

Abstract
This study investigated the volume-regulated anion channel (VRAC) of human cervical cancer SiHa cells under various culture conditions, testing the hypothesis that the progression of the cell cycle is accompanied by differential expression of VRAC activity.
Exponentially growing SiHa cells expressed VRACs, as indicated by the presence of large outwardly rectifying currents activated by hypotonic stress with the anion permeability sequence I− > Br− > Cl−. VRACs were potently inhibited by tamoxifen with an IC50 of 4.6 μm.
Fluorescence-activated cell sorting (FACS) experiments showed that 59 ± 0.5, 5 ± 0.5 and 36 ± 1.1% of unsynchronized, exponentially growing cervical cancer SiHa cells were in G0/G1, S and G2/M stage, respectively. Treatment with aphidicolin (5 μm) arrested 88 ± 1.4% of cells at the G0/G1 stage.
Arrest of cell growth in the G0/G1 phase was accompanied by a significant decrease of VRAC activity. The normalized hypotonicity-induced current decreased from 48 ± 5.2 pA pF−1 at +100 mV in unsynchronized cells to 15 ± 2.6 pA pF−1 at +100 mV in aphidicolin-treated cells. After removal of aphidicolin, culturing in medium containing 10% fetal calf serum triggered a rapid re-entry into the cell cycle and a concomitant recovery of VRAC density.
Pharmacological blockade of VRACs by tamoxifen or NPPB caused proliferating cervical cancer cells to arrest in the G0/G1 stage, suggesting that activity of this channel is critical for G1/S checkpoint progression.
This study provides new information on the functional significance of VRACs in the cell cycle clock of human cervical cancer cells.
Cells have to avoid dramatic changes of cell volume that would jeopardize the structural integrity and constancy of the intracellular milieu. Epithelial cells possess multiple volume-sensitive transport pathways leading to regulatory volume decrease (RVD) in response to hypotonic stress (reviewed in Lang et al. 1998). Different ion transport pathways have been reported to be responsible for the RVD-associated KCl loss, but in most cell types, the predominant pathway for RVD is the activation of separate volume-regulated K+ and Cl− channels (Hoffmann & Dunham, 1995; Okada, 1997). In addition to volume regulation, volume-regulated anion channels (VRACs) participate in several important physiological processes, such as osmolyte transport, metabolism, hormone release, cell migration, proliferation and differentiation (Nilius et al. 1996; Lang et al. 1998). During cell cycle progression, cells undergo a significant increase in size (especially at the G1/S transition), which perturbs cell volume homeostasis and is counterbalanced by RVD. Co-activation of K+ channels and VRACs is therefore proposed to be a necessary step for volume regulation during cell cycle progression. Several studies suggest that differential expression of K+ channels and concomitant changes in membrane potential are critical for cell cycle checkpoints (reviewed in Wonderlin & Strobl, 1996). However, comparatively little is known about the association of cell cycle regulation with VRAC activity.
Cervical cancer constitutes the second most common cancer in women worldwide (Parkin et al. 1993). This disease is strongly associated with infection by oncogenic types of human papillomaviruses (HPVs), but only a small fraction of those infected develop cancer, indicating that other factors contribute to the progression to cervical cancer (zur Hausen, 1991). Despite intensive studies, the aetiology and tumour biology of this disease are still largely unknown, and the membrane transport properties of human cervical epithelial cells have never received attention. We have demonstrated previously that KCl and organic osmolyte efflux are strongly up-regulated during human cervical carcinogenesis (Chou et al. 1995, 1997; Shen et al. 1996, 2000). Alterations in osmosensing signalling pathways were also apparent during the process of human cervical carcinogenesis (Shen et al. 1998; Chou et al. 1998).
The present study investigated VRACs in human cervical cancer cells under various culture conditions, testing the hypothesis that the progression of the cell cycle is accompanied by differential expression of VRAC activity. We demonstrate that cell cycle progression correlates with the expression of VRAC activity. Pharmacological blockade of VRACs by tamoxifen or causes proliferating cervical cancer cells to arrest in G0/G1 stage, suggesting that the activity of these channels is critical for G1/S checkpoint progression.
METHODS
Cell culture
SiHa cells, a cervical cancer cell line, were obtained from the American Type Culture Collection (Rockville, MD, USA). SiHa cells were maintained at 37oC in a CO2-O2 (5%-95%) atmosphere and cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 10% fetal calf serum (FCS; Gibco), 80 IU ml−1 penicillin and 80 μg ml−1 streptomycin (Sigma).
Chemicals and solutions
All chemicals were obtained from Sigma. NPPB, tamoxifen, clomiphene and nafoxidine were dissolved in DMSO. The final DMSO concentration in electrophysiological and cell proliferation experiments was less than 0.05%. We have shown that this concentration has no effect on current measurements and cell growth. The osmolarity of solutions was measured using a vapour pressure osmometer (Wescor 5500, Schlag, Gladbach, Germany). The standard extracellular medium was a Krebs solution containing (mm): NaCl 150, KCl 6, MgCl2 1, CaCl2 1.5, glucose 10 and Hepes 10, titrated to pH 7.4 with NaOH (osmolarity, 320 ± 5 mosmol l−1). For electrophysiological recording, the Krebs solution was replaced with another isotonic solution after seal formation. This isotonic solution contained (mm): NaCl 105, CsCl 6, MgCl2 1, CaCl2 1.5, glucose 10, Hepes 10 and mannitol 90, titrated to pH 7.4 with NaOH (320 ± 5 mosmol l−1). VRACs were activated in SiHa cells by superfusing the cells with the same solution without mannitol, resulting in a 25% hypotonicity (240 ± 5 mosmol l−1). To measure VRAC activity, the pipette solution contained (mm): CsCl 40, caesium aspartate 100, MgCl2 1, CaCl2 0.81, BAPTA 5, Na2ATP 4 and Hepes 10. In this pipette solution, the free Ca2+ concentration was buffered at 25 nm. The pipette solution was adjusted to pH 7.2 with CsOH.
