.62 Na2HPO4, 1.09 NaH2PO4 and 1.47 KH2PO4, pH 7.2) for 10 min. Fixed platelets were permeabilised by incubation for 10 min with 0.025 % (v/v) Nonidet P-40 detergent dissolved in PBS. The platelets were then incubated for 30 min with fluorescein isothiocyanate (FITC)-labelled phalloidin (1 μm) in PBS supplemented with 0.5 % (w/v) BSA. Cells were collected by centrifugation in an MSE Micro-Centaur centrifuge (MSE Scientific Instruments) for 60 s at 3000 g and resuspended in PBS. The platelets were visualised using a Leica TCS4D confocal microscope.
Materials
Fura-2 acetoxymethyl ester (fura-2 AM) was from Texas Fluorescence (Austin, TX, USA). Apyrase (grade VII), aspirin, BSA, paraformaldehyde, Nonidet P-40, FITC-labelled phalloidin, thrombin and thapsigargin (TG) were from Sigma (Poole, Dorset, UK). Ionomycin (IONO), xestospongin C (Xest C), phorbol-12-myristate-13-acetate (PMA), 1-oleoyl-2-acetyl-sn-glycerol (OAG) and Ro-31-8220 were from Calbiochem (Nottingham, UK). Jasplakinolide (JP) was from Molecular Probes (Leiden, The Netherlands). All other reagents were of analytical grade.
Statistical analysis
Analysis of statistical significance was performed using Student’s t test. For multiple comparisons, one-way analysis of variance combined with Dunnett’s test was used.
RESULTS
Thrombin-evoked receptor-operated divalent cation entry
Addition of TG (1 μm) and a low concentration of IONO (50 nM) to platelets in a Ca2+-free medium caused a transient increase in [Ca2+]i as the intracellular Ca2+ stores were depleted (Fig. 1A). A combination of TG and IONO was used since platelets are reported to possess two Ca2+ stores with high and low Ca2+ leakage rates (Doni et al. 1994; Cavallini et al. 1995) and both agents are required to ensure extensive Ca2+ store depletion. Subsequent stimulation with a maximally effective concentration of thrombin (10 U ml−1; Sage & Rink, 1987) was unable to further increase [Ca2+]i, indicating that the agonist-releasable Ca2+ stores were fully depleted (Fig. 1A). We have previously shown that depletion of intracellular Ca2+ stores in platelets induces divalent cation entry (Sargeant et al. 1992; Jenner et al. 1994), hence addition of Sr2+ (300 μm) to the external medium after depletion of the Ca2+ stores resulted in a rapid Sr2+ entry (Fig. 1B) which did not occur in non-depleted cells (Control). Addition of the physiological platelet agonist thrombin (1 or 10 U ml−1) to platelets in which Sr2+ entry had already been stimulated by complete depletion of the intracellular Ca2+ stores caused a further, concentration-dependent increase in Sr2+ entry (Fig. 1C). The peak elevations in the 340 nm/380 nm fluorescence ratio above the level attained following store-mediated Sr2+ entry were 0.44 ± 0.05 and 0.31 ± 0.04 arbitrary units after treatment with 10 or 1 U ml−1 thrombin, respectively (n = 7). The addition of thrombin (10 U ml−1) after store-mediated Ca2+ entry had been initiated by the addition of 300 μm Ca2+ to store-depleted platelets resulted in further Ca2+ entry (Fig. 1D). The additional thrombin-evoked divalent cation entry was not due to thrombin-evoked hyperpolarisation of the cells, since similar responses were elicited when the membrane potential was clamped close to the K+ reversal potential using valinomycin (5 μm; Mahaut-Smith et al. 1990; data not shown). Since addition of thrombin to TG- and IONO-treated platelets did not evoke further release of Ca2+ from the stores (Fig. 1A), the thrombin-evoked entry of Sr2+ or Ca2+ observed in addition to that evoked by store depletion suggests the existence of a novel and Ca2+ store-independent divalent cation entry pathway, stimulated following receptor occupation.
Figure 1. Capacitative and non-capacitative cation entry pathways in human platelets.
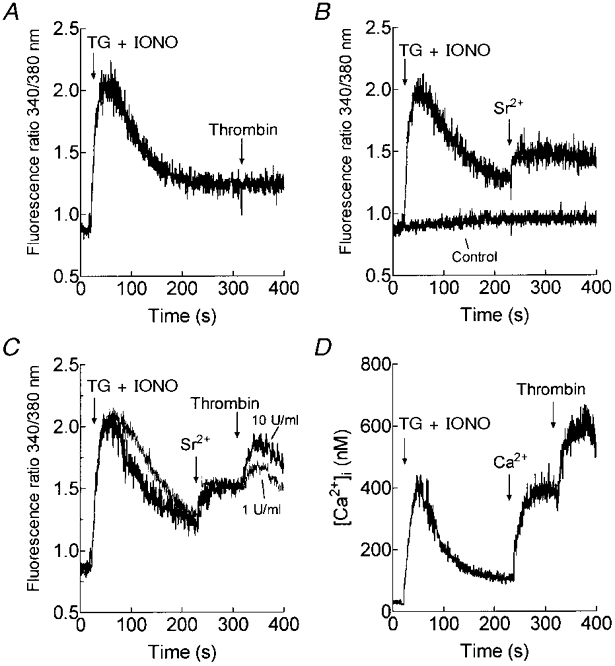
A, fura-2-loaded human platelets were stimulated with TG (1 μm) and IONO (50 nM) in Ca2+-free HBS (100 μm EGTA added) and 5 min later thrombin (10 U ml−1) was added (n = 5). B, cells were stimulated with TG and IONO or the vehicles alone (Control) in Ca2+-free HBS (100 μm EGTA added) and 3.5 min later SrCl2 (300 μm) was added (n = 7). C, cells were stimulated with TG and IONO in Ca2+-free HBS (100 μm EGTA added); 3.5 min later SrCl2 (300 μm) was added to initiate Sr2+ entry followed by thrombin (1 or 10 U ml−1; n = 7). D, cells were stimulated with TG and IONO in Ca2+-free HBS (100 μm EGTA added); 3.5 min later CaCl2 (300 μm) was added to initiate Ca2+ entry followed by thrombin (10 U ml−1; n = 6).
