

Abstract
Recent studies have shown that ryanodine and IP3 receptor (RyR/IP3R)-mediated cytosolic Ca2+ signals propagate to the mitochondria, initiating chains of events vital in the regulation of different cellular functions. However, the fraction of released Ca2+ utilized by the mitochondria during these processes has not been quantified.
To measure the amount of Ca2+ taken up by the mitochondria, we used a novel approach that involves simultaneous fluorescence imaging of mitochondrial and cytosolic [Ca2+] in permeabilized H9c2 myotubes and RBL-2H3 mast cells. Communication between sarco-endoplasmic reticulum (SR/ER) and mitochondria is maintained in these permeabilized cells, as evidenced by the large RyR/IP3R-driven mitochondrial matrix [Ca2+] and NAD(P)H signals and also by preservation of the morphology of the SR/ER-mitochondrial junctions.
Ca2+ was released from the SR/ER by addition of saturating caffeine or IP3 and subsequently thapsigargin (Tg), an inhibitor of SR/ER Ca2+ pumps. The amount of Ca2+ transmitted to the mitochondria was determined by measuring increases of global [Ca2+] in the incubation medium (cytosolic [Ca2+] ([Ca2+]c)). Mitochondrial Ca2+ uptake was calculated from the difference between [Ca2+]c responses recorded in the absence and presence of uncoupler or from [Ca2+]c elevations evoked by uncoupler or ionophore applied after complete Ca2+ mobilization from the SR/ER. [Ca2+]c increases were calibrated by adding Ca2+ pulses to the permeabilized cells.
In H9c2 cells, caffeine induced partial mobilization of SR Ca2+ and mitochondria accumulated 26% of the released Ca2+. Sequential application of caffeine and Tg elicited complete discharge of SR Ca2+ without further increase in mitochondrial Ca2+ uptake. In RBL-2H3 mast cells, IP3 by itself elicited complete discharge of the ER Ca2+ store and the increase of the ionophore-releasable mitochondrial Ca2+ content reached 50% of the Ca2+ amount mobilized by IP3+ Tg. Thus, RyR/IP3R direct a substantial fraction of released Ca2+ to the mitochondria.
Increasing evidence suggests that mitochondria are important in intracellular Ca2+ signalling (for recent reviews see Babcock & Hille, 1998; Jouaville et al. 1998; Simpson & Russell, 1998; Duchen, 1999; Rizzuto et al. 1999; Hajnoczky et al. 2000; Hüser et al. 2000). It has been shown that cytosolic calcium signals elicited by activation of IP3R or RyR are transmitted to the mitochondria (Rizzuto et al. 1993, 1998; Hajnoczky et al. 1995; Chacon et al. 1996; Drummond & Tuft, 1999). Mitochondrial Ca2+ content measured by different techniques has also been shown to be elevated in cells exposed to Ca2+ mobilizing agonists (e.g. Wendt-Gallitelli & Isenberg, 1989; Hoek et al. 1997).
An important function of mitochondrial Ca2+ signals is the control of energy metabolism. Increases in mitochondrial [Ca2+] ([Ca2+]m), coupled to elevations in [Ca2+]c, participate in activation of the respiratory chain through stimulation of Ca2+-sensitive mitochondrial dehydrogenases, thereby ensuring adequate ATP synthesis (Denton & McCormack, 1980; Hansford, 1981; Duchen, 1992; Pralong et al. 1994; Hajnoczky et al. 1995; Robb-Gaspers et al. 1998; Jouaville et al. 1999). Propagation of calcium signals to the mitochondria is also important for apoptotic cell death (Stout et al. 1998; Szalai et al. 1999). Furthermore, mitochondrial Ca2+ transport appears to modulate the spatio-temporal pattern of [Ca2+]c responses evoked by IP3, suggesting that mitochondria are also involved in the shaping of [Ca2+]c signals (Jouaville et al. 1995; Budd & Nicholls, 1996; Babcock et al. 1997; Hoth et al. 1997; Ichas et al. 1997; Simpson et al. 1997; Landolfi et al. 1998; Boitier et al. 1999; Hajnoczky et al. 1999). However, how much calcium is utilized in mitochondrial signalling during intracellular Ca2+ mobilization has not yet been measured.
Our aim was to determine the amount of Ca2+ transmitted from SR/ER to the mitochondria during RyR/IP3R-mediated [Ca2+]c signals. Since calibration of Ca2+ amounts is difficult in intact cells, we established a novel fluorescence Ca2+ imaging approach using permeabilized H9c2 myotubes and RBL-2H3 mast cells, which allowed calibration of Ca2+ release and uptake by intracellular organelles by adding known amounts of calcium. In particular, this method was suitable for estimating the relation between SR/ER release and mitochondrial Ca2+ uptake. We have already shown that the Ca2+ coupling between reticular and mitochondrial Ca2+ stores is well preserved in adherent permeabilized cells (Hajnoczky et al. 1999; Csordás et al. 1999). Furthermore, we found that the morphology of the SR/ER-mitochondrial junctions is retained in permeabilized H9c2 myotubes and RBL-2H3 mast cells. Using these experimental models we show that a large fraction (25-50%) of Ca2+ released through RyR and IP3R can be accumulated by the mitochondria.