Cell cycle synchronization
For experiments on cell synchronization, SiHa cells were seeded at a density of 2 × 104 cells cm−2 in T25 culture flasks and grown to 60–80% confluence to obtain cultures in the logarithmic growth phase (Merrill, 1998). In experiments on serum starvation, cell cultures were washed 3 times with DMEM and then cultured in DMEM supplemented with 0.1% FCS. For chemical synchronization, cultures were growth arrested by serum starvation for 2 days followed by treatment with 5 μm aphidicolin for 16 h. Therefore, the downregulation of VRAC activity is a specific effect of cell cycle arrest. Identically treated cultures grown in parallel were harvested for fluorescence-activated cell sorting (FACS) and electrophysiological analysis at the indicated time points. Synchronization experiments were repeated at least 3 times. Cell counts were performed with the aid of a haemocytometer using Trypan Blue exclusion (0.08%) to monitor cell viability.
Determination of cell cycle stage by FACS
Cellular DNA content was determined by staining cells with propidium iodide and measuring fluorescence in a Becton Dickinson FACScan (Rutherford, NJ, USA). SiHa cells were harvested by trypsinization and fixed in cool 70% ethanol for 6 h. Subsequently, the fixed cells were incubated in a solution containing 1 mg ml−1 RNase and 20 μg ml−1 propidium iodide for at least 30 min. For each cell population, 10 000 cells were analysed by FACS and the proportion in G0/G1, S and G2/M phases estimated using the Modfit cell cycle analysis program (version 2.0, Verity Software House Inc.). The percentage of cells in a specific phase of the cell cycle was determined with a propidium iodide DNA staining technique: cells were classified as being in G0/G1, G2/M and S phase depending on the intensity of the fluorescence peaks (MacFarlane & Sontheimer, 2000). FACS measurements were performed in at least three independent experiments on synchronized or unsynchronized cells.
Electrophysiological recording and data analysis
All experiments were performed at room temperature (20–23oC). The whole-cell mode of the patch-clamp technique was used to measure membrane potentials and membrane currents. Currents were monitored with an EPC-7 patch clamp amplifier (List Electronic, Germany). Patch electrodes had a resistance of between 3 and 5 MΩ. A Ag-AgCl wire was used as the reference electrode. Linear capacitance, leak currents and series resistance were compensated. The current-voltage relationship and time course of VRACs were obtained from either a ramp or a step protocol. The ramp protocol consisted of a step to −80 mV for 0.4 s, followed by a step to −150 mV for 0.1 s and a 1.3 s linear voltage ramp to +100 mV, after which the potential was stepped back to the holding potential of −20 mV. This voltage protocol was repeated every 15 s from a holding potential of −20 mV. Currents were sampled at 2 ms intervals (1024 points per record, filtered at 200 Hz). The step protocol consisted of a 1 s voltage step, applied every 15 s from a holding potential of −20 mV to test potentials from −100 to +100 mV with an increment of 20 mV. Currents were sampled at 1 ms intervals. Current density was determined by normalizing the whole-cell current to the membrane capacitance.
The permeability of various anions (X−) relative to that of Cl− (PX/PCl) was determined from the shift of the reversal potential (ΔVrev) in anion substitution experiments. In this case, an agar bridge was used to minimize the junction potential and permeability ratios were calculated from a modified Goldman-Hodgkin-Katz equation:
where [Cl−]n and [Cl−]s are the Cl− concentrations in the normal and substituted external solutions, respectively, [X−]s is the concentration of the substituting anion, F is the Faraday constant, R is the gas constant and T is absolute temperature.
Volume measurements
Cell volume was estimated by measuring the cell diameter directly from the cell image on the monitor before cell swelling and 3 min after challenge by the hypotonic solution.
Data were analysed by Winascd (designed by G. Droogmans) and by Origin (version 6.0, MicroCal Software Inc). All data are presented as means ±s.e.m. Student's paired or unpaired t tests were used for statistical analyses. Differences between values were considered significant when P < 0.05.
RESULTS
Distribution of cell cycle stages in normal growth conditions and following synchronization
We first characterized the cell cycle distribution of SiHa cervical cancer cells under various culture conditions (Table 1). SiHa cells cultured in medium supplemented with 10% FCS proliferated continuously and reached confluence within 2–3 days. The FACS measurements showed that 59, 5 and 36% of cells were in G0/G1, S and G2/M stage, respectively, in the presence of 10% FCS for 48 h. The above percentages changed to 65, 7 and 28% in the presence of 10% FCS for 72 h. Compared to growth under normal conditions, serum starvation (0.1% FCS) for 72 h significantly increased the percentage of cells arrested in G0/G1 stage to 73% (P < 0.05, unpaired t test, Table 1).
Table 1.
