

Abstract
The role of the sequence 1572-1651 in the C-terminal tail of the α1C subunit in run-down of Ca2+ channels was studied by comparing functional properties of the conventional α1C,77 channel with those of three isoforms carrying alterations in this motif.
The pore-forming α1C subunits were co-expressed with α2δ and β2a subunits in HEK-tsA201 cells, a subclone of the human embryonic kidney cell line, and studied by whole-cell and single-channel patch-clamp techniques.
Replacement of amino acids 1572-1651 in α1C,77 with 81 different amino acids leading to α1C,86 significantly altered run-down behaviour. Run-down of Ba2+ currents was rapid with α1C,77 channels, but was slow with α1C,86.
Transfer of the α1C,86 segments L (amino acids 1572-1598) or K (amino acids 1595-1652) into the α1C,77 channel yielded α1C,77L and α1C,77K channels, respectively, the run-down of which resembled more that of α1C,77. These results demonstrate that a large stretch of sequence between residues 1572 and 1652 of α1C,86 renders Ca2+ channels markedly resistant to run-down.
The protease inhibitor calpastatin added together with ATP was able to reverse the run-down of α1C,77 channels. Calpastatin expression was demonstrated in the HEK-tsA cells by Western blot analysis.
These results indicate a significant role of the C-terminal sequence 1572-1651 of the α1C subunit in run-down of L-type Ca2+ channels and suggest this sequence as a target site for a modulatory effect by endogenous calpastatin.
The voltage-gated L-type Ca2+ channel is an essential part of signal transduction systems in many cell types triggering essential processes ranging from muscle contraction (Fabiato & Fabiato, 1979; Rios & Brum, 1987) to gene expression (Sheng et al. 1990; Murphy et al. 1991; Deisseroth et al. 1996, 1998). In electrophysiological studies usually carried out by the patch-clamp technique, activity of L-type Ca2+ channels decreases when the cytoplasmic side of the channels is perfused with an artificial intracellular solution. This phenomenon is called run-down and is most pronounced in cells dialysed internally in whole-cell recording or inside-out patches (Hagiwara & Byerly, 1983; Kostyuk, 1984; McDonald et al. 1994). So far, several mechanisms have been suggested for run-down, including proteolysis (Chad & Eckert, 1985; Belles et al. 1988a; Romanin et al. 1991) and dephosphorylation (Kostyuk, 1984; Armstrong & Eckert, 1987; Ono & Fozzard, 1992; Costantin et al. 1999). Alternatively, it has been suggested that wash-out of a cytoplasmic factor is the cause of run-down (M. Kameyama et al. 1988; A. Kameyama et al. 1997). Recent studies indicate that this factor might be calpastatin (Romanin et al. 1991; M. Kameyama et al. 1998), an endogenous inhibitor of the protease calpain (Molinari & Carafoli, 1998). The actions of calpastatin appear, however, not to be mediated through inhibition of calpain, as run-down is both reversible and not affected by synthetic calpain inhibitors (Seydl et al. 1995). Run-down has also been observed with Ca2+ channel subunits heterologously expressed in oocyte (Costantin et al. 1999) and mammalian expression systems (Höfer et al. 1997). The Ca2+ channel is a heteromeric protein complex that occurs in several subunit combinations (Hofmann et al. 1994; Catterall, 1995). It is composed of the pore-forming α1C subunit and auxiliary β and α2δ subunits. The carboxyl terminus of the α1C subunit has attracted much attention (Schultz et al. 1993; Soldatov et al. 1997, 1998) because of its potential involvement in channel gating. Removal of approximately 70 % of the tail causes an increase in the opening probability of the rabbit cardiac α1C channel (Wei et al. 1994; Schmid et al. 1995) and accelerates inactivation of the human cardiac α1C as compared to the wild-type channel (Klöckner et al. 1995). Besides the carboxyl tail several structural domains may contribute to channel inactivation properties (e.g. Speatgens & Zamponi, 1999). Alternative splicing of the human α1C subunit generates multiple isoforms of the channel, including those with a structurally altered carboxyl terminal tail. Two human splice variants of the principal 2138 amino acid pore-forming α1C subunit, a ubiquitous isoform α1C,77 and a hippocampal isoform α1C,86, show differences in their carboxyl terminal tail (Soldatov, 1992, 1994). Due to alternative splicing of exons 40-42, the α1C,77 channel has 80 amino acid residues (1572-1651) in the second quarter of the 662 amino acid carboxyl tail replaced with 81 non-identical amino acids yielding the α1C,86 splice variant (Table 1). These two channel splice variants, when expressed in Xenopus oocytes, exhibit strong differences in inactivation properties (Soldatov et al. 1997). Whole-cell Ba2+ currents of the α1C,86 channel inactivate significantly faster than those through α1C,77. Furthermore, with Ca2+ as charge carrier, inactivation of the current through α1C,77 is greatly accelerated in contrast to the α1C,86 inactivation rate, which is essentially Ca2+ independent (Soldatov et al. 1997). Furthermore, we have recently reported (Kepplinger et al. 2000) that the sequence 1572-1651 in the carboxyl terminus is also important for targeting, conductance and open probability. We additionally observed (N. M. Soldatov, unpublished observations) a difference in the reduction of Ba2+ currents over time between α1C,77 and α1C,86 channels in the oocyte expression system suggesting distinct run-down properties.
Table 1.
Structure of the variable parts in the carboxyl terminal tail of α1C subunits under investigation
| α1C, 77 | IKTEGNLEQANEELRAIIKKIWKRTSMKLLDQVVPPAGDDEVTVGKFYATFL-IQEYFRKFKKRKEQGLVGKPSQRNALSL | (1572–1651) |
| α1C, 77L | ETELSSQVQYQAKEASLLERRRKSSHP | (1572–1598) |
| α1C,77K | SSHPKSSTKPNKLLSSGGSTGWVEDARALEGQVLARGCGWLGSLEERERGPHHPPLGF | (1595–1652) |
| α1C,86 | ETELSSQVQYQAKEASLLERRRKSSHPKSSTKPNKLLSSGGSTGWVEDARALEGQVLARGCGWLGSLEERERGPEHPPLGF | (1572–1652) |
Amino acid sequences of α1C,77 (1572–1651) and α1C,86 (1572–1652) are shown in the top and bottom rows, respectively. Indicated amino acids of α1C,86 replace the respective residues in the amino acid sequence of α1C,77. In α1C,77L and α1C,77K subunits, indicated segments of α1C,86 replace the respective motifs L (1572–1598) and K (1595–1651) of the α1C,77 subunit. Note that the overlapping 4 amino acid segment SSHP has been proven not to contribute to the kinetics, voltage or Ca2+ dependence of inactivation (Soldatov et al. 1998). Residues in bold are located in identical positions between α1C subunits.
In an attempt to analyse the role of amino acids 1572-1651 in the regulation of Ca2+ channel run-down, we studied functional properties of the α1C,77 and α1C,86 channels and its two sub-segmental mutants, α1C,77K and α1C,77L (Table 1, Soldatov et al. 1998) in the HEK-tsA201 mammalian expression system. The results of our study indicate that the sequence 1572-1651 in the carboxyl terminal tail of the α1C subunit determines not only channel inactivation but also run-down of L-type Ca2+ channels.