METHODS
Cell culture
H9c2 cardiac cells (obtained from American Type Culture Collection, Rockville, MD, USA) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum, 2 mm glutamine, 100 u ml−1 penicillin, 100 μg ml−1 streptomycin and 1 mm pyruvate in humidified air (5% CO2) at 37°C. RBL-2H3 mast cells (kindly provided by Clare Fewtrell) were cultured in Eagle’s minimum essential medium supplemented with 20% fetal bovine serum as described previously (Csordás et al. 1999). For imaging experiments cells were plated onto poly-d-lysine-coated glass coverslips. H9c2 myoblasts were grown to reach confluency (1 week on average) and subsequently for an additional 4-7 days to allow differentiation to myotubes. Since differentiation of H9c2 myoblasts was associated with the loss of an increasing number of cells from the coverslips and confluent cultures were required to obtain measurable [Ca2+] responses in the bath solution, cultures were used before the differentiation of every cell was completed. As such, mixed populations of multinucleated myotubes and differentiating mononucleated H9c2 cells were imaged in the present studies. The average calculated number of H9c2 and RBL cells on each coverslip was 0.11 × 106 and 1.09 × 106, respectively.
Fluorescence imaging measurements in permeabilized H9c2 and RBL-2H3 cells
Prior to use, the cells were preincubated for 30 min in extracellular medium composed of (mm): 121 NaCl, 5 NaHCO3, 10 Na-Hepes, 4.7 KCl, 1.2 KH2PO4, 1.2 MgSO4, 2 CaCl2, 10 glucose and 2% bovine serum albumin (BSA), pH 7.4 at 37°C.
For measurements of [Ca2+]m, the cells were loaded with 5 μm fura-2FF AM or 3 μm rhod-2 AM in the presence of 0.003% (w/v) pluronic acid for 50-70 min. For mitochondrial membrane potential (Δm) measurement, H9c2 cells were loaded with 100 nm tetramethylrhodamine ethyl ester (TMRE) for 15 min. TMRE (10 nm) was also present in the intracellular buffer during the measurements.
Dye-loaded cells were washed with Ca2+-free extracellular buffer composed of (mm): 120 NaCl, 20 Na-Hepes, 5 KCl, 1 KH2PO4 and 100 μm EGTA/Tris at pH 7.4. They were then permeabilized by incubation for 5 min with 15 μg ml−1 digitonin in intracellular medium composed of (mm): 120 KCl, 10 NaCl, 1 KH2PO4, 20 Tris-Hepes at pH 7.2 with 2 mm MgATP, 2 mm succinate and 1 μg ml−1 each of antipain, leupeptin and pepstatin. Intracellular medium was passed through a Chelex column to lower the ambient [Ca2+]. Medium free [Ca2+] was < 100 nm after Chelex treatment and did not exceed 300-400 nm after addition of ATP, succinate and protease inhibitors. In most of the experiments 20 μm EGTA was also present during permeabilization to maintain low [Ca2+] (< 50 nm) and EGTA-free medium was added after permeabilization. For measurements of perimembrane [Ca2+] ([Ca2+]pm) labelling of cells with Calcium Green-C18 or fura-C18 (1.5-5 μm) was carried out during permeabilization. After permeabilization, the cells were washed into fresh buffer without digitonin and incubated in the imaging chamber, at 35°C.
Changes of the global [Ca2+] in the incubation medium ([Ca2+]c) were measured by the Ca2+ tracers fura-2 free acid (FA) (2 μm) and Calcium Green FA (0.25 μm).
Measurements of [Ca2+]m, [Ca2+]pm and [Ca2+]c were carried out using a multiwavelength beamsplitter/emission filter combination and a high quantum-efficiency cooled CCD camera as described earlier (Hajnoczky & Thomas, 1997; Csordás et al. 1999). Excitation at 340 and 380 nm was used for fura-2FF, fura-2 and fura-C18, 490 nm was used for Calcium Green and Calcium Green-C18 and 545 nm was used for rhod-2.
To determine the average [Ca2+]pm and [Ca2+]m signals, mean fluorescence intensities (for rhod-2 and Calcium Green-C18) or ratios (for fura-2FF and fura-C18) were calculated for essentially all individual whole-cell areas in each run after subtraction of the background fluorescence measured at cell-free areas of the field (Fig. 3 and 4). To determine changes in [Ca2+]c, fluorescence intensities of fura-2 or Calcium Green dissolved in the incubation medium were taken from the entire field without background subtraction (Fig. 3 and 4). The Kd values of 224 nm for fura-2 and 300 nm for Calcium Green were used for calculation of [Ca2+]c.
Figure 3. Changes of perimembrane and global [Ca2+]c during Ca2+ mobilization.