Distribution (%) of SiHa cervical cancer cells at G0/G1, S and G2/M stages of the cell cycle under various culture conditions
| Cell cycle stage | 48 h with 10% FCS | 72 h with 10% FCS | 72 h with 0.1% FCS | *16 h with aphidicolin | 6 h release frome aphidicolin | 12 h release frome aphidicolin | 24 h release from aphidicolin |
|---|---|---|---|---|---|---|---|
| G0/G1 | 59 ± 0.5 | 65 ± 1.2 | 73 ± 1.1 | 88 ± 1.4 | 42 ± 1.0 | 46 ± 0.7 | 63 ± 2.8 |
| S | 5 ± 0.5 | 7 ± 0.6 | 7 ± 0.7 | 6 ± 1.7 | 22 ± 0.7 | 9 ± 1.4 | 5 ± 0.7 |
| G2/M | 36 ± 1.1 | 28 ± 0.1 | 20 ± 1.1 | 6 ± 2.0 | 36 ± 1.1 | 45 ± 0.7 | 33 ± 2.1 |
For synchronization, cultures were growth arrested by serum starvation for 2 days followed by μM aphidicolin for 16 h. Data represent means ±s.e.m. (n = 5).
Subsequently we used aphidicolin to synchronize the cell cycle. Aphidicolin can inhibit DNA synthesis and prevents cells in G0/G1 phase from entering the DNA synthesis period (Pedrali-Noy et al. 1980; Levenson & Hamlin, 1993). Serum starvation for 48 h followed by 16 h treatment with aphidicolin arrested 88% of cells in G0/G1 phase (Table 1). Six hours after removal of aphidicolin and reintroduction of medium containing 10% FCS, 22 and 36% of cells reached S and G2/M phase, compared to 6 and 6% in synchronized cells (Table 1). Moreover, 45% of cells reached G2/M phase 12 h after release of aphidicolin. After 24 h of release from aphidicolin, the distribution of cell cycle stages had become comparable to that of cells continuously growing in 10% FCS (Table 1). Thus, aphidicolin plus serum starvation blocked the cell cycle of SiHa cells more effectively than serum starvation alone and this effect was fully reversible.
Hypotonic cell swelling-induced Cl− currents in cervical cancer cells
Whole-cell voltage-clamp recordings were obtained from SiHa cervical cancer cells. Membrane currents recorded during the step protocol applied to SiHa cells in isotonic solution were small and time independent (Fig. 1A). Application of a hypotonic solution induced cell swelling as evidenced by visual inspection, which was accompanied by activation of large outwardly rectifying currents. At potentials more positive than +80 mV, the currents showed time-dependent inactivation, which was more pronounced at higher membrane potentials (Fig. 1A and B). The current-voltage relationship in hypotonic solution, obtained from the step protocols, reversed close to the theoretical equilibrium potential for Cl− (ECl=−25 mV), indicating that the volume-regulated currents were carried mainly by Cl− (Fig. 1B). The anion selectivity of the volume-activated currents in SiHa cervical cancer cells was examined using the voltage ramp protocol. At maximal current activation, the normal hypotonic solution was replaced by hypotonic solutions containing NaI or NaBr. The sequence of anion permeability, calculated from the shifts in reversal potential, was I− > Br− > Cl− (Fig. 1C, 1.4 ± 0.2: 1.3 ± 0.1:1, n = 5).
Figure 1. Volume-activated currents in SiHa cervical cancer cells.
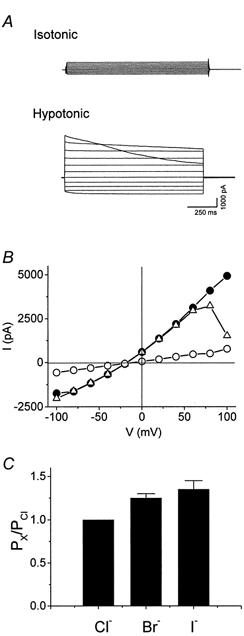
A, current traces (step protocol) were recorded in isotonic and hypotonic solutions. Horizontal lines either side of the traces represent the zero current level. B, current-voltage relationships obtained from traces in A. • and ▵, hypotonicity-induced current at the beginning and end of the voltage pulse, respectively; ^, current in isotonic solution. C, anion permeability of volume-activated currents. The anion permeability relative to that of Cl− (PX/PCl) was calculated from the shift in reversal potential as described in Methods. Each bar represents the mean ±s.e.m. (n = 5).
Tamoxifen, a well-known anti-oestrogen drug, has been shown to be a potent inhibitor of VRACs in several cell types, such as endothelial cells (Voets et al. 1995), vascular smooth muscle cells (Greenwood & Large, 1998) and colonic myocytes (Dick et al. 1999). The inhibitory effect of tamoxifen on VRACs is thought to occur via an oestrogen-independent mechanism (Hardy & Valverde, 1994). However, its effect has never been tested on the activation of VRACs in human cervical epithelial cells. To characterize VRACs further, we investigated the effect of some non-steroidal anti-oestrogen drugs on the activation of VRACs in human cervical cancer cells. As depicted in Fig. 2A and B, 10 μm tamoxifen induced a fast, potent and reversible inhibition of VRACs in SiHa cervical cancer cells. Tamoxifen and its analogue clomiphene inhibited VRACs in a dose-dependent manner with half-maximal inhibitory concentrations (IC50) of 4.6 and 15.6 μm, respectively (Fig. 2C). However, another non-steroidal oestrogen antagonist, nafoxidine, showed only a minor inhibition of VRAC activity even at concentrations of 100 μm (Fig. 2C).
Figure 2. Effect of non-steroidal oestrogen antagonists on the volume-regulated Cl− currents of human cervical cancer SiHa cells.
