

Abstract
We tested the hypothesis that reflexes arising from working respiratory muscle can elicit increases in sympathetic vasoconstrictor outflow to limb skeletal muscle, in seven healthy human subjects at rest.
We measured muscle sympathetic nerve activity (MSNA) with intraneural electrodes in the peroneal nerve while the subject inspired (primarily with the diaphragm) against resistance, with mouth pressure (PM) equal to 60 % of maximal, a prolonged duty cycle (TI/TTot) of 0.70, breathing frequency (fb) of 15 breaths min−1 and tidal volume (VT) equivalent to twice eupnoea. This protocol was known to reduce diaphragm blood flow and cause fatigue.
MSNA was unchanged during the first 1–2 min but then increased over time, to 77 ± 51 % (s.d.) greater than control at exhaustion (mean time, 7 ± 3 min). Mean arterial blood pressure (+12 mmHg) and heart rate (+27 beats min−1) also increased.
When the VT, fb and TI/TTot of these trials were mimicked with no added resistance, neither MSNA nor arterial blood pressure increased.
MSNA and arterial blood pressure also did not change in response to two types of increased central respiratory motor output that did not produce fatigue: (a) high inspiratory flow rate and fb without added resistance; or (b) high inspiratory effort against resistance with PM of 95 % maximal, TI/TTot of 0.35 and fb of 12 breaths min−1. The heart rate increased by 5–16 beats min−1 during these trials.
Thus, in the absence of any effect of increased central respiratory motor output per se on limb MSNA, we attributed the time-dependent increase in MSNA during high resistance, prolonged duty cycle breathing to a reflex arising from a diaphragm that was accumulating metabolic end products in the face of high force output plus compromised blood flow.
Reflexes arising in working limb muscles, along with augmented neural output from locomotor areas of the central nervous system, are responsible for the sympathetically mediated increase in arterial blood pressure and redistribution of cardiac output that occurs during exercise (Rowell et al. 1996). These reflexes, which are activated during muscular contraction by local factors such as mechanical deformation of nerve endings (Victor et al. 1989), accumulation of metabolic by-products in the interstitium (Sinoway et al. 1994) and vascular distension (Haouzi et al. 1999), augment sympathetic vasoconstrictor outflow to both resting and exercising skeletal muscle (Hansen et al. 1994). It is well established that group III and IV nerve fibres arising from free nerve endings in limb muscle comprise the afferent arm of this reflex (Mitchell et al. 1983).
The diaphragm is also richly innervated by group III and IV afferent nerve fibres (Hinsey et al. 1939; Duron, 1981). In anaesthetized rats, single unit discharge in group IV afferents is augmented during fatiguing, electrically induced diaphragmatic contractions (Hill, 2000). Furthermore, electrical or chemical stimulation of phrenic nerve afferents in anaesthetized animals has been shown to elicit increases in sympathetic outflow to and vasoconstriction in several vascular beds (Szulczyk et al. 1988; Hussain et al. 1991; Offner et al. 1992). However, the role played by these afferents under physiological conditions of increased diaphragmatic contraction, such as occurs during exercise or other types of hyperpnoea, has not been investigated.
In humans exercising at maximal workloads, increases and decreases in the work of the respiratory muscles caused vasoconstriction and vasodilatation in the exercising limb, and increases and decreases in noradrenaline (norepinephrine) spillover across the limb, respectively, with no coincident changes in systemic arterial blood pressure (Harms et al. 1997). These findings suggest that respiratory muscle work per se is an important determinant of vascular conductance in exercising limb muscle, at least under conditions in which cardiac output is at maximal levels. We hypothesized that reflexes arising from respiratory muscles subjected to high levels of force output with limited available blood flow will elicit increases in sympathetic vasoconstrictor outflow to skeletal muscle (Harms et al. 1997; Wetter et al. 1999). To test this hypothesis, we measured muscle sympathetic nerve activity (MSNA) using intraneural electrodes in the human at rest, while we manipulated potential influences on sympathetic outflow such as respiratory muscle force output, breathing pattern and the level of central respiratory motor output. The findings obtained under these narrowly defined experimental conditions point to a significant role for feedback reflexes evoked by fatiguing contractions of inspiratory muscles in determining sympathetic vasoconstrictor outflow to limb muscle.
METHODS
General procedures
Four women and three men aged 35 ± 10 years (range, 25- 50 years), and of normal weight (69 ± 17 kg) and height (174 ± 19 cm), served as subjects after providing written, informed consent. All subjects were normotensive and free from cardiovascular and pulmonary disease. All experimental procedures and protocols were approved by the University of Wisconsin Center for Health Sciences and the Middleton Memorial Veteran’s Hospital Human Research Review Committees and conformed with the Declaration of Helsinki. Subjects breathed through a mouthpiece with nose occluded. Airflow rates, tidal volume (VT), mouth pressure (PM) and the end-tidal partial pressure of CO2 were measured using equipment and techniques described previously (Wilson et al. 1998). A single lead electrocardiogram was continuously recorded. A diaphragmatic electromyogram (EMGdi) was obtained from surface electrodes (3M Red Dot, 3M, St Paul, MN, USA) placed over the sixth and seventh intercostal spaces in the anterior axillary line. The raw EMGdi signal was amplified and bandpass filtered from 30 to 1000 Hz (model P511; Grass Instruments, Quincy, MA, USA). Ribcage and abdominal excursions were measured using a direct current-coupled respiratory inductive plethysmograph (Respitrace; Ambulatory Monitoring, Ardsley, NY, USA). Blood pressure was measured beat by beat using the finger photoplethysmography technique (Finapres model 2300; Ohmeda, Englewood, CO, USA) and at 1 min intervals with an automated sphygmomanometer (Dinamap model 1846 SX/P; Critikon, Tampa, FL, USA).
Recordings of sympathetic nerve activity
Multiunit recordings of postganglionic muscle sympathetic nerve activity were obtained from the right peroneal nerve as described previously (Vallbo et al. 1979; Morgan et al. 1996). The neural signals were passed to a differential preamplifier, an amplifier, a bandpass filter (700- 2000 Hz) and an integrator (time constant, 100 ms; total gain, 100000). When acceptable MSNA recordings (spontaneous pulse synchronous activity with signal-to-noise ratio > 3:1) were obtained, the subject was instructed to maintain the leg in a relaxed position for the duration of the study. Segments of the neural recording that showed evidence of mechanoreceptor or α-motoneuron activity were excluded from the analysis.
Determination of maximal inspiratory pressures (MIPs)
Prior to obtaining the nerve recording, subjects were asked to perform a maximal inspiratory effort at functional residual capacity against an occluded airway. The manoeuvre was repeated several times at 1 min intervals until reproducible maximal peak values were obtained.
Experimental protocols
Protocol 1A: 60 % maximum inspiratory pressure + 0.70 duty cycle (task failure)
This trial was designed to cause fatigue of the diaphragm. The subject inspired against an added resistive load to a target PM of 60 % MIP by following a tracing of PM on an oscilloscope to a preset target for each inspiration. Throughout the trial the subjects maintained a constant respiratory frequency (fb; 15 breaths min−1) and prolonged duty cycle (inspiratory time (TI)/total time (TTot); 0.70) by listening to a computer-generated audio signal with distinct inspiratory and expiratory tones. During each inspiratory effort, subjects were instructed to: (1) maintain a square wave in PM throughout each inspiration; and (2) isolate the diaphragm using feedback from the inductance plethysmograph signal as an index of abdominal excursions. This fatiguing breathing trial was continued to the point of task failure (defined as the point at which the subject could not achieve or maintain the target PM despite verbal encouragement). Throughout the trial, subjects were instructed to avoid inadvertent contraction of non-respiratory muscles. End-tidal PCO2 was maintained at eucapnoeic levels by increasing the inspired CO2 fraction (FI,CO2), for this trial and all of the following trials.
