

Abstract
It is widely accepted that while release of amino acid neurotransmitters occurs with relatively high fidelity, peptidergic synapses require clustered bursts of action potentials for optimal transmitter release. Here we describe for the first time the occurrence and mechanisms of bursting by neurones in the subfornical organ (SFO), cells that utilize the peptide angiotensin II (ANG) in neurotransmission in autonomic pathways.
In current clamp recording of isolated SFO neurones in vitro, 53 % (n = 74) showed either spontaneous or evoked burst-like discharge patterns. Bursts typically appeared as shifts in bistable membrane potential, with action potentials superimposed on a depolarizing afterpotential (DAP). Similarly, in vivo single unit recordings of identified SFO neurones showed that 9 of 15 neurones fired in bursts.
The pattern of bursting, as well as duration of evoked DAPs was strongly dependent upon membrane potential, suggesting that the DAP contributes to burst generation. Based on our previous observation of calcium-sensing receptor (CaR)-activated bursts, we investigated the effects of NPS R-467, an allosteric agonist of the CaR, on evoked DAPs. NPS R-467 (1 μM) potentiated DAP duration throughout the voltage range tested.
We observed a dependence of evoked DAPs upon Na+ channels, as shown by sensitivity to tetrodotoxin (0.5 μM) or reduction of external [Na+] from 140 to 40 |mM. The duration of DAPs suggested that a persistent Na+ current mediates these events. Voltage-clamp analysis revealed the presence of a subthreshold sodium current, INaP.
Pharmacological blockade of INaP with 100 μM lidocaine reduced the duration of evoked DAPs, and inhibited bursting in SFO neurones. Facilitation of INaP with 10 nM anemone toxin (ATX) increased DAP duration and led to marked excitation of bursting cells. These data indicate that INaP is the main current underlying bursting in SFO neurones.
Our observations of receptor-mediated facilitation of bursting by SFO neurones represents an intriguing mechanism through which the release of the peptide neurotransmitter ANG may be regulated.
Interneuronal chemical transmission occurs through the co-ordinated release of a diffusible messenger from the presynaptic terminal, a process controlled largely through action potential-dependent influx of Ca2+. The dynamics of transmitter release depend upon the identity of the transmitter molecule (Verhage et al. 1991), the kinetics of Ca2+ accumulation within the terminal (Heidelberger et al. 1994), and presynaptic action potential waveform (Klein & Kandel, 1980; Wheeler et al. 1996; Sabatini & Regehr, 1997). Presynaptic transmitter release shows use-dependent facilitation though modulation of these parameters, as can occur during bursts of action potentials. It is now widely accepted that bursting is one mechanism through which presynaptic neurones can encode information (Lisman, 1997).
The precise pattern in which neurones fire action potentials depends upon both synaptic inputs and their intrinsic membrane properties. Bursting neurones have been described in diverse structures including the hippocampal CA1 region (Jefferys & Haas, 1982), neocortex (Chagnac-Amitai & Connors, 1989; Connors & Gutnick, 1990; Silva et al. 1991), and hypothalamic neuroendocrine cells (Hatton, 1982; Andrew & Dudek, 1983). These intrinsic output patterns arise from specialized combinations of ion channels that generate the action potential waveform and the subsequent spike afterpotentials.
Anatomical and neurochemical studies have attempted to correlate burst-firing properties with specific morphological and neurotransmitter phenotypes. Interestingly, many peptidergic neurones fire in bursts (Andrew & Dudek, 1983; Porter et al. 1998). Furthermore, hypothalamic magnocellular neuroendocrine cells have a need for burst-like output patterning for optimal vasopressin release (Bicknell & Leng, 1981). Thus it appears that bursting may be an important mechanism through which peptidergic neurones can selectively release these non-traditional chemical messengers.
Neurones in the subfornical organ (SFO), a forebrain circumventricular structure, have been shown to utilize the peptide angiotensin II (ANG) in synaptic transmission (Bains et al. 1992; Bains & Ferguson, 1995; reviewed by Ferguson & Washburn, 1998). The SFO contains dense binding sites for numerous circulating factors including peptides (McKinley et al. 1998), inflammatory mediators (Vallieres et al. 1997; Quan et al. 1998) and steroid hormones (Voisin et al. 1997). As a result of its extensive efferent projections, the SFO has been implicated in the control of autonomic functions as diverse as reproduction (Limonta et al. 1981; Donevan et al. 1989), respiration (Ferguson et al. 1989), fluid balance (Simpson & Routenberg, 1973; Smith et al. 1995) and the febrile response to infection (Takahashi et al. 1997). Although much of the current literature on the physiology of the SFO consists of in vivo studies, more recent advances have allowed investigation into the functioning of neurones in this structure through in vitro extracellular recording (Li & Ferguson, 1993a; Ferguson & Bains, 1996; Rauch et al. 1997) and now through patch clamp recording in isolated SFO neurones (Ferguson et al. 1997; Washburn et al. 1999a). However, little is known about the firing properties of SFO neurones and the specific cellular mechanisms that underlie their firing patterns. We have shown previously that agonists of the novel neuronal calcium-sensing receptor (CaR) induce plateau-like burst firing patterns, due at least in part to activation of an intracellular Ca2+-activated non-selective cation channel (NSCC) (Washburn et al. 1999b).
In this study, we describe for the first time intrinsic burst firing in SFO neurones and the ionic mechanisms underlying this behaviour. Furthermore, we show that extrinsic modulation of this activity by agonists of the CaR can have profound effects on the excitability of these specialized central nervous system neurones through the combined effects of voltage-gated persistent sodium channels and metabotropically operated NSCC channels.
METHODS
Dissociated SFO neurone preparation
SFO neurones were dissociated by the protocol previously described (Ferguson et al. 1997). Male Sprague-Dawley rats (125–150 g) were rapidly decapitated. The brains were quickly removed and immersed in ice-cold artificial cerebrospinal fluid (ACSF) of the following composition (mM): 140 NaCl, 5 KCl, 1.6 CaCl2, 1 MgCl2, 10 Hepes and 10 glucose; pH adjusted to 7.4 with NaOH. A tissue block containing the hippocampal commissure and SFO was dissected and placed in Ca2+- and Mg2+-free ACSF. All surrounding tissue was removed from the SFO with the aid of a dissection microscope, placed in Ca2+- and Mg2+-free ACSF containing 1 mg ml−1 trypsin (Sigma), and incubated in 5 % CO2 and 95 % O2 at 37°C for 30 min, with periodic gentle shaking, followed by trituration through a tuberculin syringe fitted with an 18-gauge needle. Cells were then resuspended in ACSF and plated onto plastic culture dishes (Gibco), to which they adhered rapidly. For patch experiments, all drugs and solutions were delivered through the bath at a rate of 1–2 ml min−1. All procedures conformed to the standards outlined by the Canadian Council on Animal Care and the Queen's University Animal Care Committee (authorization no. 2000–023-I).