METHODS
Materials
Enhanced green fluorescent protein (EGFP) was purchased from Clontech (Heidelberg, Germany). The cDNA of the CD8 receptor (EBO pcD Leu2) was kindly provided by Richard Horn (Thomas Jefferson University Medical School, Philadelphia, PA, USA). Tissue culture media and reagents were purchased from Life Technology, Vienna, Austria. (-)BayK 8644 was from Research Biochemicals International, Vienna, Austria and all other chemicals from Sigma, Vienna, Austria. Magnetic beads carrying antibodies against the CD8 receptor were purchased from Dynal, Hamburg, Germany.
Molecular biology
Preparation of eukaryotic expression plasmids encoding α1C,77, α1C,86, α1C,77K and α1C,77L channels
All cDNAs of the human Ca2+ channel α1C subunit used for eukaryotic transfection were prepared in the pcDNA3 vector (Invitrogen, Carlsbad, CA, USA). To incorporate Kozak sequence, the 5′-terminal RT-PCR clone 5′(2)6 (nucleotides (nt) -51-440) (Soldatov, 1992), obtained by the rapid amplification of cDNA ends (RACE) method, was subcloned into the Bluescript SK(-) vector (Stratagene, La Jolla, CA, USA) at HindIII, EcoRI sites, digested with HindIII and NcoI and ligated (160 ng) with a mixture of 18.2 pmol each of oligonucleotides 5′-AGCTTGGATCCGCCAC-3′ and 5′-CATGGTGGCGGATCCA-3′ which had been previously phosphorylated with T4 polynucleotide kinase (New England Biolabs, Beverly, MA, USA) in the presence of 10 mm ATP. Incorporation of the Kozak consensus sequence (5′-CCGCCA-3′) preceding the initiation codon was confirmed by sequence analysis. This construct was digested with HindIII at the 5′-flanking non-coding region and with MunI (408) in an open reading frame (ORF), and the resulting 430 base pair fragment was ligated into the pHLCC77 at HindIII, MunI sites to give pHLCC109. The 3′-terminal region was engineered using the cDNA hybrid (3275-6519) constructed in pBluescript SK(-) from the h2.05 cDNA (nt 3275-5267), which was supplemented at the 3′-end with a nucleotide sequence from the RT-PCR clone 3′t-12 (nt 5705-6519) obtained by the 3′-RACE extension of the ORF (Soldatov, 1992). This hybrid was cut at the 3′-UTR region with HpaI (6515) and in the vector site with XbaI, the 3′-recessed ends were filled in using the Klenow fragment of DNA polymerase I (New England Biolabs) and ligated yielding pXIc. The 3′-terminal SfuI (3341), NotI fragment of pXIc was then ligated into pHLCC109 to replace the corresponding SfuI/NotI fragment in pHLCC77B with modified 5′ and 3′ ends. Finally, the 5′→ 3′HindIII, NotI cassette of pHLCC77B was subcloned into the pcDNA3 eukaryotic expression vector (Invitrogen). The integrity of the ORF was verified by sequencing.
86pcDNA3 was prepared by replacing the SfuI (3341)/AatII (5494) fragment of pHLCC77B with the corresponding fragment of pHLCC86 (Soldatov et al. 1997) and subcloning the HindIII, NotI cassette of the obtained pHLCC86B construct into the pcDNA3 vector. To prepare 77KpcDNA3 and 77LpcDNA3, the pHLCC77K and pHLCCL plasmids (Soldatov et al. 1998), respectively, were digested with BamHI, blunt-ended using the Klenow DNA polymerase, digested with PpuMI (2760) and the resulting 3′-terminal 3.9 kb fragments were ligated into 77pcDNA3 to replace the corresponding fragment in the PpuMI (2760)/NotI (blunt-ended) cassette.
Nucleotide sequences of all PCR products, as well as ligation sites were verified using the ABI Prism dye terminator cycle sequencing kit with AmpliTaq DNA polymerase (Perkin-Elmer, Norwalk, USA).
Cell culture and transfection of HEK-tsA201 cells
HEK-tsA201 cells (kindly provided by Richard Horn, Thomas Jefferson University Medical School, Philadelphia, USA) were cultured in Dulbecco’s modified Eagle’s medium supplemented with streptomycin (100 μg ml−1), penicillin (100 U ml−1) and 10 % fetal calf serum in a humidified atmosphere (95 %) at 5 % CO2 and 37°C. Cells were used for 12-14 passages and were transferred every 4 days. Transfection was performed using SuperFect (Qiagen, Hilden, Germany). In brief, cells exhibiting confluence of about 30-50 % were transfected with 2.5 μg of total cDNA (molar ratio of α1C:β2a:α2δ:EGFP:CD8 of 1:1.6:1.4:2.5:0.7). Transfection efficiency was estimated by counting cells showing EGFP fluorescence, and was in the range 20-60 %. Furthermore, co-expression of the CD8 receptor and binding of CD8 antibody-coated beads (Dynal) was used as a visual marker to identify cells for electrophysiological experiments. Binding of the beads (range 3-15 per cell) was estimated to occur in 1-5 % of the total number of cells.
Preparation of a supernatant of HEK-tsA201 cells
The HEK-tsA201 cell pellet obtained by centrifugation at 1000 g (10 min, 4°C) was washed three times with PBS and finally resuspended in 0.4 M NaCl, 25 % (v/v) glycerol, 1 mm EDTA, 0.5 mm DTT, protease inhibitors (×1000 of benzamidine, phenylmethylsulfonyl fluoride (PMSF), pepstatin) and 20 mm Hepes-Na (pH 7.9). Then the cells were lysed by freezing (liquid N2, 1 min) and thawing (ice, 30 min). Cell debris and insoluble components were removed by centrifugation at 12 000 g (15 min, 4°C) yielding the 12 000 g supernatant.
Immunoblot procedures
Western Blot was carried out according to Towbin et al. (1979) using a monoclonal antibody against human calpastatin (Takara Shuzo Co., Ltd, Japan; Clone CSL 1-5, Yokota et al. 1991). This antibody, which recognizes domain III of calpastatin, was generated in mouse, and its sensitivity is 10 pmol l−1 according to Yokota et al. 1991. Sigma calpastatin (55 μg) or the 12 000 g supernatant (100 μg) was separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) on the gel and blotted onto a nitrocellulose membrane. Calpastatin from Sigma appears to be a crude preparation as observed in Coomassie Blue staining of the SDS gel. The membrane was then incubated with anti-calpastatin antibody, followed by incubation with peroxidase-conjugated rabbit anti-mouse IgG antibody (Sigma, Austria) and visualized using the Enhanced Chemiluminescence detection kit (Amersham, Austria). Control experiments have been carefully performed in the sole presence of the primary or secondary antibody and indicated clear specificity of the anti-calpastatin antibody.
Electrophysiology
Both whole-cell and single-channel patch-clamp recordings (Hamill et al. 1981) were obtained from successfully transfected HEK-tsA201 cells employing a List L/M EPC 7 amplifier.
Whole-cell recordings
The pipette solution contained (mm): caesium methane sulfonate 120, CaCl2 5, MgCl2 2, Hepes 10, EGTA 10, MgATP 2; pH (CsOH) 7.30. The bath solution consisted of (mm): N-methyl glucamine 154, MgCl2 1, D-glucose monohydrate 5, Hepes 10, 4-aminopyridine 5, BaCl2 15; pH (HCl) 7.40. Soft glass pipettes (microhaematocrit tubes, No. 564, Fa. Assistant, Vienna, Austria) with a resistance of 2-4 MΩ were used for whole-cell recordings. Ba2+ currents were activated by repetitive (0.2 Hz) depolarizations from a holding potential of -80 mV to test potentials (0.244 s) between -10 and +60 mV with an incremental increase of 5 or 10 mV. Current traces were filtered at 3 kHz, digitized at 8 kHz and were neither capacity nor leak current corrected in order to verify the quality of the voltage clamp. A liquid junction potential of 6 mV was determined but not taken into account. This value should be subtracted from all voltages in whole-cell recordings (Neher, 1992).