Protocol 1B: 2 % maximum inspiratory pressure + 0.70 duty cycle (no task failure)
The purpose of this protocol was to determine the effect of increased VT and TI/TTot (in the absence of fatigue) on MSNA. The subjects were asked to mimic for 2 min the breathing pattern, fb and VT used in the fatiguing trial. However, during this trial, the inspiratory resistance was removed and the work was not fatiguing. Auditory and visual feedback was provided as described above.
Protocol 2A: hyperpnoea (no task failure)
The purpose of this protocol was to determine the effect of increased central respiratory motor output (in the absence of fatigue) on MSNA by having the subject maintain, using feedback, very high airflow rates and increased velocity of inspiratory muscle shortening at high VT (1.5 l) and fb (45 breaths min−1) with a normal TI/TTot (0.4). Hyperoxia (inspired O2 fraction (FI,O2) = 0.5) was maintained during these trials to prevent the possibility of a limited oxygen supply to the working inspiratory muscles. The hyperpnoea trials were continued for 2 min.
Protocol 2B: 95 % maximum inspiratory pressure + 0.35 duty cycle (no task failure)
The purpose of this protocol was to determine the effect of near maximal central respiratory motor output (in the absence of task failure) on MSNA. The subject was asked to inspire for 2 min in hyperoxia (FI,O2= 0.50) against an added resistive load to a target PM of 95 % MIP at an fb of 12 breaths min−1 and TI/TTot of 0.35.
Protocol 2C: 2 % maximum inspiratory pressure + 0.35 duty cycle (no task failure)
The purpose of this protocol was to determine the effect of the increased VT and shortened TI/TTot (but without the added inspiratory resistance) used during the 95 % MIP trial on MSNA.
Protocol 3: 60 % maximum voluntary contraction + 0.70 duty cycle: intermittent handgrip exercise to task failure
Subjects performed rhythmic handgrip exercise at 60 % maximum voluntary contraction using visual feedback of force output on the oscilloscope. Contraction frequency (15 contractions min−1) and duty cycle (0.70) were maintained using the computer-generated audio signals. Subjects were encouraged to continue the exercise until they were unable to maintain the required force output. Throughout the trial, the subjects maintained a constant respiratory frequency and near eupnoeic (spontaneous) levels of VT.
Spontaneous breathing
At regular intervals throughout the experimental session, data were collected during 5 min of eupnoeic breathing. These eupnoeic control sessions served as the baseline control period for each of the experimental manipulations of breathing pattern and inspiratory muscle force output.
Data analysis
All tracings were recorded continuously on paper (Gould, Cleveland, OH, USA) and on videotape (Vetter, Rebersburg, PA, USA). Respiratory and cardiovascular parameters were computed as described previously (Taha et al. 1995). Bursts of MSNA were identified by visual inspection of the integrated neurogram by the same investigator (C.M.StC.). MSNA was quantified by calculating the number of bursts per minute and the total amount of minute activity (burst frequency × mean burst amplitude) expressed in arbitrary units per minute (a.u. min−1).
Mean values for MSNA, blood pressure and heart rate in each condition were compared using one-way ANOVA with repeated measures. Post hoc analyses (Tukey’s test) were performed if differences were detected. Significance was set at P < 0.05.
The arterial blood pressure and MSNA signals were signal averaged over the breath cycles. Data were digitally recorded at 128 Hz using custom data acquisition software. MSNA was advanced in time to account for nerve conduction delay using the subject’s height and an estimate of conduction velocity in the peroneal nerve of 1.11 m s−1 (Fagius & Wallin, 1980). Diastolic blood pressure was converted into a continuous signal, advancing the time by 0.2 s to correct for the propagation time from the central circulation to the finger. MSNA data were read into buffers for inspiration and expiration. The data were then normalized to breath length and resampled at 10 Hz. These data were imported into Microsoft Excel and signal averaged. Thus, the signal-averaged signal is a timed activity curve of MSNA or arterial blood pressure averaged over 15 consecutive diaphragmatic contractions and relaxations.
Values are given as means ±s.d.
RESULTS
Variability of MSNA and cardiovascular measurements over time
During seven to nine periods of eupnoeic breathing repeated at regular intervals throughout the experimental session, there were no systematic trends in MSNA, mean arterial blood pressure or heart rate over time (P > 0.05). Over all control periods of eupnoeic breathing, average values were 61 ± 6 beats min−1 for heart rate, 88 ± 8 mmHg for mean arterial blood pressure and 19 ± 6 bursts min−1 for MSNA. Nevertheless, there was some random variability over time within each of the seven subjects, with an average coefficient of variation of ±10.5 % for MSNA, ±6.1 % for mean arterial blood pressure and ±3.1 % for heart rate. Therefore, we believed it was essential to quantify the MSNA, heart rate and blood pressure responses to each of the experimental conditions by comparison with their corresponding preceding control period of eupnoeic breathing.
Time-dependent effects of increased respiratory motor output with diaphragm task failure (protocols 1A and 1B; n = 7)
Breathing at 60 % MIP with a prolonged duty cycle of 0.70 and a frequency of 15 breaths min−1 produced task failure, as manifested: (a) initially, by the inability, after the first 6–15 inspiratory efforts, to maintain the target inspiratory PM throughout the prolonged inspiration, even though the target PM could be achieved transiently at the onset of inspiration; and (b) finally, after 2.5–8 min, either the inability to reach even the target nadir inspiratory PM using the diaphragm, or an unwillingness to continue. Figure 1A and B shows polygraph records for two representative subjects who reached task failure either very quickly (2.5 min; A) or after 8 min (B) of high levels of inspiratory muscle work. There was a time-dependent increase in mean arterial blood pressure and in the frequency and amplitude of sympathetic bursts that was not evident until after 1.5–2 min of each trial. In contrast, in protocol 1B increases in fb, VT and prolonged duty cycle, which were equivalent to those in the fatiguing protocol 1A but without added inspiratory resistance, had no effect on either arterial blood pressure or MSNA (see Fig. 1C).
Figure 1. Raw data from subjects during protocols 1A and 1B.