Previous studies have reported a non-capacitative purinoceptor-activated cation entry pathway in human platelets (Mackenzie et al. 1996). To investigate whether this pathway could be responsible for the cation entry mechanism described above, through thrombin-evoked secretion of adenine nucleotides, we desensitised the P2x1 receptor by adding α,β-methylene ATP, a non-hydrolysable ATP analogue. In agreement with previous observations (Mackenzie et al. 1996; Vial et al. 1997), the selective P2x1 agonist α,β-methylene ATP (10 μm) induced a transient rise in [Ca2+]i in the presence of external calcium (n = 4; Fig. 2A). As reported previously (Vial et al. 1997), addition of α,β-methylene ATP desensitised the P2x1 purinoceptor as indicated by the lack of effect of a subsequent addition of 10 μmα,β-methylene ATP. The addition of α,β-methylene ATP to platelets in the absence of external Ca2+ (with 100 μm EGTA added) was, as expected (MacKenzie et al. 1996), without effect on [Ca2+]i (Fig. 2B). The subsequent addition of 1 mm CaCl2 was also without effect, indicating that P2 x1 receptor stimulation in the absence of external Ca2+ still results in desensitisation of the receptor, a conclusion supported by the fact that a further addition of α,β-methylene ATP also did not affect [Ca2+]i (Fig. 2B). As shown in Fig. 2C and D, prior desensitisation of the P2 x1 receptors by addition of 10 μmα,β-methylene ATP failed to inhibit thrombin-evoked Sr2+ entry in store-depleted cells (0.53 ± 0.04 arbitrary 340 nm/380 nm fluorescence units in α,β-methylene ATP-treated platelets vs. 0.50 ± 0.02 units in control; n = 4), indicating that this pathway is different from the purinoceptor-activated receptor-operated cation entry pathway. We subsequently refer to the novel entry pathway as a non-capacitative process to differentiate it from the purinoceptor-activated receptor-operated divalent cation entry pathway.
Figure 2. Effect of P2x1 receptor desensitisation on thrombin-evoked non-capacitative Sr2+ entry in human platelets.
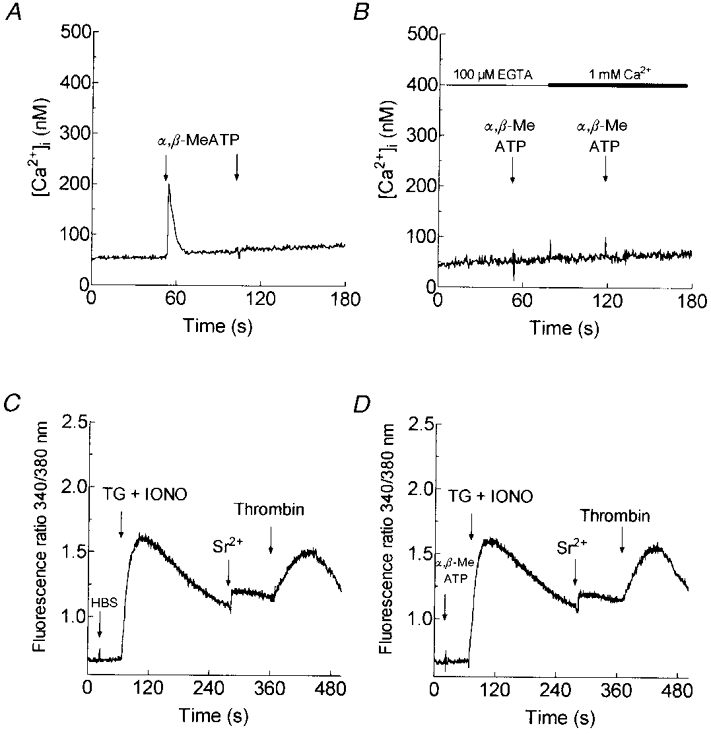
Fura-2-loaded human platelets were resuspended in HBS containing 1 mm CaCl2 (A) or in Ca2+-free HBS (100 μm EGTA added; B) and then α,β-methylene ATP (α,β-MeATP; 10 μm) was added. A second addition of 10 μmα,β-methylene ATP in the presence of 1 mm external Ca2+ demonstrates desensitisation of the P2 x1 receptor (n = 4; A). Addition of 1 mm CaCl2 after 10 μmα,β-methylene ATP and a second addition of 10 μmα,β-methylene ATP were without effect, indicating that desensitisation occurs independently of Ca2+ entry (n = 4; B). C and D, platelets were treated in Ca2+-free HBS (100 μm EGTA added) with α,β-methylene ATP (10 μm; D) or the vehicle (HBS; C) prior to stimulation with TG (1 μm) and IONO (50 nM); 3.5 min later SrCl2 (300 μm) was added followed by thrombin (10 U ml−1; n = 4).
