

Abstract
Mitochondria possess a highly permeable outer membrane and an inner membrane that was originally thought to be relatively impermeable to ions to prevent dissipation of the electrochemical gradient for protons. Although recent evidence has revealed a rich diversity of ion channels in both membranes, the purpose of these channels remains incompletely determined. Pores in the outer membrane are fundamental participants in apoptotic cell death, and this process may also involve permeability transition pores on the inner membrane. Novel functions are now being assigned to other ion channels of the inner membrane. Examples include protection against ischaemic injury by mitochondrial KATP channels and the contribution of inner membrane anion channels to spontaneous mitochondrial oscillations in cardiac myocytes. The central role of mitochondria in both the normal function of the cell and in its demise makes these channels prime targets for future research and drug development.
Chemiosmotic hypothesis and energy dissipative ion channels
In 1961, Mitchell proposed the chemiosmotic hypothesis to explain the mechanism of mitochondrial energy transduction (Mitchell, 1961). Paraphrasing the essential postulates of the hypothesis in terms of the current view gives four main features: (1) H+ translocation down its electrochemical gradient across the mitochondrial inner membrane is reversibly coupled to ATP phosphorylation through the ATP synthase (F1F0-ATPase); (2) the flow of reducing equivalents down the electron transport chain (based on the differing redox potentials of substrates in the chain) is coupled to H+ pumping from the matrix to the intermembrane space, thus establishing the large electrochemical gradient for H+ (or protonmotive force); (3) exchange- diffusion carrier proteins are present on the inner membrane to transport metabolites and selected inorganic ions into and out of the matrix, and (4) the mitochondrial inner membrane is generally impermeable to ions other than H+ movement through the ATPase.
As pointed out in the original Mitchell paper (Mitchell, 1961), the last point applies only to mitochondria with tight coupling between oxygen consumption (i.e. electron transport) and phosphorylation of ADP, and the extent of coupling would be expected to vary with the leakiness of the membrane. Since that time, techniques such as mitochondrial swelling assays, patch-clamp recordings, or reconstitution of mitochondrial membrane proteins in lipid bilayers indicate that such ‘leak’ may be mediated by a variety of inner membrane ion channels with specific ion selectivities, conductances and sensitivities to modulators. Despite the profound effect of opening these energy dissipating ion channels on mitochondrial metabolism, there is remarkably little known about their molecular structure, regulation or physiological role in intact cells. The present article explores the hypothesis that opening specific mitochondrial ion channels leads to specific functional consequences, either detrimental or beneficial to cell function. Examples of ongoing studies in our laboratory are given in support of this idea and are complemented by the other articles in this issue.
Channel types in mitochondrial membranes
A wide variety of selective ion transport pathways in mitochondria have been identified (summarized in Table 1), either through swelling or fluorescence assays in isolated mitochondria (Beavis, 1992; Garlid, 1994; Bernardi, 1999), by patch-clamp of isolated mitochondria and mitoplasts (mitochondria with ruptured outer membranes to allow access to the inner membrane) (Sorgato et al. 1987; Inoue et al. 1991; Moran et al. 1992; Sorgato & Moran, 1993), or by reconstitution of mitochondrial proteins in bilayers or proteoliposomes (Ballarin & Sorgato, 1995; Brenner et al. 2000).
Table 1.
Mitochondrial ion channels
| Location | Type | Conductance (∼150 mM salt) | Modulators/Inhibitors | Putative Role |
|---|---|---|---|---|
| Outer membrane | VDAC (porin) | 0.5–4 nS | Bax/Bak/Bcl-xL, TOM20, Ca2+, pH, ΔV, NADH, VDAC modulator | Metabolic transport, cytochrome C release/apoptosis, PTP complex |
| TOM40 (PSC) | 0.5 nS | Signal peptides | Protein transport | |
| BH proteins | — | Bax/Bid/Bik | Cytochrome C release/apoptosis | |
| Misc. | 10–307 pS | ΔV (for > 100 pS) | — | |
| Inner membrane | Ca2+ uniporter | — | Divalents, nucleotides | Ca2+ uptake |
| PTP MCC | 0.03–1.5 nS | Ca2+, ΔV, signal peptides, CsA | Protein transport | |
| PTP MMC | 0.3–1.3 nS | CsA, pH, Ca2+, thiols, Bax, ANT inhibition | Necrosis, apoptosis | |
| UCP | 75 pS | Fatty acids | Thermogenesis | |
| KCa | 295 pS | Ca2+, ΔV, ChTx | Volume regulation | |
| KATP | 9.7 pS | ATP, GTP, palmitoyl-CoA, Mg2+, Ca2+ | Volume regulation, protection, apoptosis | |
| IMACs | 45, 450 pS | ATP | (In yeast) volume regulation | |
| 15 pS (LCC) 107 pS (centum pS) | Mg2+, pH, P1, thiols, DIDS, Cationic amphiphiles | Volume regulation |
A summary of mitochondrial ion channel types identified either in isolated mitochondria, proteolipid bilayers, or in patch-clamp experiments. Detected single channel conductances have been tentatively assigned to a given type, but these assignments have not been unequivocally proven. Abbreviations as follows: VDAC (porin), voltage-dependent anion channel; TOM40 (PSC), TOM20, pore forming translocases in outer membrane; BH proteins, Bcl2 homology proteins; Misc., miscellaneous conductances reported by Moran et al. (1992); PTP, permeability transition pore; MCC, multiconductance channel; MMC, mitochondrial megachannel; UCP, uncoupling protein; IMAC inner membrane anion channel; LCC, low conductance channel; Bax, Bak, Bid, Bik, Bcl-xl, apoptosis-related proteins; ΔV, change in membrane voltage; CsA, cyclosporin A; ANT inhibition, adenine nucleotide translocase inhibition; ChTx, charybdotoxin; palmitoyl CoA, palmitoyl coenzyme A; P1, inorganic phosphate; thiols, redox-sensitive reactive thiols.
Outer membrane channels
The mitochondrial outer membrane contains one of the most extensively studied mitochondrial channels, the voltage-dependent anion channel (VDAC, or porin) (Colombini et al. 1996; Hodge & Colombini, 1997). The high permeability of VDAC in reconstitution experiments has contributed to the impression that the outer membrane plays a rather mundane role in cell physiology. However, resurgent interest in the outer membrane has been spurred by the discovery that a key event of programmed cell death (apoptosis) is the translocation of cytochrome c from the intermembrane space to the cytoplasm. A possible explanation for this is that apoptosis-inducing proteins (e.g. Bax) either form pores across the outer membrane or increase the conductance of endogenous channels like VDAC to allow passage of large molecules. Pore formation by the introduction of some Bcl2-homology proteins into lipid bilayers has been observed (Brenner et al. 2000), and there is recent evidence implicating VDAC in the permeability increase (Shimizu et al. 2000). It has also been suggested that modulation of outer membrane permeability may control the entry or exit of metabolites, perhaps playing a role in the regulation of mitochondrial energy metabolism (Rostovtseva et al. 1997; Vander Heiden et al. 2000). In addition, mitochondrial protein import involves pore-forming translocases in the outer (tom) and inner (tim) membranes which can be blocked by protein import targeting peptides (hence the name peptide-sensitive channels (Chich et al. 1991; Pelleschi et al. 1997). It is interesting to note that the large VDAC conductances seen in bilayers are not generally observed in patch-clamp experiments of intact mitochondria; rather, a variety of conductances spanning the range from 10-307 pS have been reported (Moran et al. 1992). At present, functions have not been assigned to these conductances and little is known about their regulation.
Inner membrane channels
The mitochondrial inner membrane was originally assumed to be generally impermeable to ions as a prerequisite for efficient chemiosmotic coupling. It is now clear that a number of ion channels are present and may open under particular circumstances. The major identified channels are listed in Table 1 and discussed briefly below. More detailed reviews are available for the reader interested in mitochondrial ion transport (Antonenko et al. 1991; Beavis, 1992; Brierley et al. 1994; Zoratti & Szabo, 1994; Garlid, 1996; Szewczyk, 1998; Bernardi, 1999).