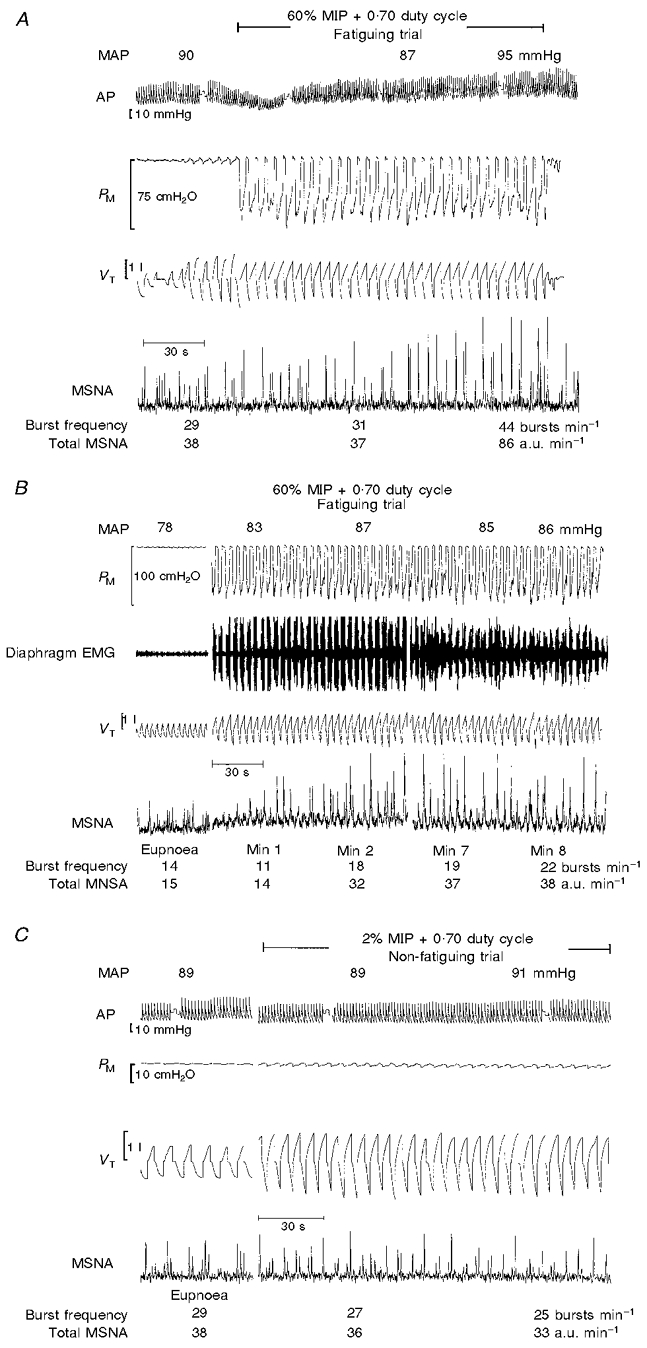
A, raw data from representive subject X who reached task failure relatively quickly (2.5 min) during protocol 1A. Note that both the frequency and amplitude of sympathetic nerve activity were unchanged during the first minute of increased diaphragm force output, and thereafter rose in a time-dependent manner. B, raw data from another representive subject Y who reached task failure after 8 min during protocol 1A. Note that the frequency and amplitude of sympathetic nerve activity were unchanged during the first 30 s, began to increase during minute (Min) 2 of the trial and increased to 157 and 153 % of baseline, respectively, by the end of minute 8. C, raw data from representative subject X during protocol 1B, which was designed to mimic the increased VT and fb, and prolonged TI/TTot during the fatiguing trial (1A) but without the added inspiratory resistance. Note that the frequency and amplitude of sympathetic activity were unchanged over the entire duration of the trial. AP, arterial blood pressure; MAP, mean arterial blood pressure; a.u., arbitrary units.
Data for all individual subjects for both protocols (1A and 1B) conducted at high VT and prolonged duty cycle are shown in Fig. 2 and group mean data in Table 1. For the task failure trials, integrated PM per breath and per minute increased to about 200 times eupnoeic levels. All seven subjects showed time-dependent increases in MSNA that averaged +77 ± 51 % (range, 26–230 %; P < 0.05; Fig. 2 and Table 1) at the time of task failure (6.8 ± 2.9 min; range, 2.5–9.0 min). In six of the seven subjects, the increases in MSNA were not evident until the second minute of the task failure trial, and the average increase in MSNA did not reach statistical significance until minute 3 (+17.5 ± 34.5 % increase; P < 0.05). In the remaining subject, MSNA increased by 11 % during the first 30 s of the trial and continued to increase to 130 % of the eupnoeic level at the end of the trial. Mean arterial blood pressure rose in all seven subjects (+12 ± 7 mmHg; range, +10 to +25 mmHg at task failure); however, these increases were highly variable among subjects and did not reach statistical significance (P = 0.16). Heart rate also increased in all subjects within the first minute and remained constant thereafter, averaging +27 ± 11 beats min−1 at task failure (P < 0.05). The MSNA, arterial blood pressure and heart rate responses during protocol 1A were found to be highly reproducible in four subjects who repeated the fatiguing trial twice, either within the same day or on different days (data not shown).
Figure 2. Data for individual subjects (n = 7) for protocols 1A and 1B.
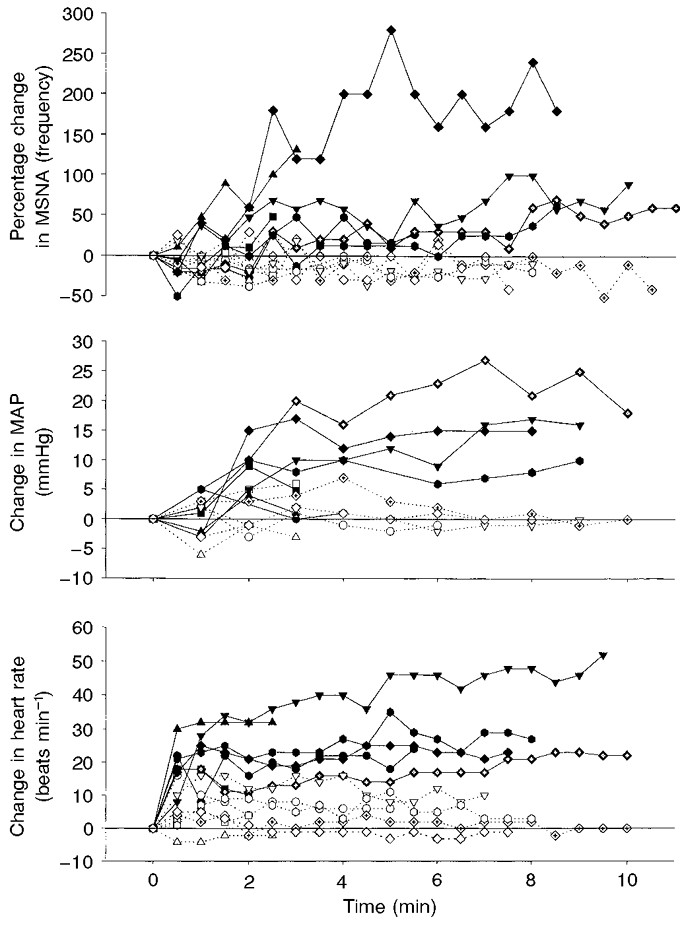
Filled symbols and continuous lines: protocol 1A, fatiguing trial at 60 % MIP + 0.70 duty cycle. Open symbols and dotted lines: protocol 1B, non-fatiguing trial at 2 % MIP + 0.70 duty cycle, which mimicked the changes in breathing pattern during the fatiguing trial. The time-dependent increase in MSNA during fatiguing breathing (protocol 1A) reached statistical significance (P < 0.05) at minute 3 of the trial. There was no change in MSNA during protocol 1B.
Table 1.
