

Abstract
Generalized epilepsy with febrile seizures plus (GEFS+) is a benign epileptic syndrome of humans. It is characterized by febrile and afebrile generalized seizures that occur predominantly in childhood and respond well to standard antiepileptic therapy. A mutation in the β1-subunit of the voltage-gated sodium channel, linked to chromosome 19q13 (GEFS+ type 1) has been found in one family. For four other families, linkage was found to chromosome 2q21-33 (GEFS+ type 2) where three genes encoding neuronal sodium channel α-subunits are located (SCN1-3A). Recently, the first two mutations were identified in SCN1A.
We introduced one of these mutations, which is highly conserved to SCN1A, into the cDNA of the gene SCN4A encoding the α-subunit of the human skeletal muscle sodium channel (hSkm1). The mutation is located in the S4 voltage sensor of domain IV, predicting substitution of histidine for the fifth of eight arginines (R1460H in hSkm1). Functional studies were performed by expressing the α-subunit alone in the mammalian tsA201 cell line using the whole-cell patch clamp technique.
Compared to wild-type (WT), mutant R1460H channels showed small defects in fast inactivation. The time course of inactivation was slightly (1.5-fold) slowed and its voltage dependence reduced, and recovery from inactivation was accelerated 3-fold. However, there was no increase in persistent sodium current as observed for SCN4A mutations causing myotonia or periodic paralysis. The activation time course of R1460H channels was slightly accelerated. Slow inactivation was slightly but significantly stabilized, confirming the importance of this region for slow inactivation.
The combination of activation and fast inactivation defects can explain the occurrence of epileptic seizures, but the effects were much more subtle than the inactivation defects described previously for mutations in SCN4A causing disease in skeletal muscle. Hence, with regard to pathological excitability, our results suggest a greater vulnerability of the central nervous system compared to muscle tissue.
Voltage-gated Na+ channels are membrane-spanning proteins responsible for the initiation and propagation of action potentials in nerve and muscle cells. In response to membrane depolarization the channels open from the resting, closed state and then inactivate spontaneously. Upon repolarization the channels recover from inactivation. The functionally important α-subunit contains four domains (I-IV) of six transmembrane segments each (S1-S6). All S4 segments contain positively charged residues conferring voltage dependence on the channel protein. There are several genes encoding different α-subunits (SCN1A-11A) that are expressed specifically in skeletal muscle (SCN4A), heart muscle (SCN5A) and neuronal tissue; four subunits (encoded by SCN1A, SCN2A, SCN3A and SCN8A) are considered to be responsible for the sodium current in brain. There are three genes for the auxiliary β-subunits (SCN1B-3B), which are all expressed in brain; the β1-subunit is also expressed in skeletal and heart muscle (reviewed by Goldin, 1999; Lehmann-Horn & Jurkat-Rott, 1999; Catterall, 2000; Morgan et al. 2000).
Ion channel disorders are rare inherited diseases providing interesting models to study dysfunction of excitability in vivo and in vitro. The first so-called ‘channelopathies’ identified were skeletal muscle diseases, the myotonias and hyperkalaemic periodic paralysis, which are sodium and chloride channel disorders. For about 20 known mutations in SCN4A, a gain of function mechanism causes hyper- or hypoexcitability in the sodium channel diseases through a defect in channel inactivation resulting in an increase in the sodium inward current, which depolarizes the sarcolemma. A small depolarization will increase whereas a large depolarization will decrease excitability. The same pathophysiological mechanism applies to one form of the long-QT syndrome (LQT type 3, mutations in SCN5A), an inherited cardiac arrhythmia (reviewed by Lehmann-Horn & Jurkat-Rott, 1999).