Protein kinase C activates non-capacitative cation entry
It is well established that thrombin activates phospholipase C in human platelets, resulting in the generation of diacylglycerol (DAG) which in turn activates PKC (Haslam et al. 1990; Sargeant et al. 1993). To investigate the possible role of PKC in the activation of non-capacitative cation entry, we assessed the effects of activators of PKC, PMA and the DAG analogue OAG. Treatment of platelets with PMA (1 μm) elicited a transient entry of both Ca2+ and Sr2+ without releasing Ca2+ from the internal stores (Fig. 3A and B). The initial peak elevations in [Ca2+]i or 340 nm/380 nm fluorescence ratio above the basal level after treatment with PMA (1 μm) were 149 ± 41 nM and 0.42 ± 0.05 arbitrary units, respectively (n = 6). Moreover, addition of PMA (1 μm) to platelets where Sr2+ entry had already been stimulated by treatment with TG (200 nM) resulted in further Sr2+ entry (Fig. 3C and D) of similar magnitude to that stimulated by thrombin (Fig. 1C). The peak elevation in the 340 nm/380 nm fluorescence ratio above the level attained following store-mediated Sr2+ entry after treatment with PMA was 0.46 ± 0.07 arbitrary units (n = 6). Similar results were obtained with 100 μm OAG (not shown). Treatment of platelets with OAG (100 μm) elicited a transient elevation in cytosolic Ca2+ or Sr2+, reaching a peak elevation in [Ca2+]i or 340 nm/380 nm fluorescence ratio above the basal level of 137 ± 34 nM and 0.38 ± 0.04 arbitrary units, respectively (n = 6). In addition, in platelets in which Sr2+ entry had been activated by Ca2+ store depletion, OAG (100 μm) evoked additional Sr2+ entry (0.05 ± 0.05 340 nm/380 nm fluorescence arbitrary units above the level attained by store-mediated Sr2+ entry; n = 6). These results indicate that the activation of PKC stimulates non-capacitative cation entry in human platelets.
Figure 3. Effect of PMA on Ca2+ and Sr2+ entry in human platelets.
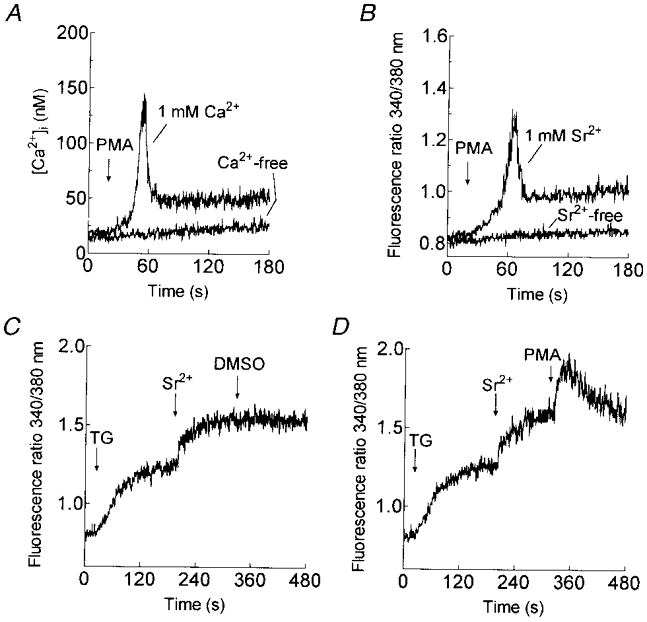
A and B, platelets were treated with 1 μm PMA in HBS containing 1 mm Ca2+ (A) or 1 mm Sr2+ (B; n = 6). The same experiments were performed in the absence of external Ca2+ (A) or Sr2+ (B; n = 6). C and D, cells were treated with TG (200 nM) in Ca2+-free HBS (100 μm EGTA added) and 3 min later SrCl2 (final concentration, 300 μm) was added followed by PMA (1 μm, D) or the vehicle (DMSO) alone (C; n = 6).
Time-dependent effect of PKC on cation entry
Previous studies in Xenopus oocytes have shown that PKC activation initially potentiates Ca2+ entry while more prolonged activation abolishes this process (Petersen & Berridge, 1994). We therefore investigated the time-dependent effect of PKC activation on store-mediated cation entry in human platelets. Pretreatment of platelets at 37°C with PMA (1 μm) for 5 min significantly inhibited TG-induced Sr2+ entry by 39.4 ± 7.7 % (P < 0.01; n = 6; Fig. 4A). Preincubation of platelets with PMA for 30 min abolished Sr2+ entry (P < 0.001; n = 6; Fig. 4A). Similarly, pretreatment of platelets with OAG (100 μm) for 5 min reduced TG-evoked Sr2+ entry by 54.0 ± 12.5 % (P < 0.01; n = 6) while pretreatment for 30 min completely inhibited Sr2+ entry (P < 0.001; n = 6; Fig. 4B). Treatment of platelets with PMA or OAG had no effect on the TG-evoked release of Ca2+ from the intracellular stores, indicating that Ca2+ storage was unaffected by PKC activation (data not shown).
Figure 4. Time-dependent effect of PKC activation on cation entry.
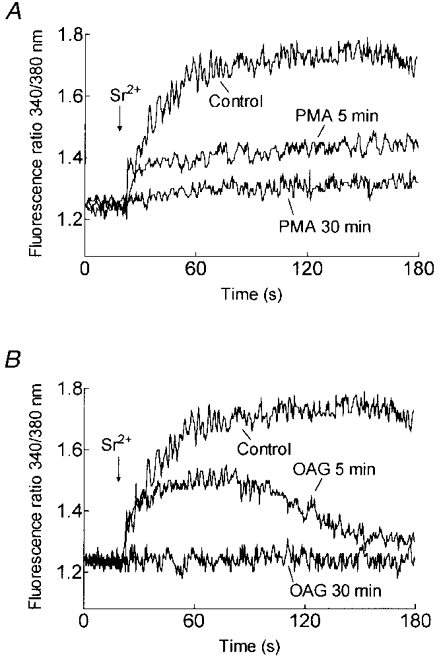
A, platelets were preincubated at 37 °C for 5 or 30 min in the absence (Control) or presence of PMA (1 μm). Cells were then treated with TG (200 nM) and 3 min later SrCl2 (final concentration, 300 μm) was added (n = 6). B, platelets were preincubated at 37 °C for 5 or 30 min in the absence (Control) or presence of OAG (100 μm). Cells were then treated with TG (200 nM) and 3 min later SrCl2 (final concentration, 300 μm) was added (n = 6).