Electrophysiological techniques
Whole-cell patch clamp recordings were obtained using micropipettes pulled using a P-87 Flaming/Brown pipette puller (Sutter). Tip resistances were 2–6 MΩ when filled with a solution containing (mM): 140 potassium gluconate, 0.1 CaCl2, 2 MgCl2, 1.1 EGTA, 10 Hepes and 2 Na2ATP, with the pH adjusted to 7.25 with KOH. For voltage clamp experiments isolating the subthreshold Na+ currents (INaP), K+ currents were blocked using an intracellular solution containing (mM): 135 CsCl, 0.1 CaCl2, 2 MgCl2, 1.1 EGTA, 10 Hepes, 2 Na2ATP, 1 tetraethylammonium chloride and 1 4-aminopyridine, with the pH adjusted to 7.25 with CsOH. Following the establishment of a gigaohm seal, the whole-cell configuration was obtained with a brief pulse of suction. The control bath solution consisted of ACSF of the following composition (mM): 140 NaCl, 3 KCl, 1.6 CaCl2, 1 MgCl2, 10 Hepes and 10 glucose; pH adjusted to 7.4 with NaOH. External solutions for Na+ channel isolation also contained 100 μM Cd2+ to block calcium channels. Drugs were dissolved in ACSF and applied directly through the bath perfusion system. Nifedipine was first dissolved in dimethyl sulphoxide (DMSO), and diluted into ACSF such that the final concentration of DMSO was 0.01 %, which had no effect on firing patterns. Nifedipine was protected from light at all times. ω-Conotoxin GVIA (CTX; Alomone) was prepared in ACSF with 0.1 % cytochrome c. NPS R-467, a selective CaR agonist, was a generous gift from NPS Pharmaceuticals (Salt Lake City, UT, USA).
Patch-clamp recordings were processed using a List EPC-7 amplifier, filtered at 3 kHz with an 8-pole Bessel filter, digitized using the CED 1401 Plus interface at 5 kHz (20 kHz for Na+ channel experiments), and stored on computer for off-line analysis. Data were collected using the EPC/Signal (episode-based capture) or Spike2 (continuous recording) packages (CED, Cambridge, UK).
In vivo electrophysiology
Male Sprague-Dawley rats (150–400 g) were anaesthetized with sodium pentobarbitone (65 mg kg−1i.p.), with intravenous supplements as required. Animals were then placed in a stereotaxic frame, and a small burr hole was made in the skull such that electrodes could be advanced into the region of the SFO according to the co-ordinates of Paxinos & Watson (1982). Extracellular single-unit recordings were obtained using glass micropipettes filled with 1 M NaCl (tip diameter < 1 μM, resistance 10–25 MΩ). At the conclusion of the experiment, animals were perfused with 0.9 % saline followed by 10 % formalin through the left ventricle of the heart. All recording sites were verified histologically as falling within the boundaries of the SFO.
RESULTS
In the current-clamp recording configuration, SFO neurones show a variety of spontaneous firing patterns, with approximately 53 % (39/74 cells tested in continuous recording mode) showing either spontaneous or evoked periods of burst-like activity. This firing pattern consisted of clustered action potentials surrounded by periods of quiescence, observed in both loose patch extracellular (Fig. 1A; n = 7) and conventional whole-cell current clamp (mean burst duration 11.4 ± 2.5 s; Fig. 1B; n = 32). In order to confirm that SFO neurones can fire in bursts in situ, we undertook a study of in vivo single unit recordings in antidromically identified SFO neurones projecting to the paraventricular nucleus of the hypothalamus (PVN). We found that 9 of 15 SFO neurones showed periods of burst firing (burst duration 2.4 ± 0.5 s; Fig. 1C), confirming that SFO neurones do in fact burst in vivo, substantiating our observations of intrinsic bursting in vitro.
Figure 1. Subfornical organ neurones fire bursts of action potentials.

A, in vitro extracellular (loose patch) recording showing burst firing by an isolated SFO neurone. B, whole-cell recording of the same SFO neurone as in A, indicating that bursting is maintained in this configuration. Note the bistable membrane potential variation between quiet and burst periods. C, in vivo single unit recording of bursting activity by an antidromically identified paraventricular nucleus of the hypothalamus (PVN)-projecting SFO neurone in an anaesthetized rat.
Spontaneous bursts typically appeared as either oscillatory periods of firing, or more typically, as shifts in bistable membrane potential properties resulting in plateau-type depolarizations. In bursting cells, spontaneous bistable plateau depolarizations generally followed a slow depolarization to spike threshold, after which subsequent action potentials were superimposed on a DAP (Fig. 2A). Similarly, bursts could be evoked by brief positive current pulses that initiated the DAP (Fig. 2A and B). The pattern of firing activity was found to be strongly dependent upon membrane potential, as cells fired in bursts around resting potential, but could be converted to nearly tonic or nearly quiescent by depolarization or hyperpolarization, respectively (Fig. 3). It is important to note that in all cells showing bistable burst-like activity, bursts could be initiated by pulses sufficient to initiate a DAP, but not all cells that showed DAPs showed spontaneous bursting. The rapid fluctuation in instantaneous firing frequency was typical of bursting cells.
Figure 2. Spontaneous and evoked bursts are superimposed on depolarizing afterpotentials (DAPs).
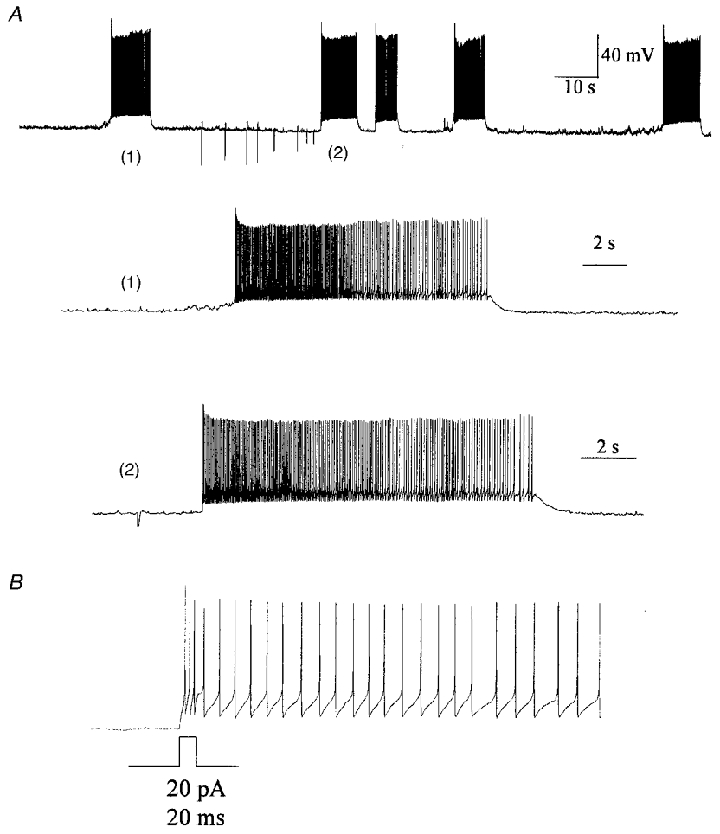
A, current-clamp recording of a phasically firing SFO neurone showing spontaneous (1) and current-evoked (2) bursts. Spontaneous bursts were triggered by a slow depolarization to action potential threshold, near which the membrane potential remained until the rapid cessation and return to resting potential. B, small current pulses (20 pA, 20 ms) can trigger a burst event consisting of a rapid DAP with superimposed action potentials.