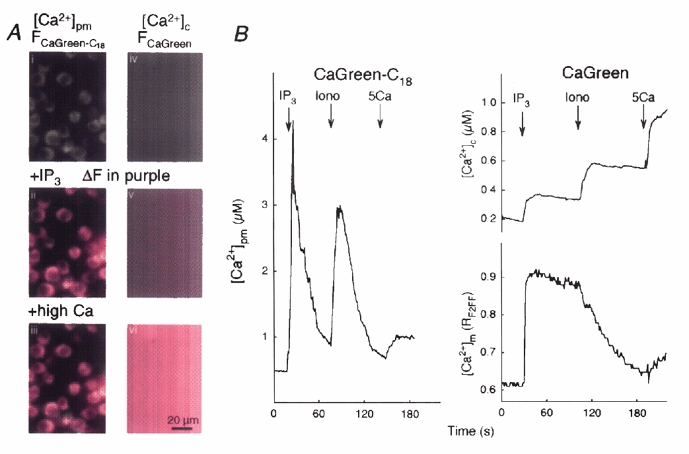
A, i and iv, grey-scale images show the fluorescence of Calcium Green-C18 attached to permeabilized RBL-2H3 mast cells (left) and Calcium Green added to the medium (right). Increases of the fluorescence (ΔF) evoked by IP3-induced Ca2+ release (ii and v) and a saturating dose of CaCl2 (1.5 mm, high calcium, iii and vi) are shown in purple. B, time course of the [Ca2+]pm responses evoked by IP3 (12.5 μm), ionomycin (Iono, 10 μm) and Ca2+ (5 μm, 5Ca) are shown on the left. Time courses of [Ca2+]c recorded simultaneously with [Ca2+]m are shown on the right.
Figure 4. Effect of Ca2+ mobilization from SR on [Ca2+]c, [Ca2+]m and Δm.
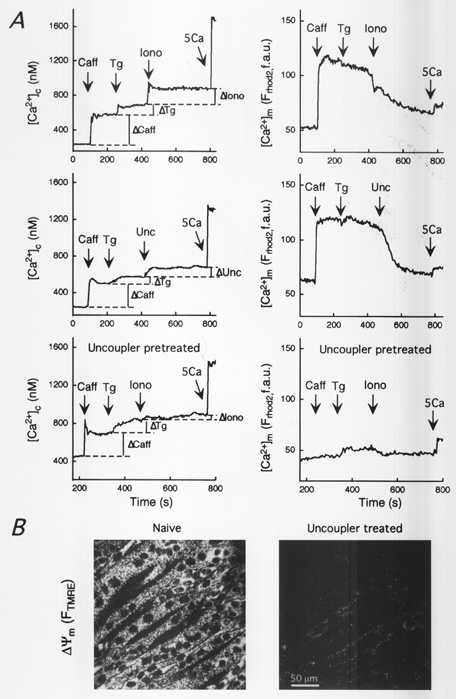
A, time courses of [Ca2+]m and [Ca2+]c recorded in permeabilized H9c2 myotubes exposed to caffeine (Caff, 20 mm), thapsigargin (Tg, 2 μm), ionomycin (Iono, 10 μm, top) or uncoupler (Unc: FCCP/oligomycin, 5 μg ml−1 each, middle). The experiment in the top panel was repeated on uncoupler-pretreated cells (lower panel). [Ca2+]m and [Ca2+]c were measured using compartmentalized rhod-2 and fura-2FA (2 μm) dissolved in the incubation medium, respectively. For calculations of Ca2+ amounts a calibrating pulse of Ca2+ (CaCl2, 5 μm) was applied at the end of each run. B, confocal images of TMRE-loaded permeabilized H9c2 myotubes before (left) and after (right) exposure to uncoupler. The fluorescence intensity of TMRE reflecting Δm is shown using grey-scale.
Confocal imaging of Δm and two-photon imaging of NAD(P)H were carried out using a BioRad-MRC1024/2P imaging system fitted to an Olympus-IX70 inverted microscope. For confocal imaging a Kr/Ar-ion laser source (568 nm excitation) was used whereas two-photon (2P) imaging was carried out using a pulsed femtosecond laser system (Millennia V/Tsunami, tuned to 720-740 nm, 80 fs pulses) and non-descanned detectors for recording the fluorescence signal.
Experiments were carried out with at least four to five different cell preparations, and 25-100 cells were monitored in each experiment. Traces represent single cell responses unless it is indicated otherwise. The data are shown as means ±s.e.m. Significance of differences from the relevant controls was calculated by Student’s t test.
Fura-2, fura-2FF, Calcium Green and rhod-2 were obtained from Teflabs (Austin, TX, USA) and Calcium Green-C18 and fura-C18 were from Molecular Probes (Eugene, OR, USA).
Electron microscopy
Intact and permeabilized cells were fixed in 2% glutaraldehyde, 1% tannic acid, 0.1 m sodium cacodylate at pH 7.4 for 2 h at room temperature. The fixed cells were then carefully detached using a rubber-tipped cell scraper. All samples were washed three times in 0.1 m sodium cacodylate, pH 7.4, and then post-fixed in 1% OsO4 at room temperature in the same buffer. All samples were then washed three times in distilled water and stained with 1% uranyl acetate and pelleted in 2% agarose. The pellets were dehydrated in graded steps of acetone and embedded into Spurrs resin. Sections (80 nm thick) were cut on a Reichert Ultracut E microtome and stained using uranyl acetate and sodium bismuth. The sections were examined with a Hitachi 7000 scanning transmission electron microscope, and micrographs were obtained using Kodak 4489 film. Electron micrographs are typical of data obtained in two experiments.