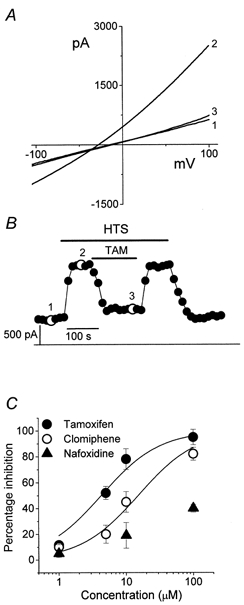
A, representative recordings of volume-regulated Cl− currents from the ramp protocol. Trace 1, basal membrane current recorded in isotonic solution; traces 2 and 3, currents recorded after perfusion with hypotonic solution in the absence or presence of 10 μm tamoxifen, respectively. B, time course of membrane currents activated at +100 mV. Data points were obtained from the voltage ramp protocol, which was applied every 15 s. The numbered points correspond to the current traces recorded in A. Horizontal bars indicate application of hypotonic solution (HTS) or 10 μm tamoxifen (TAM). Horizontal line, zero current level. C, dose-response curves for the inhibition of the volume-regulated Cl− currents by non-steroidal oestrogen antagonists, measured at +100 mV. Each point represents the mean ±s.e.m. (n = 4).
Downregulation of VRACs in G0/G1 stage of the cell cycle
To investigate VRAC activity during the cell cycle, we compared the activity of VRACs in synchronized and unsynchronized cells (Fig. 3). The unsynchronized cells had a small background current density in the isotonic medium, averaging −14 ± 2.0 pA pF−1 at −100 mV and +22 ± 3.4 pA pF−1 at +100 mV (n = 25). For SiHa cells synchronized in G0/G1 phase, the background current density was −18 ± 2.4 pA pF−1 at −100 mV and +21 ± 3.6 pA pF−1 at +100 mV, respectively (n = 22). There was no significant difference in background isotonic current density between these two groups of cells (P > 0.05, unpaired t test).
Figure 3. Downregulation of volume-regulated Cl− current in growth-arrested cells.
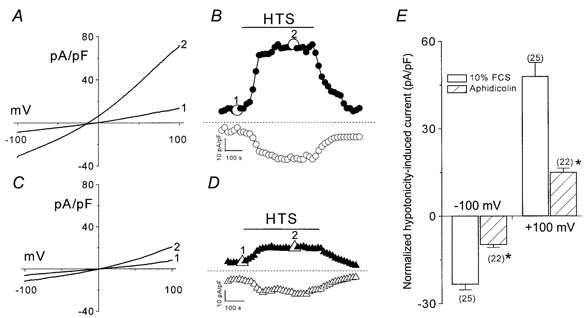
A and C, representative recordings of volume-regulated Cl− currents of SiHa cervical cancer cells during unsynchronized, exponential growth (A) and growth arrest by aphidicolin (C) from the ramp protocol. Trace 1, basal membrane current recorded in isotonic solution; trace 2, currents recorded after perfusion with hypotonic solution. B and D, time course of membrane currents activated at +100 mV (filled symbols) or −100 mV (open symbols). Data points were obtained from the voltage ramp protocol, which was applied every 15 s. The numbered points in B and D correspond to the current traces recorded in A and C, respectively. Horizontal bars indicate application of hypotonic solution (HTS). Horizontal line, zero current level. E, normalized currents activated by hypotonicity measured at −100 or +100 mV in synchronized or unsynchronized SiHa cells. The number of cells examined is indicated in parentheses beside each bar. 10% FCS, unsynchronized cells cultured with 10% fetal calf serum. Aphidicolin, cell synchronization in G0/G1 phase by incubation with 5 μm aphidicolin. *P < 0.0001 by unpaired t test.
For unsynchronized cells growing in 10% FCS, exposure to hypotonicity induced a fast activating and outward rectifying current (Fig. 3A and B). The normalized hypotonicity-induced current was used to compare VRAC activity in various culture conditions. It was defined as the difference in current density between isotonic and hypotonic solutions and was expressed per unit membrane capacitance. For cells cultured in 10% FCS, the normalized hypotonicity-induced current was 48 ± 5.2 pA pF−1 at +100 mV and −23 ± 2.3 pA pF−1 at −100 mV. VRAC activity was still present in arrested cells (Fig. 3C and D), but the current density was significantly decreased to 15 ± 2.6 pA pF−1 at +100 mV and −9.9 ± 1.7 pA pF−1 at −100 mV (P < 0.0001, unpaired t test; Fig. 3E). However, other fundamental characteristics of VRACs were similar to those of proliferating cells, such as inactivation at potentials greater than +80 mV, the weak field strength anion selectivity I− > Br− > Cl− and sensitivity to the Cl− channel inhibitors tamoxifen and NPPB (data not shown). Therefore, these results demonstrate that cell cycle arrest in G0/G1 is accompanied by a significant drop in VRAC activity. Acute administration of aphidicolin alone up to 20 μm showed no effect on VRAC activity. Therefore, the downregulation of VRAC activity is a specific effect of cell cycle arrest. In response to hypotonic stress, cells swelled to a diameter 30 ± 3 and 31 ± 2% (n = 12) above the original size within 3 min for synchronized and unsynchronized cells, respectively. This rules out the possibility that the down-regulation of VRACs in growth-arrested cells was due to the absence of full swelling. In addition, it should be mentioned that different cell swelling can be ruled out in patch clamp experiments because due to dialysis with the pipette solution the cells remain swollen although the current reaches a constant value (for detailed discussion see Voet et al. 1999).
Recovery of VRAC activity after cell cycle re-entry
Subsequently, we tested whether the activity of VRACs could be recovered after release from G0/G1 arrest. We chose SiHa cells which were initially incubated with 5 μm aphidicolin for 16 h and then cultured for 12 h with 10% FCS after removal of aphidicolin. Under these conditions, 46, 9 and 45% of cells were in G0/G1, S and G2/M phases, respectively (Table 1). For these cells, the normalized current density induced by hypotonicity increased markedly to −23 ± 4.2 pA pF−1 at −100 mV and +36 ± 5.1 pA pF−1 at +100 mV (n = 43), both of which were significantly different from the VRAC activity of arrested cells (P < 0.01, unpaired t test) but were not significantly different from those of normally proliferating cells (P > 0.1, unpaired t test). Therefore, continuation of the cell cycle beyond the G1/S checkpoint is accompanied by an increase in VRAC activity.