Group mean data
| MSNA (% change) | ΔMAP (mmHg) | ΔHeart rate (beats min−1) | VT (1) | fb (breaths min−1) | T1/TTot | Peak PμM (cm H2O) | ∫PM | ∫PM× fb | VT/TI (1 s−1) | Pco2 (mmHg) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Eupnoea | 0 | 0 | 0 | 0.673 ± 0.196 | 13.3 ± 4.6 | 0.38 ± 0.03 | −0.68 ± 0.28 | −0.68 ± 0.39 | −7.52 ± 4.18 | 0.36 ± 0.06 | 37.8 ± 2.3 |
| Protocol 1A | |||||||||||
| Min 1 | −3.2 ± 37.3 | 0.9 ± 4.1 | 20.6 ± 9.5* | 1.056 ± 0.282† | 14.9 ± 0.1 | 0.71 ± 0.05† | −68.3 ± 18.0* | −140.9 ± 25.5* | 2073 ± 404* | 0.38 ± 0.11 | 38.2 ± 5.0 |
| Min 2 | 22.2 ± 33.6 | 10.1 ± 4.4 | 22.1 ± 9.6* | 1.107 ± 0.295† | 15.0 ± 0.2 | 0.73 ± 0.05† | −70.4 ± 17.9* | −145.9 ± 29.4* | −2177 ± 444* | 0.38 ± 0.11 | 40.2 ± 4.6 |
| End | 76.9 ± 51.1* | 12.4 ± 7.4 | 27.0 ± 11.3* | 1.044 ± 0.360† | 15.1 ± 0.5 | 0.71 ± 0.05† | −71.8 ± 17.5* | −131.3 ± 27.4* | −1970 ± 414* | 0.37 ± 0.13 | 41.3 ± 4.5 |
| Protocol 1B | |||||||||||
| Min 1 | −5.5 ± 21.4 | 1.0 ± 4.2 | 6.0 ± 5.4 | 1.106 ± 0.315† | 15.0 ± 1.4 | 0.64 ± 0.05† | −2.0 ± 0.9 | −1.7 ± 0.8† | −25.1 ± 11.2† | 0.43 ± 0.12 | 40.1 ± 4.4 |
| Min 2 | −7.7 ± 18.6 | 1.4 ± 4.2 | 5.7 ± 5.9 | 1.134 ± 0.288† | 15.0 ± 1.2 | 0.65 ± 0.03† | −2.2 ± 0.9 | −1.7 ± 0.8† | −26.3 ± 10.8† | 0.43 ± 0.09 | 41.0 ± 5.5 |
| End | −5.5 ± 19.0 | 1.1 ± 3.6 | 4.1 ± 4.7 | 1.114 ± 0.267† | 14.9 ± 1.1 | 0.66 ± 0.05† | −2.1 ± 0.7 | −1.9 ± 0.8† | −27.4 ± 11.4† | 0.42 ± 0.10 | 40.6 ± 4.4 |
| Protocol 2A | |||||||||||
| Min 1 | −12.1 ± 37.2 | 3.8 ± 1.2† | 20.5 ± 5.9† | 1.221 ± 0.055† | 42.5 ± 2.3† | 0.49 ± 0.01† | −11.0 ± 1.9† | −4.9 ± 0.5† | −206.1 ± 31.6† | 1.78 ± 0.17† | 38.0 ± 6.5 |
| Min 2 | 0.4 ± 18.6 | 8.0 ± 3.3† | 16.3 ± 8.9† | 1.237 ± 0.083† | 42.6 ± 2.6† | 0.49 ± 0.01† | −10.2 ± 2.7† | −4.7 ± 1.3† | −199.0 ± 62.1† | 1.79 ± 0.20† | 38.1 ± 4.9 |
| Protocol 2B | |||||||||||
| Min 1 | −9.8 ± 28.5 | −1.8 ± 7.5 | 6.3 ± 7.8 | 0.933 ± 0.218 | 12.0 ± 0.1 | 0.38 ± 0.08 | −94.7 ± 27.1† | −116.0 ± 32.3† | −1379 ± 380† | 0.49 ± 0.05† | 38.5 ± 3.6† |
| Min 2 | 4.4 ± 25.1 | 0.5 ± 5.1 | 9.3 ± 5.0 | 0.970 ± 0.215† | 12.0 ± 0.1 | 0.38 ± 0.08 | −99.3 ± 28.6† | −125.0 ± 30.4† | −1503 ± 348† | 0.51 ± 0.04† | 40.5 ± 4.4 |
| Protocol 2C | |||||||||||
| Min 1 | −15.7 ± 16.6 | −0.3 ± 4.5 | −0.7 ± 2.1 | 1.270 ± 0.404 | 12.1 ± 0.1 | 0.35 ± 0.04 | −1.9 ± 0.6 | −1.7 ± 1.1 | −20.2 ± 13.0† | 0.67 ± 0.09† | 36.1 ± 0.4 |
| Min 2 | 2.9 ± 6.0 | 0.3 ± 2.1 | −0.7 ± 2.5 | 1.187 ± 0.215 | 11.9 ± 0.3 | 0.34 ± 0.08 | −2.0 ± 0.9 | −1.9 ± 1.1 | −23.4 ± 13.8† | 0.68 ± 0.14† | 39.0 ± 4.1 |
Values are means ± S.D. Protocol 1A: 60% MIP + 0.70 duty cycle, fatiguing (n = 7). Protocol 1B: 2% MIP + 0.70 duty cycle, non-fatiguin (n = 7). Protocol 2A: hyperpnoean, non-fatiguing (n = 6). Protocol 2B: 95% MIP + 0.35 duty cycle, non-fatiguing (n = 4). Protocol 2C: 2% MIP + 0.35 duty cycle, non-fatiguin (n = 3).
Significantly different from eupnoea, and from non-fatiguing trial at 2% MIP + 0.70 duty cycle (protocol 1B; P < 0.05).
Significantly different from eupnoea (P < 0.05). End-tidal Po2 averaged 110 mmHg for protocols 1A and 1B and 350 mmHg for protocols 2A, 2B and 2C. ∫PM, integrated mouth pressure; ∫PM × fb, average ∫PM per breath multiplied by breathing frequency.
Increases in VT and duty cycle without added resistance or task failure (protocol 1B) generated 4-fold increases in integrated PM per breath and per minute and in the nadir of inspiratory pressure. In contrast to the trials with task failure, these trials had no effect on MSNA, arterial blood pressure or heart rate in any of the seven subjects (Fig. 2 and Table 1).
Effects of increased respiratory motor output (high flow rate and breathing frequency, low pressure, normal duty cycle) without diaphragm task failure (protocol 2A; n = 6)
Inspiratory muscle effort was significantly increased relative to eupnoea, as noted by the more negative nadir of inspiratory pressure (−10 vs.−1 cmH2O). Integrated PM per breath increased to 7 times that in eupnoea and per minute to 25 times the eupnoeic level (Table 1). A raw polygraph record from a representative subject is shown in Fig. 3. MSNA remained unchanged in all subjects over the 2 min of these trials. This hyperpnoeic (non-task failure) breathing caused significant increases in heart rate (+16 ± 9 beats min−1) and in mean arterial blood pressure (+8 ± 3 mmHg), which were evident within the first minute of increased respiratory effort and sustained over the 2 min period (both P < 0.05; Table 1).
Figure 3. Raw data from representative subject X during the hypernoeic protocol.
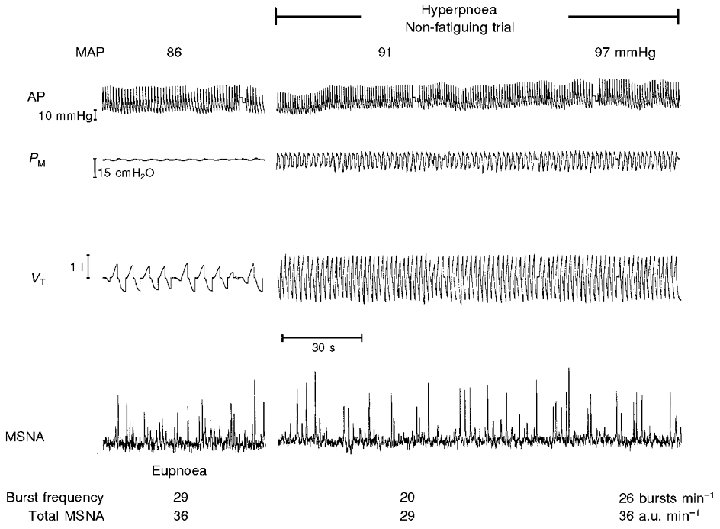
Protocol 2A, high flow rate and breathing frequency without task failure. Note that the frequency and amplitude of sympathetic activity were unchanged over the 2 min duration of the trial.