Ca2+ uniporter
The Ca2+ uniporter is believed to play a central role in matching changes in energetic workload to stimulation of mitochondrial metabolism. As part of a complex Ca2+ handling cycle, the normal function of the uniporter is to transport Ca2+ down its electrochemical gradient into the mitochondrial matrix during periods of elevated cytoplasmic Ca2+. Ca2+-regulated dehydrogenases (pyruvate, isocitrate, and α-ketoglutarate dehydrogenases) are then stimulated to increase the production of reducing equivalents by the Krebs cycle (McCormack et al. 1990; Hansford, 1994). The single channel equivalent of the uniporter has not been clearly identified, but the channels are thought to be regulated by divalent ions and nucleotides and are blocked by Ruthenium Red (Gunter & Gunter, 1994; Litsky et al. 1997). Ca2+ transport also appears to be co-operatively stimulated by high Ca2+ (Kroner, 1986; Saris & Kroner, 1990). In addition to stimulating respiration, mitochondrial Ca2+ influx can be a significant cellular Ca2+ buffer, effectively integrating the response to transient Ca2+ stimuli (Hajnóczky et al. 1995; Zhou et al. 1998; Brandes & Bers, 1999) and loading the mitochondrial Ca2+ store. A recent proposal suggests that Ca2+ release from this store may be triggered to act as an intracellular signalling mechanism (Ichas et al. 1997).
Although not shown in Table 1, a second Ruthenium Red-sensitive rapid Ca2+ uptake mode (RaM) has been described (Gunter et al. 1998). Since its kinetics and Mg2+ sensitivity differ from the classical Ca2+ uniporter, it is possible that this pathway involves a new type of Ca2+ conductive ion channel.
Permeability transition pore (PTP)
The generalized inner membrane permeability increase induced by Ca2+ described in isolated mitochondria is thought to be mediated by the opening of a large non-selective ion channel capable of passing solutes up to 1500 MW (Hunter et al. 1976; Kroemer et al. 1998; Crompton, 1999; Duchen, 1999). An in-depth discussion of this topic is presented by Crompton in this volume (Crompton, 2000). Leading candidates for the single channel underlying this transition are the mitochondrial megachannel (MMC; Szabo & Zoratti, 1992) and the multiconductance channel (MCC; Kinnally et al. 1996). Many features of MMC and MCC are similar to those of the permeability transition of intact mitochondria (in lieu of a definitive assignment, they will be referred to collectively as the permeability transition pore (PTP) herein). Common properties include induction of opening under high matrix Ca2+ loads, oxidative stress, depolarization, inhibition by Mg2+, ADP, cyclosporin A, and modulation by ligands of the adenine nucleotide translocase (ANT; e.g. bongkrekic acid and atractyloside). PTP openings are characterized by a large number of subconductance states, suggestive of active multimerization of channel proteins in the membrane (Zorov et al. 1992). The channels are also modulated by thiol redox state, pH, free radicals, Bax, and in the case of MCC, by mitochondrial signal peptides (Kushnareva et al. 1999). Alterations of the properties of MCC by a tim23 mutation and block of the channel by a tim23 antibody suggest MCC may be a component of the mitochondrial protein import machinery (Lohret et al. 1997).
It is presently unknown whether the PTP plays a role in the normal function of mitochondria, but there is strong evidence that it contributes to cellular injury during ischaemia and reperfusion (Lemasters et al. 1999) and may initiate cytochrome c release to trigger apoptosis (for a review of the role of PTP in apoptosis see: Green & Reed, 1998; Crompton, 1999). The link between PTP opening and cytochrome c release is a subject of current controversy (Goldstein et al. 2000; Hajnóczky, 2000) and may depend on the type of apoptotic stimulus.
UCP
The uncoupling protein (UCP or thermogenin) of brown fat mitochondria is a prime example of an energy dissipating pathway, as it is responsible for non-shivering thermogenesis. In addition to UCP1, the isoform responsible for heat production in brown adipose tissue, two other widely distributed isoforms have been cloned (UCP2 and UCP3; (Diehl & Hoek, 1999; Ricquier et al. 1999). UCP is discussed in detail in the article by Ricquier in this volume (Ricquier & Bouillaud, 2000). Recent evidence suggests that UCP- mediated uncoupling involves fatty acid cycling, with transport of the anionic fatty acid accompanied by protonation/deprotonation and net H+ transport. While in the case of UCP1, heat production is the primary end, it has been proposed that the presence of UCP2 and UCP3 in other tissues may aid in optimizing the efficiency of energy metabolism by partially uncoupling the mitochondria (Jezek, 1999). This explanation is based on thermodynamic considerations, which suggest that the optimal ATP flow is achieved when the conductance of oxidative phosphorylation is matched to the conductance of the workload (Stucki, 1980). Optimal conductance matching is achieved at coupling ratios less than 1. Additionally, mild uncoupling of mitochondria has previously been suggested to be protective, perhaps by altering the rate of production of free radicals (Starkov, 1997; Korshunov et al. 1998). While the carrier-mediated transport of anions by UCP would be too slow to appear in patch-clamp recordings, a 75 pS anion channel attributed to UCP1 was recently reported, leading to speculation that under some conditions the carrier may show channel-like properties (Huang & Klingenberg, 1996).
KATP
Potassium-selective ion transport is a well-known feature of isolated mitochondria (Garlid, 1996; Bernardi, 1999) and its ATP dependence and sensitivity to K+ channel openers and sulphonylurea inhibitors has led to the suggestion that a KATP channel similar to the one present on the plasmalemma of many cells exists on the inner mitochondrial membrane (Paucek et al. 1992; Beavis et al. 1993; Szewczyk et al. 1993). This was strongly supported by the patch-clamp studies of Inoue et al. (1991), who identified a channel in mitoplast membranes whose gating properties, sensitivity to K+ channel openers, and inhibition by ATP resembled KATP channels of the surface membrane, albeit with a much smaller single channel conductance (∼10 pS in 100 mm KCl). The channel was inhibited by 100 μm ATP and blocked by glibenclamide and 4-aminopyridine.
Work from the laboratory of Garlid showed that diazoxide, an effective opener of pancreatic KATP channels, was approximately 1000-fold more potent for mitochondrial KATP channels (mitoKATP) than the cardiac sarcolemmal KATP isoform in a reconstituted system (Garlid et al. 1997).
While the physiological modulators of mitoKATP are unknown, reconstituted channels are inhibited by ATP, ADP and palmitoyl- or oleyl-CoA. The inhibited channel can be activated by GTP and GDP (Yarov-Yarovoy et al. 1997). Although the patch-clamp studies suggested that the ATP inhibitory site was on the matrix face (Inoue et al. 1991) other evidence suggests that the ATP inhibitory site is on the cytoplasmic face (Yarov-Yarovoy et al. 1997).
The functional role and pharmacology of mitoKATP will be discussed further in the section on mitoKATP.
It is useful to note here that another glibenclamide-sensitive cation conductance that is selective for Na+ over K+ has been reported in EDTA-treated mitochondria (Szewczyk et al. 1996; not shown in Table 1). The single channel correlate has not been identified, but this conductance is blocked by Ruthenium Red and is modulated by Mg2+ (Kapus et al. 1990).
KCa
The most recent addition to the list of inner membrane ion channels is the Ca2+-activated K+ channel (KCa) (Siemen et al. 1999). The properties of mitochondrial KCa were similar to those of BK channels in the plasmalemma – they showed sensitivity to charybdotoxin and activation by voltage and Ca2+. The physiological role of mitochondrial KCa is unknown, but it could potentially be involved in volume regulation during times of increased matrix Ca2+ load.
Inner membrane anion channel (IMAC)
There is substantial evidence from studies of mitochondrial swelling that a partially anion-selective channel is present in mitochondria (Garlid & Beavis, 1986; Beavis & Garlid, 1987). The channel was activated by matrix Mg2+ depletion or alkalinization, and was inhibited by a wide variety of cationic amphiphiles, including Ca2+ channel blockers, antiarrhythmics, β-adrenergic antagonists, local anaesthetics, tricyclic antidepressants, and anticonvulsants.
Two prime candidates for IMAC have been suggested from electrophysiological studies. In the first patch-clamp study of the mitochondrial inner membrane, a 107 pS channel with a permeability ratio of 4.5 for chloride to potassium was detected (Sorgato et al. 1987), and this channel comprises the main conductance of the inner membrane. The 107 pS channel, also referred to as the centum pS channel, has an open probability that is steeply dependent on voltage and is predominantly closed at the negative potentials of energized mitochondria (ΔΨ >150 mV). In this initial study, since no pH dependence was observed, the authors concluded that the 107 pS was probably not the single channel equivalent of IMAC. However, subsequent investigations have revealed properties of a similar channel in brown fat mitochondria that closely match the IMAC of swelling assays, including inhibition by propranolol, dihydropyridines and the nucleotide analogue Cibacron Blue (Beavis, 1992; Borecky et al. 1997).
Another 15 pS anion-selective channel displaying many of the same properties as IMAC has been described by Antonenko et al. (1991). This channel was activated by matrix alkalinization and blocked by amiodarone, propranolol and tributyltin.
Several other anion selective conductances have been described, including ATP-sensitive anion channels found in yeast inner membranes (Ballarin et al. 1995). At present, nothing is known about their physiological role and distribution in other species.