Three forms of idiopathic epilepsies have been identified as arising from ion channel disorders. Autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) is caused by point mutations in the α4-subunit of a neuronal nicotinic ACh receptor (Steinlein et al. 1995), benign familial neonatal convulsions (BFNC) are caused by mutations in two voltage-gated potassium channels (Biervert et al. 1998; Charlier et al. 1998; Singh et al. 1998) and generalized epilepsy with febrile seizures plus (GEFS+) is a sodium channel disorder (Wallace et al. 1998; Escayg et al. 2000). GEFS+ is a benign childhood-onset epileptic syndrome featuring different forms of febrile and afebrile seizures (Scheffer & Berkovic, 1997; Singh et al. 1999). Five large families with autosomal dominant inheritance of GEFS+ have been described so far, showing linkage to chromosome 19q13 in one case (GEFS+ type 1; Wallace et al. 1998) and to chromosome 2q21-33 in the other four (GEFS+ type 2; Baulac et al. 1999; Moulard et al. 1999; Pfeiffer et al. 1999; Lopez-Cendes et al. 2000). For the chromosome 19-linked GEFS+ family, a mutation has been found in SCN1B predicting substitution of tryptophan for one of two cysteine residues (C121W) that stabilize the secondary structure of the functionally important extracellular loop of the β1-subunit. Although this auxiliary subunit is also expressed in skeletal muscle, only epileptic seizures but no myotonia were reported for affected individuals. Functional studies in Xenopus oocytes revealed a loss of β1-subunit function, resulting in a decreased rate of inactivation, which increased the sodium inward current (Wallace et al. 1999).
The neuronal sodium channel α-subunit genes SCN1-3A are located at the GEFS+ locus on chromosome 2q21-33. Recently, two mutations have been identified in SCN1A in two of the linked families (Escayg et al. 2000). Both mutations are located in functionally important regions, the S4 voltage sensors in domains II and IV (T875M in II/S4 and R1648H in IV/S4). The voltage sensor IV/S4 has been shown previously to be important for channel inactivation and mutations therein cause paramyotonia congenita, one of the sodium channel disorders in skeletal muscle (Chahine et al. 1994; Lerche et al. 1996; Lehmann-Horn & Jurkat-Rott, 1999). The sodium channel α-subunits expressed in skeletal muscle and brain are highly conserved in the functionally important regions of the channel proteins and their kinetic behaviour, known from various functional studies of the human and rat isoforms, is very similar (Goldin, 1999), but the human SCN1A gene has not been cloned and functionally expressed so far. The kinetic behaviour of the rat brain type I sodium channel (encoded by the rat SCN1A gene) has been described recently (Smith & Goldin, 1998).
In order to study the functional consequences of a mutation causing epilepsy and also to compare the defects to mutations found in the myotonias, we introduced the SCN1A mutation R1648H in the voltage sensor IV/S4 into the same conserved region of SCN4A (R1460H) and expressed wild-type (WT) and mutant α-subunits in tsA201 cells. As expected, the gain of function was much smaller than that found for the myotonia-causing mutations located within the same channel region.
METHODS
Mutagenesis and transfection
Site-directed mutagenesis to introduce the mutation R1460H was performed using a PCR-based strategy. The mutants were reassembled in the pRC/CMV plasmid (Invitrogen) for transfection into the mammalian cell line tsA201 using a standard calcium phosphate transfection method. A CD8 cDNA-containing plasmid was cotransfected in order to allow identification of transfected cells using anti-CD8 antibody-coated microbeads (Dynabeads M450, Dynal; Lerche et al. 1997).
Electrophysiology and data analysis
Standard whole-cell recording was performed using an EPC-7 amplifier (EPC7, List). The pipette solution contained (mm): 105 CsF, 35 NaCl, 10 EGTA and 10 Hepes (pH 7.4). The bath solution contained (mm): 150 NaCl, 2 KCl, 1.5 CaCl2, 1 MgCl2 and 10 Hepes (pH 7.4). For some experiments, solutions with internal CsCl instead of CsF were used (Lerche et al. 1997). Sodium currents in transfected cells for WT and mutant channels ranged between 2.5 and 15 nA. The maximal voltage error due to residual series resistance was < 5 mV. Leakage and capacitative currents were automatically subtracted using a prepulse protocol (-P/4). Currents were filtered at 3 or 10 kHz and digitized at 20 or 50 kHz using pCLAMP software (Axon Instruments). Measurements were performed at room temperature (21-23°C). For some experiments, the temperature was adjusted to 14.5-15.5°C via a water bath. All data were analysed using a combination of pCLAMP, Excel (Microsoft) and ORIGIN software (MicroCal). For statistical evaluation, Student’s t test was applied. All data are shown as means ±s.e.m.