Micrographs were collected in all areas of the sections that showed mitochondria and SR/ER. Before measurements, the shape and distribution of organelles were inspected in every micrograph and the organelles visible in more than one section were marked to avoid double counting of SR/ER-mitochondrial interfaces. The minimum distance between the mitochondrial outer membrane and the nearest SR/ER membrane was measured for all mitochondria included in images.
RESULTS
Ca2+ release responses and mitochondrial calcium signals evoked by maximal activation of RyR and IP3R
First we monitored the Ca2+ release evoked by maximal activation of RyR with caffeine (20 mm) in H9c2 myotubes. As shown in Fig. 1A, caffeine induced a large increase in perimembrane [Ca2+] ([Ca2+]pm) measured with a lipophilic Ca2+ probe, fura-C18, anchored to cellular membranes (Fig. 1A, i and ii; green to red shift in the overlaid images). The [Ca2+]pm increase was transient as released Ca2+ was diluted in the bulk cytosolic medium (Fig. 1A, v). Figure 1A also shows that the magnitude of the caffeine-induced [Ca2+]pm spike was as large as the [Ca2+]pm elevation caused by addition of 10 μm CaCl2. The mitochondrial [Ca2+] ([Ca2+]m) response elicited by caffeine was measured by rhod-2 compartmentalized in the mitochondria (Hajnoczky et al. 1995, 1999; Csordás et al. 1999). Figure 1A shows that [Ca2+]m displayed a large increase (iii and iv; shown in red) that was synchronized to the upstroke of the [Ca2+]pm response and was sustained (Fig. 1A, v).
Figure 1. Fluorescence imaging of [Ca2+]pm and [Ca2+]m and two-photon imaging of NAD(P)H in permeabilized H9c2 myotubes and RBL-2H3 mast cells.
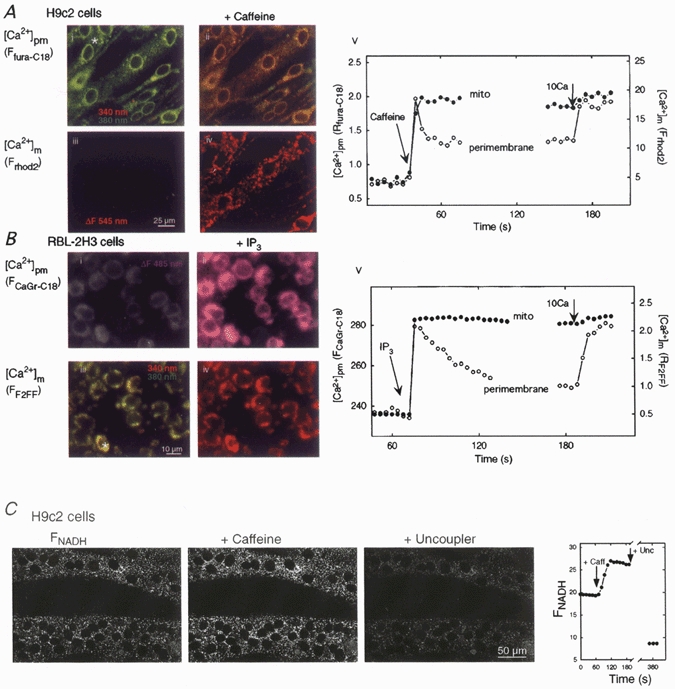
A, fluorescence images of H9c2 myotubes before and after caffeine addition (20 mm) using fura-C18 (i and ii) and compartmentalized rhod-2 (iii and iv) to obtain simultaneous measurements of [Ca2+]pm and [Ca2+]m. B, fluorescence images of RBL-2H3 mast cells before and after stimulation with IP3 (12.5 μm) using Calcium Green-C18 (i and ii) and compartmentalized fura-2FF (iii and iv). The corresponding time courses of [Ca2+]pm and [Ca2+]m during RyR/IP3R stimulation and Ca2+ (10μm CaCl2) addition are shown in v. Fluorescence signals were calculated for the total extranuclear area of the cells marked with an asterisk. Frhod2 and FCalcium Green-C18 are shown as fluorescence arbitrary units (f.a.u.). Fura-C18 (A, i and ii) and fura-2FF (B, iii and iv) fluorescence are shown as overlays of images collected at 340 nm (red, increases from Ca2+) and 380 nm excitation (green, decreases from Ca2+). Rhod-2 (A, iii and iv) and Calcium Green-C18 (B, i and ii) fluorescence is shown in grey-scale and the fluorescence changes (ΔF) are visualized in red and purple, respectively. C, two-photon imaging of the NAD(P)H response to caffeine in permeabilized H9c2 myotubes. The grey images show the NAD(P)H fluorescence in two multinucleated permeabilized myotubes before stimulation (left), after stimulation with caffeine (20 mm, middle) and after treatment with the uncoupler carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP; 5 μg ml−1, right). The uncoupler was used to stimulate mitochondrial oxidation. Because of the absence of mitochondria in the nuclear matrix, the nuclei are visible as dark circles. Time course of the FNAD(P)H signal (in f.a.u.) was calculated for the total extranuclear area of the cell shown in the upper part of the images.