Since IMAC only appears to conduct ions under alkaline matrix conditions and low divalent concentrations, it is unclear what its physiological role is or if it opens under pathophysiological conditions. It has been suggested to be an important safeguard against mitochondrial swelling as it is poised to counteract the influx of cations (Beavis, 1992). Recent work in our laboratory suggests that IMAC may be activated under metabolic stress in intact cardiomyocytes, as discussed below.
Metabolic oscillations in cardiomyocytes Spatiotemporal heterogeneity of mitochondrial metabolism
The vectorial orientation of enzyme catalytic sites required for the chemiosmotic model of oxidative phosphorylation is a prime illustration of how structural organization is essential for normal function. Even in the simplest case of aqueous extracts, spatiotemporal patterns of product formation have been observed (Hess & Boiteux, 1971; Rapp, 1979) and the level of complexity increases as structural and kinetic compartmentation is introduced. Examples include the localization of glycolytic enzymes near surface ion channels (Weiss & Lamp, 1987), the association of multiple enzymes at the contact sites between the mitochondrial inner and outer membranes (Marzo et al. 1998; Vyssokikh et al. 1999) and the compartmentalization of the pathways of oxidative metabolism in the mitochondrial cristae and matrix. This organization is particularly clear in cardiac cells, where mitochondria take up 30-40 % of the intracellular volume, and are distributed along the myofilaments poised to meet the huge energy demands of the contractile apparatus. While the structure of the mitochondrial network it easily resolved using fluorescent markers, little is known about how it is functionally organized and what factors underly a co-ordinated metabolic response.
Insight into these issues has been gained by studying metabolic oscillation in isolated cardiomyocytes. Originally identified electrophysiologically by the observation of oscillation in sarcolemmal KATP currents when myocytes were deprived of substrate (O’Rourke et al. 1994, 1995), we have subsequently focused on the mechanism of mitochondrial redox oscillation associated with this response (Romashko et al. 1998; O’Rourke et al. 1999). Changes in mitochondrial redox potential can be monitored using the native fluorescence of endogenous flavoproteins (480 nm excitation/530 nm emission). This fluorescence arises primarily from FAD bound to the mitochondrial dehydrogenase enzymes containing lipoamide dehydrogenase (Fig. 1), and the redox state of the FAD/FADH2 pair is in equilibrium with mitochondrial NAD+/NADH (flavoprotein oxidation is largely blocked by rotenone, which inhibits NADH dehydrogenase (O’Rourke et al. 1999)). Flavoprotein fluorescence increases when the mitochondrial matrix is oxidized (e.g. in the presence of an uncoupler). ΔΨ can also be recorded simultaneously using the lipophilic cation tetramethylrhodamine ethyl ester (TMRE; excitation, 547: emission, ∼605 nm). In the context of the theme of this article, the opening of an inner membrane ion channel would be expected to cause net oxidation of the matrix (increased flavoprotein fluorescence), as well as a decrease in ΔΨ (decreased matrix TMRE fluorescence) if the supply of reducing equivalents is not enough to match the increased flux through the electron transport chain.
Figure 1. Mitochondrial redox and membrane potentials.
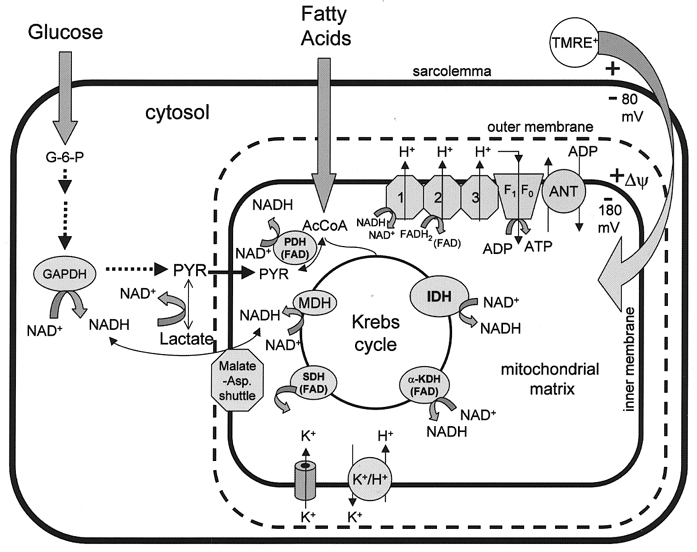
The redox state of the cell depends on the rates of production and oxidation of reducing equivalents in the cytoplasm and in the mitochondrial matrix. FAD-linked dehydrogenases in the matrix including pyruvate dehydrogenase (PDH), α-ketoglutarate dehydrogenase (α-KGDH) and succinate dehydrogenase (SDH) fluoresce green (excitation 480 nm/emission peak ≈520 nm) when oxidized. These flavoproteins are in equilibrium with the mitochondrial NADH/NAD+ redox couple. NADH is also fluorescent, emitting blue light (peak ≈450 nm) with ultraviolet excitation (350 nm). Mitochondrial inner membrane potential (ΔΨ) can be assessed by the distribution of the lipophilic cations like tetramethylrhodamine ethyl ester (TMRE), which emits red fluorescence (excitation 540 nm/emission 605 nm). The opening of an inner membrane ion channel (e.g. mitoKATP) leads to acceleration of NADH oxidation by the electron transport chain and partial dissipation of the proton gradient through activation of K+-H+ exchange. A net change in ΔΨ or redox potential occurs if NADH production and proton pumping cannot match the increase in energy dissipation.
Oscillations induced by metabolic stress in cardiomyocytes are characterized by periodic rapid oxidation of mitochondria flavoproteins and the collapse of ΔΨ. Such oscillations can occur in single chains of mitochondria without affecting neighbouring areas in the same focal plane (Romashko et al. 1998). An example of the spatiotemporal pattern of these mitochondrial transitions is shown in Fig. 2, in which a large ‘supercluster’ of mitochondria repeatedly switches between the polarized and depolarized state. The co-ordinated transition among mitochondria in the cluster without a change in others nearby implicitly suggests that there are physical connections between them, in accord with the idea that subpopulations of mitochondria are connected in units that behave like protonophoric cables (Amchenkova et al. 1988). In other cells, we have observed the fast collapse and recovery of ΔΨ in 1-2 μm regions of the cell without synchronization of neighbouring mitochondria, demonstrating that the smallest independent oscillator is the individual mitochondrion.
Figure 2. Regional depolarization of mitochondria.
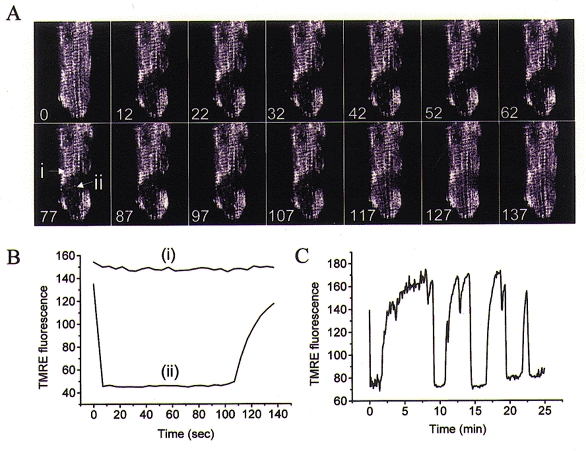
A, loss of ΔΨ in a substrate-deprived guinea-pig ventricular myocyte can be restricted to ‘superclusters’ of mitochondria, as shown for this series of images of TMRE fluorescence (100 nM TMRE loading). The timing of the images in seconds is denoted on each frame. B, rapid loss and slow recovery of TMRE fluorescence in the cluster (region ii) and unchanging signal in a neighbouring region (i; as shown in A) illustrate the independent behaviour of adjacent mitochondria. C, the full time course of multiple slow oscillations of ΔΨ in the supercluster.
Thus, the cardiac mitochondrial network can be viewed as a network of potential oscillators that are normally synchronized, but can act independently under conditions of metabolic stress. Variable coupling between these oscillators contributes to higher order structures like superclusters or cell-wide reponses. In addition to physical connections between mitochondria, diffusible factors may also influence synchronization. In this regard, we have also observed propagating waves of flavoprotein oxidation that can travel not only within a cell, but also between myocytes connected by an intercalated disc (Romashko et al. 1998). Similar heterogeneity of metabolism, presumed to be mediated by the opening of the PTP, has been reported by others in response to various stressors including photo-oxidation (Huser et al. 1998) and triggering by spontaneous Ca2+ release in local regions of the cell (Ichas et al. 1997; Duchen et al. 1998; Lemasters et al. 1998), leading us to investigate the involvement of inner membrane ion channels.