RESULTS
The mutation R1460H in the skeletal muscle sodium channel α-subunit predicts the substitution of histidine for the fifth of eight positively charged amino acids in the S4 voltage sensor of domain IV (IV/S4). The almost complete conservation of IV/S4 and R1460 among the SCN1A and SCN4A gene products and other known voltage-gated sodium channel α-subunits is shown in Fig. 1A. The R1460H mutation was engineered into the cDNA of the SCN4A gene, and WT or mutant plasmids were transfected into tsA201 cells. Families of normalized whole-cell sodium currents for WT and mutant channels elicited by various depolarizing voltage steps from a holding potential of -140 mV are shown in Fig. 1B. In order to look for differences in gating between WT and R1460H channels that may explain the occurrence of epileptic seizures, the kinetics and voltage dependence of activation, deactivation, fast and slow inactivation were determined.
Figure 1. Mutation R1460H in segment IV/S4.
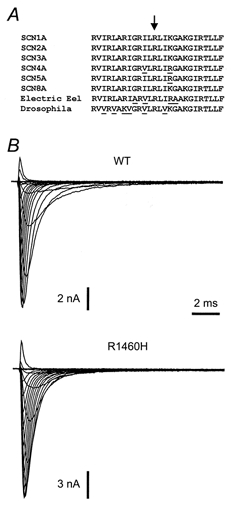
A, amino acid sequence comparison of the IV/S4 segment of various sodium channel α-subunits derived from different genes. Sequence changes are underlined. Residue R1648 (SCN1A numbering) corresponding to R1460 (SCN4A numbering) is marked by an arrow. B, representative whole-cell sodium current families recorded from cells transfected with either WT or mutant channel cDNA.
Fast inactivation
The time course of fast inactivation was fitted to a second order exponential function. The fast time constant, τh, accounted for > 95% of the current amplitude for both WT and R1460H. There was a small but significant slowing of fast inactivation at depolarized potentials and a marked decrease in its voltage dependence in the R1460H mutant (Fig. 2A). The loss of voltage dependence has already been described for other mutations in IV/S4 and may indicate an uncoupling of inactivation from activation (Chahine et al. 1994; Lerche et al. 1996; Mitrovic et al. 1999). The most distinct difference between WT and R1460H channels was found in the recovery from inactivation, measured at -100, -120 and -140 mV after a 100 ms depolarization to 0 mV. Its time course was well fitted to a first order exponential function, yielding the time constant for recovery, τrec. For R1460H, τrec was decreased by about 3-fold at -100 mV (Fig. 2B). The steady-state fast inactivation was shifted slightly towards more hyperpolarized potentials (Fig. 2C).
Figure 2. Parameters of fast inactivation.
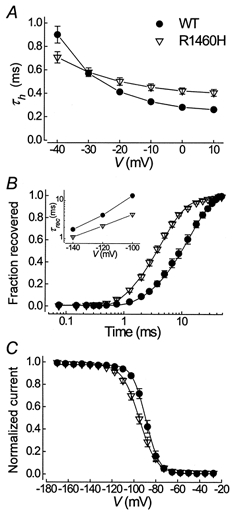
A, voltage dependence of the inactivation time constant, τh. Values at 0 mV were 0.28 ± 0.01 for WT vs. 0.42 ± 0.03 ms for R1460H; n = 8, 14; P < 0.001. B, recovery from inactivation at -100 mV. Lines are fits to a first order exponential function with recovery time constants (τrec) of 13.7 ± 1.9 vs. 4.4 ± 0.3 ms; n = 7, 11; P < 0.001; and an initial delay of 1.0 ± 0.1 vs. 0.59 ± 0.04 ms, P < 0.001, for WT and R1460H, respectively. The inset shows the voltage dependence of τrec, n = 4-11. C, steady-state inactivation was determined using 300 ms prepulses to the potentials indicated, followed by a short test pulse to -20 mV. Lines are fits to a standard Boltzmann function: I/Imax= 1/(1 + exp[(V–V0.5)/kV]), where V0.5 is the voltage of half-maximal inactivation and kV is a slope factor. V0.5 was -89.0 ± 1.4 vs. -96.0 ± 1.6 mV; n = 7, 16; P < 0.02; kV was 5.5 ± 0.3 vs. 7.2 ± 0.2 mV, P < 0.001, for WT and R1460H, respectively.