Desensitisation of PKC by incubation with PMA (1 μm) for 30 min almost abolished non-capacitative divalent cation entry stimulated by a maximal concentration of thrombin (10 U ml−1; Fig. 5). Sr2+ entry evoked by 10 U ml−1 thrombin was inhibited by 91.5 ± 3.8 % (P < 0.001; n = 6; Fig. 5). Consistent with the results presented above, prolonged stimulation of PKC with PMA for 30 min also inhibited TG plus IONO-induced Sr2+ entry without affecting Ca2+ release from the intracellular stores (Fig. 5).
Figure 5. Desensitisation of PKC inhibits non-capacitative Sr2+ entry in human platelets.
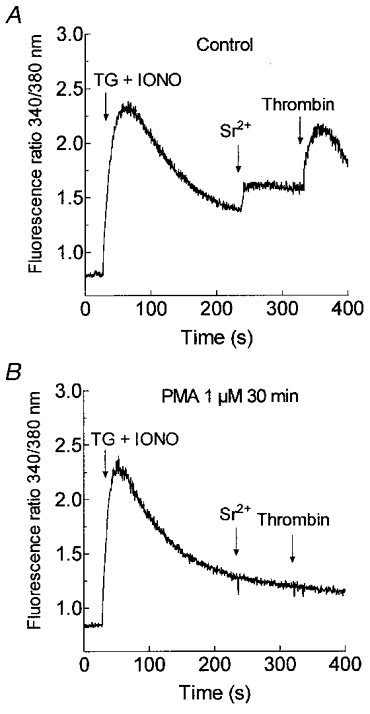
Cells were pretreated for 30 min at 37 °C with 1 μm PMA (B) or the vehicle (DMSO) alone (Control; A). The platelets were then stimulated with TG (1 μm) and IONO (50 nM) in Ca2+-free HBS (100 μm EGTA added) and 3.5 min later SrCl2 (300 μm) was added followed by thrombin (10 U ml−1; n = 6).
These findings indicate that although activation of PKC initially leads to stimulation of non-capacitative cation entry, continued activation or desensitisation of this protein kinase inhibits both store-mediated and non-capacitative divalent cation entry.
Effect of Ro-31-8220 on non-capacitative cation entry
In further support of a role for PKC in the thrombin-evoked non-capacitative cation entry, we observed that treatment of platelets for 5 min with the PKC inhibitor Ro-31-8220 (3 μm), which abolished PMA-evoked Sr2+ entry (P < 0.001; n = 6; Fig. 6A), significantly inhibited thrombin-evoked non-capacitative Sr2+ entry by 89 ± 6 % (P < 0.001; n = 6; Fig. 6B). Treatment of human platelets with Ro-31-8220 did not alter the elevation in [Ca2+]i evoked by TG and IONO indicating that storage of Ca2+ in the intracellular stores was unaffected by this compound. This is in agreement with the lack of effect of PKC activation on Ca2+ storage described above. In addition, treatment of platelets with Ro-31-8220 had no effect on store depletion-evoked Sr2+ entry (Fig. 6B), indicating that this compound is not a non-selective cation channel blocker.
Figure 6. Effect of PKC inhibition on non-capacitative Sr2+ entry in human platelets.
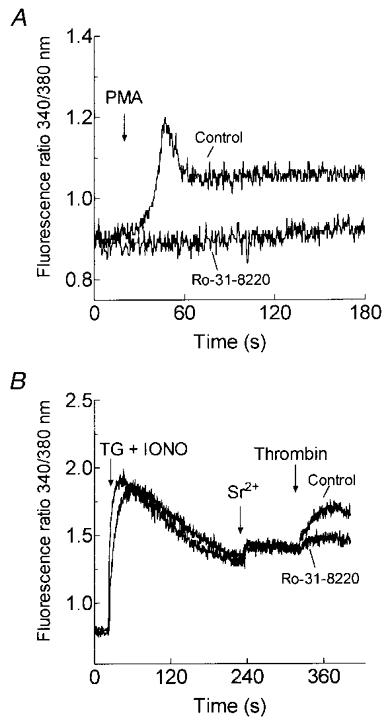
A, cells were pretreated for 5 min at 37 °C with 3 μm Ro-31-8220 or the vehicle (DMSO) alone (Control). The platelets were then treated with 1 μm PMA in HBS containing 1 mm Sr2+(n = 6). B, cells were pretreated for 5 min at 37 °C with 3 μm Ro-31-8220 or the vehicle (Control). The platelets were then stimulated with TG (1 μm) and IONO (50 nM) in Ca2+-free HBS (100 μm EGTA added) and 3.5 min later SrCl2 (300 μm) was added followed by thrombin (10 U ml−1; n = 6).
Effect of jasplakinolide on divalent cation entry
It has recently been proposed that SMCE is mediated by a reversible trafficking and coupling of the endoplasmic reticulum with the plasma membrane in several cell types (Patterson et al. 1999; Yao et al. 1999), including human platelets (Rosado et al. 2000). In this model, cortical actin filaments play an inhibitory role, as demonstrated by treatment with JP, a cell-permeant peptide that induces polymerisation and stabilisation of actin filaments (Cooper, 1987). JP has been shown to block SMCE, without altering receptor-evoked Ca2+ release, by formation of a cortical actin barrier at the plasma membrane, so preventing close association between the plasma membrane and the endoplasmic reticulum and thus providing evidence that association between the endoplasmic reticulum and the plasma membrane is required for the activation of SMCE (Patterson et al. 1999; Rosado et al. 2000; Rosado & Sage, 2000a,c). We have previously shown that treatment of human platelets with 10 μm JP for 30 min induces full actin polymerisation (Rosado et al. 2000). This action correlates with the formation of a dense cortical layer of actin filaments (Fig. 7D), which contrasts with the more restricted cortical actin distribution in resting platelets (Fig. 7B). If non-capacitative cation entry was directly stimulated by receptor occupation via PKC activation, one would expect that this pathway might be insensitive to JP treatment. In contrast, activation of the store-mediated (capacitative) cation entry, which requires coupling between the endoplasmic reticulum and the plasma membrane, is, as might be expected, blocked by JP (Rosado et al. 2000). JP treatment did not alter either thrombin or PMA-evoked activation of non-capacitative Sr2+ entry (Fig. 7A, C, E and F; n = 6-7) or thrombin-evoked non-capacitative Ca2+ entry (Fig. 7G and H; n = 7). In contrast, in agreement with our previous studies (Rosado et al. 2000), treatment of human platelets with JP essentially abolished store depletion-induced Sr2+ and Ca2+ entry but had no effect on the release of Ca2+ from the intracellular stores (Fig. 7F and H; n = 7). These findings suggest that the novel non-capacitative cation entry in human platelets is stimulated by direct activation of plasma membrane divalent cation permeable channels and does not involve interaction between the plasma membrane and another organelle. In addition, these data indicate that the non-capacitative cation entry and store-mediated Ca2+ entry pathways are independently regulated, since the non-capacitative pathway operates normally under conditions in which SMCE is blocked.