Figure 3. SFO neurone bursting is membrane potential dependent.

The current clamp traces show variation in firing patterns of an SFO neurone by changing membrane potential. At hyperpolarized potentials (−73 mV) this neurone was nearly quiescent, but showed few burst events. Near resting potential (−62 mV), this cell fired in regular bursts, as shown in the middle panel. Depolarizing the cell (−56 mV) with positive current shifted the cell to a tonic firing mode. Scale bars in the lower trace apply to all current clamp records.
Our observation of DAP-triggered bursts led us to study the intrinsic characteristics of SFO neurone DAPs evoked by depolarizing current pulses in an additional 22 cells. The duration of the DAP was calculated as the time required for membrane potential to return to baseline following termination of the current pulse. Out of 96 cells tested in the current clamp configuration, 42 showed obvious DAPs greater than the time required for passive return to baseline membrane potential; that is, a period of time greater than that expected when compared with the passive membrane time constant of 55.0 ± 4.4 ms (n = 55), calculated from the voltage response to negative current pulses. Following our observation that membrane potential had a prominent effect on the discharge patterns of SFO neurones, we investigated the effects of membrane potential on current-evoked DAPs. The initial membrane potential had a significant effect on DAP duration, as shown in Fig. 4. The duration of the DAP (measured from the end of the current pulse until return to baseline membrane potential) increased significantly with depolarization, ranging from 1.2 ± 0.1 s at −80 mV, to 7.8 ± 1 s at −50 mV (P < 0.01; ANOVA followed by a Bonferroni post hoc test).
Figure 4. Evoked DAPs are dependent upon initial membrane potential.

Examples of DAPs (indicated by shaded areas) evoked by 500 ms depolarizing current pulses from holding potentials of −70, −60 and −50 mV. Note the marked extension of the DAP with more depolarized membrane potentials, eventually leading to a burst event at −50 mV. The membrane potential effect is significant, and occurs through the voltage range tested (*P < 0.01 compared with −80 mV; one-way ANOVA followed by Bonferroni post hoc test).
Based on our previous findings that following activation of the neuronal CaR SFO neurones would fire in burst-like plateau potentials, we studied the potential for CaR-mediated modulation of the DAP as one mechanism through which this bursting could be initiated. We found that 1 μM NPS R-467, a specific CaR agonist (Hammerland et al. 1998; Washburn et al. 1999b) significantly potentiated the DAP duration throughout the voltage range tested (P < 0.01; ANOVA; see Fig. 5). We also observed a change in shape of the DAP following CaR activation. As seen in Fig. 5A, in addition to increasing the duration of the DAP, the amplitude was also increased, although it appeared to be due to the addition of a slower component to the DAP (arrowhead), as shown in the shaded regions of the DAP (Fig. 5A). These data, together with our previous report indicating no modification of voltage-gated conductances by NPS R-467 (Washburn et al. 1999b), suggest that CaR-mediated potentiation of the DAP occurs through activation of the NSCC, which sums together with the current underlying the control DAP (INaP; see below) to provide the drive to burst, as shown in Fig. 6A. The CaR-mediated bursts typically showed a first spike with a lower activation threshold than subsequent spikes (indicated by the arrow). Blockade of Na+ channels with tetrodotoxin (TTX; 0.5 μM) eliminated bursts, leaving behind the residual CaR-mediated depolarization. During the delivery of depolarizing current pulses, we observed that the pulse had to be of sufficient amplitude and duration to evoke at least a single fast action potential in order to evoke a DAP (Fig. 6B). That is, subthreshold current pulses could not initiate a DAP, and showed only a passive, monotonic decline back to baseline membrane potential with a time course of the order of the passive membrane time constant for that cell. This observation suggested that activation of sodium channels are required to generate a DAP. This hypothesis was further tested pharmacologically through Na+ channel blockade by TTX (0.5 μM; Fig. 6B). TTX completely blocked the DAP generated by current pulses that would normally generate fast action potentials, even when the pulse amplitude was twice that required to generate a DAP under control conditions (n = 6). Furthermore, reduction of extracellular Na+ from 140 to 40 mM abolished the DAP (Fig. 6C; n = 4). We tested the possibility that Ca2+ channels could contribute to the DAP by blocking N-type channels with the irreversible blocker CTX (100 nM; after 5 min of treatment), a dose that blocks approximately 80 % of the net inward Ca2+ current (D. L. S. Washburn & A. V. Ferguson, unpublished observations). This treatment failed to block current-evoked DAPs (Fig. 6D; n = 3), indicating that N-type Ca2+ channels do not contribute significantly. We also tested the role of dihydropyridine-sensitive L-type channels by blocking with nifedipine (100 μM; n = 4). This had no significant effect on DAP duration (data not shown). The observations that at least a single spike is required (and is sufficient) to generate a DAP and that TTX or Na+ reduction completely prevented DAP generation suggested the involvement of Na+ channels in the generation and maintenance of the DAP. The relatively long duration of the DAP suggests that a persistent Na+ current (INaP) probably mediates these events.
Figure 5. Activation of the CaR potentiates evoked DAPs.
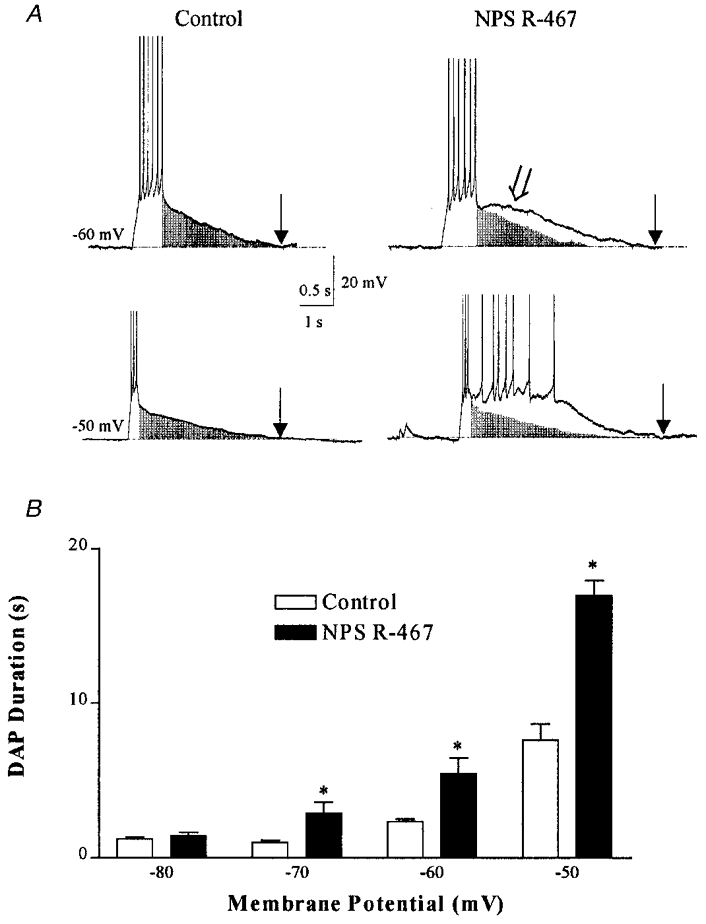
A, examples of current-evoked DAPs under control (left) and after 2 min in 1 μM NPS R-467 (right) from −60 and −50 mV. The shaded area indicates the size and shape of the control DAP. Note that in addition to increasing the duration of the DAP, NPS R-467 induced a region of active depolarization (arrowhead). Arrows indicate return to baseline membrane potential. These results are summarized in the histogram (B). *DAPs of significantly different duration from control (P < 0.01; two-way ANOVA, followed by Bonferroni post hoc test).