Both the slowing of fast inactivation and the acceleration of its recovery increased excitability, as has previously been shown for SCN4A mutations causing myotonia. However, the most important finding with SCN4A mutations causing disease in skeletal muscle was an increase in the persistent sodium current depolarizing the muscle fibre membrane (Cannon, 1997; Lehmann-Horn & Jurkat-Rott, 1999). As can be seen in Fig. 1B, there was no increase in persistent current for R1460H compared to WT channels. We determined the persistent sodium current (Iss) at the end of a 70 ms depolarizing test pulse to -20 mV relative to the peak current (Ipeak) using both a CsF and a CsCl intracellular solution (the latter was used previously to analyse myotonia-causing mutations, e.g. Mitrovic et al. 1999). There was no significant difference between WT and R1460H channels in either solution (Iss/Ipeak at -20 mV for WT vs. R1460H in CsF: 0.6 ± 0.2 vs. 0.7 ± 0.3%; n = 7, 16; in CsCl: 0.7 ± 0.1 vs. 0.6 ± 0.4%; n = 7, 3).
Activation and deactivation
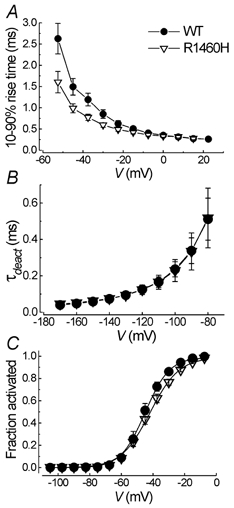
For a better time resolution, the kinetics of activation and deactivation were determined at 15°C. For quantification of the activation time course, the 10-90% rise time was calculated. There was a significant acceleration of the activation time course for R1460H channels between -52.5 and -30 mV (Fig. 3A). In contrast, the deactivation time course was almost identical for the two clones (Fig. 3B). For the steady-state activation curve we found a significant difference in slope (Fig. 3C).
Figure 3. Activation and deactivation parameters.
Slow inactivation
Entry into, recovery from and steady-state slow inactivation were determined as shown in Fig. 4. Slow inactivation was slightly stabilized for R1460H channels, as revealed by a hyperpolarizing shift in steady-state slow inactivation, a more complete slow inactivation and a decreased rate of its recovery.
Figure 4. Parameters of slow inactivation.
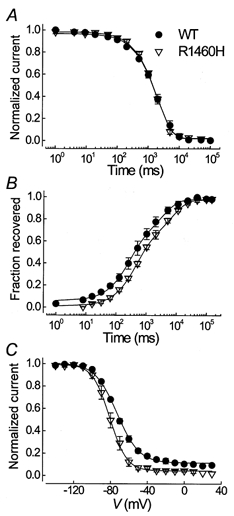
A, entry into slow inactivation at 0 mV. Cells were held at -100 mV, depolarized to 0 mV for increasing durations as indicated on the abscissa, repolarized for 100 ms to -100 mV to let the channels recover from fast inactivation and then depolarized again to -10 mV to determine the fraction of slow inactivated channels. The lines represent fits to a first order exponential function with the following time constants: 1.9 ± 0.3 for WT vs. 2.2 ± 0.1 s for R1460H; n = 3, 6; P > 0.05. B, recovery from slow inactivation measured at -100 mV after a 30 s conditioning pulse to 0 mV. Curves were best fitted to a second order exponential function with the following slow recovery time constants: τsrec1= 0.31 ± 0.11 vs. 0.46 ± 0.06 s, τsrec2= 4.2 ± 1.8 vs. 7.5 ± 1.3 s; relative amplitude of τsrec1= 54 ± 1 vs. 56 ± 4%; n = 3, 5; P > 0.05, for WT and R1460H, respectively. C, steady-state slow inactivation was determined using 30 s prepulses to potentials indicated on the abscissa, followed by a 20 ms repolarizing pulse to the holding potential of -140 mV to let the channels recover from fast inactivation, and a short test pulse to -20 mV. The data were fitted to a standard Boltzmann function: V0.5= -73.0 ± 1.1 for WT vs. -80.9 ± 2.1 mV for R1460H, n = 4, P < 0.02.
Effects of pH on R1460H
Protonation of the substituted histidine residue in R1460H is pH dependent. For another arginine to histidine mutation in IV/S4 causing paramyotonia congenita (R1448H), protonation at low pH restores normal gating (Chahine et al. 1994). Since residue 1460 is only accessible from the intracellular side of the membrane (Yang et al. 1996), we examined the gating of R1460H at intracellular pH 6.2 and 8.5. There were no significant differences in comparison to pH 7.4 in all the parameters presented in Fig. 2–4. Hence, the functional alterations of R1460H are caused by steric effects of the histidine side chain rather than by the decrease in charge.