Single-channel recordings
Ba2+ currents through single Ca2+ channels were recorded in the cell-attached and inside-out configuration (Höfer et al. 1997). Cell potentials were set to approximately 0 mV by the use of a high K+, low Cl− extracellular solution that contained (mm): L-aspartic acid 110, KCl 20, MgCl2 2, Hepes 20, EGTA 2; pH (KOH) 7.35. The pipette solution consisted of (mm): BaCl2 96, Hepes 5; pH (NaOH) 7.40. The dihydropyridine Ca2+ channel activator (-)BayK 8644 (2.5 μm) was included in the pipette solution to facilitate channel activity in the cell-attached and inside-out configurations. Pipettes (GC150F-7.5F) were fabricated from borosilicate glass (Clark Medical Instruments, Pangbourne, UK) and had resistances of 4-6 MΩ. Sigmacote was used to reduce pipette capacitance. Single-channel currents of the channels were evoked by repetitive depolarizations (0.66 Hz) applied for 0.487 s from a holding potential of -80 mV to 0 mV (α1C,77) and 10 mV (α1C,86, α1C,77K and α1C,77L). Single-channel traces were filtered at 1 kHz and digitized at 4 kHz. To recover channel activity from the usual run-down in inside-out patches, 2 mm Na2ATP and 2 U ml−1 calpastatin (P-0787, Sigma, Munich, Germany) were added to the bath solution following patch excision. All experiments were performed at room temperature.
Analysis of electrophysiological data
Estimation of single Ca2+ channel activity was primarily based on the determination of the time course of mean channel activity NPo (N is the total number of channels, Po is the open probability; Schmid et al. 1995) determined for each depolarizing voltage pulse defined as a sweep. For this, single-channel sweeps are idealized using the 50 % threshold method. The mean current I during one sweep of depolarization is determined and divided by the unitary current amplitude i to yield NPo.
Statistics
Results are presented as means ±s.e.m. for the number of experiments usually given in parentheses. Student’s two-tailed t test was used for statistical comparison considering differences statistically significant at P < 0.05.
RESULTS
Two human neuronal splice variants of α1C, α1C,77 and α1C,86 (see Table 1), were transiently expressed together with β2a and α2δ subunits in HEK-tsA201 cells. Electrophysiological characteristics of the whole-cell Ba2+ currents were initially determined, and the study was then extended to single-channel currents, which allowed for a further characterization of run-down, potentially determined by the carboxyl tail sequence 1572-1651. To narrow structures within this sequence, the two segmental mutants α1C,77L and α1C,77K were additionally studied in which segments of 27 (L) and 58 (K) amino acids, respectively, of α1C,86 replace the corresponding residues in the 80 amino acid sequence of α1C,77 (Table 1).
Whole-cell characteristics of the α1C,77, α1C,86, α1C,77K and α1C,77L channels
Figure 1 depicts an overview of whole-cell currents recorded with 15 mm Ba2+ as charge carrier. Representative current traces obtained by depolarizing voltage pulses between -5 and +55 mV for α1C,77, α1C,86, α1C,77K and α1C,77L are shown in Fig. 1A, B, E and F, respectively. Ba2+ currents through the α1C,77 channel exhibited a remarkably slower inactivation compared to those of the α1C,86, α1C,77K and α1C,77L channels. The corresponding current-voltage relationships (Fig. 1C and G) further revealed that the maximum of activation was shifted by ∼10 mV to more positive values for α1C,86, α1C,77K as well as α1C,77L channels culminating at about 30-35 mV. Inactivation of Ba2+ currents (at 25 mV) through the α1C,77 channel was better fitted monoexponentially (τ= 412 ± 64 ms, n = 8), while that through α1C,86 exhibited a biexponential decay rate (τ1 = 35 ± 12 ms; τ2 = 216 ± 72, n = 6; Fig. 1D). Similar to α1C,86, both α1C,77K and α1C,77L channels exhibited rapid inactivation, where inactivation of both currents was best fitted by two exponentials (α1C,77K: τ1 = 10 ± 3 ms, τ2 = 60 ± 10 ms, n = 5; α1C,77L: τ1 = 25 ± 8 ms, τ2 = 113 ± 32 ms, n = 4; Fig. 1H) and was slightly faster compared to that of the α1C,86 channel. These main differences between α1C,77 and α1C,86, α1C,77K as well as α1C,77L channels are consistent with those found in the oocyte expression system (Soldatov et al. 1997, 1998).
Figure 1. Whole-cell characteristics of the α1C,77, α1C,86, α1C,77K and α1C,77L channels.
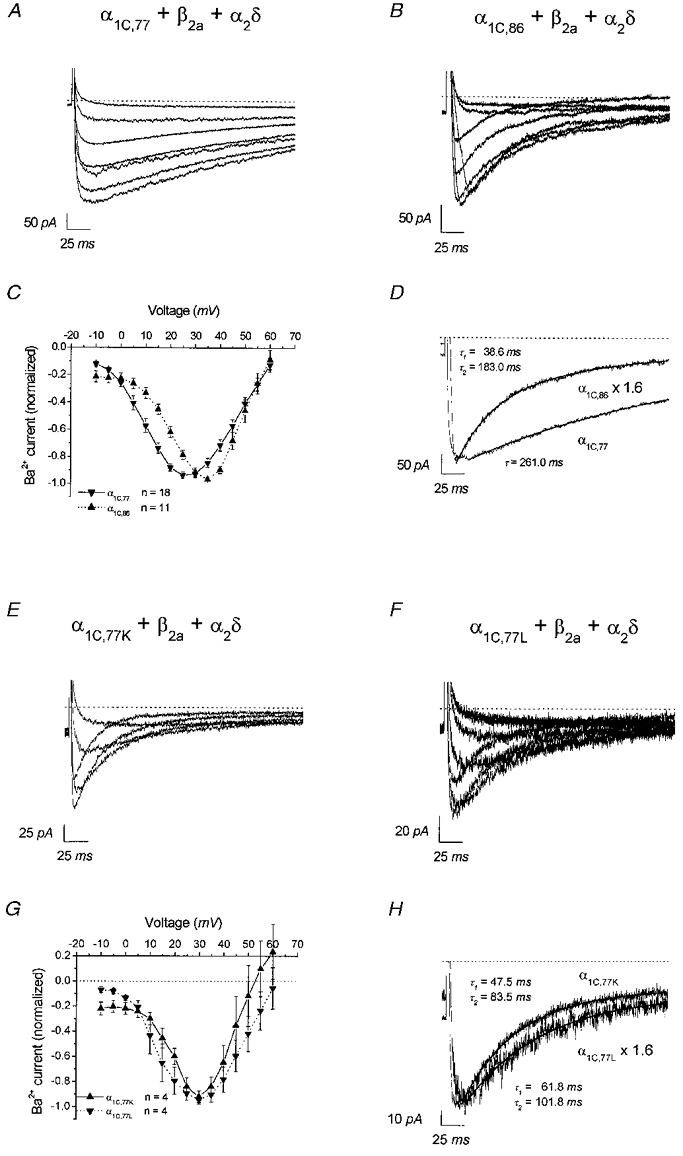
Representative whole-cell current traces with 15 mm Ba2+ as charge carrier obtained with α1C,77 (A), α1C,86 (B), α1C,77K (E) and α1C,77L (F) channels by repetitive (0.2 Hz) depolarizations from a holding potential of -80 mV to test potentials between -5 and 55 mV with an incremental increase of 10 mV. C and G, corresponding averaged current-voltage relationships derived from the indicated number of experiments, each normalized to maximum current. D and H, whole-cell traces recorded from α1C,77 and α1C,86 (D) as well as α1C,77K and α1C,77L channels (H) at a test potential of 25 mV were scaled with respect to the peak current size. Current decays were fitted by mono-exponential (α1C,77) and double-exponential (α1C,86, α1C,77K and α1C,77L) functions yielding the indicated time constants. Dotted lines in A and B, and D–H denote zero current level.