Effects of mitochondrial ion channel inhibitors
To determine if the properties of the observed mitochondrial transitions induced by substrate deprivation were consistent with those of known mitochondrial ion channels, we tested the effects of a variety of conditions and pharmacological agents. In contradistinction to the properties of the mitochondrial permeability transition observed in isolated mitochondria, metabolic oscillations induced by substrate deprivation were independent of mitochondrial Ca2+ overload. Although Ca2+ cycling is impaired during the phase of oxidation in stimulated myocytes (O’Rourke et al. 1994), depletion of SR Ca2+ with EGTA in the intracellular solution does not alter the incidence of oscillation. Ruthenium Red, which should eliminate mitochondrial Ca2+ uptake, also does not block the transitions. Furthermore, cyclosporin A, a known inhibitor of the PTP, did not suppress oscillations (O’Rourke et al. 1999).
Interestingly, reported inhibitors of IMAC do show significant effects on the oscillations. Figure 3 shows the effect of PK11195, a peripheral benzodiazepine receptor inhibitor that blocks IMAC in isolated mitochondria and also the 107 pS channel seen in patch-clamp recordings. PK11195 (200 μm) suppressed the fast redox transitions in a reversible and reproducible manner. Similar suppression was observed for other inhibitors of IMAC (e.g. amiodarone, amitriptylline, tributyltin, propranolol); but, as for PK11195, concentrations higher than those used in isolated mitochondria or patch-clamp studies (Beavis, 1992) were required (usually >100 μm). This difference in potency in intact cell studies, which is common to most pharmacological attempts to correlate mitochondrial ion channels with a response (see also the section on mitoKATP), may be due to diffusional barriers present in the intact cell. A note of caution is warranted, however, since non-specific inhibitory effects of amphipathic compounds on metabolism have been reported (Hirsch et al. 1989; Fromenty et al. 1990). Nevertheless, the common effect of structurally different IMAC inhibitors, and the lack of influence of agents known to influence PTP, supports the hypothesis that inner membrane anion channels underlie mitochondrial oscillations in substrate-deprived myocytes.
Figure 3. Suppression of oscillations by a benzodiazepine receptor ligand.
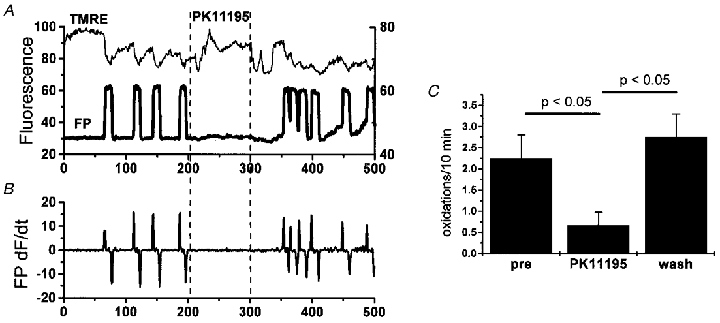
A, exposure to PK11195 (200 μm applied between dashed lines) reversibly suppressed oscillations in ΔΨ (TMRE signal) and flavoprotein redox (FP signal; thick line) as derived from time course image analysis. B, taking the first derivative of the redox signal (FP dF/dt) facilitates analysis of the frequency of transitions by eliminating slow baseline changes. Counting fast transitions that exceed a threshold (in this case 20 % of peak + dF/dt) permits quantitative determination of drug efficacy C, the number of oxidations over a period of 10 min in the presence of PK11195 was significantly suppressed (n = 12 cells) compared with before and after application.
As mentioned above, it has been proposed that anion efflux through IMAC may be a safeguard against excessive matrix swelling. Mitochondrial anion efflux has also recently been implicated in hypoxic preconditioning (Vanden Hoek et al. 1998). It remains to be determined if the redox transitions are somehow beneficial to the cell during metabolic stress; however, as we have shown previously (O’Rourke et al. 1994), these metabolic oscillations profoundly influence the excitability of the cardiac cell through the activation of sarcolemmal KATP channels. On the scale of the whole heart, spatiotemporal variation in excitability would have a decidedly detrimental effect by increasing the dispersion of repolarization, thus setting the stage for arrhythmias. Important future goals are to determine whether the oscillations described here are present in the whole heart during ischaemia/reperfusion and to find out if they can be pharmacologically modulated.
MitoKATP and protection against ischaemia/reperfusion injury
As described earlier, the existence of a KATP channel in the mitochondrial inner membrane is supported by studies of isolated mitochondria or reconstituted proteoliposomes (Paucek et al. 1992; Beavis et al. 1993; Szewczyk et al. 1993; Garlid, 1996; Garlid et al. 1997) and patch-clamp of mitoplasts (Inoue et al. 1991). With the large electrochemical driving force for K+ entry into the matrix, mitoKATP opening should lead to net K+ uptake and subsequent osmotic swelling (assuming that anion transport accompanies the cation). This enhanced K+ influx is counterbalanced by K+-H+ exchange, resulting in concomitant dissipation of the H+ gradient. Thus, mitoKATP is part of a volume regulatory K+ cycle, with energy expenditure as a consequence (Garlid, 1996). While the physiological role of mitochondrial volume regulation has not been fully elucidated, changes in mitochondrial volume influence the rate of oxidative metabolism (Halestrap, 1989) and thus could represent a metabolic control mechanism. By analogy with the proposed role of UCP (Jezek, 1999), optimization of respiration (Stucki, 1980) may also be the result of the partial uncoupling induced by mitoKATP opening.
In addition to volume homeostasis, recent experiments have revealed a new physiological role for mitoKATP in protecting cells from ischaemia and reperfusion injury. In particular, ischaemic preconditioning appears to depend critically on the opening of mitoKATP. Ischaemic preconditioning is a self-protective mechanism of the heart in which the injury caused by a long ischaemia is blunted if preceded by short ‘preconditioning’ ischaemic periods (Murry et al. 1986). KATP channels have been implicated in preconditioning because blockers like glibenclamide attenuate protection and KATP openers (cromakalin, etc.) mimic the protection (see references within (Gross & Fryer, 1999; Grover & Garlid, 2000). The mechanism of protection was assumed to be through the energy sparing effect of opening sarcolemmal KATP channels (sarcKATP). sarcKATP are activated by energy depletion and render the myocytes inexcitable, thus suppressing energy consuming reactions such as Ca2+ cycling. However, this explanation was called into question with the finding that low doses of openers that had little effect on action potential shortening still conferred protection (Yao & Gross, 1994; Grover et al. 1995) and that artificially preventing ischaemic action potential shortening did not eliminate preconditioning (Grover et al. 1996).
To test whether mitoKATP rather than sarcolemmal KATP could be an effector of protection, differences in the pharmacological selectivity of the two isoforms were exploited. By comparing cardiac sarcolemmal and mitochondrial KATP channels reconstituted in proteoliposomes, it was shown that diazoxide was approximately 1000 times more potent in opening the mitochondrial isoform (K1/2 0.8 vs. 840 μm for sarcKATP (Garlid et al. 1997)). Furthermore, the effect of diazoxide could be blocked by 5-hydroxydecanoate (5-HD) with a Ki of 45-85 μm. 5-HD is a compound reported to be an ischaemia-selective KATP inhibitor that can block preconditioning (Auchampach et al. 1992). Glibenclamide (Ki 1-6 μm) and 5-HD block mitoKATP in a state-dependent manner, having no effect on channels activated by removing ATP, but inhibiting channels activated by GTP or K+ channel openers in the presence of ATP (Jaburek et al. 1998).
In parallel with the biochemical studies of mitoKATP, Garlid et al. (1997) examined the effects of diazoxide and 5-HD on ischaemic injury in Langendorff-perfused rat hearts. It was found that at concentrations shown to have no effect on sarcKATP (1-100 μm), diazoxide significantly protected hearts against ischaemic injury. Furthermore, 5-HD (100 μm) eliminated the protective effect of preconditioning.
The opening of mitoKATP by diazoxide was detected for the first time in intact cardiomyocytes as a reversible increase in the oxidation of flavoproteins, using the same fluorescence method described in the preceding section (Liu et al. 1998; Fig. 4). Importantly, at concentrations of up to 100 μm (with a K½ of 27 μm), diazoxide had no effect on sarcolemmal KATP currents in intact cells and 5-HD inhibited the diazoxide effect. The latter finding was an important confirmation, since we have subsequently shown that 5-HD selectively inhibits mitoKATP (Sato et al. 1998). Using the same methodology, we have characterized the pharmacological profile of a number of KATP openers and inhibitors, and have been able to identify compounds which specifically act on either the mitochondrial or sarcolemmal channels (Fig. 5).
Figure 4. Selective activation of mitoKATP by diazoxide in cardiomyocytes.