DISCUSSION
Subtle differences in activation and fast inactivation of the voltage-gated sodium channel were identified for a mutation causing a benign form of human epilepsy. According to the genetic data (Escayg et al. 2000), there is no doubt that this mutation in SCN1A is causative for the disease since it cosegregates perfectly with the phenotype and is localized in an important functional region of a gene that is essential for excitability in nervous tissue. Here, we demonstrate that this mutation has functional consequences that can account for the occurrence of hyperexcitability. One might argue that we did not use the right gene for our functional studies. However, there are three important arguments that prompted us to perform the studies with the same mutation in SCN4A. First, all functionally important regions of SCN1A and SCN4A are highly conserved, as is the voltage sensor IV/S4 (Fig. 1). Second, known functional studies of neuronal and skeletal muscle channels only showed small differences in gating (Goldin, 1999). Third, and most important for us, the use of an established expression system, SCN4A and tsA201 cells, had the advantage that we could directly compare our results to those of previous studies concerning sodium channelopathies of skeletal muscle from our and other laboratories, in particular to those for mutations also located in IV/S4 (Chahine et al. 1994; Lerche et al. 1996; Cannon, 1997; Mitrovic et al. 1999; Lehmann-Horn & Jurkat-Rott, 1999). Nevertheless, the data presented here must be interpreted with some caution until they have been confirmed in the human SCN1A gene in studies including coexpression of the β1- and β2-subunits, which have considerable effects on inactivation of the brain sodium channels (Smith & Goldin, 1998; see also the discussion about different expression systems and the β-subunit below).
The effects of R1460H on fast inactivation presented here are very small compared to those described for other mutations in SCN4A or SCN5A causing disease in skeletal or heart muscle (Cannon, 1997; Lehmann-Horn & Jurkat-Rott, 1999). For example, the inactivation time constant was found to be increased 3- to 6-fold for other mutations in IV/S4 causing paramyotonia congenita (R1448H/C/P: Chahine et al. 1994; Mitrovic et al. 1999). Here, τh was increased only 1.5-fold. Moreover, the most important and the most consistent finding for almost all known mutations in SCN4A and SCN5A is an increased persistent sodium current leading to a permanent sodium inward current, which depolarizes the cell membrane. This will decrease the threshold to elicit an action potential and therefore cause hyperexcitability. A very large persistent current in combination with a defect in slow inactivation will cause an even larger depolarization that will inactivate the sodium channels and therefore result in paralysis (Cannon, 1997). For R1460H in contrast, there was definitely no increase in persistent sodium current.
The main mechanism of the R1460H mutation causing hyperexcitability may be a combination of the faster activation time course and the 3-fold acceleration of recovery from inactivation. This will shorten both the period of depolarization needed to elicit an action potential and the refractory period after an action potential. In contrast to a persistent sodium current, neither mechanism should influence the resting membrane potential, which probably would be fatal when occurring in neurons in contrast to skeletal or heart muscle fibres. An acceleration of sodium channel activation has not been described as a disease-causing mechanism so far.
The subtle alterations in channel gating found in this study for the R1460H mutation are in line with two previous findings concerning sodium channel gating and epilepsy. First, the β1-subunit mutation described by Wallace and colleagues (Wallace et al. 1998) also shows a small defect in inactivation and causes epilepsy but no myotonia, although SCN1B is also expressed in skeletal muscle. Second, a transgenic model introducing a SCN2A mutation in the mouse yields severe status epilepticus, although the inactivation defect was also very small compared to other known mutations (Kearny et al. 1998).
The corresponding mutation to R1460H has already been studied in the rat brain IIa sodium channel for other purposes, before it was known that this mutation can cause epilepsy (R1638H; Kühn & Greeff, 1999). The results are difficult to compare to those presented here, since in the other study the α-subunit was expressed alone in Xenopus oocytes. Isolated expression of brain or skeletal muscle sodium channel α-subunits in oocytes results in a large decrease in the rate of inactivation. This is due to the occurrence of a second slow gating mode of inactivation that is not seen upon coexpression of the β1-subunit. In contrast, isolated expression of α-subunits in HEK293 or tsA201 cells does not significantly alter channel kinetics. The difference between the two expression systems is probably due to an endogenous β1-subunit in HEK cells (Moran et al. 2000). The slow gating mode observed in oocytes seemed to be favoured by R1638H, resulting in a slowing of the inactivation time course.