Figure 7. Effect of jasplakinolide on non-capacitative cation entry in human platelets.
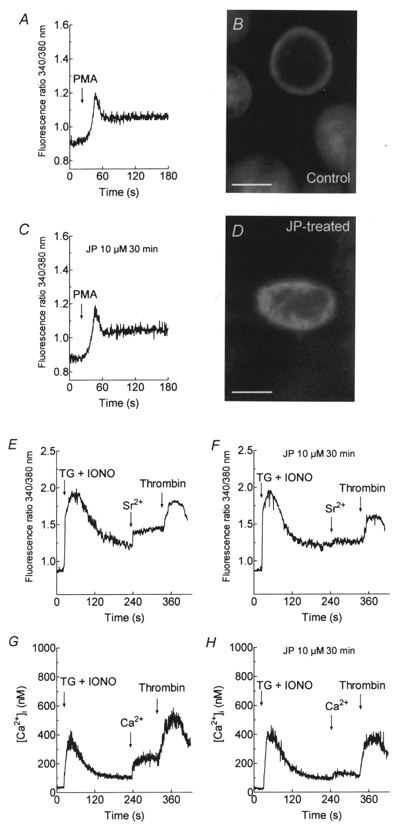
A-D, platelets were pretreated for 30 min at 37 °C with 10 μm JP (C and D; n = 6) or the vehicle (ethanol; A and B; n = 6). Cells were stimulated with PMA (1 μm) in HBS containing 1 mm Sr2+. B and D show, respectively, confocal images of F-actin labelled with FITC-conjugated phalloidin in control or JP-treated human platelets. Scale bars represent 1 μm. E-H, cells were pretreated at 37 °C with 10 μm JP (F and H) or the vehicle (ethanol; E and G) for 30 min. The platelets were then treated with TG (1 μm) and IONO (50 nM) in Ca2+-free HBS (100 μm EGTA added) and 3 min later SrCl2 (final concentration, 300 μm; E and F) or CaCl2 (final concentration 300 μm; G and H) was added followed by thrombin (10 U ml−1; n = 7).
Effect of xestospongin C on divalent cation entry
Further evidence for the independent regulation of the store-mediated and non-capacitative divalent cation entry pathways was obtained using the inhibitor of inositol 1,4,5-trisphosphate receptor (InsP3R) function, Xest C (Gafni et al. 1997). We have previously shown that Xest C treatment, which blocks thrombin-evoked release of Ca2+ from intracellular stores, also blocks SMCE in TG-treated platelets (Rosado & Sage, 2000c). Treatment with 20 μm Xest C for 30 min completely inhibited SMCE in platelets treated with TG and IONO to maximally deplete the agonist-releasable Ca2+ stores (Fig. 8A and B; n = 4). Addition of a maximally effective concentration of thrombin (10 U ml−1) to Xest C-treated platelets in the absence of external Ca2+ (100 μm EGTA added) was without effect on [Ca2+]i, indicating complete blockade of thrombin-evoked release of Ca2+ from intracellular stores (Fig. 8C). TG plus IONO were able to increase [Ca2+]i in Xest C-treated platelets, confirming that the intracellular Ca2+ stores were intact (Fig. 8C). However, addition of thrombin (10 U ml−1) to Xest C-treated cells in the presence of 1 mm external Ca2+ still evoked a rise in [Ca2+]i (n = 6; Fig. 8D), even though both thrombin-evoked release of Ca2+ from intracellular stores (Fig. 8C) and SMCE (Fig. 8B) were blocked.
Figure 8. Thrombin-induced non-capacitative Ca2+ entry is independent of store depletion.
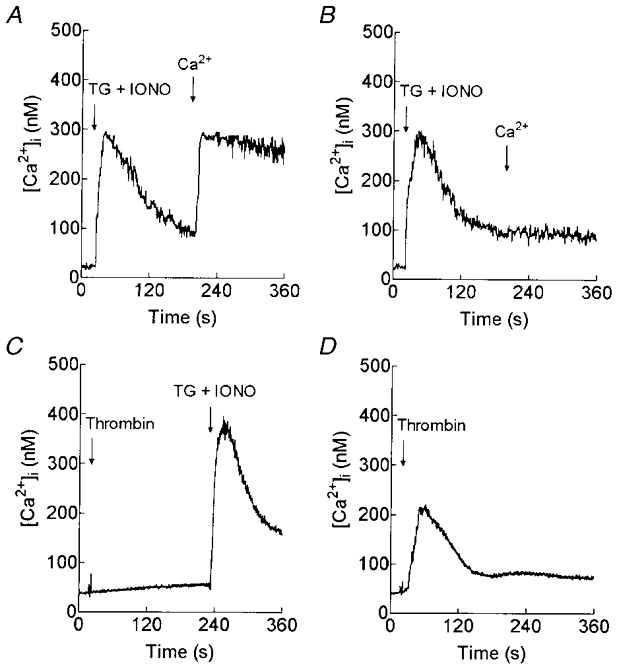
Fura-2-loaded human platelets were pretreated for 30 min at 37 °C with 20 μm Xest C (B-D) or the vehicle (DMSO) alone (A). In A and B, cells were then stimulated with TG (1 μm) and IONO (50 nM) in Ca2+-free HBS (100 μm EGTA added) and 3 min later CaCl2 (final concentration, 200 μm) was added to initiate Ca2+ entry (n = 4). C, cells were stimulated with 10 U ml−1 thrombin in Ca2+-free HBS (100 μm EGTA added) and 3.5 min later TG (1 μm) and IONO (50 nM) were added (n = 5). D, cells were stimulated with 10 U ml−1 thrombin in the presence of 1 mm external Ca2+ (n = 6).