Effects of near maximal inspiratory pressures without task failure (protocol 2B; n = 4)
Data from these trials were discarded from three of the seven subjects due to changes in the baseline level of the MSNA recording and/or loss of the nerve recording caused by an inability to maintain the leg in a stable position during these very forceful breathing manoeuvres. Two minutes of breathing at 95 % MIP with a shortened duty cycle of 0.35 and slightly below normal fb required an 180-fold increase in integrated PM per breath and a 200-fold increase in integrated PM per minute. Figure 4A shows a polygraph record from a representative subject in whom no increase in MSNA was apparent over the 2 min of this trial. The responses of all subjects who participated in this trial are shown in Fig. 4B. Three of the four subjects showed no change in MSNA during these trials (also see below). The heart rate increased within the first minute of increased diaphragm force output in three of the four subjects and mean arterial blood pressure was variable. The breathing pattern of protocol 2B was mimicked in protocol 2C in three of the four subjects without any added inspiratory resistance. These changes in breathing pattern alone caused no systematic changes in MSNA, blood pressure or heart rate (see Table 1).
Figure 4. Raw data from a representative subject and data for individual subjects for protocols 2B and 2C.
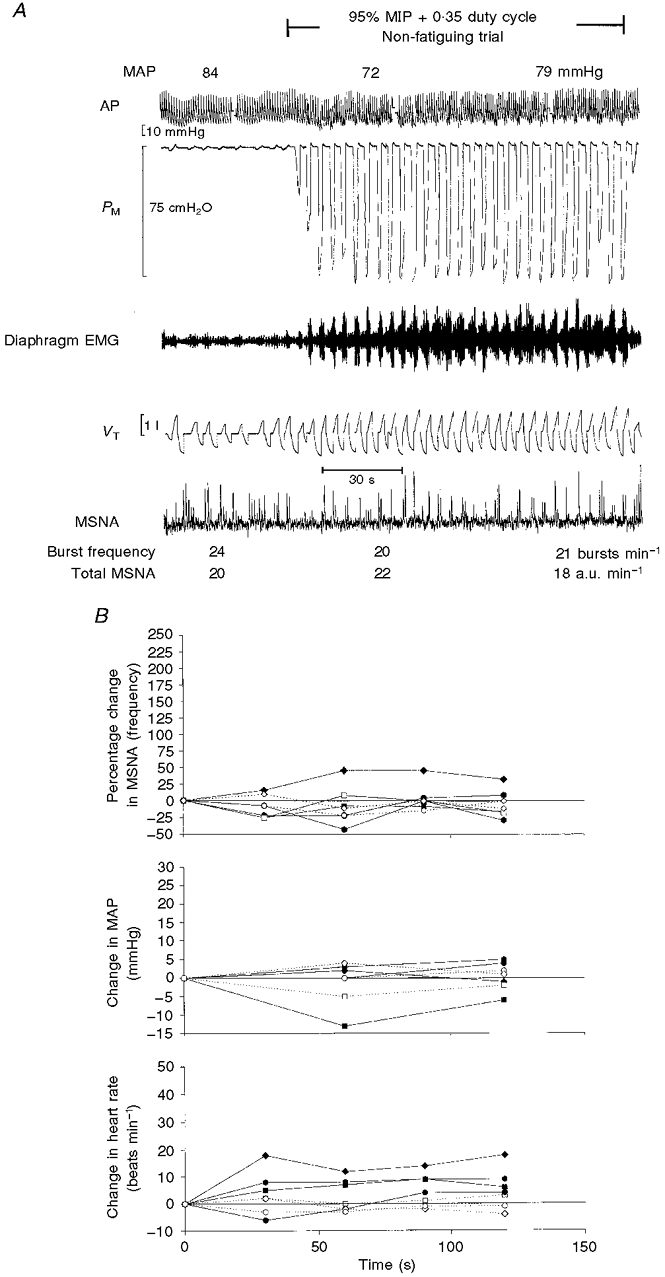
A, raw data from representative subject X during protocol 2B (near maximal inspiratory pressures without task failure). Note that the frequency and amplitude of sympathetic activity were unchanged over the 2 min duration of the trial. B, data for individual subjects for protocol 2B (95 % MIP + 0.35 duty cycle; filled symbols; n = 4) and protocol 2C (2 % MIP + 0.35 duty cycle; open symbols; n = 3). In one of four subjects, MSNA (bursts min−1) increased by 40 % by the end of the first minute of the trial. This same subject showed a 230 % increase in MSNA during the fatiguing trial (protocol 1A; see Fig. 2, ♦).
One subject did show an increase in MSNA during protocol 2B, which was highly reproducible over two trials on different test days. This increase in MSNA was substantially less than the increase caused by the fatiguing breathing trials (protocol 1A) with prolonged duty cycle and task failure (+40 % vs.+230 % of baseline MSNA). Like other subjects, this subject did not show an increase in MSNA during the control trial at the same VT, fb and duty cycle (protocol 2C). This finding suggests that the 40 % increase in MSNA produced by the near-maximal inspiratory pressure trial was due to factors associated with the high inspiratory effort rather than the changes in breathing pattern. This subject showed the greatest increase in heart rate and the most negative inspiratory pressures (−121 cmH2O). This subject also showed, uniquely, an increase in MSNA during the high flow rate trial (+30 % MSNA); however, as for all other subjects, MSNA in this subject did not increase during the first 2 min of the fatiguing trial at 60 % MIP.
Timing of MSNA during the breath cycle
The temporal patterns of diastolic blood pressure and MSNA within a breath, as derived from cycle triggered, signal-averaged data over 15 breaths, early and late in the diaphragm fatiguing trial, are shown in Fig. 5. Two representative subjects are shown: subject 1 with an increase in MSNA of 50 % greater than eupnoea during the fatigue trial and an increase in mean arterial blood pressure of 10 mmHg, which was slightly below the group mean for all seven subjects, and subject 2 who had the largest (> 200 %) increase in MSNA and mean arterial blood pressure (+ 20–25 mmHg) of all subjects. A characteristic common to all fatiguing or non-fatiguing trials and to all subjects was that MSNA was always lowest during most or at least the latter 2/3 of inspiration and highest during expiration, especially the latter 2/3 of expiration. The difference between the two subjects shown in Fig. 5 in terms of the time-dependent effect of the diaphragm fatiguing trials was that in subject 1 (as in 5 other subjects) the increase in MSNA occurred primarily during expiration and occasionally also during the beginning of inspiration, whereas in subject 2 the higher minute MSNA over time occurred throughout inspiration and expiration.
Figure 5. Temporal relationships of breathing with MSNA.