From a biophysical point of view, our results confirm the importance of the voltage sensor in domain IV for fast channel inactivation (Chahine et al. 1994; Chen et al. 1996; Yang et al. 1996; Lerche et al. 1996; Mitrovic et al. 1999; Kühn & Greeff, 1999; Cha et al. 1999; Horn et al. 2000). The effects on activation have not been described so far for other mutations in IV/S4 and suggest a definite role of IV/S4 in channel activation. However, the faster inactivation time course at these potentials may also influence the current rise time. The stabilization of slow inactivation observed for R1460H channels confirms an important role of IV/S4 in slow inactivation and extends recent results of Mitrovic et al. (2000). The authors proposed a model in which mutations near the midpoint on one side of the putative IV/S4 α-helix enhance slow inactivation. R1460 is located on the same side of the helix just below the important region between residues A1453 and V1458.
We expect that more mutations in different sodium channel genes associated with epilepsy will be found in the future. It will be interesting to study these defects in comparison with disease-causing mutations in other sodium channel genes. This might establish the view presented here that such subtle alterations in gating are sufficient to cause hyperexcitability in the brain, whereas larger defects are needed to cause disease in skeletal or heart muscle.
Acknowledgments
We thank Ms Christine Heuschmid for secretarial help. This work was supported by the Deutsche Forschungsgemeinschaft (DFG Le1030/5-1) and the Interdisziplinares Zentrum für Klinische Forschung/Bundesministerium für Bildung und Forschung (IZKF Ulm/BMBF project B8).
References
- Baulac S, Gourfinkel-An I, Picard F, Rosenberg-Bourgin M, Prud’homme J-F, Baulac M, Brice A, Leguern E. A second locus for familial generalized epilepsy with febrile seizures plus maps to chromosome 2q21-q33. American Journal of Human Genetics. 1999;65:1078–1085. doi: 10.1086/302593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- Cannon SC. From mutation to myotonia in sodium channel disorders. Neuromuscular Disorders. 1997;7:241–249. doi: 10.1016/s0960-8966(97)00430-6. [DOI] [PubMed] [Google Scholar]
- Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Cha A, Ruben PC, George AL, Jr, Fujimoto E, Bezanilla F. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron. 1999;22:73–87. doi: 10.1016/s0896-6273(00)80680-7. [DOI] [PubMed] [Google Scholar]
- Chahine M, George AL, Zhou M, Ji S, Sun W, Barchi RL, Horn R. Na+ channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, Leppert M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nature Genetics. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- Chen LQ, Santarelli V, Horn R, Kallen RG. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. Journal of General Physiology. 1996;108:549–556. doi: 10.1085/jgp.108.6.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, Leguern E, Moulard B, Chaigne D, Buresi C, Malafosse A. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nature Genetics. 2000;24:343–345. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- Goldin AL. Diversity of mammalian voltage-gated sodium channels. Annals of the New York Academy of Sciences. 1999;868:38–50. doi: 10.1111/j.1749-6632.1999.tb11272.x. [DOI] [PubMed] [Google Scholar]
- Horn R, Ding S, Gruber HJ. Immobilizing the moving parts of voltage-gated ion channels. Journal of General Physiology. 2000;116:461–476. doi: 10.1085/jgp.116.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearny JA, Plummer N, Smith MR, Kapur J, Goldin AL, Meisler MH. Seizures in transgenic mice associated with expression of a mutated sodium channel with altered inactivation kinetics. Society for Neuroscience Abstracts. 1998;24:1820. [Google Scholar]
- Kühn FJP, Greeff NG. Movement of voltage sensor S4 in domain 4 is tightly coupled to sodium channel fast inactivation and gating charge immobilization. Journal of General Physiology. 1999;114:167–183. doi: 10.1085/jgp.114.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann-Horn F, Jurkat-Rott K. Voltage-gated ion channels and hereditary disease. Physiological Reviews. 1999;79:1317–1372. doi: 10.1152/physrev.1999.79.4.1317. [DOI] [PubMed] [Google Scholar]