Figure 6. Bursts and DAPs are dependent upon a sodium current.
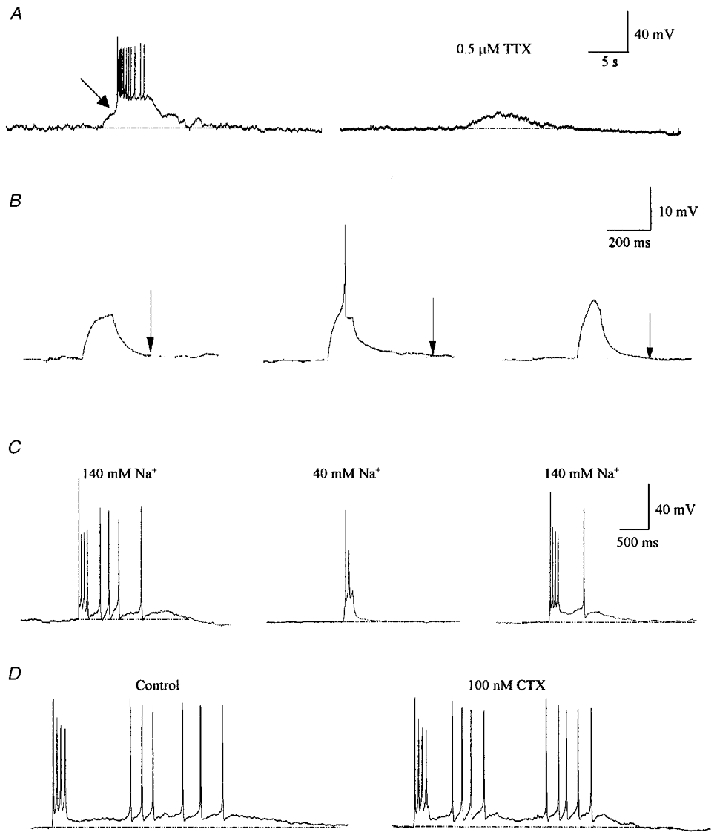
A, bursts induced by NPS R-467 are inhibited by TTX, as shown in this example. NPS R-467 (1 μM) caused a slow depolarization (left) which eventually reached a low threshold for a rapid depolarization (indicated by the arrow) leading to a burst superimposed on a DAP. Pretreatment with 0.5 μM TTX (right) prevented generation of the burst, leaving only the NPS R-467-mediated slow depolarization. B, subthreshold depolarizing current pulses (10 pA) that fail to elicit an action potential do not generate a DAP (left trace), whereas a pulse large enough (15 pA) to generate a single action potential is sufficient to activate a DAP (middle trace, action potential truncated). TTX abolishes evoked DAPs, even when pulses reach potentials greater than spike threshold, as shown through the delivery of larger current pulses (25 pA). C, reduction of extracellular Na+ from 140 mM (left) to 40 mM (middle) by replacement with NMDG reduced the DAP almost entirely. The DAP returned with reinstitution of 140 mM Na+ (right), indicating that Na+ is the major charge carrier underlying the DAP. D, blocking N-type Ca2+ channels with 100 nM CTX does not attenuate the current-evoked DAP.
We next examined the properties of sodium currents in SFO neurones through voltage clamp experiments. Activation of the rapidly activating, rapidly inactivating Na+ current was studied through 100 ms voltage steps from −100 to −15 mV in 5 mV increments (Fig. 7Ai and iii). We found that the persistent current, INaP, was more readily isolated during slow depolarizing voltage ramps (20 mV s−1, −80 to −20 mV; Fig. 7Aii and iii; n = 6). During these voltage ramps, the resultant current showed a region of negative slope conductance, indicative of an inward current, activating near −75 mV and reaching a peak near −55 mV (see Fig. 7Aii). This current was blocked by TTX, confirming its identity as a voltage-gated Na+ current (not shown). The TTX-sensitive current remained activated for as long as 2 s under slow ramp conditions, confirming that it is of the ‘persistent’ rather than the classic rapidly inactivating Na+ channel type. Inactivation properties of both the classical and subthreshold persistent Na+ currents were determined through a protocol in which a series of inactivating prepulses of variable potential (−100 to 0 mV; 150 ms) were delivered prior to a test pulse to −20 mV (Fig. 7B i). The relative peak currents were fitted to a Boltzmann function (Fig. 7Bii), from which values of −52.5 mV and −6.8 mV−1 were obtained for V50 and slope, respectively (r2= 0.99; n = 4), for the peak rapidly inactivating current. Similarly, we obtained values of −37.9 mV and −6.1 mV−1 for V50 and slope, respectively (r2= 0.99; n = 4), for the persistent current measured 12 ms after onset of the test potential. We then tested the ability to block INaP selectively with 100 μM lidocaine (lignocaine). The peak current was attenuated by 64 ± 5 % by 100 μM lidocaine (Fig. 7C; n = 4) which was selective for INaP at this dose, as it did not affect upstroke velocity of spontaneous or evoked action potentials (n = 4; data not shown).
Figure 7. Properties of fast and persistent sodium currents in SFO neurones.