Run-down of whole-cell and single-channel currents of the α1C,77 and α1C,86 channels
In the course of whole-cell experiments, it appeared that the run-down behaviour of these channels was different (Fig. 2). The α1C,77 channel exhibited a remarkably faster run-down compared to the α1C,86 subunit. The peak current of α1C,77 declined to about 32 ± 8 % (n = 8) within 5 min in contrast to 78 ± 13 % (n = 7) that remained during the same time period with α1C,86 (Fig. 2A, P < 0.01 at 4 min). Consistently, in double-electrode voltage-clamp experiments on oocytes the α1C,86 channel showed a 3 ± 3 % (n = 3) decrease in IBa after 27-39 min compared to 27 ± 10 % (n = 4) of the α1C,77 isoform after 22-38 min (N. M. Soldatov, unpublished observations). Run-down was characterized by a decrease of the peak current, whereas the rate of inactivation of whole-cell currents was not changed during run-down in either the α1C,77 or α1C,86 channel (Fig. 2B). Furthermore, the amount of run-down was not correlated with the size of the initial peak current for either channel (Fig. 2C), indicating that a smaller peak current did not necessarily correlate with a slower run-down.
Figure 2. Time dependence of whole-cell Ba2+ currents through α1C,77 and α1C,86 channels.
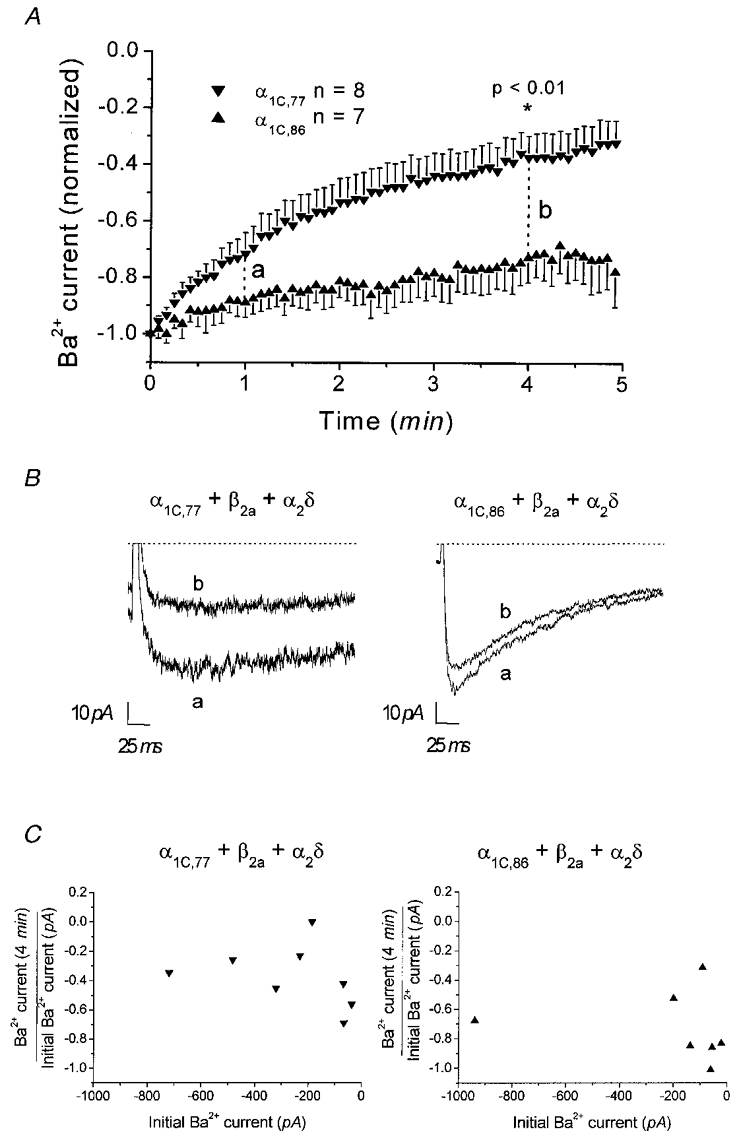
A, peak currents generated by repetitive depolarizations to 25 mV (α1C,77) and 35 mV (α1C,86). Comparison of currents at 4 min indicated a significant (P < 0.01) difference in current size between the α1C,77 and α1C,86 channels. B, representative current traces recorded for α1C,77 and α1C,86 channels at 1 and 4 min as indicated in A. Dotted lines denote zero current level. C, the initial current (0 min) measured in each individual experiment for the respective channel is plotted against the normalized current reached after 4 min.
The difference in run-down behaviour between the α1C,77 and α1C,86 channels was also visible in single-channel experiments following the formation of an inside-out patch (Fig. 3). Ca2+ channel activity (NPo) of the α1C,77 channel decreased rapidly within 1 min (Fig. 3A). In contrast, the single-channel activity of α1C,86 remained largely unchanged for 3 min after patch excision and subsequently declined slowly (Fig. 3B). Corresponding single-channel traces for α1C,77 and α1C,86 are shown in Fig. 3C and D, respectively. The inset in Fig. 3A depicts the mean run-down behaviour of α1C,77 and α1C,86 channels yielding Ca2+ channel activities normalized to those of the cell-attached patch of 3.1 ± 0.4 % (n = 12) and 65.0 ± 19.1 % (n = 6), respectively, when determined within 1.5-2.5 min after patch excision. Analysis of channel activity at a later time point (4.5-5.5 min) revealed 0 % (n = 12) of α1C,77, whereas 6.1 ± 3.3 % (n = 5) of α1C,86 activity was still left (not shown).
Figure 3. Time dependence of single-channel currents of α1C,77 and α1C,86 channels in cell-attached and inside-out patch configurations.
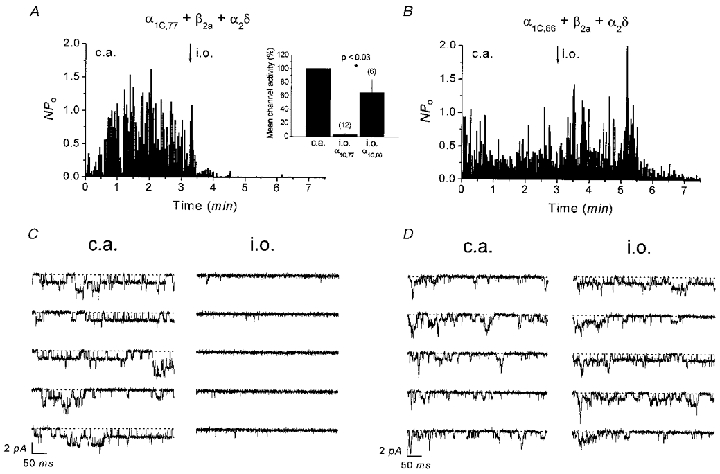
Average channel activity (NPo) of α1C,77 (A) and α1C,86 channels (B) recorded in the cell-attached (c.a.) patch followed by inside-out (i.o.) patch configuration. Inset in A shows mean channel activity reached 1.5-2.5 min after patch excision normalized to the previous activity in the cell-attached patch. Consecutive single-channel traces corresponding to the experiments in A and B are depicted in C and D both in the cell-attached and inside-out patch. Dotted lines indicate zero current level.