- Lerche H, Mitrovic N, Dubowitz V, Lehmann-Horn F. Paramyotonia congenita: the R1448P Na+ channel mutation in adult human skeletal muscle. Annals of Neurology. 1996;39:599–608. doi: 10.1002/ana.410390509. [DOI] [PubMed] [Google Scholar]
- Lerche H, Peter W, Fleischhauer R, Pika-Hartlaub U, Malina T, Mitrovic N, Lehmann-Horn F. Role in fast inactivation of the IV/S4-S5 loop of the human muscle Na+ channel probed by cysteine mutagenesis. Journal of Physiology. 1997;505:345–352. doi: 10.1111/j.1469-7793.1997.345bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes-Cendes I, Scheffer IE, Berkovic SF, Rousseau M, Andermann E, Rouleau GA. A new locus for generalized epilepsy with febrile seizures plus maps to chromosome 2. American Journal of Human Genetics. 2000;66:698–701. doi: 10.1086/302768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovic N, George AL, Jr, Horn R. Role of domain 4 in sodium channel slow inactivation. Journal of General Physiology. 2000;115:707–717. doi: 10.1085/jgp.115.6.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovic N, George AL, Jr, Rüdel R, Lehmann-Horn F, Lerche H. Mutant channels contribute < 50% to Na+ current in paramyotonia congenita muscle. Brain. 1999;122:1085–1092. doi: 10.1093/brain/122.6.1085. [DOI] [PubMed] [Google Scholar]
- Moran O, Nizzari M, Conti F. Endogenous expression of the β1A sodium channel subunit in HEK-293 cells. FEBS Letters. 2000;473:132–134. doi: 10.1016/s0014-5793(00)01518-0. [DOI] [PubMed] [Google Scholar]
- Morgan K, Stevens EB, Shah B, Cox PJ, Dixon AK, Lee K, Pinnock RD, Hughes J, Richardson PJ, Mizuguchi K, Jackson AP. β3: an additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proceedings of the National Academy of Sciences of the USA. 2000;97:2308–2313. doi: 10.1073/pnas.030362197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulard B, Guipponi M, Chaigne D, Mouthon D, Buresi C, Malafosse A. Identification of a new locus for generalized epilepsy with febrile seizures plus (GEFS+) on chromosome 2q24-q33. American Journal of Human Genetics. 1999;65:1396–1400. doi: 10.1086/302621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer A, Thompson J, Charlier C, Otterud B, Varvil T, Pappas C, Barnitz C, Gruenthal K, Kuhn R, Leppert M. A locus for febrile seizures (FEB3) maps to chromosome 2q23–24. Annals of Neurology. 1999;46:671–678. doi: 10.1002/1531-8249(199910)46:4<671::aid-ana20>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain. 1997;120:479–490. doi: 10.1093/brain/120.3.479. [DOI] [PubMed] [Google Scholar]
- Singh NA, Charlier C, Stauffer D, Dupont BR, Leach RJ, Melis R, Ronen GM, Bjerre I, Quattlebaum T, Murphy JV, McHarg ML, Gagnon D, Rosales TO, Peiffer A, Anderson E, Leppert M. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nature Genetics. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- Singh R, Scheffer IE, Crossland K, Berkovic SF. Generalized epilepsy with febrile seizures plus: a common childhood-onset genetic epilepsy syndrome. Annals of Neurology. 1999;45:75–81. doi: 10.1002/1531-8249(199901)45:1<75::aid-art13>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Smith RD, Goldin AL. Functional analysis of the rat I sodium channel in Xenopus oocytes. Journal of Neuroscience. 1998;18:811–820. doi: 10.1523/JNEUROSCI.18-03-00811.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinlein OK, Mulley JC, Propping P, Wallace RH, Phillips HA, Sutherland GR, Scheffer IE, Berkovic SF. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nature Genetics. 1995;11:201–203. doi: 10.1038/ng1095-201. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Wang DW, Singh R, Scheffer IE, George AL, Jr, Phillips HA, Saar K, Reis A, Johnson EW, Sutherland GR, Berkovic SF, Mulley JC. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel β1 subunit gene SCN1B. Nature Genetics. 1998;19:366–370. doi: 10.1038/1252. [DOI] [PubMed] [Google Scholar]
- Yang N, George AL, Horn R. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]