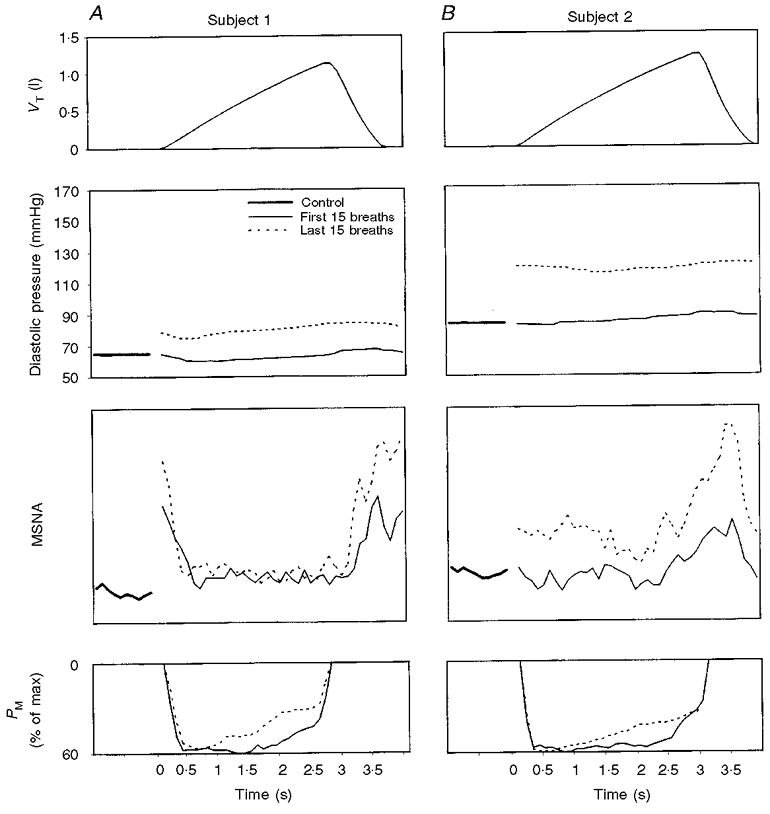
A and B show examples for 2 subjects of signal averaging of VT, diastolic blood pressure and MSNA over 15 breaths during the initial and the final minute of fatiguing trials conducted at 60 % of maximum PM with a duty cycle of 0.7 and frequency of 15 breaths min−1 (see text). The plot for control eupnoeic conditions refers to signal-averaged data over 15 consecutive 4 s periods of spontaneous breathing immediately prior to each of the fatigue trials. During the fatiguing trials, MSNA frequency (bursts min−1) in subject 1 was 25 % less than control in minute 1 and 50 % greater than control in minute 3; in subject 2 MSNA frequency was 15 % greater than control in minute 1 and 225 % greater than control in minute 8 (see Fig. 2).
DISCUSSION
We observed a time-dependent increase in MSNA in the resting lower limb when very high levels of inspiratory muscle force output were generated using a prolonged duty cycle that limited muscle perfusion. Increases in tidal volume or breath duty cycle or in central respiratory motor output in the absence of fatigue did not affect MSNA; therefore, we attribute these changes in MSNA to a reflex arising in fatiguing respiratory muscles which increases sympathetic outflow to the resting limb.
Influence of increased central respiratory motor output on MSNA
During the fatiguing trials in our subjects it is likely that central respiratory motor output increased over time, giving rise to recruitment of additional motor units so that force output could be maintained as fatigue ensued. Nevertheless, several lines of evidence argue against an important role for ‘central command’ in causing the sympathetic activation associated with these manoeuvres. First, during the fatiguing task, integrated PM and EMGdi measurements showed that respiratory motor output was quite high beginning with the first diaphragm contraction, yet there was no significant increase in MSNA until at least the second minute of the trial. Again, we acknowledge that the central respiratory output also probably increased progressively as the diaphragm became more fatigued and additional motor units were recruited.
Second, with one notable exception (see Fig. 4B), MSNA was unchanged during the high resistive breathing trials (95 % MIP with short duty cycle), which evoked near-maximum recruitment of the diaphragm without incurring fatigue. The purpose of these trials was to mimic the high levels of central respiratory motor output that might be expected at the end of the fatiguing trial. The exceptional subject showed a rapid (< 30 s) and reproducible increase in MSNA in response to non-fatiguing, resistive breathing at 95 % MIP. This subject also generated the most negative pressures (−121 cmH2O) and showed the largest increase in heart rate (+19 beats min−1) among all subjects. While the 40 % increase in MSNA was small in comparison with the 230 % increase this subject showed during the diaphragm fatiguing trial, it does suggest that extreme levels of central respiratory motor output per se may influence MSNA during heavy respiratory muscle work.
Third, an increase in both the rate and amplitude of central respiratory motor output with voluntary increases in inspiratory flow rate and rate of rise of EMGdi to near maximum levels – but without fatigue – was also without significant effect on sympathetic outflow. It is noteworthy that all trials that increased central inspiratory motor output (attained by either high resistance or by high flow rate) produced immediate and sustained increases in heart rate. The current concept is that such changes are manifestations of the inhibitory effect of ‘central command’ on cardiac vagal outflow (Mark et al. 1985).
Finally, in none of our trials of high central respiratory motor output – including the latter portions of the diaphragm fatiguing trials when overall MSNA was increasing – did we observe a temporal association between inspiratory effort and sympathetic outflow. Limited evidence from humans who performed voluntary limb muscle contractions would predict a synchronization of motor output and sympathetic bursts in circumstances where ‘central command’ exerted a significant influence over MSNA. Victor et al. (1995) demonstrated that brief, static forearm contractions coincided in time with MSNA bursts only when ‘central command’ was very high, such as occurred with a handgrip of 75 % of maximum force output or with maximum effort during neuromuscular blockade, when force output was very small.
We did not see such congruence in our experiments. Rather, MSNA was inhibited at high lung volumes during inspiration and increased at low lung volumes during expiration. This respiration-linked MSNA pattern held for all experimental conditions, i.e. fatigue vs. no fatigue of the diaphragm, a high vs. low central respiratory motor output, and prolonged vs. normal breath duty cycle. It is important to note that, in our experiments, high central respiratory output (‘central command’) coincided with inspiration and lung inflation, during which MSNA was markedly attenuated rather than enhanced, apparently because the inhibitory influences from peripheral feedback mechanisms are more important than central respiratory motor output in causing respiratory modulation of MSNA in the intact human or cat (Boczek-Funcke et al. 1992; Seals et al. 1993; St Croix et al. 1999).
Influence of reflexes from contracting diaphragm muscle on MSNA
Our data suggest that reflexes arising in the fatiguing diaphragm have a powerful influence on MSNA to resting skeletal muscle. The time-dependent 77 ± 51 % increase in MSNA during resistive breathing occurred and persisted even in the face of inhibitory feedback from systemic baroreceptors due to increases in arterial blood pressure (+10 to +25 mmHg mean arterial blood pressure). Scherrer et al. (1990) demonstrated that inhibitory feedback from baroreceptors attenuated the time-dependent increase in MSNA during forearm muscle fatigue. These time-dependent increases in MSNA, arterial blood pressure and heart rate were similar to those during fatigue of the forearm muscle (protocol 3, data not shown), which was also produced via rhythmic handgrip contractions at high force output and prolonged duty cycle. The effects of this forearm protocol on MSNA confirm those previously reported using quite different duty cycles for handgrip (Scherrer et al. 1990; Hansen et al. 1994; Vissing et al. 1998).
The afferent arm of the ‘exercise pressor’ reflex arising in rhythmically contracting limb skeletal muscle is comprised of thinly myelinated type III and unmyelinated type IV fibres (McCloskey & Mitchell, 1972; Adreani et al. 1997) (see Introduction). The diaphragm is also richly innervated by group III and group IV afferent nerve fibres. Multifibre recordings from small diameter phrenic afferents show increased activity during induced diaphragmatic fatigue in anaesthetized cats (Balzamo et al. 1992; Jammes & Balzamo, 1992). Most relevant to the present findings is the recent work of Hill (2000), who recorded single unit discharge from type III and IV afferents in the diaphragm in anaesthetized rats and electrically stimulated the phrenic nerve. At the onset of phrenic nerve stimulation, neither type III nor IV afferents changed their discharge; however, as phrenic stimulation continued and diaphragmatic tension was reduced due to fatigue, type IV afferent discharge increased more than 2-fold and type III discharge was reduced.