Run-down of whole-cell and single-channel currents of α1C,77L and α1C,77K
To further pinpoint the molecular determinants for run-down within the sequence 1572-1651, we measured decline of the peak current over time in whole-cell recordings of α1C,77K and α1C,77L (Fig. 4A, see Table 1). Within 4 min in the whole-cell configuration, peak currents of both channels decreased to about half or even less of their initial value (α1C,77K: 53.2 ± 1.9 %, n = 5; α1C,77L: 36.1 ± 16.8, n = 3) with no substantial change in their inactivation kinetics (Fig. 4B). Consistently, single Ca2+ channel activity of both α1C,77K (Fig. 5A) and α1C,77L (Fig. 5B) mutants exhibited a significant run-down following inside-out patch formation leading to a decrease in the mean channel activity to 23.0 ± 6.2 % (n = 6) and 8.4 ± 4.7 % (n = 12), respectively (Fig. 5A, inset), within 1.5-2.5 min after patch excision. At a later time point (4.5-5.5 min), almost no activity was visible for either α1C,77K (0.6 ± 0.3 %, n = 6) or α1C,77L (0.7 ± 0.5 %, n = 6) channels. Thus, these results indicate that the run-down behaviour of the α1C,77K and α1C,77L channels resembled more that of the α1C,77 channel in both whole-cell and single-channel experiments.
Figure 4. Time dependence of whole-cell currents of α1C,77K and α1C,77L channels.
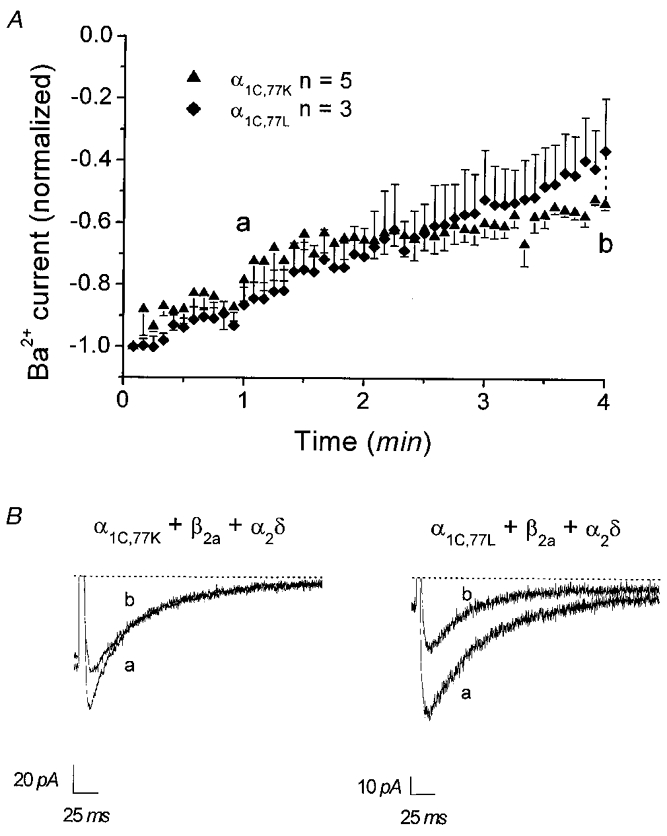
A, peak currents normalized to initial currents are shown as dependent on time for α1C,77K and α1C,77L channels recorded at test potentials of 35 mV. Comparison of currents at 4 min indicated no significant (P > 0.05) difference in current size between the α1C,77K and α1C,77L channels. B, representative current traces recorded for α1C,77K and α1C,77L channels at 1 and 4 min as indicated in A. Dotted lines denote zero current level.
Figure 5. Time dependence of single-channel Ba2+ currents through α1C,77K and α1C,77L channels in cell-attached and inside-out patch configurations.
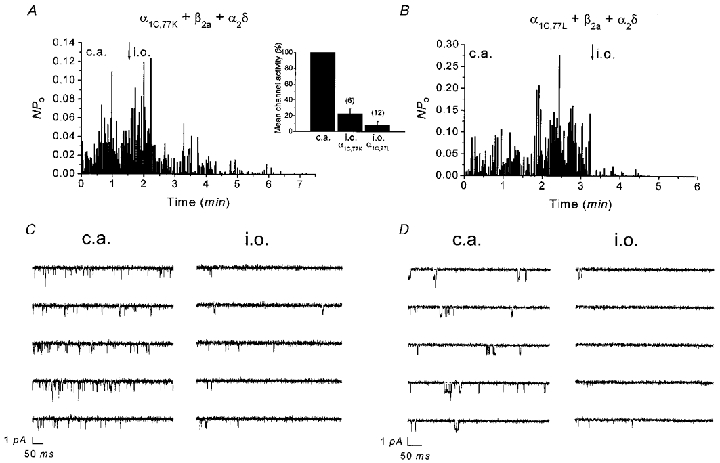
Average channel activity (NPo) of α1C,77K (A) and α1C,77L (B) channels recorded in the cell-attached (c.a.) patch followed by inside-out (i.o.) patch configuration. Inset in A shows mean channel activity reached 1.5-2.5 min after patch excision normalized to the preceding activity in the cell-attached patch. Consecutive single-channel traces corresponding to the experiments in A and B are depicted in C and D both in the cell-attached and inside-out patch. Single channel openings are visible as downward deflections.
Effect of calpastatin on single Ca2+ channel activity of α1C,77, α1C,77L and α1C,86 in the inside-out patch
Previous experiments have shown that run-down of L-type Ca2+ channels is reversed by the addition of calpastatin plus ATP to the intracellular face of the inside-out patch (Romanin et al. 1991; Seydl et al. 1995; A. Kameyama et al. 1998; M. Kameyama et al. 1998). Here we investigated the effect of calpastatin when applied to the α1C,77 and α1C,86 channels (Fig. 6). The complete run-down of the α1C,77 channel in the inside-out patch was largely reversed by addition of calpastatin plus ATP (Fig. 6A), excluding proteolysis as a mechanism of run-down. A qualitatively similar result was obtained with the α1C,77L channel (data not shown). The α1C,86 channel showed, as demonstrated before, almost no run-down within 1.5 min following inside-out patch formation and the remaining channel activity was only slightly increased by calpastatin + ATP (Fig. 6B). Single-channel traces corresponding to α1C,77 and α1C,86 channel experiments are shown in Fig. 6C and D, respectively. Figure 6E illustrates that 56.7 ± 9.3 % (n = 9) of the previously observed cell-attached α1C,77 channel activity recovered by the addition of calpastatin + ATP to the inside-out patch, while the activity of the α1C,86 channel reached 97.3 ± 14.0 % (n = 3) under the same conditions.
Figure 6. Effect of calpastatin + ATP on the activity of single α1C,77 and α1C,86 channels in the inside-out patch.