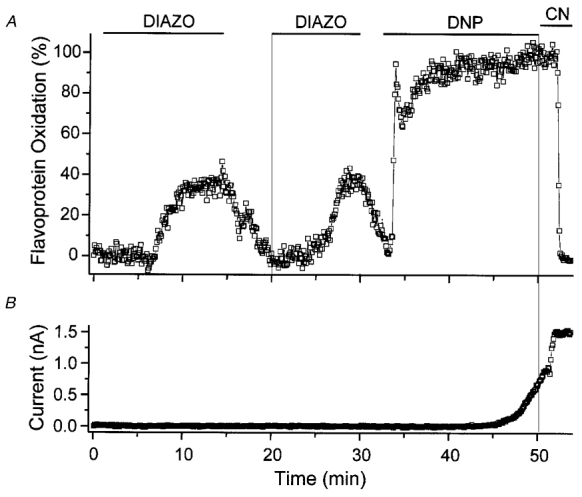
A, diazoxide causes reversible and reproducible oxidation of mitochondrial flavoproteins (Diazo; 100 μm). The fluorescence signal can be calibrated by maximally oxidizing (with the uncoupler 2,4-dinitrophenol; DNP) and reducing the mitochondria (with the cytochrome oxidase inhibitor cyanide; CN). B, in the same experiment, diazoxide did not activate sarcolemmal KATP current. Severe metabolic inhibition with DNP or CN activated sarcolemmal KATP, confirming the presence of these channels. From Liu et al. (1998).
Figure 5. Summary of the selectivity of KATP channel openers and blockers.
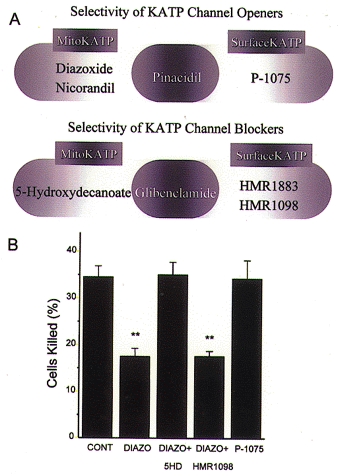
A, based on studies of intact myocytes using methods similar to that shown in Fig. 4, the selective mitoKATP agonists diazoxide and nicorandil have been identified, while pinacidil activates both mitochondrial and surface isoforms. The pinacidil derivative P-1075 selectively activates sarcolemmal KATP in intact myocytes. B, cellular protection against simulated ischaemia is conferred by diazoxide, but not P-1075, and is blocked by 5-HD, but not HMR-1098, thus supporting a mechanism involving mitoKATP rather than sarcKATP.
The link between mitoKATP and protection in cardiomyocytes was demonstrated by examining the extent of cell killing in response to simulated ischaemia using a cell pelleting method (Liu et al. 1998). As summarized in Fig. 5, protection was afforded only by compounds capable of activating mitoKATP, and this protection was inhibited only by compounds effective at blocking mitoKATP (Sato et al. 2000). Compounds selective for sarcolemmal channels have no such actions and serve as convincing evidence that sarcolemmal channels play much less of a role in protection.
Studies of intact hearts have confirmed the hypothesis that mitoKATP is fundamentally involved in cardioprotection. Protection can be mimicked by even a short exposure to diazoxide prior to the long ischaemia and preconditioning induced by brief ischaemia can be blocked by 5-HD or glibenclamide, which have no effects on their own (see references in Gross & Fryer, 1999). The inability of the surface-selective blocker HMR1883 to further eliminate protection supports a mitochondrial vs. sarcolemmal site of action (Fryer et al. 2000).
The obvious question remains as to how the opening of an energy dissipating ion channel could protect cells against ischaemic injury. Available evidence suggests several possible mechanisms. First, as discussed above, mitochondrial matrix swelling induced by mitoKATP opening may improve mitochondrial energy production. Recent evidence supports the hypothesis that the pharmacological opening of mitoKATP improves oxidative phosphorylation during ischaemia and reperfusion and that this effect is inhibited by 5-HD or glibenclamide (Tanonaka et al. 1999; Iwai et al. 2000; Miura et al. 2000).
A second protective influence of mitoKATP may be an effect on Ca2+ handling. Holmuhamedov et al. (1999) showed that diazoxide or pinacidil decreased the rate of Ca2+ uptake by isolated mitochondria and a similar effect was observed in intact neonatal myocytes. 5-HD inhibited the latter effect. A decrease in the driving force for Ca2+ entry due to a 10-15 mV depolarization of ΔΨ was thought to be responsible.
A third mechanism is supported by recent work demonstrating that the opening of mitoKATP channels may alter the rate of mitochondrial reactive oxygen species (ROS) production and contribute to cardioprotection. An early increase in ROS production during hypoxic preconditioning in embryonic myocytes was noted in several studies (Vanden Hoek et al. 1998, 2000; Yao et al. 1999) and both protection and ROS production were inhibited by 5-HD, the thiol reductant 2-mercaptoproionyl glycine or the mitochondrial site III inhibitor myxothiazol. This suggests that mitochondria were the source of ROS production and that the opening of mitoKATP could stimulate ROS accumulation. ROS production may in turn activate PKC, which is a known component of cardioprotective pathways, and may also influence mitoKATP activity (Sato et al. 1998; Sasaki et al. 2000). Wang & Ashraf (1999) reported that diazoxide induced PKC translocation and protection in Langendorff-perfused rat hearts and these responses could be blocked by PKC inhibitors. In contrast, Miura et al. (2000) reported that the PKC inhibitor calphostin C could block adenosine- but not diazoxide-mediated cardioprotection. Thus, the chain of events linking mitoKATP activation, ROS production and PKC activation remains incompletely defined.
Both mechanistic and structure-function studies of mitoKATP will require a breakthrough in efforts to clone the channel. Thus far, putative channel subunits have been only partially purified, but support the notion that the channel is composed of sulfonylurea receptor (SUR)-like and inward rectifier-like components, although their sizes were smaller than their surface membrane counterparts (Grover & Garlid, 2000). Although it has been reported that antibodies against Kir6.1 immunolocalize to mitochondria (Suzuki et al. 1997), a recent effort to knock out Kir6.1 channels by dominant negative suppression in cardiac cells had no effect on the mitochondrial response to diazoxide (Seharaseyon et al. 2000), challenging the suggestion that Kir6.1 is a subunit of mitoKATP.
The recent explosion of interest in mitoKATP as a trigger and/or mediator of cellular protection against ischaemia- reperfusion injury has motivated efforts to develop potent and selective openers of this channel. New compounds with nanomolar affinities for mitoKATP and little effect on vascular or sarcolemmal KATP have recently been developed and these agents appear to confer protection without the potentially arrhythmogenic consequences of opening sarcKATP (Rovnyak et al. 1997; Grover, 2000).
SUMMARY
Novel imaging and patch-clamp techniques are now being applied to identify and characterize new mitochondrial ion channel types and the agents that modulate them. These methods complement the myriad studies of isolated mitochondria that have convincingly identified selective conductance pathways in a membrane formerly thought to be largely impermeable to ions. We are just beginning to define the functional roles of these channels and are finding that they are essential for both preserving the function of the cell and in killing it. In addition to the more widely known Ca2+ and PTP channels, we have found that different physiological responses can be initiated by the opening of specific channels. In the case of metabolic oscillation, which could be detrimental in terms of whole heart arrhythmias, inner membrane anion channels may be involved. In contrast, mitoKATP opening apparently protects both isolated cells and the intact heart against ischaemic injury. Undoubtedly, new types of channels are likely to be identified in mitochondrial membranes and functional assignments will be made for known channels of previously undefined purpose. Progress will be accelerated by future definition of the molecular structure of these channels, which will aid in the development of new drugs to treat or prevent disease.
Acknowledgments
I would like to thank Dr Eduardo Marbán and the postdoctoral fellows with whom I have collaborated. This research was supported by NIH Grants R01 HL-54598 (to B.O’R.) and R01 HL-44065 (to E.M.).