Similar measurements of [Ca2+]pm and [Ca2+]m were also carried out in permeabilized RBL-2H3 mast cells exposed to IP3. Saturating doses of IP3 (12.5 μm) elicited a [Ca2+]pm rise as measured by Calcium Green-C18 (Fig. 1B, i and ii; the fluorescence increase (ΔF) visualized in purple) and the [Ca2+]pm signal was transient (Fig. 1B, v). [Ca2+]m was measured in RBL-2H3 mast cells using fura-2FF as described previously (Csordás et al. 1999). All cells in the field showed a [Ca2+]m rise in association with the [Ca2+]pm spikes (Fig. 1B, iii and iv; green to red shift in the overlaid images). The corresponding time course shows a sustained IP3-induced increase in [Ca2+]m (Fig. 1B, v). Sustained elevations of [Ca2+]m were not due to saturation of the compartmentalized Ca2+ tracers, since addition of 2-5 mm CaCl2 following maximal stimulation of RyR/IP3R resulted in substantial further increases in [Ca2+]m (data not shown).
Several mitochondrial dehydrogenases are activated by elevated [Ca2+]m and this activation can be monitored fluorometrically through changes in pyridine nucleotide redox state (e.g. Duchen, 1992; Pralong et al. 1994; Hajnóczky et al. 1995). Figure 1C shows that the caffeine-induced [Ca2+]m signal resulted in an increase in NAD(P)H fluorescence in permeabilized H9c2 myotubes, reflecting the Ca2+-dependent dehydrogenase activation. The redox response appeared throughout the cell, apart from the nuclear matrix. These data suggest that regulation of intramitochondrial metabolism by RyR-driven [Ca2+]m signals is retained in permeabilized H9c2 cells. Measurement of Ca2+ signal-dependent NAD(P)H responses was not feasible in RBL-2H3 mast cells owing to the very small NAD(P)H fluorescence in these cells (G. Csordás & G. Hajnoczky, unpublished data).
Close junctions between SR/ER and mitochondrial membranes are thought to be the sites where RyR/IP3R-mediated Ca2+ signals are relayed to the mitochondria (Rizzuto et al. 1993, 1998; Csordás et al. 1999). We used electron microscopy to study whether the morphology of the contacts between SR/ER and mitochondria is preserved in adherent permeabilized cells. Previous electron microscopy studies have shown close contacts in intact RBL-2H3 cells and cardiomyocytes (Wilson et al. 1998; Sharma et al. 2000) and preservation of the integrity of intracellular membranes in adherent cardiac cells and hepatocytes permeabilized with low concentrations of digitonin (e.g. Altschuld et al. 1985; Renard-Rooney et al. 1993). Figure 2 shows electron micrographs of H9c2 cells (A) and RBL-2H3 cells (B) before (left) and after (right) permeabilization. In intact as well as in permeabilized H9c2 cells two types of SR/ER morphology were observed: polymorph structures with ample intraluminal space mostly in smaller cells (Fig. 2A, i and ii) and linear stacks with narrow lumen particularly in the multinucleated myotubes (Fig. 2A, iii and iv). Mitochondria appeared as tubular and oval structures in H9c2 cells. Strikingly, most of the mitochondria exhibited close junctions with SR (shown by arrows). Although the intercristal regions of the mitochondria were dilated in permeabilized cells, the close junctions between mitochondria and SR/ER did not appear to be affected by permeabilization of the adherent cells. Less than 100 nm distance was measured between mitochondria and SR/ER for 30 out of 42 mitochondria (71%) and 45 out of 69 mitochondria (65%) in intact and permeabilized H9c2 cells, respectively. The remaining ∼30% of mitochondria could also have junctions with SR/ER since we analysed only one section of most of the mitochondria. Furthermore, the distance distribution of the contacts in permeabilized H9c2 cells was not different from that observed in intact cells (Fig. 2C), suggesting that permeabilization did not yield an increase of the space between SR/ER and mitochondrial membranes.
Figure 2. Morphology of the junctions between SR/ER and mitochondria in intact and permeabilized H9c2 and RBL-2H3 cells.