DISCUSSION
The activation of Ca2+ entry following receptor occupation is a signalling process of great physiological relevance. Receptor-operated Ca2+ entry might occur by several different mechanisms (Putney, 1990; Sage, 1992). These include a receptor-operated channel formed by the subunits of the receptor protein, a channel closely linked with the receptor via a GTP-binding protein, a second messenger-operated channel gated by a diffusible molecule generated by receptor occupation and a store-mediated (or capacitative) mechanism, where Ca2+ entry is controlled by the filling state of the intracellular Ca2+ stores (Putney, 1990). In many non-excitable cells the latter pathway is a major mechanism for Ca2+ entry. Here we describe the existence of a new receptor-operated non-capacitative divalent cation entry pathway in human platelets, which are a good model for the study of Ca2+ signalling in normal human non-excitable cells.
This non-capacitative cation entry pathway can be activated by physiological agonists, such as thrombin, which activate PKC. Our data using activators and inhibitors of PKC indicate that the activity of this enzyme is required for the non-capacitative divalent cation entry pathway to operate. Our findings suggest that activation of PKC initially leads to stimulation of a non-capacitative divalent cation entry pathway that would add to the well-established store-mediated cation entry in these cells (Sargeant et al. 1992). However, in agreement with previous studies (Petersen & Berridge, 1994), continued activation of PKC inhibited both store-mediated and non-capacitative divalent cation entry in platelets. This inhibition may prevent a dangerously high [Ca2+]i being reached during prolonged cell stimulation.
Our results showing the stimulation of Ca2+ entry by the PKC activators PMA and OAG contrast with an earlier study in which OAG and the phorbol ester 12-O-tetradecanoyl phorbol-13-acetate (TPA) were reported not to affect [Ca2+]i in quin-2-loaded human platelets (Rink et al. 1983). The failure to detect changes in [Ca2+]i in response to PKC activators in this earlier study may reflect the low concentrations used (15-20 nM), the much higher cytosolic Ca2+ buffering power after the introduction of approximately 1 mm quin-2, or both of these factors.
A number of receptor-operated non-capacitative Ca2+ entry pathways have been reported including those activated by ADP and ATP in platelets (Mackenzie et al. 1996), vasopressin in rat A7r5 smooth muscle cells (Byron & Taylor, 1995) and hepatocytes (Kass et al. 1994), and carbachol in PC12 cells (Clementi et al. 1992) and exocrine avian nasal gland cells (Shuttleworth & Thompson, 1996). In addition, several intracellular messengers have been identified as regulators of non-capacitative divalent cation entry pathways, including DAG (Helliwell & Large, 1997), PKC (Oike et al. 1993) and arachidonic acid (Van Delden et al. 1993; Broad et al. 1999; Shuttleworth & Thompson, 1999). Our results indicate that thrombin-evoked non-capacitative divalent cation entry in platelets is mediated by activation of PKC, since direct activation of PKC using PMA or the DAG analogue OAG stimulated the pathway and the PKC inhibitor Ro-31-8220 blocked the thrombin-evoked entry. In addition, we have previously reported that arachidonic acid does not mediate rises in [Ca2+]i in human platelets in the presence of the cyclo-oxygenase inhibitors aspirin (used throughout the present study) or indomethacin (Vindlacheruvu et al. 1991), indicating that a hydrolysis product of DAG is unlikely to be responsible for the effect. Taken together, these results indicate that PKC mediates the regulation of receptor-operated non-capacitative divalent cation entry in human platelets.
The novel divalent cation entry pathway described here is regulated independently of the filling state of the intracellular Ca2+ stores and thus is different from the well-established SMCE mechanism, since the PKC inhibitor Ro-31-8220 reduced non-capacitative cation entry without modifying store-mediated entry and, in addition, the non-capacitative pathway could operate under conditions in which SMCE was completely inhibited (in JP- or Xest C-treated platelets). Further evidence comes from the finding that thrombin evoked non-capacitative cation entry alongside store-mediated cation entry when the Ca2+ stores were completely depleted, so that thrombin could not induce further Ca2+ release. In addition we found that PKC activators could stimulate the non-capacitative pathway without releasing Ca2+ from the intracellular stores.
The non-capacitative cation entry pathway is independent of the purinoceptor-activated receptor-operated Ca2+ entry pathway previously reported to be mediated by P2x1 receptors in platelets (Mackenzie et al. 1996). Firstly, prior desensitisation of the P2 x1 receptors by addition of 10 μmα,β-methylene ATP (Vial et al. 1997), a non-hydrolysable ATP analogue, had no effect on the non-capacitative cation entry subsequently evoked by thrombin. Secondly, the magnitude of divalent cation entry evoked by thrombin was comparable with that of store depletion-evoked entry and substantially larger than the reported cation entry evoked by P2 x1 receptor stimulation (Mackenzie et al. 1996). Thirdly, the occupation of purinoceptors by secreted ADP would be expected to evoke the release of Ca2+ from internal stores as well as the activation of divalent cation entry through the P2 x1 receptor, which was not the case when platelets were stimulated with PMA.