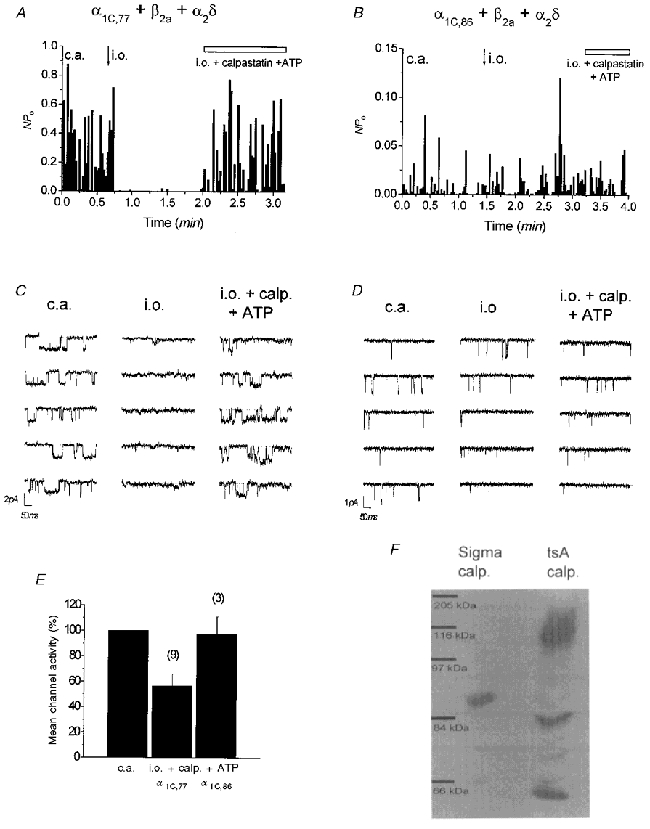
Time course of average channel activity (NPo) of α1C,77 (A) and α1C,86 (B) in the cell-attached (c.a.) and inside-out (i.o.) patch before and after addition of calpastatin (2 U ml−1) + Na2ATP (1 mm). Single-channel activity of α1C,77 almost completely recovered from run-down in the inside-out (i.o.) patch, whereas almost no effect was observed on channel activity of α1C,86, which was resistant to run-down in the i.o. patch. C and D depict single-channel traces for the corresponding experiments in A and B, recorded under the indicated conditions. Single channel openings are visible as downward deflections. E, mean channel activity of α1C,77 and α1C,86 in the presence of calpastatin + ATP, normalized to the preceding channel activity in the cell-attached configuration. F, representative Western blot analysis (n = 4) of Sigma calpastatin and HEK-tsA201 cell 12000 g supernatant using an anti-calpastatin antibody.
To test whether calpastatin might be involved as an endogenous regulator of L-type Ca2+ channel activity in HEK-tsA201 cells, we examined its presence in these cells using an anti-calpastatin antibody. Figure 6F presents immunoblots of Sigma rabbit skeletal muscle calpastatin used in the above experiments and of a 12000 g supernatant prepared from HEK-tsA201 cells. Both lanes show the presence of specific bands indicative of calpastatin. Skeletal muscle calpastatin exhibited a relative molecular weight of 88 kDa. HEK-tsA201 calpastatin appeared at molecular sizes of 116, 84 and 64 kDa. The occurrence of calpastatin in various sizes from different tissues has been described (Molinari & Carafoli, 1997) as well as their efficacy in recovery of Ca2+ channels from run-down (M. Kameyama et al. 1998).
In summary, the run-down behaviour of the segmental mutants α1C,77K and α1C,77L resembled more that of the α1C,77 channel, whereas inactivation properties of these mutated channels were similar to that of the α1C,86 channel. Recovery from Ca2+ channel run-down was obtained with calpastatin, the natural occurrence of which was also confirmed in HEK-tsA201 cells.
DISCUSSION
In this study, we characterized the functional expression of the human L-type Ca2+ channel splice variants α1C,77 and α1C,86 as well as their segmental mutants α1C,77K and α1C,77L in the mammalian cell line HEK-tsA201. Our results strongly suggest that the sequence 1572-1651 in the carboxyl terminal tail of α1C is important for the run-down phenomenon of L-type Ca2+ channels.
Electrophysiological features of α1C splice variants and segmental mutants in whole-cell and single-channel experiments
The α1C,77 channel exhibited a significantly slower inactivation than α1C,86 and the two segmental mutants α1C,77K and α1C,77L in both whole-cell and single-channel experiments. These data substantiated the role of the sequence 1572-1651 and of its L and K segments (see Table 1) as molecular determinants of voltage-dependent inactivation (Soldatov et al. 1998).
Molecular determinants of run-down
Conventional L-type Ca2+ channels are subject to run-down in both whole-cell (Belles et al. 1988b) and inside-out configurations (Cavalie et al. 1983; Romanin et al. 1991). However, L-type Ca2+ channels have been found in hippocampal neurones (Kavalali & Plummer, 1997), which show almost no run-down under cell-free conditions. Similarly, anomalous L-type channels in motoneurones are resistant to run-down (Hivert et al. 1999). Run-down has been reported to occur on the α1C subunit (Hao et al. 1998; Costantin et al. 1999) suggesting that the site of regulation that is the cause of run-down is located on the pore-forming subunit itself. In accordance, the results presented here imply that the sequence 1572-1651 in the carboxyl terminal tail of the α1C subunit is one molecular determinant of run-down. The whole-cell experiments performed revealed an essentially slower run-down of the α1C,86 channel compared to the conventional α1C,77. As peak currents of α1C,86 were usually smaller than those observed with α1C,77, a faster run-down might be correlated with larger currents independent of the structural difference between α1C,77 and α1C,86. However, no correlation was found between the current size and the amount of run-down for α1C,77 and α1C,86 channels. Consistently, α1C,77 and α1C,86 channels exhibited a significantly different run-down in oocytes. The whole-cell data were substantiated by single-channel experiments in which IBa through α1C,86 showed a significantly slowed run-down within 1.5-2.5 min following patch excision, in contrast to a rapid run-down of α1C,77 within the same time period. As run-down was not completely abolished, other structures within the α1C subunit might additionally contribute to this process. The qualitatively similar results obtained in whole-cell and single-channel configurations indicated that the presence of the Ca2+ agonist (-)BayK 8644 in single-channel experiments exerted no main effect on run-down behaviour. In both whole-cell and single-channel experiments the segmental mutants α1C,77K and α1C,77L showed a similar and substantial run-down which resembled more that of the α1C,77 channel, indicating that a large stretch of the 81 amino acid sequence of the α1C,86 subunit is required to markedly reduce run-down of L-type Ca2+ channels. As this sequence also determines channel inactivation (Soldatov et al. 1997, 1998), both channel properties may be interdependent. However, the segmental mutants in contrast to the α1C,86 channel displayed a fast run-down, though they all showed rapid Ba2+ current inactivation. In addition, the inactivation rate did not change during run-down. Thus, the mechanism of run-down seems to be different from that governing channel inactivation.
Mechanism of run-down
Dephosphorylation and wash-out of a regulatory factor have been suggested as prime mechanisms responsible for channel run-down. Reversal of run-down by protein kinase A (PKA) is controversial. While Ono & Fozzard (1992) reported a clear recovery from run-down, Costantin et al. (1999) observed an effect only in a subset of patches and Yazawa et al. (1997) found a recovery of channel activity essentially independent of PKA. The functionally important PKA phosphorylation site (De Jongh et al. 1996; Gao et al. 1997) is not present within the sequence 1572-1651 in the α1C,77 channel, whereas a putative motif (R/KRXS) is found in the α1C,86 channel within the L segment (amino acids 1592-1595). However, a role of this putative PKA site in the sensitivity of Ca2+ channels to run-down is rather unlikely as the α1C,77L contains this motif and showed a rapid run-down similar to the α1C,77 channel.