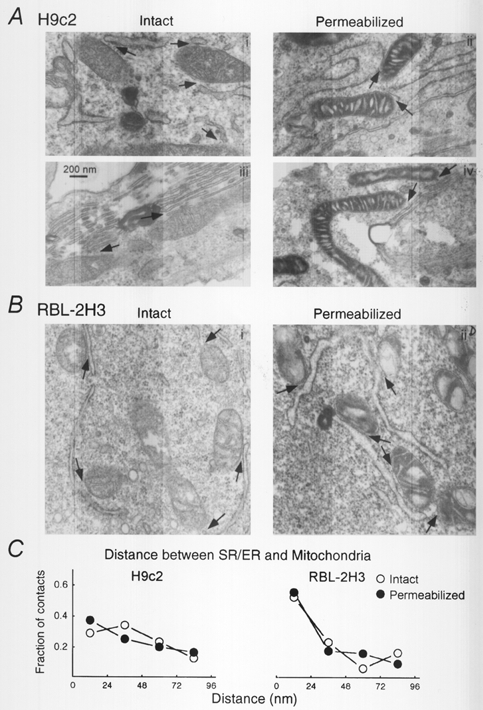
Electron micrographs of H9c2 myotubes (A) and RBL-2H3 mast cells (B). In each case intact cells are shown in the left and permeabilized cells are shown in the right panels. Two pairs of micrographs of H9c2 cells are shown to illustrate the two typical patterns of ER morphology observed in these cells. Arrows indicate the close junctions between SR/ER and mitochondria. C, distance distribution of the junctions between SR/ER and mitochondria. Close appositions of SR/ER and mitochondrial membranes (< 100 nm distance) were sorted into four groups, < 24, 24-48, 48-72, 72-96 nm distance, and the number of contacts in each group was normalized to the total number of contacts (H9c2: intact, 42 contacts; permeabilized, 67 contacts; RBL-2H3: intact, 62 contacts; permeabilized, 64 contacts).
In RBL-2H3 cells, the majority of the mitochondria were oval shaped and similar to the H9c2 cells most of the mitochondria appeared in close association with ER (Fig. 2B). In intact cells 43 out of 56 mitochondria (77%), and in permeabilized cells 50 out of 66 mitochondria (76%) appeared at less than 100 nm distance from ER and no major difference was found between the distance distribution of the contacts in the two conditions (Fig. 2C, right). Interestingly, over 50% of the contacts exhibited less than 24 nm distance between ER and mitochondrial membranes in RBL-2H3 cells, whereas only 30% did in H9c2 cells, suggesting that the local coupling is particularly close in RBL-2H3 cells (Fig. 2C). Taken together, these electron microscopy data demonstrated preservation of the morphology of the contacts between SR/ER and mitochondria in permeabilized H9c2 and RBL-2H3 cells. Although the intramitochondrial morphology was affected by permeabilization, the RyR/IP3R-mediated [Ca2+]m and NAD(P)H signals suggest that mitochondrial calcium regulation was retained in the permeabilized cells.
In summary, the rapid and large increases of [Ca2+]m show the efficient propagation of RyR- and IP3R-mediated Ca2+ signals to the mitochondria in permeabilized H9c2 myotubes and RBL-2H3 mast cells. Preservation of the close contacts between SR/ER and mitochondria in permeabilized H9c2 and RBL-2H3 cells provides the basis of local calcium signal transmission. Furthermore, the RyR-dependent NAD(P)H responses show that propagation of calcium signals to the mitochondria results in activation of the mitochondrial effectors in permeabilized cells. Also the rise in [Ca2+]m following maximal stimulation of RyR/IP3R was sustained in both cell types suggesting that mitochondria retained the accumulated Ca2+. As such, the amount of Ca2+ releasable from the mitochondria during the sustained phase of [Ca2+]m signals can be used as a measure of the Ca2+ taken up during RyR/IP3R activation.
Measurements of global [Ca2+] in the cytosolic compartment
To determine the amount of Ca2+ discharged from the mitochondria we designed an approach to measure released Ca2+ in the cytosolic buffer. As shown in Fig. 1, Ca2+ release from the stores yielded transient elevations of [Ca2+] to the micromolar range in the vicinity of membranes. By contrast, Ca2+ release was anticipated to yield small changes of global [Ca2+]c, owing to the cell permeabilization-induced expansion of the cytosolic space. To increase the changes in global [Ca2+] we used confluent cultures. Under these conditions 170 ± 3 (n = 4) or 340 ± 15 μg protein (n = 2) of permeabilized H9c2 myotubes or RBL-2H3 mast cells was used. Since the size of the SR/ER Ca2+ store was reported to be in the range of 2 to > 20 nmol (mg protein)−1 in permeabilized cells (e.g. Altschuld et al. 1985; Biden et al. 1986; Kindman & Meyer, 1993), we estimated that caffeine or IP3 may release at least 0.5 nmol Ca2+ from the confluent cultures to the 1 ml volume of buffer in the imaging chamber. We also postulated that a relatively low Ca2+ binding ratio in the cytosolic buffer may partially compensate for the increased cytosolic volume resulting from cell permeabilization. To establish a calibration for [Ca2+] in the cytosolic buffer taking into account Ca2+ buffering we added known amounts of Ca2+ to give a total Ca2+ load of 5 μm, and calibrated the effective change in [Ca2+]c achieved using fura-2. The [Ca2+]c increase was 680 ± 23 nm (n = 30), suggesting that the Ca2+ binding ratio is approximately 6. Although cytosolic Ca2+ binding ratios have not been reported for H9c2 myotubes or RBL-2H3 cells, in cardiac muscle the global cytosolic Ca2+ binding ratio is between 50 and 128 at submicromolar [Ca2+] (for review see Neher, 1995). Thus, a weaker Ca2+ buffering in the cytosolic buffer may facilitate intracellular Ca2+ mobilization to evoke measurable increases of [Ca2+] in the expanded cytosolic space. Since propagation of RyR/IP3R-mediated Ca2+ signals to the mitochondria is likely to occur at the SR/ER mitochondrial junctions and the ability of Ca2+ buffers to move between the junctions and the global cytosolic space is not known, it is not clear whether dilution of the cytosol also affects Ca2+ signalling to the mitochondria. One clue to this point is that propagation of IP3R-mediated Ca2+ signals to the mitochondria is relatively insensitive to increases in global [Ca2+]c buffering in RBL-2H3 mast cells (Csordás et al. 1999). Taken together, on the basis of these data we predicted that intracellular Ca2+ mobilization from confluent cell cultures yields a measurable increase in the bulk [Ca2+]c.