The results presented here are consistent with those reported recently in T3-65 clonal human embryonic kidney (HEK293) cells stably transfected to express the human homologue of the Drosophila transient receptor potential channel, TRP3 (Ma et al. 2000). In these cells, an increase in DAG levels brought about by application of DAG lipase inhibitors, or treatment with DAG analogues, stimulated TRP3-mediated Sr2+ entry. Although speculative, there is a potential relationship between the results presented here and the presence of TRP channels in human platelets (D. Molin, E. den Dekker, G. Breikers, R. van Oerle, J. W. Akkerman & J. W. A. Heemskerk, unpublished observations; Rosado & Sage, 2000c). These channels may be activated following occupation of G-protein-coupled receptors by physiological agonists which activate phospholipase C and therefore PKC, thereby generating non-capacitative Ca2+ entry alongside that initiated following the release of Ca2+ from the intracellular stores.
Acknowledgments
This work was supported by grant 051560 from the Wellcome Trust. J.A.R. is supported by Junta de Extremadura Consejería de Educación Ciencia y Tecnología and Fondo Social Europeo. We thank Austin Hockaday for assistance with confocal microscopy.
References
- Berridge MJ. Capacitative calcium entry. Biochemical Journal. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad LM, Cannon TR, Taylor CW. A non-capacitative pathway activated by arachidonic acid is the major Ca2+ entry mechanism in rat A7r5 smooth muscle cells stimulated with low concentrations of vasopressin. The Journal of Physiology. 1999;517:121–134. doi: 10.1111/j.1469-7793.1999.0121z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byron KL, Taylor CW. Vasopressin stimulation of Ca2+ mobilization, two bivalent cation entry pathways and Ca2+ efflux in A7r5 rat smooth muscle cells. The Journal of Physiology. 1995;485:455–468. doi: 10.1113/jphysiol.1995.sp020742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallini L, Coassin M, Alexandre A. Two classes of agonist-sensitive Ca2+ stores in platelets, as identified by their differential sensitivity to 2,5-di-(tert-butyl)-1,4-benzohydroquinone and thapsigargin. Biochemical Journal. 1995;310:449–452. doi: 10.1042/bj3100449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementi E, Scheer H, Zacchetti D, Fasolato C, Pozzan T, Meldolesi J. Two separate plasma membrane Ca2+ carriers participate in receptor-mediated Ca2+ influx in rat hepatocytes. Journal of Biological Chemistry. 1992;267:2164–2172. [PubMed] [Google Scholar]
- Cooper JA. Effects of cytochalasin and phalloidin on actin. Journal of Cell Biology. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doni MG, Cavallini L, Alexandre A. Ca2+ influx in platelets: activation by thrombin and by the depletion of the stores. Effect of cyclic nucleotides. Biochemical Journal. 1994;303:599–605. doi: 10.1042/bj3030599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafni J, Munsch JA, Lam TH, Catlin MC, Costa LG, Molinski TF, Pessah IN. Xestospongins: potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron. 1997;19:723–733. doi: 10.1016/s0896-6273(00)80384-0. [DOI] [PubMed] [Google Scholar]
- Graf E, Verma AK, Gorski JP, Lopaschuk G, Niggli V, Zurini M, Carofoli E, Penniston JT. Molecular properties of calcium-pumping ATPase from human erythrocytes. Biochemistry. 1982;21:4511–4516. doi: 10.1021/bi00261a049. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Haslam RJ, Williams KA, Davis W, Sherwood J, VanDerMeulen J. Roles of GTP-binding proteins and protein kinase C in signal transduction in the platelet. Advances in Second Messenger and Phosphoprotein Research. 1990;24:364–369. [PubMed] [Google Scholar]
- Helliwell RM, Large WA. α1-Adrenoceptor activation of a non-selective cation current in rabbit portal vein by 1,2-diacyl-sn-glycerol. The Journal of Physiology. 1997;499:417–428. doi: 10.1113/jphysiol.1997.sp021938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner S, Farndale RW, Sage SO. Effects of Ca2+ store depletion and refilling with Ca2+, Sr2+ or Ba2+ on tyrosine phosphorylation and Mn2+ entry in human platelets. Biochemical Journal. 1994;303:337–339. doi: 10.1042/bj3030337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass GEN, Chow SC, Gahm A, Webb D-L, Berggren P-O, Llopis J, Orrenius S. Two separate plasma membrane Ca2+ carriers participate in receptor-mediated Ca2+ influx in rat hepatocytes. Biochimica et Biophysica Acta. 1994;1223:226–233. doi: 10.1016/0167-4889(94)90230-5. [DOI] [PubMed] [Google Scholar]
- Ma H-T, Patterson RL, vanRossum DB, Birmbaumer L, Mikoshiba K, Gill DL. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 2000;287:1647–1651. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- Mackenzie AB, Mahaut-Smith MP, Sage SO. Activation of receptor-operated cation channels via P2x1 not P2T purinoceptors in human platelets. Journal of Biological Chemistry. 1996;271:2879–2881. doi: 10.1074/jbc.271.6.2879. [DOI] [PubMed] [Google Scholar]
- Mahaut-Smith MP, Rink TJ, Collins SC, Sage SO. Voltage-gated potassium channels and the control of membrane potential in human platelets. The Journal of Physiology. 1990;428:723–735. doi: 10.1113/jphysiol.1990.sp018237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteith GR, Roufogalis BD. The plasma membrane calcium pump – a physiological perspective on its regulation. Cell Calcium. 1995;18:459–470. doi: 10.1016/0143-4160(95)90009-8. [DOI] [PubMed] [Google Scholar]