As depicted in Fig. 4A, 88% (19/22) of synchronized cells had a VRAC current density below 20 pA pF−1 at +100 mV and the same percentage of cells were arrested in G0/G1 phase. Accordingly, we used 20 pA pF−1 as the cut-off point to analyse the distribution of VRAC activity during cell cycle re-entry. After removal of aphidicolin and culture for 12 h in 10% FCS, the percentage of cells in G0/G1 phase was reduced to 46% and, concomitantly, only 44% (19/43) of cells had a VRAC current density below 20 pA pF−1 (Fig. 4B). These data suggest that a correlation between cell cycle phase and VRAC activity exists, with VRAC activity decreasing in G0/G1 and increasing upon progression through S and G2/M.
Figure 4. Recovery of activity of volume-regulated Cl− current following cell cycle re-entry.
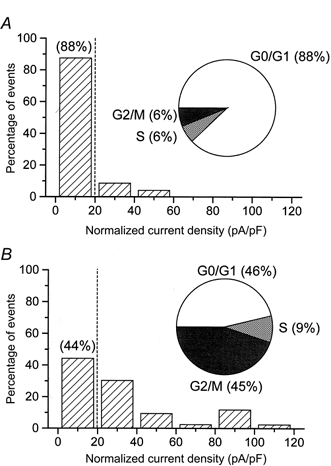
Distribution of normalized currents activated by hypotonicity measured at +100 mV in synchronized cells (A) or in cells re-entering the cell cycle (B). The histograms were constructed with a bin width of 20 pA pF−1 and the inset shows the distribution of cells within the cell cycle, measured in parallel experiments by FACS. The vertical dashed line is the cut-off point for convenience of comparison. The cell numbers in A and B are 25 and 43, respectively.
Pharmacological blockade of VRACs causes proliferating cervical cancer cells to arrest in G0/G1 stage
In the next series of experiments, we investigated whether potent VRAC blockers such as tamoxifen and NPPB can inhibit the proliferation of cervical cancer cells and, if so, whether the inhibitory effect on cell growth is due to arrest in the G0/G1 phase. Figure 5A and C shows the dose-dependent inhibition of the proliferation of SiHa cervical cancer cells by tamoxifen and NPPB. SiHa cells were seeded initially at a density of 1 × 105 cells ml−1 and counted 24 and 48 h after incubation with various concentrations of tamoxifen or NPPB. After 48 h incubation, 5 μm tamoxifen, which blocks 55% of VRAC activity, inhibited proliferation by 55–60%. Moreover, 10 μm tamoxifen, which blocks 80% of VRAC activity, completely abolished proliferation. Importantly, the viability of cells grown in the presence of tamoxifen was not different from that of control cells, suggesting that tamoxifen inhibited cell growth rather than causing cell death (viability 48 h after seeding: control, 94 ± 2%; 10 μm tamoxifen, 91 ± 3%; 60 μm NPPB, 90 ± 0.8%; n = 3). FACS measurements showed that 73 ± 5 and 90 ± 2.5% of SiHa cells were arrested in G0/G1 phase after treatment with 5 and 10 μm tamoxifen, respectively. In contrast, in the absence of tamoxifen only 58 ± 3% of cells were in G0/G1 (Fig. 5B). NPPB, another potent VRAC inhibitor, exerted similar effects to tamoxifen on both cell proliferation and cell cycle arrest in G0/G1 phase (Fig. 5C and D). These results indicate that pharmacological blockade of VRACs by tamoxifen or NPPB causes proliferating cervical cancer cells to arrest in G0/G1, suggesting that the activity of this channel is critical for G1/S checkpoint progression.
Figure 5. Blockade of volume-regulated Cl− current by tamoxifen and NPPB induces growth arrest of proliferating SiHa cervical cancer cells in G0/G1 phase.
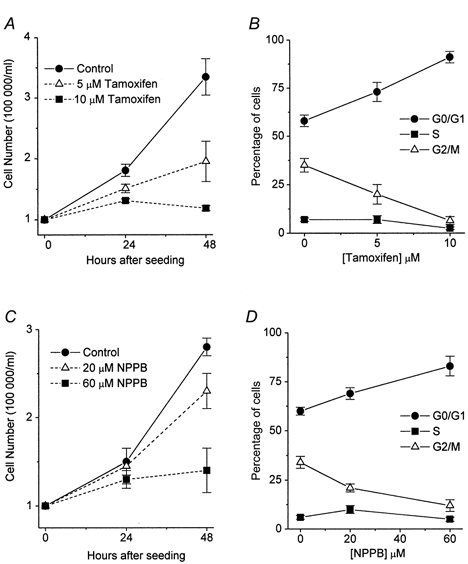
A and C, inhibition of the proliferation of SiHa cells by tamoxifen and NPPB. Cell were seeded at a density of 1 × 105 ml−1, and counted 24 and 48 h after incubation with various concentrations of tamoxifen or NPPB. B and D, the simultaneous FACS measurement for the distribution of cells in the different cell cycle phases (G0/G1, S and G2/M) after 48 h incubation with various concentrations of tamoxifen or NPPB. Each point in the curves represents the mean ±s.e.m. (n = 3).