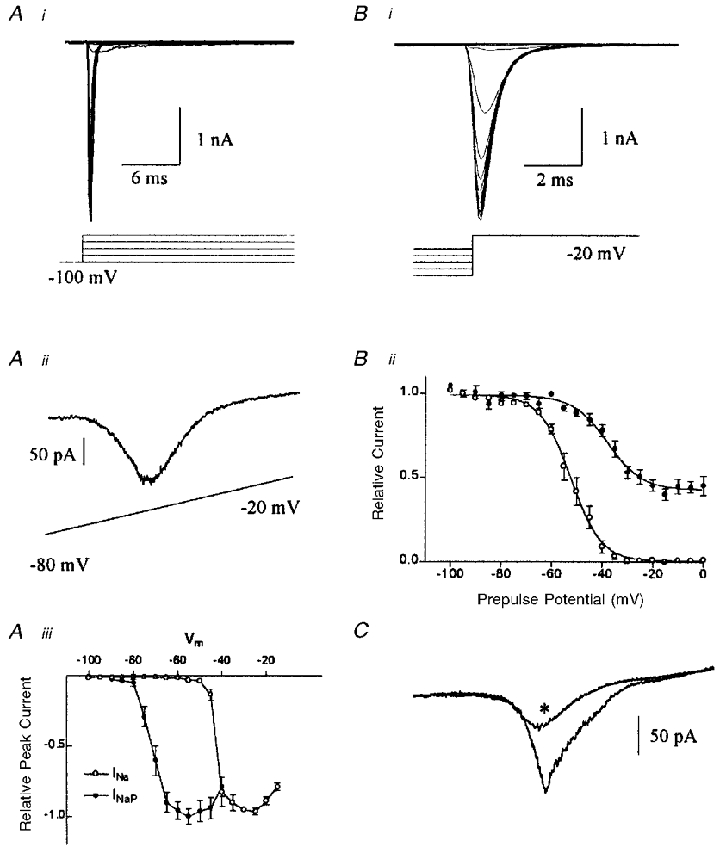
A, activation of SFO Na+ currents. Ai, sodium currents elicited from voltage steps from −100 to −15 mV in 5 mV increments, as in the voltage protocol shown below. Note the rapid activation and inactivation kinetics. Aii, persistent Na+ current activated by slow (20 mV s−1) depolarizing voltage ramps from −80 to −20 mV. Note the relatively negative activation threshold (near −75 mV), and peak current near −55 mV. Aiii, plot of the relative peak inward Na+ currents for both the fast (^) and persistent (•) Na+ currents (obtained from ramp experiments). B, voltage dependence of inactivation of Na+ currents. Bi, currents obtained from a test pulse at −20 mV following 150 ms conditioning pulses ranging from −100 to 0 mV in 5 mV increments (voltage prepulse protocol shown below the current trace). Bii, inactivation plot of normalized current against prepulse potential, fitted to a Boltzmann function (V50=−52.5 mV, slope =−6.8 mV−1, r2= 0.98, n = 4 for fast Na+ current (peak current; ^) and V50=−37.9 mV, slope =−6.1 mV−1, r2= 0.99, n = 4 for persistent Na+ current (•), measured 12 ms after onset of test pulse). C, persistent Na+ current is sensitive to 100 μM lidocaine. Currents activated from depolarizing voltage ramps (−100 to 0 mV, 14 mV s−1) under control and lidocaine (*) conditions. The peak inward current was reduced by 64.0 ± 5 % (n = 5).
We next examined the role of INaP in the regulation of currents underlying bursting by determining the effects of lidocaine on evoked DAPs and spontaneously bursting cells. Application of 100 μM lidocaine reduced the duration of evoked DAPs by 65 ± 5 % (Fig. 8A; n = 4). In spontaneously bursting cells lidocaine caused a cessation of high-frequency bursting, leaving only spontaneous solitary spikes (Fig. 8B). Conversely, we attempted to induce burst firing in quiescent or irregularly firing cells through two positive modulators of INaP, anemone toxin (ATX, 10–50 nM; Mantegazza et al. 1998; Fig. 9) and veratridine (100 μM; data not shown), a more effective, although less specific modulator. We found that both treatments increased the excitability of all cells tested, although true bursting was difficult to induce, as spontaneous or evoked spikes usually led to prolonged depolarization, especially in cells tested with veratridine. At lower doses (10 nM), ATX increased the duration of evoked DAPs by 32 ± 12 % (n = 4). To assess to the affects of ATX on INaP directly, cells that were shown to respond to ATX in current clamp were also tested in voltage clamp ramp experiments. Treatment with 10 nM ATX increased the inward current in the voltage range consistent with INaP (Fig. 9A, inset; n = 3). Note that the current is not prominent under basal conditions, as these recordings were made with potassium gluconate internal solutions for current clamp recording. Together, these data indicate that the DAP is mediated largely by a persistent sodium current, and that this current plays an important role in generating the output patterning of SFO neurones.
Figure 8. Lidocaine attenuates evoked DAPs and inhibits spontaneous bursting.

A, sequential current evoked (20 pA, 50 ms) DAPs under control (left) and 100 μM lidocaine (right) conditions. DAPs marked with an asterisk (*) are shown at greater resolution in the inset. Note the prominent reduction in DAP duration by lidocaine. B, spontaneous bursting (upper panel) is blocked by lidocaine (lower panel, same cell). Spontaneous single action potentials are not blocked by lidocaine, but do fail to elicit afterpotentials leading to burst discharge.
Figure 9. Anemone toxin potentiates evoked DAPs and excites bursting cells.

A, current-evoked DAPs (20 pA 500 ms) under control (upper panel) and 10 nM anemone toxin (ATX; lower panel) conditions, during which the DAP duration is increased by 32 ± 12 % (n = 4). Arrow indicates termination of the DAP. Inset: ATX potentiates the inward current (*) evoked from depolarizing voltage ramps (20 mV s−1). B, current clamp record of a spontaneously bursting cell under control (top) and 10 nM ATX (bottom) conditions. ATX lead to a marked increase in firing frequency accompanied by a small depolarization of baseline membrane potential (n = 4).
DISCUSSION
In this study we have described for the first time, the ability of SFO neurones to fire in periods of burst-like activity in isolated cells in vitro, and confirmed in vivo, as observed from recordings obtained from anaesthetized rats. Interestingly, bursts recorded in vivo appeared to be of shorter duration than those in vitro. This is probably due to termination of the burst by inhibitory GABAergic inputs, as have been described both in vivo (Osaka et al. 1992), and in an in vitro slice preparation (Inenaga et al. 1995). It is also possible that the whole-cell recording configuration could dialyse intracellular mediators (e.g. Ca2+) that may be responsible for repolarizing the membrane potential, thus lengthening burst duration. Furthermore, we have shown that bursting can be potentiated by agonists of the CaR. In bursting cells, brief pulses of depolarizing current could initiate prolonged bursts, suggesting that the ionic basis of these events ultimately depends upon a sustained, voltage-gated current. This finding, combined with our observation that both the evoked DAPs and CaR-mediated busts were completely abolished by TTX, suggested the involvement of voltage-gated Na+ channels in the generation of the DAP. Given the extended duration of the DAP, we hypothesized that a persistent Na+ current, INaP, would underlie these events. This hypothesis was tested through three complementary methods: (1) replacement of extracellular Na+ with the impermeant N-methyl-D-glucamine, (2) pharmacological antagonism of INaP by 100 μM lidocaine, a blocker of persistent sodium channels, and (3) potentiation of INaP by ATX or veratridine, activators of persistent sodium channels. In voltage clamp experiments where depolarizing voltage ramps were delivered, we found that the majority of SFO neurones tested showed a region of negative slope conductance, indicative of INaP that was indeed sensitive to TTX and lidocaine. We observed that bursts were initiated by a single spike that triggered a DAP upon which subsequent spikes were superimposed, resulting in a bistable membrane potential.
The mechanisms of CaR-mediated bursting were examined by determining the effects of CaR activation on the DAP. While we found that NPS R-467 significantly increased the DAP duration, there was no observed effect on INaP (Washburn et al. 1999b), suggesting that multiple conductances must be involved in the process of burst generation. In fact, it appears that positive modulation by the CaR agonist serves to depolarize the cell to a voltage range in which bursting may occur through activation of INaP.