Our protocol to produce diaphragmatic fatigue was based on the findings of Bellemare et al. (1983) and Buchler et al. (1985) in anaesthetized dogs, who used phrenic nerve electrical stimulation. They showed that combining high force output with a prolonged breathing duty cycle (i.e. high tension time index of the diaphragm or TTDI) compromised blood flow to the diaphragm, presumably because a rising abdominal pressure during inspiration compressed feed arteries to the diaphragm. When this high TTDI was imposed with a normal breathing frequency, sufficient time was not allowed during brief periods of relaxation to restore the diaphragm blood flow to levels that were adequate to meet the increased metabolic requirements. While we have no direct evidence of diaphragmatic fatigue in our study, we did observe an inability of our subjects to maintain a square wave of pressure development throughout inspiration early in trials conducted at high TTDI and eventually subjects had difficulty in achieving even the target nadir PM. These signs of ‘task failure’ were previously shown to coincide first with changes in the frequency spectrum of the EMGdi consistent with impending fatigue (Bellemare et al. 1983) and later with reductions in transdiaphragmatic pressure in response to phrenic nerve stimulation (Yan et al. 1992). Based on our monitoring of abdominal displacement during each inspiration, we believe that the diaphragm was maintained as the primary inspiratory muscle throughout our trials, although it is also likely that accessory muscles were recruited to some extent in an attempt to maintain the target nadir in PM as fatigue progressed and the perception of inspiratory effort intensified.
We considered the possibility that muscle pain and/or mental stress might have contributed to the observed increase in MSNA during fatiguing contractions of the diaphragm. Previous investigators have shown that MSNA increases during application of some, but not all, types of noxious stimuli that produce equivalent levels of pain perception (Hjemdahl et al. 1989; Nordin & Fagius, 1995). To our knowledge, the effects of muscle pain on MSNA have not been investigated systematically. However, the observation in patients with metabolic myopathies that contraction-induced increases in MSNA resolve after exercise in the face of persistent pain argues against muscle pain as a major determinant of exercise-induced sympathetic activation (Vissing et al. 1998). Some mental stress protocols (e.g. mental arithmetic, colour word test) have been shown to increase muscle sympathetic outflow to the leg in some individuals (Hjemdahl et al. 1989; Anderson et al. 1987). In our study, certainly the diaphragmatic contractions required intense effort and some subjects reported sensations of muscle discomfort towards the termination of the fatiguing trial. On the other hand, our subjects were free to interrupt the resistive breathing trials whenever they wished and alveolar CO2 was not allowed to rise nor O2 to fall so that sensory inputs arising from chemoreceptors which commonly invoke sensations of ‘smothering’ were avoided. Furthermore, none of our subjects complained specifically about mental stress produced by the experimental protocols, perhaps in part because they concentrated primarily on achieving the target nadir PM and were not really aware of their task failure until the termination of the trials, at which time they had difficulty consistently producing the 60 % MIP pressure. Therefore, although we cannot absolutely rule out an effect of mental stress we consider it unlikely that such distress contributed significantly to the activation of MSNA during the development of diaphragmatic fatigue.
In summary, our findings show a time-dependent increase in lower limb MSNA in response to fatiguing contractions of the diaphragm induced by a combination of high force output and compromised perfusion. Furthermore, increased central inspiratory motor output and/or high force output by the diaphragm were not sufficient by themselves to elicit an increase in MSNA. The specific causes of this apparent reflex effect from the fatiguing diaphragm and its influence on vascular resistance in the resting or exercising limb await further study.
Acknowledgments
This work was supported by the National Heart, Lung and Blood Institute (grants R01–15469 and R29-57401) and (in part) by a research fellowship from the American Heart Association of Wisconsin and the Hazel Mae Mayer Foundation (to C.M.StC.). We thank A. Jacques, D. Puleo and D. F. Pegelow for technical assistance.
References
- Adreani CM, Hill JM, Kaufman MC. Responses of group III and IV muscle afferents to dynamic exercise. Journal of Applied Physiology. 1997;82:1811–1817. doi: 10.1152/jappl.1997.82.6.1811. [DOI] [PubMed] [Google Scholar]
- Anderson EA, Wallin BG, Mark AL. Dissociation of sympathetic nerve activity in arm and leg muscle during mental stress. Hypertension. 1987;9(suppl. III):114–119. doi: 10.1161/01.hyp.9.6_pt_2.iii114. [DOI] [PubMed] [Google Scholar]
- Balzamo E, Lagier-Tessonnier F, Jammes Y. Fatigue-induced changes in diaphragmatic afferents and cortical activity in the cat. Respiration Physiology. 1992;90:213–226. doi: 10.1016/0034-5687(92)90082-8. [DOI] [PubMed] [Google Scholar]
- Bellemare F, Wight D, Lavigne CM, Grassino A. Effect of tension and timing of contraction on the blood flow of the diaphragm. Journal of Applied Physiology. 1983;54:1597–1606. doi: 10.1152/jappl.1983.54.6.1597. [DOI] [PubMed] [Google Scholar]
- Boczek-Funcke A, Häbler H-J, Jänig W, Michealis M. Respiratory modulation of the activity in sympathetic neurones supplying muscle, skin and pelvic organs in the cat. The Journal of Physiology. 1992;449:333–361. doi: 10.1113/jphysiol.1992.sp019089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchler B, Magder S, Kastardis H, Jammes Y, Roussos C. Effects of pleural pressure and abdominal pressure on diaphragmatic blood flow. Journal of Applied Physiology. 1985;58:691–697. doi: 10.1152/jappl.1985.58.3.691. [DOI] [PubMed] [Google Scholar]
- Duron B. Intercostal and diaphragmatic muscle afferents. In: Hornbein TR, editor. Regulation of Breathing. New York: Marcel Dekker; 1981. pp. 473–540. part 1. [Google Scholar]
- Fagius J, Wallin BG. Sympathetic reflex latencies and conduction velocities in normal man. Journal of the Neurological Sciences. 1980;47:443–448. doi: 10.1016/0022-510x(80)90098-2. [DOI] [PubMed] [Google Scholar]
- Hansen J, Thomas GD, Jacobsen TN, Victor RG. Muscle metaboreflex triggers parallel sympathetic activation in exercising and resting human skeletal muscle. American Journal of Physiology. 1994;266:H2508–2514. doi: 10.1152/ajpheart.1994.266.6.H2508. [DOI] [PubMed] [Google Scholar]