First, parallel measurements of [Ca2+]pm and [Ca2+]c were carried out in permeabilized RBL-2H3 mast cells using Calcium Green-C18 and Calcium Green FA, respectively. Using Calcium Green-C18 the IP3-induced [Ca2+] elevation appeared as a large fluorescence increase over the cells (Fig. 3A, ii; ΔF is shown in purple), whereas using Calcium Green a small fluorescence increase was measured all over the image (Fig. 3A, v). Incubation with Calcium Green-C18 and Calcium Green was set so that the maximal fluorescence increase evoked by CaCl2 (1.5 mm) was similar using these two Ca2+ tracers (Fig. 3A, iii and vi). As Calcium Green-C18 was attached to cellular membranes the fluorescence increase caused by saturating [Ca2+] was visible above the cells, whereas the [Ca2+] rise detected by Calcium Green was homogeneous. In addition to the difference in magnitude and spatial pattern between IP3-induced [Ca2+]pm and [Ca2+]c responses, the time courses revealed that IP3 and Ca2+ ionophore (ionomycin)-dependent [Ca2+]pm signals appeared as spikes, whereas the [Ca2+]c signals were sustained (Fig. 3B). It is noteworthy that the calibrating Ca2+ pulse (CaCl2 5 μm) added after ionomycin caused very similar increases in [Ca2+]pm and [Ca2+]c (Fig. 3B left vs. upper right) underscoring that the difference in the [Ca2+] change evoked by IP3 was not due to differences in the sensitivity of the two probes to Ca2+. Simultaneous measurements of [Ca2+]c and [Ca2+]pm carried out using Calcium Green and compartmentalized fura-2FF show that the IP3-induced [Ca2+]c signal was associated with a rapid and large [Ca2+]m rise (right panel). Addition of ionomycin caused a decay of [Ca2+]m and simultaneous rise in [Ca2+]c. [Ca2+]c increases were calibrated with Ca2+ pulses added at the end of each run. In permeabilized H9c2 myotubes [Ca2+]c and [Ca2+]m were measured using fura-2FA dissolved in the incubation medium and compartmentalized rhod-2, respectively. The pattern of [Ca2+]c signals evoked by intracellular Ca2+ mobilization in H9c2 myotubes was essentially the same as it was in RBL-2H3 mast cells (Fig. 4, see below).
Collectively, these data demonstrated that Ca2+ release in permeabilized H9c2 myotubes and RBL-2H3 mast cells can be measured by monitoring global [Ca2+]c. The sustained temporal pattern of the [Ca2+]c changes and simple calibration of the [Ca2+]c signals give an opportunity to determine the amount of Ca2+ mobilized from different intracellular stores.
Quantification of mitochondrial Ca2+ uptake
Figure 4A shows [Ca2+]c and [Ca2+]m signals during mobilization of the SR and mitochondrial Ca2+ stores in H9c2 myotubes. Maximal activation of the RyR with caffeine yielded rapid and prolonged elevations in [Ca2+]c and [Ca2+]m (top and middle). It is assumed that the initial rise of [Ca2+]c over the plateau level was due to the fact that the image was focused on the cells and so a fraction of the Ca2+ tracer sensed the initial [Ca2+] burst in the vicinity of the Ca2+ stores. The amount of Ca2+ released by caffeine was determined using the plateau level (marked by dashed lines). To establish complete depletion of the reticular stores we added Tg, which caused an increase in [Ca2+]c, whereas [Ca2+]m remained unchanged (top and middle). Then a supramaximal dose of ionomycin (10 μm) was added to release Ca2+ from all the remaining non-acidic membrane compartments. After complete depletion of the reticular stores, Ca2+ released by ionomycin can be considered to be of mitochondrial origin in most cell types (Hoth et al. 1997). In support of this point, the ionomycin-induced [Ca2+]c rise was associated with a decrease in [Ca2+]m (upper right), and pretreatment with uncoupler (FCCP/oligomycin 5 μg ml−1 each) that dissipates Δm (Fig. 4B, Δm was measured using TMRE), the driving force for mitochondrial Ca2+ accumulation, markedly reduced ionomycin-induced Ca2+ release after complete discharge of the SR by caffeine + Tg (Fig. 4A; upper left vs. lower left; statistics are shown in Fig. 5A). Alternatively, uncoupler was used to release Ca2+ from the mitochondria after caffeine-induced Ca2+ mobilization from the SR in H9c2 cells (Fig. 4A, middle). Addition of uncoupler after ionomycin or vice versa did not cause a significant decrease in [Ca2+]m (not shown). Furthermore, Fig. 4A shows that uncoupler eliminated propagation of the [Ca2+]c signal to the mitochondria (lower right).