Ionic mechanisms underlying neuronal burst firing
The intrinsic ability of neurones to fire in bursts of action potentials has been described in a variety of CNS neurones and has been attributed to several different mechanisms. While there are undoubtedly many processes contributing to this type of co-ordinated excitability, one of the common elements is the presence of prominent DAP. Studies have attempted to understand the exact ionic basis of these excitatory events and have implicated NSCCs (Fraser & MacVicar, 1996), Ca2+ (Kobayashi et al. 1997), K+ conductances (Li & Hatton, 1997; Ghamari-Langroudi & Bourque, 1998), and Na+ channels (Guatteo et al. 1996). Here we show that a persistent Na+ current, INaP, is present in SFO neurones, and underlies the fast DAP. This conclusion is supported by several lines of evidence, including the observations that: (1) a single action potential is sufficient and essential to the activation of DAPs and initiation of bursts; (2) the evoked DAP is completely blocked by TTX; and (3) SFO neurones have persistent Na+ currents that remain active for periods long enough to account for DAPs of the duration observed here. Slow depolarizing voltage ramps were found to activate a negative sloped conductance that activated near −75 mV, inactivated incompletely at even more depolarized potentials, and was sensitive to TTX and lidocaine, verifying the presence of a subthreshold, persistent Na+ current, or INaP. In current clamp experiments lidocaine abolished the tendency to burst (although spontaneous solitary action potentials still occurred), and blocked current-evoked DAPs. Although others have shown that decreasing repolarizing conductances can contribute to DAPs (Li & Hatton, 1997), the presence of regular action potentials and spike afterhyperpolarizations argues against a decrease in K+ channel conductance in this case.
These data indicate the intrinsic ability of a population of SFO neurones to produce bursts of action potentials. This, combined with our previous observation that activation of the CaR induced periods of burst firing led us to investigate the potential for the CaR to facilitate bursting through modulation of the DAP and the intrinsic conductances which underlie the DAP. In cells that showed current-evoked DAPs, we found that bath application of NPS R-467 significantly potentiated the DAP duration. Interestingly, in addition to the observed increased time to baseline, activation of the CaR changed the shape of the DAP such that there was often a region of active re-depolarization on the DAP rather than only extending the time required to decay fully. This finding agrees with our earlier reports that NPS R-467 activates a NSCC in SFO neurones, and that this current, in part, underlies the burst-like plateau potentials induced by activation of the CaR (Washburn et al. 1999b). In this study, we show that application of NPS R-467 after blockade of Na+ channels with TTX, leads only to a slow depolarization without the generation of a stable plateau, suggesting that the CaR-mediated activation of the NSCC results in the depolarization to the INaP threshold, which can then initiate a burst. This demonstrates that while the TTX-sensitive current that mediates DAPs is required for intrinsic burst firing of SFO neurones, the CaR-mediated potentiation of burst firing relies upon the secondary activation of INaP subsequent to the depolarization carried by the NSCC directly regulated by the CaR. This type of interaction between both voltage and metabotropically gated ion channels represents an interesting and novel mechanism for the regulation of neuronal firing by extracellular messengers. Interestingly, metabotropic alteration of Na+ channel function has been identified as a mechanism through which neuromodulators can influence target neurones (Dascal & Lotan, 1991; West et al. 1991; Astman et al. 1998).
Physiological relevance of bursting in SFO neurones
The physiological implications of burst firing in SFO neurones are not fully understood at this point, although there are some intriguing possibilities. Amongst the known targets of SFO neurones are the autonomic and neurosecretory neurones of the paraventricular nucleus of the hypothalamus (PVN). One conceivable situation is that extrinsic modulation of the output of SFO neurones projecting to PVN could result in state-dependent plasticity in the neural circuits governing autonomic output. Bursting by the presynaptic SFO neurone may explain the short-term changes in synaptic efficacy in this pathway observed in single-unit neuronal recordings obtained in the PVN during SFO stimulation (Ferguson & Washburn, 1998).
A more likely possibility is that of the potential for preferential neurotransmitter release, as it has been suggested that SFO neurones use both classical amino acid (L-glutamate) and peptide (ANG) neurotransmitters. In vivo electrophysiological studies have identified both long- and short-latency excitatory monosynaptic inputs from SFO neurones to PVN autonomic neurones (Bains et al. 1992; Li & Ferguson, 1993b; Bains & Ferguson, 1995). While the long-latency effects are abolished by the ANG AT1 receptor antagonist losartan (Li & Ferguson, 1993b; Bains & Ferguson, 1995), the short-latency synaptic effects were unaltered by such treatment. These data suggest that the SFO-PVN synapse is capable of using both peptidergic (slow) and classical (rapid) neurotransmitters. Although it was not possible to directly ascertain the chemical identity of the neurones recorded from in this study, the relative proportions of bursting cells (approximately 50 %) and ANG-immunoreactive SFO neurones, for which estimates range from approximately 40 to 65 % (Lind et al. 1984, 1985), are comparable.
Perhaps the most meaningful physiological correlate of SFO bursting, however, is the observation of frequency-dependent effects of electrical stimulation of the SFO in elevating blood pressure, reported by Ferguson & Renaud (1984), where it was shown that the hypertensive effects were maximal at a stimulation frequency of 10 Hz. Similarly, stimulation of the SFO at 10 Hz also elicits drinking behaviour in the rat (Smith et al. 1995). In the present study, it was observed that the peak instantaneous frequency during a single burst event was approximately 10 Hz. This sets up the potential for individual SFO neurones to be able to utilize peptidergic and/or amino acid neurotransmitters, and regulate their release through appropriately patterned discharge. It is well understood that single action potentials are sufficient to release glutamate with reasonable probability, but a more sophisticated process is involved in the release of peptide neurotransmitters. While the exact mechanism of post-synaptic excitation by ANG in the PVN is unknown, there is considerable evidence in the literature on which to base a model. Studies from our laboratory have shown ANG inhibition of a prominent PVN transient outward K+ conductance (IA) (Li & Ferguson, 1996), probably of dendritic origin. This, together with the observations of mixed ionotropic glutamate receptor-mediated synaptic potentials (Daftary et al. 1998), sets up the possibility of the ANG-sensitive IA acting as a dendritic excitation filter, as suggested by Schoppa & Westbrook (1999). In this manner, ANG released by bursting SFO neurones could then relieve the inhibitory IA‘filter’, facilitating the propagation of slow NMDA receptor potentials, thus resulting in excitation of PVN neurones, and ultimately leading to autonomic outputs.
The SFO is ideally situated outside the blood-brain barrier, to continuously monitor changes in the constituents of the circulating plasma, and modulate its neuronal output to specific effector regions accordingly. The ability of neurones within the SFO to dramatically change their firing properties due to specific receptor-mediated changes in membrane properties may represent an essential feature of these cells which confers on them their unique ability to produce appropriate physiological responses to such a diversity of homeostatic challenges.
Acknowledgments
This work was funded by grants to A.V.F. from the Medical Research Council of Canada and NPS Pharmaceuticals Inc. D.L.S.W. is funded by scholarships from the Heart and Stroke Foundation and Medical Research Council of Canada.