Wash-out of a regulatory factor has been suggested as the second mechanism of run-down. It has been reported that the ubiquitous protein calpastatin is an important cytoplasmic factor regulating L-type Ca2+ channel activity (Romanin et al. 1991; Seydl et al. 1995; M. Kameyama et al. 1998; Hao et al. 1999). Calpastatin has been also detected in oocytes (Lorca et al. 1991), and as shown here in HEK-tsA201 cells, where overexpression of calpastatin was found to increase L-type Ca2+ channel activity (K. Leitner & C. Romanin, unpublished observations). Indeed, the activity of the α1C,77 channel subjected to run-down following inside-out patch formation was recovered by addition of calpastatin + ATP to the intracellular face of the membrane. Disturbances of the calpain-calpastatin system have been claimed to be related to a number of pathological conditions such as cardiac ischaemia, stroke and brain trauma (Molinari & Carafoli, 1997), suggesting a physiological role of endogenous calpastatin in the regulation of Ca2+ channel activity.
Run-down has been reported to occur without changes in the gating currents (Josephson & Varadi, 1996; Costantin et al. 1999) suggesting a disruption of the linkage between the voltage sensor and the opening of the ionic gate (Costantin et al. 1999). Thus it is tempting to speculate that the sequence 1572-1651 that determines run-down properties represents the target site for the modulatory effects of calpastatin + ATP, which then restore coupling of the ionic gate to the voltage sensor, resulting in channel opening upon depolarization. A calmodulin-binding IQ region (1624-1635) within the sequence 1572-1651 has been recently reported as a critical site for Ca2+-induced inactivation (Peterson et al. 1999; Qin et al. 1999; Zühlke et al. 1999). Hence, a cross-talk between Ca2+ channel regulation by calpastatin + ATP and Ca2+ appears possible and remains to be investigated in future studies.
In summary, the amino acid sequence 1572-1651 in the carboxyl terminal tail of the α1C subunit is critical for channel inactivation, and independently represents an important structure for L-type Ca2+ channel run-down.
Acknowledgments
We thank N. Klugbauer, F. Hofmann (Munich) and V. Flockerzi (Heidelberg) for the gifts of clones of β2a and α2δ subunits. This work was supported by the Austrian Science Foundation P12803 to T.S., P12728 to C.R., SFB Biomembranes F715 and P12667 to K.G., NB 7000 to C.R. and AHA to N.M.S. We wish to thank Badia AlBanna, Sabine Buchegger, Ingrid Gegenleitner and Bettina Kenda for their excellent technical assistance.
References
- Armstrong DL, Eckert R. Voltage-dependent Ca2+ channels that must be phosphorylated to respond to membrane depolarization. Proceedings of the National Academy of Sciences of the USA. 1987;84:2518–2522. doi: 10.1073/pnas.84.8.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belles B, Hescheler J, Trautwein W, Blomgren K, Karlsson JO. A possible physiological role of the Ca-dependent protease calpain and its inhibitor calpastatin on the Ca2+ current in guinea pig myocytes. Pflügers Archiv. 1988a;412:554–556. doi: 10.1007/BF00582548. [DOI] [PubMed] [Google Scholar]
- Belles B, Malecot CO, Hescheler J, Trautwein W. Run-down’ of the Ca current during whole-cell recordings in guinea pig heart cells: role of phosporylation and intracellular Ca2+ Pflügers Archiv. 1988b;411:353–360. doi: 10.1007/BF00587713. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and function of voltage-gated ion channels. Annual Review of Biochemistry. 1995;64:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- Cavalie A, Ochi R, Pelzer D, Trautwein W. Elementary currents through Ca2+ channels in guinea pig myocytes. Pflügers Archiv. 1983;398:284–297. doi: 10.1007/BF00657238. [DOI] [PubMed] [Google Scholar]
- Chad JE, Eckert R. An enzymatic mechanism for Ca2+ current inactivation in dialysed Helix neurons. The Journal of Physiology. 1985;378:31–51. doi: 10.1113/jphysiol.1986.sp016206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantin JL, Qin N, Waxham MN, Birnbaumer L, Stefani E. Complete reversal of run-down in rabbit cardiac Ca2+ channels by patch-cramming in Xenopus oocytes; partial reversal by protein kinase A. Pflügers Archiv. 1999;437:888–894. doi: 10.1007/s004240050859. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- De Jongh KS, Murphy BJ, Colvin AA, Hell JW, Takahashi M, Catterall WA. Specific phosphorylation of a site in the full-length form of the α1 subunit of the cardiac L-type Ca2+ channel by adenosine 3′,5′-cyclic monophosphate-dependent protein kinase. Biochemistry. 1996;35:10392–10402. doi: 10.1021/bi953023c. [DOI] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Ca2+ and cardiac excitation-contraction coupling. Annual Review of Physiology. 1979;41:473–484. doi: 10.1146/annurev.ph.41.030179.002353. [DOI] [PubMed] [Google Scholar]
- Gao T, Yatani A, Dell’Acqua ML, Sako H, Green SA, Dascal N, Scott JD, Hosey MM. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron. 1997;19:185–196. doi: 10.1016/s0896-6273(00)80358-x. [DOI] [PubMed] [Google Scholar]
- Hagiwara S, Byerly L. The Ca2+ channel. Trends in Neurosciences. 1983;6:189–193. [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch clamp techniques for high resolution current recording from cells and cell free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hao L-Y, Kameyama A, Kameyama M. A cytoplasmic factor, calpastatin and ATP together reverse run-down of Ca2+ channel activity in guinea-pig heart. The Journal of Physiology. 1999;514:687–699. doi: 10.1111/j.1469-7793.1999.687ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao L-Y, Kameyama A, Kuroki S, Nishimura S, Kameyama M. Run-down of L-type Ca2+ channels occurs on the α1 subunit. Biochemical and Biophysical Research Communications. 1998;247:844–850. doi: 10.1006/bbrc.1998.8886. [DOI] [PubMed] [Google Scholar]
- Hivert B, Luvisetto S, Navangione A, Tottene A, Pietrobon D. Anomalous L-type Ca2+ channels of rat spinal motoneurones. Journal of General Physiology. 1999;113:679–693. doi: 10.1085/jgp.113.5.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höfer GF, Hohentanner K, Baumgartner W, Groschner K, Klugbauer N, Hofmann F, Romanin C. Intracellular Ca2+ inactivates L-type Ca2+ channels with a Hill coefficient of ∼1 and an inhibition constant of ∼4 μM by reducing channel’s open probability. Biophysical Journal. 1997;73:1857–1865. doi: 10.1016/S0006-3495(97)78216-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann F, Biel M, Flockerzi V. Molecular basis for Ca2+ channel diversity. Annual Review of Neuroscience. 1994;17:399–418. doi: 10.1146/annurev.ne.17.030194.002151. [DOI] [PubMed] [Google Scholar]
- Josephson IR, Varadi G. The beta subunit increases Ca2+ currents and gating charge movements of human cardiac L-type Ca2+ channels. Biophysical Journal. 1996;70:1285–1292. doi: 10.1016/S0006-3495(96)79685-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameyama A, Hao L-Y, Takano E, Kameyama M. Characterization and partial purification of the cytoplasmic factor that maintains cardiac Ca2+ channel activity. Pflügers Archiv. 1998;435:338–343. doi: 10.1007/s004240050520. [DOI] [PubMed] [Google Scholar]