DISCUSSION
This is the first study to demonstrate a direct correlation between VRAC activity and cell cycle progression. Human cervical cancer SiHa cells, grown in normal culture conditions, expressed VRACs as shown by the presence of large outwardly rectifying currents induced by hypotonic stress. The VRACs had an anion permeability sequence of I− > Br− > Cl− and were inhibited by tamoxifen with an IC50 of 4.6 μm. Arrest of cell growth in G0/G1 phase was accompanied by a marked decrease of VRAC current density. Most importantly, VRAC activity recovered upon re-entry into the cell cycle. Pharmacological blockade of VRACs by tamoxifen or NPPB caused proliferating SiHa cells to arrest in G0/G1, suggesting that the activity of this channel is involved in G1/S checkpoint progression. However, we cannot exclude the contribution of other Cl− channels, which might be of importance in other cell types (Schlichter et al. 1996; Rouzaire-Dubois & Dubois, 1998).
What is the functional significance of the differential expression of VRACs during cell cycle progression? One possibility is the involvement of VRACs in cell volume regulation. Proliferating cells usually have higher rates of metabolism, mitosis and migration, compared with cells in growth arrest. Growth, mitosis and migration will all perturb cell volume homeostasis. The maintenance of cell volume homeostasis is a fundamental property of mammalian cells, and all cells possess mechanisms to regulate their volume during osmotic challenge. Therefore, some beneficial and necessary mechanisms are proposed to be activated in the processes of cellular proliferation and growth. The close linkage of cell volume homeostasis, cell growth and proliferation suggests that VRACs play an important role in the processes of the cell cycle clock. Modulation of the factors controlling cell volume would influence cell proliferation. For example, Cl− channel blockers as well as K+ channel blockers increase the cell volume and decrease the proliferative rate of neuroblastoma cells (Rouzaire-Dubois & Dubois, 1998). Furthermore, during the G1/S transition, cells prepare for entry into S phase and are committed to several tasks, including uptake of amino acids, metabolic substrates and material for the synthesis of proteins, and the processing of cell cycle regulatory signals (Wonderlin & Strobl, 1996). VRACs have been reported to function as a potential transport pathway for metabolic compounds (e.g. amino acids) which are required for proliferation in many cell types (Kirk et al. 1992; Chou et al. 1997). Moreover, cell cycle rate might be maximal at a slightly acid intracellular pH. For example, intracellular alkalinization accounts for the inhibition of proliferation in astrocytes (Pappas et al. 1994). VRACs are also possibly involved in pH-regulatory steps and their inhibition may induce cell alkalinization (Sakai et al. 1999). From the viewpoint of metabolism and pH regulation, it is therefore suggested that upregulation of VRAC activity is necessary for passing through the G1/S transition.
Cl− channels have been implicated in the proliferative response of particular cell types. Cl− channel blockage inhibited cell proliferation of rat microglia (Schlichter et al. 1996), neuroblastoma cells (Rouzaire-Dubois & Dubois, 1998), endothelial cells (Voets et al. 1995) and glioma cells (Ullrich & Sontheimer, 1996). However, the Cl− channel blockers 4-acetamido-4′-isocyanatostilbene-2,2′-disulphonate (SITS) and 4,4′-diisothiocyanatostilbene-2,2′-disulphonate (DIDS) enhanced the proliferation of Schwann cells cultured from newborn rat sciatic nerve (Wilson & Chiu, 1993). In lymphocytes, Cl− permeability varied with cell cycle phase, being low in G0 and S phase and increasing in G1/S (Bubien et al. 1990). The expression of a glioma-specific Cl− channel depended on cell cycle stage and was proposed to be linked to cytoskeletal changes (Ullrich & Sontheimer, 1997). Alterations of VRAC activity have also been associated with muscle differentiation (Voets et al. 1997). In addition to Cl− channels, K+ channels are associated with cell proliferation. K+ channel inhibitors such as 4-aminopyridine and tetraethylammonium suppressed proliferation of lymphocytes in a dose-dependent manner (DeCoursey et al. 1984). Blockade of voltage-activated K+ channels also led to a decrease in the proliferation of melanoma cells (Nilius & Wohlrab, 1992), breast cancer cells (Woodfork et al. 1995), and several types of neurons (Pappas et al. 1994). Although the exact role that channel activity plays in cell proliferation and cell cycle progression is still a mystery, it has been proposed that changes in channel activity result in both short-term modulation of pre-existing channel proteins and long-term changes in gene expression (Premack & Gardner, 1991).
VRACs have been described in many mammalian and non-mammalian cell types (Okada, 1997). However, they have not yet been identified at the molecular level and little information is available on specific high-affinity ligands for VRACs. A search for pharmacological tools that bind to VRACs with high affinity may therefore be useful for the purification and molecular identification of these channels as well as for their functional characterization. The non-steroidal anti-oestrogens tamoxifen and clomiphene are used primarily for the treatment of breast cancer and female infertility, respectively. They belong to the triphenylethylene class of compounds derived from the same stilbene nucleus as diethylstilbestrol. The present study reveals that tamoxifen and clomiphene inhibit the VRACs of cervical cancer cells in a dose-dependent manner with IC50 values of 4.6 and 15.6 μm, respectively. We have shown previously that NPPB, SITS and DIDS inhibited VRACs of human cervical cancer cells with IC50 values of 50, 100 and 500 μm, respectively (Chou et al. 1998; Shen et al. 1999). Tamoxifen and clomiphene are therefore more potent than these well-known Cl− channel inhibitors. In the present study, we also found that pharmacological blockade of VRACs by tamoxifen or NPPB caused a dose-dependent inhibition of proliferation of cervical cancer cells. More importantly, the inhibitory effect of these agents on proliferation was via growth arrest in G0/G1 phase, indicating that VRAC activity is required for G1/S checkpoint progression.