References
- Andrew RD, Dudek FE. Burst discharge in mammalian neuroendocrine cells involves an intrinsic regenerative mechanism. Science. 1983;221:1050–1052. doi: 10.1126/science.6879204. [DOI] [PubMed] [Google Scholar]
- Astman N, Gutnick MJ, Fleidervish IA. Activation of protein kinase C increases neuronal excitability by regulating persistent Na+ current in mouse neocortical slices. Journal of Neurophysiology. 1998;80:1547–1551. doi: 10.1152/jn.1998.80.3.1547. [DOI] [PubMed] [Google Scholar]
- Bains JS, Ferguson AV. Paraventricular nucleus neurons projecting to the spinal cord receive excitatory input from the subfornical organ. American Journal of Physiology. 1995;268:R625–633. doi: 10.1152/ajpregu.1995.268.3.R625. [DOI] [PubMed] [Google Scholar]
- Bains JS, Potyok A, Ferguson AV. Angiotensin II actions in paraventricular nucleus: functional evidence for a neurotransmitter role in efferents originating in subfornical organ. Brain Research. 1992;599:223–229. doi: 10.1016/0006-8993(92)90395-p. [DOI] [PubMed] [Google Scholar]
- Bicknell RJ, Leng G. Relative efficiency of neural firing patterns for vasopressin release in vitro. Neuroendocrinology. 1981;33:295–299. doi: 10.1159/000123248. [DOI] [PubMed] [Google Scholar]
- Chagnac-Amitai Y, Connors BW. Synchronized excitation and inhibition driven by intrinsically bursting neurons in neocortex. Journal of Neurophysiology. 1989;62:1149–1162. doi: 10.1152/jn.1989.62.5.1149. [DOI] [PubMed] [Google Scholar]
- Connors BW, Gutnick MJ. Intrinsic firing patterns of diverse neocortical neurons [see comments] Trends in Neurosciences. 1990;13:99–104. doi: 10.1016/0166-2236(90)90185-d. [DOI] [PubMed] [Google Scholar]
- Daftary SS, Boudaba C, Szabo K, Tasker JG. Noradrenergic excitation of magnocellular neurons in the rat hypothalamic paraventricular nucleus via intranuclear glutamatergic circuits. Journal of Neuroscience. 1998;18:10619–10628. doi: 10.1523/JNEUROSCI.18-24-10619.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dascal N, Lotan I. Activation of protein kinase C alters voltage dependence of a Na+ channel. Neuron. 1991;6:165–175. doi: 10.1016/0896-6273(91)90131-i. [DOI] [PubMed] [Google Scholar]
- Donevan SD, Van Vugt DA, Ferguson AV. Subfornical organ activation stimulates luteinizing hormone secretion in the rat. Brain Research. 1989;488:398–402. doi: 10.1016/0006-8993(89)90738-5. [DOI] [PubMed] [Google Scholar]
- Ferguson AV, Bains JS. Electrophysiology of the circumventricular organs. Frontiers in Neuroendocrinology. 1996;17:440–475. doi: 10.1006/frne.1996.0012. [DOI] [PubMed] [Google Scholar]
- Ferguson AV, Beckmann LM, Fisher JT. Effects of subfornical organ stimulation on respiration in the anesthetized rat. Canadian Journal of Physiology and Pharmacology. 1989;67:1097–1101. doi: 10.1139/y89-173. [DOI] [PubMed] [Google Scholar]
- Ferguson AV, Bicknell RJ, Carew MA, Mason WT. Dissociated adult rat subfornical organ neurons maintain membrane properties and angiotensin responsiveness for up to 6 days. Neuroendocrinology. 1997;66:409–415. doi: 10.1159/000127266. [DOI] [PubMed] [Google Scholar]
- Ferguson AV, Renaud LP. Hypothalamic paraventricular nucleus lesions decrease pressor responses to subfornical organ stimulation. Brain Research. 1984;305:361–364. doi: 10.1016/0006-8993(84)90443-8. [DOI] [PubMed] [Google Scholar]
- Ferguson AV, Washburn DLS. Angiotensin II: a peptidergic neurotransmitter in central autonomic pathways. Progress in Neurobiology. 1998;54:169–192. doi: 10.1016/s0301-0082(97)00065-8. [DOI] [PubMed] [Google Scholar]
- Fraser DD, MacVicar BA. Cholinergic-dependent plateau potential in hippocampal CA1 pyramidal neurons. Journal of Neuroscience. 1996;16:4113–4128. doi: 10.1523/JNEUROSCI.16-13-04113.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghamari-Langroudi M, Bourque CW. Caesium blocks depolarizing after-potentials and phasic firing in rat supraoptic neurones. The Journal of Physiology. 1998;510:165–175. doi: 10.1111/j.1469-7793.1998.165bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guatteo E, Franceschetti S, Bacci A, Avanzini G, Wanke E. A TTX-sensitive conductance underlying burst firing in isolated pyramidal neurons from rat neocortex. Brain Research. 1996;741:1–12. doi: 10.1016/s0006-8993(96)00866-9. [DOI] [PubMed] [Google Scholar]
- Hammerland LG, Garrett JE, Hung BCP, Levinthal C, Nemeth EF. Allosteric activation of the Ca2+ receptor expressed in Xenopus laevis oocytes by NPS 467 or NPS 568. Molecular Pharmacology. 1998;53:1083–1088. [PubMed] [Google Scholar]
- Hatton GI. Phasic bursting activity of rat paraventricular neurons in the absence of synaptic transmission. The Journal of Physiology. 1982;327:273–284. doi: 10.1113/jphysiol.1982.sp014231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger R, Heinemann C, Neher E, Matthews G. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature. 1994;371:513–515. doi: 10.1038/371513a0. [DOI] [PubMed] [Google Scholar]
- Inenaga K, Nagatomo T, Honda E, Ueta Y, Yamashita H. GABAergic inhibitory inputs to subfornical organ neurons in rat slice preparations. Brain Research. 1995;705:85–90. doi: 10.1016/0006-8993(95)01149-8. [DOI] [PubMed] [Google Scholar]
- Jefferys JG, Haas HL. Synchronized bursting of CA1 hippocampal pyramidal cells in the absence of synaptic transmission. Nature. 1982;300:448–450. doi: 10.1038/300448a0. [DOI] [PubMed] [Google Scholar]
- Klein M, Kandel ER. Mechanism of calcium current modulation underlying presynaptic facilitation and behavioral sensitization in Aplysia. Proceedings of the National Academy of Sciences of the USA. 1980;77:6912–6916. doi: 10.1073/pnas.77.11.6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Inoue T, Matsuo R, Masuda Y, Hidaka O, Kang Y, Morimoto T. Role of calcium conductances on spike afterpotentials in rat trigeminal motoneurons. Journal of Neurophysiology. 1997;77:3273–3283. doi: 10.1152/jn.1997.77.6.3273. [DOI] [PubMed] [Google Scholar]
- Li Z, Ferguson AV. Angiotensin II responsiveness of rat paraventricular and subfornical organ neurons in vitro. Neuroscience. 1993a;55:197–207. doi: 10.1016/0306-4522(93)90466-s. [DOI] [PubMed] [Google Scholar]
- Li Z, Ferguson AV. Subfornical organ efferents to the paraventricular nucleus utilize angiotensin as a neurotransmitter. American Journal of Physiology. 1993b;265:R302–309. doi: 10.1152/ajpregu.1993.265.2.R302. [DOI] [PubMed] [Google Scholar]