- Kameyama A, Yazawa K, Kaibara M, Onzo K, Kameyama M. Run-down of the cardiac Ca2+ channel: Characterization and restoration of channel activity by cytoplasmic factors. Pflügers Archiv. 1997;433:547–556. doi: 10.1007/s004240050313. [DOI] [PubMed] [Google Scholar]
- Kameyama M, Kameyama A, Nakayama T, Kaibara M. Tissue extract recovers cardiac Ca2+ channels from ‘run-down. Pflügers Archiv. 1988;412:328–330. doi: 10.1007/BF00582516. [DOI] [PubMed] [Google Scholar]
- Kameyama M, Kameyama A, Takano E, Maki M. Run-down of the cardiac L-type Ca2+ channel: partial restoration of channel activity in cell-free patches by calpastatin. Pflügers Archiv. 1998;435:344–349. doi: 10.1007/s004240050521. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Hwang KS, Plummer MR. cAMP-dependent enhancement of dihydropyridine-sensitive Ca2+ channel availability in hippocampal neurons. Journal of Neuroscience. 1997;17:5334–5348. doi: 10.1523/JNEUROSCI.17-14-05334.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepplinger KJF, Kahr H, Förstner G, Sonnleitner M, Schindler H, Schmidt T, Groschner K, Soldatov NM, Romanin C. A sequence in the carboxy-terminus of the α1C subunit important for targeting, conductance and open probability of L-type Ca2+ channels. FEBS Letters. 2000;477:161–169. doi: 10.1016/s0014-5793(00)01791-9. [DOI] [PubMed] [Google Scholar]
- Klöckner U, Mikala G, Varadi M, Varadi G, Schwartz A. Involvement of the carboxyl-terminal region of the α1 subunit in voltage-dependent inactivation of cardiac Ca2+ channels. Journal of Biological Chemistry. 1995;270:17306–17310. doi: 10.1074/jbc.270.29.17306. [DOI] [PubMed] [Google Scholar]
- Kostyuk PG. Intracellular perfusion of nerve cells and its effects on membrane currents. Physiological Reviews. 1984;64:435–454. doi: 10.1152/physrev.1984.64.2.435. [DOI] [PubMed] [Google Scholar]
- Lorca T, Galas S, Fesquet D, Devault A, Cavadore JC, Doree M. Degradation of the proto-oncogene product p39mos is not necessary for cyclin proteolysis and exit from meiotic metaphase: requirement for a Ca2+-calmodulin dependent event. EMBO Journal. 1991;10:2087–2093. doi: 10.1002/j.1460-2075.1991.tb07741.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald TF, Pelzer S, Trautwein W, Pelzer D. Regulation and modulation of Ca2+ channels in cardiac, skeletal and smooth muscle cells. Physiological Reviews. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- Molinari M, Carafoli E. Calpain: A cytosolic proteinase active at the membranes. Journal of Membrane Biology. 1997;156:1–8. doi: 10.1007/s002329900181. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Worley PF, Barbaran JM. L-type voltage-sensitive Ca2+ channels mediate synaptic activation of immediate early genes. Neuron. 1991;7:625–635. doi: 10.1016/0896-6273(91)90375-a. [DOI] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potential in patch clamp experiments. Methods in Enzymology. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- Ono K, Fozzard HA. Phosphorylation restores activity of L-type Ca2+ channels after rundown in inside-out patches from rabbit cardiac cells. The Journal of Physiology. 1992;454:673–688. doi: 10.1113/jphysiol.1992.sp019286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BZ, DeMaria CD, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type Ca2+ channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- Quin N, Olcese R, Bransby M, Lin T, Birnbaumer L. Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proceedings of the National Academy of Sciences of the USA. 1999;96:2435–2438. doi: 10.1073/pnas.96.5.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios E, Brum G. Involvement of dihydropyridine receptors in excitation-contraction coupling in skeletal muscle. Nature. 1987;325:717–720. doi: 10.1038/325717a0. [DOI] [PubMed] [Google Scholar]
- Romanin C, Grösswagen P, Schindler H. Calpastatin and nucleotides stabilize cardiac Ca2+ channel activity in excised patches. Pflügers Archiv. 1991;418:86–92. doi: 10.1007/BF00370456. [DOI] [PubMed] [Google Scholar]
- Schmid R, Seydl K, Baumgartner W, Groschner K, Romanin C. Trypsin increases availability and open probability of cardiac L-type Ca2+ channels without affecting inactivation induced by Ca2+ Biophysical Journal. 1995;69:1847–1857. doi: 10.1016/S0006-3495(95)80055-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz D, Mikala G, Yatani A, Engle DB, Iles DE, Segers B, Sinke RJ, Weghuis DO, Klockner U, Wakamori M. Cloning, chromosomal localization, and functional expression of the α1 subunit of the L-type voltage-dependent Ca2+ channel from normal human heart. Proceedings of the National Academy of Sciences of the USA. 1993;90:6228–6232. doi: 10.1073/pnas.90.13.6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seydl K, Karlsson JO, Dominik A, Gruber H, Romanin C. Action of calpastatin in prevention of cardiac L-type Ca2+ channel run down cannot be mimicked by synthetic calpain inhibitors. Pflügers Archiv. 1995;429:503–510. doi: 10.1007/BF00704155. [DOI] [PubMed] [Google Scholar]
- Sheng M, McFadden G, Greenberg ME. Membrane depolarization and Ca2+ induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron. 1990;4:571–582. doi: 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- Spaetgens RL, Zamponi GW. Multiple structural domains contribute to voltage-dependent inactivation of rat brain α1E Ca2+ channels. Journal of Biological Chemistry. 1999;274:22428–22436. doi: 10.1074/jbc.274.32.22428. [DOI] [PubMed] [Google Scholar]
- Soldatov NM. Molecular diversity of L-type Ca2+ channel transcripts in human fibroblasts. Proceedings of the National Academy of Sciences of the USA. 1992;89:4628–4632. doi: 10.1073/pnas.89.10.4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldatov NM. Genomic structure of human L-type Ca2+ channel. Genomics. 1994;22:77–87. doi: 10.1006/geno.1994.1347. [DOI] [PubMed] [Google Scholar]
- Soldatov NM, Oz M, O’Brien KA, Abernethy DR, Morad M. Molecular determinants of L-type Ca2+ channel inactivation. Journal of Biological Chemistry. 1998;273:957–963. doi: 10.1074/jbc.273.2.957. [DOI] [PubMed] [Google Scholar]
- Soldatov NM, Zühlke RD, Bouron A, Reuter H. Molecular structures involved in L-type Ca2+ channel inactivation. Journal of Biological Chemistry. 1997;272:3560–3566. doi: 10.1074/jbc.272.6.3560. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon L. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proceedings of the National Academy of Sciences of the USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Neely A, Lacerda AE, Olcese R, Stefani E, Perez-Reyes E, Birnbaumer L. Modification of Ca2+ channel activity by deletions at the carboxyl terminus of the cardiac α1 subunit. Journal of Biological Chemistry. 1994;269:1635–1640. [PubMed] [Google Scholar]
- Yazawa K, Kameyama A, Yasui K, Li J-M, Kameyama M. ATP regulates cardiac Ca2+ channel activity via a mechanism independent of protein phosphorylation. Pflügers Archiv. 1997;433:557–562. doi: 10.1007/s004240050314. [DOI] [PubMed] [Google Scholar]
- Yokota H, Katayama M, Hino F, Kato I, Takano E, Maki M, Hatanaka M, Murachi T. Direct measurement of calpastatin subtypes by sandwich enzyme immunoassay using monoclonal antibodies. Molecular and Cellular Probes. 1991;5:261–269. doi: 10.1016/0890-8508(91)90047-n. [DOI] [PubMed] [Google Scholar]
- Zühlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type Ca2+ channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]