- Oike M, Kitamura K, Kuriyama H. Protein kinase C activates the non-selective cation channel in the rabbit portal vein. Pflügers Archiv. 1993;424:159–164. doi: 10.1007/BF00374607. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Store depletion and calcium influx. Physiological Reviews. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Patterson RL, vanRossum DB, Gill DL. Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell. 1999;98:487–499. doi: 10.1016/s0092-8674(00)81977-7. [DOI] [PubMed] [Google Scholar]
- Petersen CCH, Berridge MJ. The regulation of capacitative calcium entry by calcium and protein kinase C in Xenopus oocytes. Journal of Biological Chemistry. 1994;269:32246–32253. [PubMed] [Google Scholar]
- Putney JW., Jr Capacitative calcium entry revisited. Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- Putney JW., Jr TRP, inositol 1,4,5-trisphosphate receptors, and capacitative calcium entry. Proceedings of the National Academy of Sciences of the USA. 1999;96:14669–14671. doi: 10.1073/pnas.96.26.14669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink TJ, Sage SO. Stimulated calcium efflux from fura-2-loaded human platelets. The Journal of Physiology. 1987;393:513–524. doi: 10.1113/jphysiol.1987.sp016837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink TJ, Sage SO. Calcium signalling in human platelets. Annual Review of Physiology. 1990;52:431–439. doi: 10.1146/annurev.ph.52.030190.002243. [DOI] [PubMed] [Google Scholar]
- Rink TJ, Sanchez A, Hallam TJ. Diacylglycerol and phorbol ester stimulate secretion without raising cytoplasmic free calcium in human platelets. Nature. 1983;305:317–319. doi: 10.1038/305317a0. [DOI] [PubMed] [Google Scholar]
- Rosado JA, Jenner S, Sage SO. A role for the actin cytoskeleton in the initiation and maintenance of store-mediated calcium entry in human platelets. Journal of Biological Chemistry. 2000;275:7527–7533. doi: 10.1074/jbc.275.11.7527. [DOI] [PubMed] [Google Scholar]
- Rosado JA, Sage SO. The actin cytoskeleton and store-mediated calcium entry. The Journal of Physiology. 2000a;526:221–229. doi: 10.1111/j.1469-7793.2000.t01-2-00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado JA, Sage SO. Farnesylcysteine analogues inhibit store-regulated Ca2+ entry in human platelets: evidence for involvement of small GTP-binding proteins and actin cytoskeleton. Biochemical Journal. 2000b;346:183–192. [PMC free article] [PubMed] [Google Scholar]
- Rosado JA, Sage SO. IP3 receptors couple with hTrp1 channels when Ca2+ stores are depleted in human platelets. Biochemical Journal. 2000c;351:429–437. [Google Scholar]
- Sage SO. Three routes for receptor-mediated Ca2+ entry. Current Biology. 1992;2:312–314. doi: 10.1016/0960-9822(92)90885-e. [DOI] [PubMed] [Google Scholar]
- Sage SO. Calcium entry mechanisms in human platelets. Experimental Physiology. 1997;82:807–823. doi: 10.1113/expphysiol.1997.sp004066. [DOI] [PubMed] [Google Scholar]
- Sage SO, Reast R, Rink TJ. ADP evokes biphasic Ca2+ influx in fura-2-loaded human platelets. Evidence for Ca2+ entry regulated by the intracellular Ca2+ store. Biochemical Journal. 1990;265:675–680. doi: 10.1042/bj2650675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage SO, Rink TJ. Kinetic differences between thrombin-induced and ADP-induced calcium influx and release from internal stores in fura-2-loaded human platelets. Biochemical and Biophysical Research Communications. 1986;136:1124–1129. doi: 10.1016/0006-291x(86)90450-x. [DOI] [PubMed] [Google Scholar]
- Sage SO, Rink TJ. The kinetics of changes in intracellular calcium concentration in fura-2-loaded human platelets. Journal of Biological Chemistry. 1987;262:16364–16369. [PubMed] [Google Scholar]
- Sargeant P, Clarkson WD, Sage SO, Heemskerk JWM. Calcium influx evoked by store depletion in human platelets is more susceptible to cytochrome P-450 inhibitors than receptor-mediated calcium entry. Cell Calcium. 1992;13:565–576. doi: 10.1016/0143-4160(92)90035-q. [DOI] [PubMed] [Google Scholar]
- Sargeant P, Farndale RW, Sage SO. ADP- and thapsigargin-evoked Ca2+ entry and protein-tyrosine phosphorylation are inhibited by the tyrosine kinase inhibitors genistein and methyl-2,5-dihydroxycinnamate in fura-2-loaded human platelets. Journal of Biological Chemistry. 1993;268:18151–18156. [PubMed] [Google Scholar]
- Shuttleworth TJ, Thompson JL. Evidence for a non-capacitative Ca2+ entry during [Ca2+]i oscillations. Biochemical Journal. 1996;316:819–824. doi: 10.1042/bj3160819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuttleworth TJ, Thompson JL. Discriminating between capacitative and arachidonate-activated Ca2+ entry pathways in HEK293 cells. Journal of Biological Chemistry. 1999;274:31174–31178. doi: 10.1074/jbc.274.44.31174. [DOI] [PubMed] [Google Scholar]
- VanDelden C, Foti M, Lew DP, Krause K-H. Ca2+ and Mg2+ regulation of inositol 1,4,5-triphosphate binding in myeloid cells. Journal of Biological Chemistry. 1993;268:12443–12448. [PubMed] [Google Scholar]
- Vial C, Hechler B, Leon C, Cazenave JP, Gachet C. Presence of P2x1purinoceptors in human platelets and megakaryoblastic cell lines. Thrombosis and Haemostasis. 1997;78:1500–1504. [PubMed] [Google Scholar]
- Vindlacheruvu RR, Rink TJ, Sage SO. Lack of evidence for a role for the lipoxygenase pathway in increases in cytosolic calcium evoked by ADP and arachidonic acid in human platelets. FEBS Letters. 1991;292:196–200. doi: 10.1016/0014-5793(91)80866-2. [DOI] [PubMed] [Google Scholar]
- Yao Y, Ferrer-Montiel AV, Montal M, Tsien RY. Activation of store-operated Ca2+ current in Xenopus oocytes requires SNAP-25 but not a diffusible messenger. Cell. 1999;98:475–485. doi: 10.1016/s0092-8674(00)81976-5. [DOI] [PubMed] [Google Scholar]