In conclusion, this study provides new and important information on the functional significance of VRACs in the cell cycle clock of human cervical cancer cells.
Acknowledgments
Meng-Ru Shen holds a Swire Scholarship supported by John Swire & Sons Ltd. Thomas Voets is a postdoctoral fellow of the FWO. We thank Ch. Maertens (Leuven) for helpful discussion, and J. Prenen, M. Crabbé, H. Van Weijenbergh and M. Schuermans for their skilful technical assistance and help with the cell cultures. This work was supported by the Belgian Federal Government, the Flemish Government and the Onderzoeksraad KU Leuven (GOA 99/07, FWO G.0237.95, FWO G.0214.99, FWO G.0136.00; Interuniversity Poles of Attraction Program, Prime Ministers Office IUAP Nr.3P4/23, and C.O.F./96/22-069), by ‘Levenslijn’ (7.0021.99) and a grant from the ‘Alphonse and Jean Forton – Koning Boudewijn Stichting’ R7115 B0 and by the European Commission (BMH4-CT96-0602).
References
- Bubien JK, Kirk LK, Rado TA, Frizzell RA. Cell cycle dependence of chloride permeability in normal and cystic fibrosis lymphocytes. Science. 1990;248:1416–1419. doi: 10.1126/science.2162561. [DOI] [PubMed] [Google Scholar]
- Chou CY, Shen MR, Chen TM, Huang KE. Volume-activated taurine transport is differentially activated in human cervical cancer HT-3 cells, but not activated in HPV-immortalized Z 183A and normal cervical epithelial cells. Clinical and Experimental Pharmacology and Physiology. 1997;24:935–939. doi: 10.1111/j.1440-1681.1997.tb02722.x. [DOI] [PubMed] [Google Scholar]
- Chou CY, Shen MR, Hsu KS, Huang HY, Lin HC. Involvement of PKC-α in regulatory volume decrease responses and activation of volume-sensitive chloride channels in human cervical cancer HT-3 cells. Journal of Physiology. 1998;512:435–448. doi: 10.1111/j.1469-7793.1998.435be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CY, Shen MR, Wu SN. Volume-sensitive chloride channels associated with human cervical carcinogenesis. Cancer Research. 1995;55:6077–6083. [PubMed] [Google Scholar]
- DeCoursey TE, Chandy G, Gupta S, Cahalan MD. Voltage-gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature. 1984;307:465–468. doi: 10.1038/307465a0. [DOI] [PubMed] [Google Scholar]
- Dick GM, Kong ID, Sanders KM. Effects of anion channel antagonists in canine colonic myocytes: comparative pharmacology of Cl−, Ca2+ and K+ currents. British Journal of Pharmacology. 1999;127:1819–1831. doi: 10.1038/sj.bjp.0702730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood IA, Large WA. Properties of a Cl− current activated by cell swelling in rabbit portal vein vascular smooth muscle cells. American Journal of Physiology. 1998;275:H1524–1532. doi: 10.1152/ajpheart.1998.275.5.H1524. [DOI] [PubMed] [Google Scholar]
- Hardy SP, Valverde MA. Novel plasma membrane action of estrogen and antiestrogens revealed by their regulation of a large conductance chloride channel. FASEB Journal. 1994;8:760–765. doi: 10.1096/fasebj.8.10.8050676. [DOI] [PubMed] [Google Scholar]
- Hoffmann EK, Dunham PB. Membrane mechanisms and intracellular signalling in cell volume regulation. International Review of Cytology. 1995;161:173–262. doi: 10.1016/s0074-7696(08)62498-5. [DOI] [PubMed] [Google Scholar]
- Kirk K, Ellory JC, Young JD. Transport of organic substrates via a volume-activated channel. Journal of Biological Chemistry. 1992;267:23475–23478. [PubMed] [Google Scholar]
- Lang F, Busch GL, Ritter M, Volkl H, Waldegger S, Gulbins E, Haussinger D. Functional significance of cell volume regulatory mechanisms. Physiological Reviews. 1998;78:247–306. doi: 10.1152/physrev.1998.78.1.247. [DOI] [PubMed] [Google Scholar]
- Levenson V, Hamlin JL. A general protocol for evaluating the specific effects of DNA replication inhibitors. Nucleic Acids Research. 1993;21:3997–4004. doi: 10.1093/nar/21.17.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane SN, Sontheimer H. Changes in ion channel expression accompany cell cycle progression of spinal chord astrocytes. Glia. 2000;30:39–48. doi: 10.1002/(sici)1098-1136(200003)30:1<39::aid-glia5>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Merrill GF. Cell synchronization. Methods in Cell Biology. 1998;57:229–249. [PubMed] [Google Scholar]
- Nilius B, Eggermont J, Voets T, Droogmans G. Volume-activated Cl− channels. General Pharmacology. 1996;27:1131–1140. doi: 10.1016/s0306-3623(96)00061-4. [DOI] [PubMed] [Google Scholar]
- Nilius B, Wohlrab W. Potassium channels and regulation of proliferation of human melanoma cells. Journal of Physiology. 1992;445:537–548. doi: 10.1113/jphysiol.1992.sp018938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y. Volume expansion-sensing outward-rectifier Cl− channel: fresh start to the molecular identity and volume sensor. American Journal of Physiology. 1997;273:C755–789. doi: 10.1152/ajpcell.1997.273.3.C755. [DOI] [PubMed] [Google Scholar]