References
- Amchenkova AA, Bakeeva LE, Chentsov YS, Skulachev VP, Zorov DB. Coupling membranes as energy-transmitting cables. I. Filamentous mitochondria in fibroblasts and mitochondrial clusters in cardiomyocytes. Journal of Cell Biology. 1988;107:481–495. doi: 10.1083/jcb.107.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonenko YN, Kinnally KW, Tedeschi H. Identification of anion and cation pathways in the inner mitochondrial membrane by patch clamping of mouse liver mitoplasts. Journal of Membrane Biology. 1991;124:151–158. doi: 10.1007/BF01870459. [DOI] [PubMed] [Google Scholar]
- Auchampach JA, Grover GJ, Gross GJ. Blockade of ischaemic preconditioning in dogs by the novel ATP dependent potassium channel antagonist sodium 5-hydroxydecanoate. Cardiovascular Research. 1992;26:1054–1062. doi: 10.1093/cvr/26.11.1054. [DOI] [PubMed] [Google Scholar]
- Ballarin C, Sorgato MC. An electrophysiological study of yeast mitochondria. Evidence for two inner membrane anion channels sensitive to ATP. Journal of Biological Chemistry. 1995;270:19262–19268. doi: 10.1074/jbc.270.33.19262. [DOI] [PubMed] [Google Scholar]
- Beavis AD. Properties of the inner membrane anion channel in intact mitochondria. Journal of Bioenergetics and Biomembranes. 1992;24:77–90. doi: 10.1007/BF00769534. [DOI] [PubMed] [Google Scholar]
- Beavis AD, Garlid KD. The mitochondrial inner membrane anion channel. Regulation by divalent cations and protons. Journal of Biological Chemistry. 1987;262:15085–15093. [PubMed] [Google Scholar]
- Beavis AD, Lu Y, Garlid KD. On the regulation of K+ uniport in intact mitochondria by adenine nucleotides and nucleotide analogs. Journal of Biological Chemistry. 1993;268:997–1004. [PubMed] [Google Scholar]
- Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiological Reviews. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Borecky J, Jezek P, Siemen D. 108-pS channel in brown fat mitochondria might be identical to the inner membrane anion channel. Journal of Biological Chemistry. 1997;272:19282–19289. [PubMed] [Google Scholar]
- Brandes R, Bers DM. Analysis of the mechanisms of mitochondrial NADH regulation in cardiac trabeculae. Biophysics Journal. 1999;77:1666–1682. doi: 10.1016/S0006-3495(99)77014-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner C, Cadiou H, Vieira HL, Zamzami N, Marzo I, Xie Z, Leber B, Andrews D, Duclohier H, Reed JC, Kroemer G. Bcl-2 and Bax regulate the channel activity of the mitochondrial adenine nucleotide translocator. Oncogene. 2000;19:329–336. doi: 10.1038/sj.onc.1203298. [DOI] [PubMed] [Google Scholar]
- Brierley GP, Baysal K, Jung DW. Cation transport systems in mitochondria: Na+ and K+ uniports and exchangers. Journal of Bioenergetics and Biomembranes. 1994;26:519–526. doi: 10.1007/BF00762736. [DOI] [PubMed] [Google Scholar]
- Chich JF, Goldschmidt D, Thieffry M, Henry JP. A peptide-sensitive channel of large conductance is localized on mitochondrial outer membrane. European Journal of Biochemistry. 1991;196:29–35. doi: 10.1111/j.1432-1033.1991.tb15781.x. [DOI] [PubMed] [Google Scholar]
- Colombini M, Blachly-Dyson E, Forte M. VDAC, a channel in the outer mitochondrial membrane. Ion Channels. 1996;4:169–202. doi: 10.1007/978-1-4899-1775-1_5. [DOI] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochemical Journal. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Crompton M. Mitochondial intermembrane junctional complexes and their role in cell death. The Journal of Physiology. 2000;529:11–21. doi: 10.1111/j.1469-7793.2000.00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl AM, Hoek JB. Mitochondrial uncoupling: role of uncoupling protein anion carriers and relationship to thermogenesis and weight control ‘the benefits of losing control. Journal of Bioenergetics and Biomembranes. 1999;31:493–506. doi: 10.1023/a:1005452624640. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. The Journal of Physiology. 1999;516:1–17. doi: 10.1111/j.1469-7793.1999.001aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Leyssens A, Crompton M. Transient mitochondrial depolarizations reflect focal sarcoplasmic reticular calcium release in single rat cardiomyocytes. Journal of Cell Biology. 1998;142:975–988. doi: 10.1083/jcb.142.4.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromenty B, Fisch C, Labbe G, Degott C, Deschamps D, Berson A, Letteron P, Pessayre D. Amiodarone inhibits the mitochondrial beta-oxidation of fatty acids and produces microvesicular steatosis of the liver in mice. Journal of Pharmacological and Experimental Therapeutics. 1990;255:1371–1376. [PubMed] [Google Scholar]
- Fryer RM, Eells JT, Hsu AK, Henry MM, Gross GJ. Ischemic preconditioning in rats: role of mitochondrial K(ATP) channel in preservation of mitochondrial function. American Journal of Physiology. 2000;278:H305–312. doi: 10.1152/ajpheart.2000.278.1.H305. [DOI] [PubMed] [Google Scholar]
- Garlid KD. Mitochondrial cation transport: a progress report. Journal of Bioenergetics and Biomembranes. 1994;26:537–42. doi: 10.1007/BF00762738. [DOI] [PubMed] [Google Scholar]
- Garlid KD. Cation transport in mitochondria-the potassium cycle. Biochimica et Biophysica Acta. 1996;1275:123–126. doi: 10.1016/0005-2728(96)00061-8. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Beavis AD. Evidence for the existence of an inner membrane anion channel in mitochondria. Biochimica et Biophysica Acta. 1986;853:187–204. doi: 10.1016/0304-4173(87)90001-2. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D’Alonzo AJ, Lodge NJ, Smith MA, Grover GJ. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circulation Research. 1997;81:1072–108. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invarient. Nature Cell Biology. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281(5381):1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Gross GJ, Fryer RM. Sarcolemmal versus mitochondrial ATP-sensitive K+ channels and myocardial preconditioning. Circulation Research. 1999;84:973–979. doi: 10.1161/01.res.84.9.973. [DOI] [PubMed] [Google Scholar]
- Grover GJ. International Society for Heart Research Meeting. Louisville, USA: 2000. KATP channel openers. http://www.ishrworld.org. [Google Scholar]
- Grover GJ, D’Alonzo AJ, Dzwonczyk S, Parham CS, Darbenzio RB. Preconditioning is not abolished by the delayed rectifier K+ blocker dofetilide. American Journal of Physiology. 1996;271:H1207–1214. doi: 10.1152/ajpheart.1996.271.3.H1207. [DOI] [PubMed] [Google Scholar]
- Grover GJ, D’Alonzo AJ, Parham CS, Darbenzio RB. Cardioprotection with the KATPopener cromakalim is not correlated with ischemic myocardial action potential duration. Journal of Cardiovascular Pharmacology. 1995;26:145–152. doi: 10.1097/00005344-199507000-00023. [DOI] [PubMed] [Google Scholar]
- Grover GJ, Garlid KD. ATP-Sensitive potassium channels: a review of their cardioprotective pharmacology. Journal of Molecular and Cellular Cardiology. 2000;32:677–695. doi: 10.1006/jmcc.2000.1111. [DOI] [PubMed] [Google Scholar]
- Gunter KK, Gunter TE. Transport of calcium by mitochondria. Journal of Bioenergetics and Biomembranes. 1994;26:471–485. doi: 10.1007/BF00762732. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Buntinas L, Sparagna GC, Gunter KK. The Ca2+ transport mechanisms of mitochondria and Ca2+ uptake from physiological-type Ca2+ transients. Biochimica et Biophysica Acta. 1998;1366:5–15. doi: 10.1016/s0005-2728(98)00117-0. [DOI] [PubMed] [Google Scholar]
- Hajnóczky G, Csordás G, Madesh M, Pacher P. The machinery of local Ca2+ signalling between sarco-endoplasmic reticulum and mitochondria. The Journal of Physiology. 2000;529:69–81. doi: 10.1111/j.1469-7793.2000.00069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnóczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. The regulation of the matrix volume of mammalian mitochondria in vivo and in vitro and its role in the control of mitochondrial metabolism. Biochimica et Biophysica Acta. 1989;973:355–382. doi: 10.1016/s0005-2728(89)80378-0. [DOI] [PubMed] [Google Scholar]
- Hansford RG. Physiological role of mitochondrial Ca2+ transport. Journal of Bioenergetics and Biomembranes. 1994;26:495–508. doi: 10.1007/BF00762734. [DOI] [PubMed] [Google Scholar]