- Li Z, Ferguson AV. Electrophysiological properties of paraventricular magnocellular neurons in rat brain slices: modulation of IA by angiotensin II. Neuroscience. 1996;71:133–145. doi: 10.1016/0306-4522(95)00434-3. [DOI] [PubMed] [Google Scholar]
- Li Z, Hatton GI. Reduced outward K+ conductances generate depolarizing after-potentials in rat supraoptic nucleus neurones. The Journal of Physiology. 1997;505:95–106. doi: 10.1111/j.1469-7793.1997.095bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limonta P, Maggi R, Giudici D, Martini L, Piva F. Role of the subfornical organ (SFO) in the control of gonadotropin secretion. Brain Research. 1981;229:75–84. doi: 10.1016/0006-8993(81)90747-2. [DOI] [PubMed] [Google Scholar]
- Lind RW, Swanson LW, Ganten D. Angiotensin II immunoreactive pathways in the central nervous system of the rat: evidence for a projection from the subfornical organ to the paraventricular nucleus of the hypothalamus. Clinical and Experimental Hypertension. 1984;6:1915–1920. doi: 10.3109/10641968409046101. A. [DOI] [PubMed] [Google Scholar]
- Lind RW, Swanson LW, Ganten D. Organization of angiotensin II immunoreactive cells and fibers in the rat central nervous system. Neuroendocrinology. 1985;40:2–24. doi: 10.1159/000124046. [DOI] [PubMed] [Google Scholar]
- Lisman JE. Bursts as a unit of neural information: making unreliable synapses reliable. Trends in Neuroscience. 1997;20:38–43. doi: 10.1016/S0166-2236(96)10070-9. [DOI] [PubMed] [Google Scholar]
- McKinley MJ, Allen AM, Burns P, Colvill LM, Oldfield BJ. Interaction of circulating hormones with the brain: the roles of the subfornical organ and the organum vasculosum of the lamina terminalis. Clinical and Experimental Pharmacology and Physiology. 1998;25(suppl.):S61–67. doi: 10.1111/j.1440-1681.1998.tb02303.x. [DOI] [PubMed] [Google Scholar]
- Mantegazza M, Franceschetti S, Avanzini G. Anemone toxin (ATX II)-induced increase in persistent sodium current: effects on the firing properties of rat neocortical pyramidal neurones. The Journal of Physiology. 1998;507:105–116. doi: 10.1111/j.1469-7793.1998.105bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osaka T, Yamashita H, Koizumi K. Inhibitory inputs to the subfornical organ from the AV3V: involvement of GABA. Brain Research Bulletin. 1992;29:581–587. doi: 10.1016/0361-9230(92)90126-i. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. New York: Academic Press; 1982. [Google Scholar]
- Porter JT, Cauli B, Staiger JF, Lambolez B, Rossier J, Audinat E. Properties of bipolar VIPergic interneurons and their excitation by pyramidal neurons in the rat neocortex. European Journal of Neuroscience. 1998;10:3617–3628. doi: 10.1046/j.1460-9568.1998.00367.x. [DOI] [PubMed] [Google Scholar]
- Quan N, Whiteside M, Herkenham M. Time course and localization patterns of interleukin-1β messenger RNA expression in brain and pituitary after peripheral administration of lipopolysaccharide. Neuroscience. 1998;83:281–293. doi: 10.1016/s0306-4522(97)00350-3. [DOI] [PubMed] [Google Scholar]
- Rauch M, Schmid HA, deVente J, Simon E. Electrophysiological and immunocytochemical evidence for a cGMP-mediated inhibition of subfornical organ neurons by nitric oxide. Journal of Neuroscience. 1997;17:363–371. doi: 10.1523/JNEUROSCI.17-01-00363.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. Journal of Neuroscience. 1997;17:3425–3435. doi: 10.1523/JNEUROSCI.17-10-03425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoppa NE, Westbrook GL. Regulation of synaptic timing in the olfactory bulb by an A-type potassium current [see comments] Nature Neuroscience. 1999;2:1106–1113. doi: 10.1038/16033. [DOI] [PubMed] [Google Scholar]
- Silva LR, Amitai Y, Connors BW. Intrinsic oscillations of neocortex generated by layer 5 pyramidal neurons. Science. 1991;251:432–435. doi: 10.1126/science.1824881. [DOI] [PubMed] [Google Scholar]
- Simpson JB, Routenberg A. Subfornical organ: site of drinking elicitation by angiotensin II. Science. 1973;181:1172–1174. doi: 10.1126/science.181.4105.1172. [DOI] [PubMed] [Google Scholar]
- Smith PM, Beninger RJ, Ferguson AV. Subfornical organ stimulation elicits drinking. Brain Research Bulletin. 1995;38:209–213. doi: 10.1016/0361-9230(95)00088-v. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Smith PM, Ferguson AV, Pittman QJ. Circumventricular organs and fever. American Journal of Physiology. 1997;273:R1690–1695. doi: 10.1152/ajpregu.1997.273.5.R1690. [DOI] [PubMed] [Google Scholar]
- Vallieres L, Lacroix S, Rivest S. Influence of interleukin-6 on neural activity and transcription of the gene encoding corticotrophin-releasing factor in the rat brain: an effect depending upon the route of administration. European Journal of Neuroscience. 1997;9:1461–1472. doi: 10.1111/j.1460-9568.1997.tb01500.x. [DOI] [PubMed] [Google Scholar]
- Verhage M, McMahon HT, Ghijsen WE, Boomsma F, Scholten G, Wiegant VM, Nicholls DG. Differential release of amino acids, neuropeptides, and catecholamines from isolated nerve terminals. Neuron. 1991;6:517–524. doi: 10.1016/0896-6273(91)90054-4. [DOI] [PubMed] [Google Scholar]
- Voisin DL, Simonian SX, Herbison AE. Identification of estrogen receptor-containing neurons projecting to the rat supraoptic nucleus. Neuroscience. 1997;78:215–228. doi: 10.1016/s0306-4522(96)00551-9. [DOI] [PubMed] [Google Scholar]
- Washburn DLS, Beedle AM, Ferguson AV. Inhibition of subfornical organ neuronal potassium channels by vasopressin. Neuroscience. 1999a;93:349–359. doi: 10.1016/s0306-4522(99)00125-6. [DOI] [PubMed] [Google Scholar]
- Washburn DLS, Smith PM, Ferguson AV. Control of neuronal excitability by an ion sensing receptor. European Journal of Neuroscience. 1999b;11:1947–1954. doi: 10.1046/j.1460-9568.1999.00619.x. [DOI] [PubMed] [Google Scholar]
- West JW, Numann R, Murphy BJ, Scheuer T, Catterall WA. A phosphorylation site in the Na+ channel required for modulation by protein kinase C. Science. 1991;254:866–868. doi: 10.1126/science.1658937. [DOI] [PubMed] [Google Scholar]
- Wheeler DB, Randall A, Tsien RW. Changes in action potential duration alter reliance of excitatory synaptic transmission on multiple types of Ca2+ channels in rat hippocampus. Journal of Neuroscience. 1996;16:2226–2237. doi: 10.1523/JNEUROSCI.16-07-02226.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]