- Pappas CA, Ullrich N, Sontheimer H. Reduction of glial proliferation by K+ channel blockers is mediated by changes in pHi. Neuroreport. 1994;6:193–196. doi: 10.1097/00001756-199412300-00049. [DOI] [PubMed] [Google Scholar]
- Parkin DM, Pisani P, Ferlay J. Estimates of the worldwide incidence of eighteen major cancers in 1985. International Journal of Cancer. 1993;54:594–606. doi: 10.1002/ijc.2910540413. [DOI] [PubMed] [Google Scholar]
- Pedrali-Noy G, Spadari S, Miller-Faures A, Miller AO, Kruppa J, Koch G. Synchronization of HeLa cell cultures by inhibition of DNA polymerase alpha with aphidicolin. Nucleic Acids Research. 1980;8:377–387. doi: 10.1093/nar/8.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premack BA, Gardner A. Role of ion channels in lymphocytes. Journal of Clinical Immunology. 1991;11:225–238. doi: 10.1007/BF00918180. [DOI] [PubMed] [Google Scholar]
- Rouzaire-Dubois B, Dubois JM. K+ channel block-induced mammalian neuroblastoma cell swelling: a possible mechanism to influence proliferation. Journal of Physiology. 1998;510:93–102. doi: 10.1111/j.1469-7793.1998.093bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai H, Nakamura F, Kuno M. Synergetic activation of outwardly rectifying Cl− currents by hypotonic stress and external Ca2+ in murine osteoclasts. Journal of Physiology. 1999;515:157–168. doi: 10.1111/j.1469-7793.1999.157ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlichter LC, Sakellaropoulos G, Ballyk B, Pennefather PS, Phipps DJ. Properties of K+ and Cl− channels and their involvement in proliferation of rat microglial cells. Glia. 1996;17:225–236. doi: 10.1002/(SICI)1098-1136(199607)17:3<225::AID-GLIA5>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Shen MR, Chou CY, Ellory JC. Volume-sensitive KCl cotransport associated with human cervical carcinogenesis. Pflügers Archiv. 2000;440:751–760. doi: 10.1007/s004240000338. [DOI] [PubMed] [Google Scholar]
- Shen MR, Chou CY, Hsu KF, Hsu KS, Wu ML. Modulation of volume-sensitive Cl− channel and cell volume by actin filaments and microtubules in human cervical cancer HT-3 cells. Acta Physiologica Scandinavica. 1999;167:215–225. doi: 10.1046/j.1365-201x.1999.00611.x. [DOI] [PubMed] [Google Scholar]
- Shen MR, Chou CY, Wu ML, Huang KE. Differential osmosensing signalling pathways and G-protein involvement in human cervical cells with different tumor potential. Cellular Signalling. 1998;10:113–120. doi: 10.1016/s0898-6568(97)00115-0. [DOI] [PubMed] [Google Scholar]
- Shen MR, Wu SN, Chou CY. Volume-sensitive chloride channels in the primary culture cells of human cervical carcinoma. Biochimica et Biophysica Acta. 1996;1315:138–144. doi: 10.1016/0925-4439(95)00115-8. [DOI] [PubMed] [Google Scholar]
- Ullrich N, Sontheimer H. Biophysical and pharmacological characterization of chloride currents in human astrocytoma cells. American Journal of Physiology. 1996;270:C1511–1521. doi: 10.1152/ajpcell.1996.270.5.C1511. [DOI] [PubMed] [Google Scholar]
- Ullrich N, Sontheimer H. Cell cycle-dependent expression of a glioma-specific chloride current: proposed link to cytoskeletal changes. American Journal of Physiology. 1997;273:C1290–1297. doi: 10.1152/ajpcell.1997.273.4.C1290. [DOI] [PubMed] [Google Scholar]
- Voets T, Droogmans G, Raskin G, Eggermont J, Nilius B. Reduced intracellular ionic strength as the initial trigger for activation of endothelial volume-regulated anion channels. Proceedings of the National Academy of Sciences of the USA. 1999;96:5298–5303. doi: 10.1073/pnas.96.9.5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets T, Szücs G, Droogmans G, Nilius B. Blockers of volume-activated Cl− currents inhibit endothelial cell proliferation. Pflügers Archiv. 1995;431:132–134. doi: 10.1007/BF00374387. [DOI] [PubMed] [Google Scholar]
- Voets T, Wei L, DeSmet P, VanDriessche W, Eggermont J, Droogmans G, Nilius B. Downregulation of volume-activated Cl− currents during muscle differentiation. American Journal of Physiology. 1997;272:C667–674. doi: 10.1152/ajpcell.1997.272.2.C667. [DOI] [PubMed] [Google Scholar]
- Wilson GF, Chiu SY. Mitogenic factors regulate ion channels in Schwann cells cultured from newborn rat sciatic nerve. Journal of Physiology. 1993;470:501–520. doi: 10.1113/jphysiol.1993.sp019872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonderlin WF, Strobl JS. Potassium channels, proliferation and G1 progression. Journal of Membrane Biology. 1996;154:91–107. doi: 10.1007/s002329900135. [DOI] [PubMed] [Google Scholar]
- Woodfork KA, Wonderlin WF, Peterson VA, Strobl SJ. Inhibition of ATP-sensitive potassium channels causes reversible cell-cycle arrest of human breast cancer cells in tissue culture. Journal of Cellular Physiology. 1995;162:163–171. doi: 10.1002/jcp.1041620202. [DOI] [PubMed] [Google Scholar]
- zurHausen H. Human papillomavirus in the pathogenesis of anogenital cancer. Virology. 1991;184:9–13. doi: 10.1016/0042-6822(91)90816-t. [DOI] [PubMed] [Google Scholar]