- Hess B, Boiteux A. Oscillatory phenomena in biochemistry. Annual Review of Biochemistry. 1971;40:237–258. doi: 10.1146/annurev.bi.40.070171.001321. [DOI] [PubMed] [Google Scholar]
- Hirsch JD, Beyer CF, Malkowitz L, Beer B, Blume AJ. Mitochondrial benzodiazepine receptors mediate inhibition of mitochondrial respiratory control. Molecular Pharmacology. 1989;35:157–163. [PubMed] [Google Scholar]
- Hodge T, Colombini M. Regulation of metabolite flux through voltage-gating of VDAC channels. Journal of Membrane Biology. 1997;157:271–279. doi: 10.1007/s002329900235. [DOI] [PubMed] [Google Scholar]
- Holmuhamedov EL, Wang L, Terzic A. ATP-sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. The Journal of Physiology. 1999;519:347–360. doi: 10.1111/j.1469-7793.1999.0347m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SG, Klingenberg M. Chloride channel properties of the uncoupling protein from brown adipose tissue mitochondria: a patch-clamp study. Biochemistry. 1996;35:16806–16814. doi: 10.1021/bi960989v. [DOI] [PubMed] [Google Scholar]
- Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium-treated mitochondria. Journal of Biological Chemistry. 1976;251:5069–5077. [PubMed] [Google Scholar]
- Huser J, Rechenmacher CE, Blatter LA. Imaging the permeability pore transition in single mitochondria. Biophysical Journal. 1998;74:2129–2137. doi: 10.1016/S0006-3495(98)77920-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89:1145–1153. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Inoue I, Nagase H, Kishi K, Higuti T. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature. 1991;352:244–247. doi: 10.1038/352244a0. [DOI] [PubMed] [Google Scholar]
- Iwai T, Tanonaka K, Koshimizu M, Takeo S. Preservation of mitochondrial function by diazoxide during sustained ischaemia in the rat heart. British Journal of Pharmacology. 2000;129:1219–1227. doi: 10.1038/sj.bjp.0703148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaburek M, Yarov-Yarovoy V, Paucek P, Garlid KD. State-dependent inhibition of the mitochondrial KATP channel by glyburide and 5-hydroxydecanoate. Journal of Biological Chemistry. 1998;273:13578–13582. [PubMed] [Google Scholar]
- Jezek P. Fatty acid interaction with mitochondrial uncoupling proteins. Journal of Bioenergetics and Biomembranes. 1999;31:457–466. doi: 10.1023/a:1005496306893. [DOI] [PubMed] [Google Scholar]
- Kapus A, Szaszi K, Kaldi K, Ligeti E, Fonyo A. Ruthenium red inhibits mitochondrial Na+ and K+ uniports induced by magnesium removal. Journal of Biological Chemistry. 1990;265:18063–18066. [PubMed] [Google Scholar]
- Kinnally KW, Lohret TA, Campo ML, Mannella CA. Perspectives on the mitochondrial multiple conductance channel. Journal of Bioenergetics and Biomembranes. 1996;28:115–123. doi: 10.1007/BF02110641. [DOI] [PubMed] [Google Scholar]
- Korshunov SS, Korkina OV, Ruuge EK, Skulachev VP, Starkov AA. Fatty acids as natural uncouplers preventing generation of O2.- and H2O2 by mitochondria in the resting state. FEBS Letters. 1998;435:215–218. doi: 10.1016/s0014-5793(98)01073-4. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annual Reviews in Physiology. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- Kroner H. Ca2+ ions, an allosteric activator of calcium uptake in rat liver mitochondria. Archives of Biochemistry and Biophysics. 1986;251:525–535. doi: 10.1016/0003-9861(86)90360-7. [DOI] [PubMed] [Google Scholar]
- Kushnareva YE, Campo ML, Kinnally KW, Sokolove PM. Signal presequences increase mitochondrial permeability and open the multiple conductance channel. Archives of Biochemistry and Biophysics. 1999;366:107–115. doi: 10.1006/abbi.1999.1190. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Qian T, Bradham CA, Brenner DA, Cascio WE, Trost LC, Nishimura Y, Nieminen AL, Herman B. Mitochondrial dysfunction in the pathogenesis of necrotic and apoptotic cell death. Journal of Bioenergetics and Biomembranes. 1999;31:305–319. doi: 10.1023/a:1005419617371. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Qian T, Elmore SP, Trost LC, Nishimura Y, Herman B, Bradham CA, Brenner DA, Nieminen AL. Confocal microscopy of the mitochondrial permeability transition in necrotic cell killing, apoptosis and autophagy. Biofactors. 1998;8:283–285. doi: 10.1002/biof.5520080316. [DOI] [PubMed] [Google Scholar]
- Litsky ML, Pfeiffer DR. Regulation of the mitochondrial Ca2+ uniporter by external adenine nucleotides: the uniporter behaves like a gated channel which is regulated by nucleotides and divalent cations. Biochemistry. 1997;36:7071–7080. doi: 10.1021/bi970180y. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sato T, O’Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation. 1998;97:2463–2469. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- Lohret TA, Jensen RE, Kinnally KW. Tim23, a protein import component of the mitochondrial inner membrane, is required for normal activity of the multiple conductance channel, MCC. Journal of Cell Biology. 1997;137:377–386. doi: 10.1083/jcb.137.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiological Reviews. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- Marzo I, Brenner C, Zamzami N, Susin SA, Beutner G, Brdiczka D, Remy R, Xie ZH, Reed JC, Kroemer G. The permeability transition pore complex: a target for apoptosis regulation by caspases and bcl-2-related proteins. Journal of Experimental Medicine. 1998;187:1261–1271. doi: 10.1084/jem.187.8.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–148. doi: 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- Miura T, Liu Y, Kita H, Ogawa T, Shimamoto K. Roles of mitochondrial ATP-sensitive K channels and PKC in anti-infarct tolerance afforded by adenosine A1 receptor activation. Journal of the American College of Cardiology. 2000;35:238–245. doi: 10.1016/s0735-1097(99)00493-3. [DOI] [PubMed] [Google Scholar]
- Moran O, Sciancalepore M, Sandri G, Panfili E, Bassi R, Ballarin C, Sorgato MC. Ionic permeability of the mitochondrial outer membrane. European Biophysical Journal. 1992;20:311–319. doi: 10.1007/BF00196590. [DOI] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- O’Rourke B, Ramza BM, Marban E. Oscillations of membrane current and excitability driven by metabolic oscillations in heart cells. Science. 1994;265:962–966. doi: 10.1126/science.8052856. [DOI] [PubMed] [Google Scholar]
- O’Rourke B, Ramza BM, Romashko DN, Marban E. Metabolic oscillations in heart cells. In: Sideman S, Beyar R, editors. Molecular and Subcellular Cardiology: Effects on Structure and Function. Vol. 382. New York: Plenum Press; 1995. pp. 165–174. [Google Scholar]
- O’Rourke B, Romashko DN, Marban E. Subcellular heterogeneity of energy metabolism and K, ATP current oscillation in cardiac myocytes. In: Kurachi Y, Lazdunski M, Jan L, editors. Potassium Ion Channels: Molecular Structure, Function, and Diseases. Vol. 46. San Diego: Academic Press; 1999. pp. 449–467. Current Topics in Membranes. [Google Scholar]
- Paucek P, Mironova G, Mahdi F, Beavis AD, Woldegiorgis G, Garlid KD. Reconstitution and partial purification of the glibenclamide-sensitive, ATP-dependent K+ channel from rat liver and beef heart mitochondria. Journal of Biological Chemistry. 1992;267:26062–26069. [PubMed] [Google Scholar]
- Pelleschi M, Henry JP, Thieffry M. Inactivation of the peptide-sensitive channel from the yeast mitochondrial outer membrane: properties, sensitivity to trypsin and modulation by a basic peptide. Journal of Membrane Biology. 1997;156:37–44. doi: 10.1007/s002329900185. [DOI] [PubMed] [Google Scholar]
- Rapp PE. An atlas of cellular oscillators. Journal of Experimental Biology. 1979;81:281–306. doi: 10.1242/jeb.81.1.281. [DOI] [PubMed] [Google Scholar]
- Ricquier D, Bouillaud F. Mitochondrial uncoupling proteins: from mitochondria to the regulation of energy balance. The Journal of Physiology. 2000;529:3–10. doi: 10.1111/j.1469-7793.2000.00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricquier D, Miroux B, Cassard-Doulcier AM, Levi-Meyrueis C, Gelly C, Raimbault S, Bouillaud F. Contribution to the identification and analysis of the mitochondrial uncoupling proteins. Journal of Bioenergetics and Biomembranes. 1999;31:407–418. doi: 10.1023/a:1005488105076. [DOI] [PubMed] [Google Scholar]