- Haouzi P, Hill JM, Lewis BK, Kaufman MP. Responses of group III and IV muscle afferents to distension of the peripheral vascular bed. Journal of Applied Physiology. 1999;87:545–553. doi: 10.1152/jappl.1999.87.2.545. [DOI] [PubMed] [Google Scholar]
- Harms CA, Babcock MA, McClaran SR, Pegelow DF, Nickele GA, Nelson WB, Dempsey JA. Respiratory muscle work compromises leg blood flow during maximal exercise. Journal of Applied Physiology. 1997;82:1573–1583. doi: 10.1152/jappl.1997.82.5.1573. [DOI] [PubMed] [Google Scholar]
- Hill JM. Discharge of group IV phrenic afferent fibers increases during diaphragmatic fatigue. Brain Research. 2000;856:240–244. doi: 10.1016/s0006-8993(99)02366-5. [DOI] [PubMed] [Google Scholar]
- Hinsey JC, Hare K, Philips RA. Sensory components of the phrenic nerve of the cat. Proceedings of the Society of Experimental Biology and Medicine. 1939;41:411–414. [Google Scholar]
- Hjemdahl P, Fagius J, Freyschuss U, Wallin BG, Daleskog M, Bohlin G, Perski A. Muscle sympathetic activity and norepinephrine release during mental challenge in humans. American Journal of Physiology. 1989;257:E654–664. doi: 10.1152/ajpendo.1989.257.5.E654. [DOI] [PubMed] [Google Scholar]
- Hussain S, Chatillon A, Comtois A, Roussos C, Magder S. Chemical activation of thin fibre phrenic afferents. II. Cardiovascular responses. Journal of Applied Physiology. 1991;70:77–86. doi: 10.1152/jappl.1991.70.1.77. [DOI] [PubMed] [Google Scholar]
- Jammes Y, Balzamo E. Changes in afferent and efferent phrenic activities with electrically induced diaphragmatic fatigue. Journal of Applied Physiology. 1992;73:894–902. doi: 10.1152/jappl.1992.73.3.894. [DOI] [PubMed] [Google Scholar]
- McCloskey DI, Mitchell JH. Reflex cardiovascular and respiratory responses originating in exercising muscle. The Journal of Physiology. 1972;224:173–186. doi: 10.1113/jphysiol.1972.sp009887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark AL, Victor RG, Nerhed C, Wallin BG. Microneurographic studies of the mechanisms of sympathetic nerve responses to static exercise in humans. Circulation Research. 1985;57:461–469. doi: 10.1161/01.res.57.3.461. [DOI] [PubMed] [Google Scholar]
- Mitchell JH, Kaufman MP, Iwamoto GA. The exercise pressor reflex: its cardiovascular effects, afferent mechanisms, and central pathways. Annual Review of Physiology. 1983;45:229–242. doi: 10.1146/annurev.ph.45.030183.001305. [DOI] [PubMed] [Google Scholar]
- Morgan BJ, Crabtree DC, Puleo DS, Badr MS, Toiber F, Skatrud JB. Neurocirculatory consequences of abrupt change in sleep state in humans. Journal of Applied Physiology. 1996;80:1627–1636. doi: 10.1152/jappl.1996.80.5.1627. [DOI] [PubMed] [Google Scholar]
- Nordin M, Fagius J. Effect of noxious stimulation on sympathetic vasoconstrictor outflow to human muscles. The Journal of Physiology. 1995;489:885–894. doi: 10.1113/jphysiol.1995.sp021101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offner B, Dembowsky K, Czachurski J. Characteristics of sympathetic reflexes evoked by electrical stimulation of phrenic nerve afferents. Journal of the Autonomic Nervous System. 1992;41:103–112. doi: 10.1016/0165-1838(92)90132-z. [DOI] [PubMed] [Google Scholar]
- Rowell LB, O’Leary DS, Kellogg DL., Jr . Integration of cardiovascular control systems in dynamic exercise. In: Rowell L, Shepherd J, editors. Handbook of Physiology, Exercise: Regulation and Integration of Multiple Systems. Bethesda, MD,USA: American Physiological Society; 1996. pp. 770–838. section 12, part 2 chap. 17. [Google Scholar]
- St Croix CM, Satoh M, Morgan BJ, Skatrud JB, Demspey JA. Role of respiratory motor output in within-breath modulation of muscle sympathetic nerve activity in humans. Circulation Research. 1999;85:457–469. doi: 10.1161/01.res.85.5.457. [DOI] [PubMed] [Google Scholar]
- Scherrer U, Pryor SL, Bertocci LA, Victor RG. Arterial baroreflex buffering of sympathetic activation during exercise-induced elevations in arterial pressure. Journal of Clinical Investigation. 1990;86:1855–1861. doi: 10.1172/JCI114916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals DR, Suwarno NO, Joyner MJ, Iber C, Copeland JG, Dempsey JA. Respiratory modulation of muscle sympathetic nerve activity in intact and lung denervated humans. Circulation Research. 1993;72:440–454. doi: 10.1161/01.res.72.2.440. [DOI] [PubMed] [Google Scholar]
- Sinoway LI, Smith MB, Enders B, Leuenberger U, Dzwonczyk T, Gray K, Whisler S, Moore RL. Role of diprotonated phosphate in evoking muscle reflex responses in cats and humans. American Journal of Physiology. 1994;267:H770–778. doi: 10.1152/ajpheart.1994.267.2.H770. [DOI] [PubMed] [Google Scholar]
- Szulczyk A, Szulczyk P, Zywuszko B. Analysis of reflex activity in cardiac sympathetic nerve induced by myelinated phrenic nerve afferents. Brain Research. 1988;447:109–115. doi: 10.1016/0006-8993(88)90970-5. [DOI] [PubMed] [Google Scholar]
- Taha BH, Simon PM, Dempsey JA, Skatrud JB, Iber C. Respiratory sinus arrythmia in humans: an obligatory role for vagal feedback from the lungs. Journal of Applied Physiology. 1995;78:638–645. doi: 10.1152/jappl.1995.78.2.638. [DOI] [PubMed] [Google Scholar]
- Vallbo A, Hagbarth K-E, Torebjork HE, Wallin BG. Somatosensory, proprioceptive, and sympathetic activity in human peripheral nerves. Physiological Reviews. 1979;59:919–957. doi: 10.1152/physrev.1979.59.4.919. [DOI] [PubMed] [Google Scholar]
- Victor RG, Rotto DM, Pryor SL, Kaufman MP. Stimulation of renal sympathetic activity by static contraction: evidence for mechanoreceptor-induced reflexes from skeletal muscle. Circulation Research. 1989;64:592–599. doi: 10.1161/01.res.64.3.592. [DOI] [PubMed] [Google Scholar]
- Victor RG, Secher NH, Lyson T, Mitchell JH. Central command increases muscle sympathetic nerve activity during intense intermittent isometric exercise in humans. Circulation Research. 1995;76:127–131. doi: 10.1161/01.res.76.1.127. [DOI] [PubMed] [Google Scholar]
- Vissing J, Vissing SF, MacLean DA, Saltin B, Quistorff B, Haller RG. Sympathetic activation in exercise is not dependent on muscle acidosis. Journal of Clinical Investigation. 1998;101:1654–1660. doi: 10.1172/JCI555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetter TJ, Harms CA, Nelson WB, Pegelow DF, Dempsey JA. Influence of respiratory muscle work on VO2 and leg blood flow during submaximal exercise. Journal of Applied Physiology. 1999;87:643–651. doi: 10.1152/jappl.1999.87.2.643. [DOI] [PubMed] [Google Scholar]
- Wilson CR, Manchanda S, Crabtree D, Skatrud JB, Dempsey JA. An induced blood pressure rise does not alter upper airway resistance in sleeping humans. Journal of Applied Physiology. 1998;84:269–276. doi: 10.1152/jappl.1998.84.1.269. [DOI] [PubMed] [Google Scholar]
- Yan S, Gauthier AP, Similowski T, Macklem PT, Bellemare F. Evaluation of human diaphragm contractibility using mouth pressure twitches. American Review of Respiratory Disease. 1992;145:1064–1069. doi: 10.1164/ajrccm/145.5.1064. [DOI] [PubMed] [Google Scholar]