- Romashko DN, Marban E, O’Rourke B. Subcellular metabolic transients and mitochondrial redox waves in heart cells. Proceedings of the National Academy of Sciences of the USA. 1998;95:1618–1623. doi: 10.1073/pnas.95.4.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtseva T, Colombini M. VDAC channels mediate and gate the flow of ATP: implications for the regulation of mitochondrial function. Biophysical Journal. 1997;72:1954–1962. doi: 10.1016/S0006-3495(97)78841-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovnyak GC, Ahmed SZ, Ding CZ, Dzwonczyk S, Ferrara FN, Humphreys WG, Grover GJ, Santafianos D, Atwal KS, Baird AJ, McLaughlin LG, Normandin DE, Sleph PG, Traeger SC. Cardioselective antiischemic ATP-sensitive potassium channel (KATP) openers. 5. Identification of 4-(N-aryl)-substituted benzopyran derivatives with high selectivity. Journal of Medicinal Chemistry. 1997;40:24–34. doi: 10.1021/jm9605905. [DOI] [PubMed] [Google Scholar]
- Saris NE, Kroner H. Regulation of Ca2+ fluxes in rat liver mitochondria by Ca2+. Effects on Ca2+ distribution. Journal of Bioenergetics and Biomembranes. 1990;22:81–90. doi: 10.1007/BF00762847. [DOI] [PubMed] [Google Scholar]
- Sasaki N, Sato T, Ohler A, O’Rourke B, Marban E. Activation of mitochondrial ATP-dependent potassium channels by nitric oxide. Circulation. 2000;101:439–445. doi: 10.1161/01.cir.101.4.439. [DOI] [PubMed] [Google Scholar]
- Sato T, O’Rourke B, Marban E. Modulation of mitochondrial ATP-dependent K+ channels by protein kinase C. Circulation Research. 1998;83:110–114. doi: 10.1161/01.res.83.1.110. [DOI] [PubMed] [Google Scholar]
- Sato T, Sasaki N, Seharaseyon J, O’Rourke B, Marban E. Selective pharmacological agents implicate mitochondrial but not sarcolemmal K(ATP) channels in ischemic cardioprotection. Circulation. 2000;101:2418–2423. doi: 10.1161/01.cir.101.20.2418. [DOI] [PubMed] [Google Scholar]
- Seharaseyon J, Ohler A, Sasaki N, Fraser H, Sato T, Johns DC, O’Rourke B, Marban E. Molecular composition of mitochondrial ATP-sensitive potassium channels probed by viral Kir gene transfer. Journal of Molecular and Cellular Cardiology. 2000;32 doi: 10.1006/jmcc.2000.1226. in the Press. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Tsujimoto Y. Proapoptotic BH3-only Bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proceedings of the National Academy of Sciences of the USA. 2000;97:577–582. doi: 10.1073/pnas.97.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemen D, Loupatatzis C, Borecky J, Gulbins E, Lang F. Ca2+-activated K channel of the BK-type in the inner mitochondrial membrane of a human glioma cell line. Biochemical and Biophysical Research Communications. 1999;257:549–554. doi: 10.1006/bbrc.1999.0496. [DOI] [PubMed] [Google Scholar]
- Sorgato MC, Keller BU, Stuhmer W. Patch-clamping of the inner mitochondrial membrane reveals a voltage- dependent ion channel. Nature. 1987;330:498–500. doi: 10.1038/330498a0. [DOI] [PubMed] [Google Scholar]
- Sorgato MC, Moran O. Channels in mitochondrial membranes: knowns, unknowns, and prospects for the future. Critical Reviews in Biochemistry and Molecular Biology. 1993;28:127–171. doi: 10.3109/10409239309086793. [DOI] [PubMed] [Google Scholar]
- Starkov AA. Mild’ uncoupling of mitochondria. Bioscience Reports. 1997;17:273–279. doi: 10.1023/a:1027380527769. [DOI] [PubMed] [Google Scholar]
- Stucki JW. The optimal efficiency and the economic degrees of coupling of oxidative phosphorylation. European Journal of Biochemistry. 1980;109:269–283. doi: 10.1111/j.1432-1033.1980.tb04792.x. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Kotake K, Fujikura K, Inagaki N, Suzuki T, Gonoi T, Seino S, Takata K. Kir6.1: a possible subunit of ATP-sensitive K+ channels in mitochondria. Biochemical and Biophysical Research Communications. 1997;241:693–697. doi: 10.1006/bbrc.1997.7891. [DOI] [PubMed] [Google Scholar]
- Szabo I, Zoratti M. The mitochondrial megachannel is the permeability transition pore. Journal of Bioenergetics and Biomembranes. 1992;24:111–117. doi: 10.1007/BF00769537. [DOI] [PubMed] [Google Scholar]
- Szewczyk A. The intracellular potassium and chloride channels: properties, pharmacology and function. Molecular Membrane Biology. 1998;15:49–58. doi: 10.3109/09687689809027518. [DOI] [PubMed] [Google Scholar]
- Szewczyk A, Mikolajek B, Pikula S, Nalecz MJ. Potassium channel openers induce mitochondrial matrix volume changes via activation of ATP-sensitive K+ channel. Polish Journal of Pharmacology. 1993;45:437–443. [PubMed] [Google Scholar]
- Szewczyk A, Pikula S, Wojcik G, Nalecz MJ. Glibenclamide inhibits mitochondrial K+ and Na+ uniports induced by magnesium depletion. International Journal of Biochemistry and Cell Biology. 1996;28:863–871. doi: 10.1016/1357-2725(96)00040-4. [DOI] [PubMed] [Google Scholar]
- Tanonaka K, Taguchi T, Koshimizu M, Ando T, Morinaka T, Yogo T, Konishi F, Takeo S. Role of an ATP-sensitive potassium channel opener, YM934, in mitochondrial energy production in ischemic/reperfused heart. Journal of Pharmacological and Experimental Therapeutics. 1999;291:710–716. [PubMed] [Google Scholar]
- Vanden Hoek TL, Becker LB, Shao Z, Li C, Schumacker PT. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. Journal of Biological Chemistry. 1998;273:18092–18098. doi: 10.1074/jbc.273.29.18092. [DOI] [PubMed] [Google Scholar]
- Vanden Hoek TL, Becker LB, Shao ZH, Li CQ, Schumacker PT. Preconditioning in cardiomyocytes protects by attenuating oxidant stress at reperfusion. Circulation Research. 2000;86:541–548. doi: 10.1161/01.res.86.5.541. [DOI] [PubMed] [Google Scholar]
- Vanden Heiden MG, Chandel NS, Li XX, Schumacker PT, Colombini M, Thompson CB. Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proceedings of the National Academy of Sciences of the USA. 2000;97:4666–4671. doi: 10.1073/pnas.090082297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyssokikh MY, Goncharova NY, Zhuravlyova AV, Zorova LD, Kirichenko VV, Krasnikov BF, Kuzminova AE, Melikov KC, Melik-Nubarov NS, Samsonov AV, Belousov VV, Prischepova AE, Zorov DB. Biochemistry. Vol. 64. Moscow: 1999. Proteinaceous complexes from mitochondrial contact sites; pp. 390–398. [PubMed] [Google Scholar]
- Wang Y, Ashraf M. Role of protein kinase C in mitochondrial KATP channel-mediated protection against Ca2+ overload injury in rat myocardium. Circulation Research. 1999;84:1156–1165. doi: 10.1161/01.res.84.10.1156. [DOI] [PubMed] [Google Scholar]
- Weiss JN, Lamp ST. Glycolysis preferentially inhibits ATP-sensitive K+ channels in isolated guinea pig cardiac myocytes. Science. 1987;238:67–69. doi: 10.1126/science.2443972. [DOI] [PubMed] [Google Scholar]
- Yao Z, Gross GJ. Effects of the KATPchannel opener bimakalim on coronary blood flow, monophasic action potential duration, and infarct size in dogs. Circulation. 1994;89:1769–1775. doi: 10.1161/01.cir.89.4.1769. [DOI] [PubMed] [Google Scholar]
- Yao Z, Tong J, Tan X, Li C, Shao Z, Kim WC, Vanden Hoek TL, Becker LB, Head CA, Schumacker PT. Role of reactive oxygen species in acetylcholine-induced preconditioning in cardiomyocytes. American Journal of Physiology. 1999;277:H2504–2509. doi: 10.1152/ajpheart.1999.277.6.H2504. [DOI] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, Paucek P, Jaburek M, Garlid KD. The nucleotide regulatory sites on the mitochondrial KATP channel face the cytosol. Biochimica et Biophysica Acta. 1997;1321:128–136. doi: 10.1016/s0005-2728(97)00051-0. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Matlib MA, Bers DM. Cytosolic and mitochondrial Ca2+ signals in patch clamped mammalian ventricular myocytes. The Journal of Physiology. 1998;507:379–403. doi: 10.1111/j.1469-7793.1998.379bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti M, Szabo I. Electrophysiology of the inner mitochondrial membrane. Journal of Bioenergetics and Biomembranes. 1994;26:543–553. doi: 10.1007/BF00762739. [DOI] [PubMed] [Google Scholar]
- Zorov DB, Kinnally KW, Tedeschi H. Voltage activation of heart inner mitochondrial membrane channels. Journal of Bioenergetics and Biomembranes. 1992;24:119–124. doi: 10.1007/BF00769538. [DOI] [PubMed] [Google Scholar]