Figure 5. Quantification of Ca2+ redistribution between intracellular Ca2+ stores during Ca2+ mobilization.
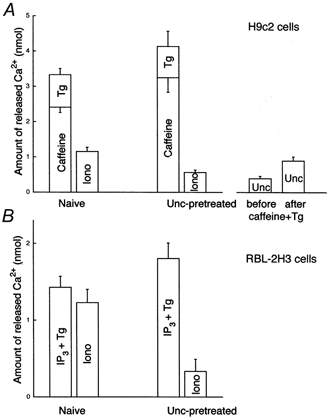
Amounts of Ca2+ released upon addition of caffeine (20 mm), Tg (2 μm) or IP3+ Tg (12.5 μm IP3, 2 μm Tg), and ionomycin (Iono,10 μm) in naive and uncoupler (Unc, FCCP + oligomycin)-pretreated H9c2 myotubes (A) and RBL-2H3 mast cells (B).
To calculate the amount of Ca2+ released from SR by caffeine + Tg and from mitochondria by ionomycin or uncoupler, the [Ca2+]c rises were calibrated with application of known amounts of Ca2+. In our experiments the relation between the Δ[Ca2+]c and amounts of Ca2+ added was linear in the range of changes caused by mobilization of intracellular stores, and thus we used the following equation to express Ca2+ uptake/release in terms of nanomoles of Ca2+:
where ΔCa2+x (nmol) is the amount of Ca2+ to be determined, Δ[Ca2+]cx is the corresponding change in the [Ca2+]c, ΔCa2+cal is the amount of Ca2+ used for calibration (nmol) and Δ[Ca2+]ccal is the increase in [Ca2+]c caused by the calibrating Ca2+ pulse.
Figure 5 shows the fractions of Ca2+ released by the different manipulations in H9c2 myotubes (Fig. 5A) and in RBL-2H3 mast cells (Fig. 5B). The amount of Ca2+ released by caffeine + Tg was 3.32 ± 0.17 nmol (n = 18; 20 nmol (mg protein)−1) in naive and 4.13 ± 0.40 nmol (n = 7) in uncoupler-pretreated H9c2 myotubes, respectively. As uncoupler prevents mitochondrial Ca2+ uptake (e.g. Fig. 4) the uncoupler-dependent increase in the [Ca2+]c response elicited by caffeine + Tg (P < 0.05) was used as a measure of mitochondrial Ca2+ accumulation.
The uncoupler-dependent increase in Ca2+ release evoked by caffeine + Tg was associated with a decrease in the residual Ca2+ released by subsequent addition of ionomycin (Fig. 5A; 1.15 ± 0.12 vs. 0.56 ± 0.06 nmol Ca2+ in naive and uncoupler-pretreated cells, n = 13 and 7, P < 0.01). The uncoupler-dependent decrease in ionomycin-induced Ca2+ release was also used as a measure of mitochondrial sequestration of Ca2+ mobilized from the SR, though uncoupler pretreatment could also attenuate ionomycin-induced Ca2+ release by mobilization of Ca2+ that had been present in the mitochondria prior to Ca2+ release. Thus, we also measured the amount of Ca2+ released by uncoupler added before and after caffeine + Tg-induced Ca2+ release. Mitochondrial uptake of released Ca2+ was expected to increase the uncoupler-sensitive store. Uncoupler-induced Ca2+ release was larger after mobilization of the SR store and the increase was similar to the change of the ionomycin-induced release (0.89 ± 0.11 vs. 0.39 ± 0.07 nmol in caffeine + Tg-treated and in naive cells, n = 5, P < 0.01). Based on these measurements the fraction of Ca2+ taken up by the mitochondria during Ca2+ mobilization from the SR was ∼15% (19.2, 14.3 and 12.1% as calculated from the changes in the caffeine + Tg, ionomycin and uncoupler-induced [Ca2+]c responses, respectively).
Importantly, caffeine added by itself mobilized 70-80% of the Tg-sensitive store in H9c2 myotubes and Tg-induced release of the residual Ca2+ was poorly transmitted to the mitochondria (Fig. 4A). Figure 5A shows that the mitochondrial Ca2+ uptake calculated from the uncoupler-dependent increase in the caffeine-induced [Ca2+]c response was as large as it was when Tg was also added (0.85 vs. 0.79 nmol). From the uncoupler-dependent increment of the caffeine-induced [Ca2+]c response, mitochondria were calculated to accumulate 26% of the mobilized Ca2+. Taken together, these results demonstrated that a large fraction of the Ca2+ released via RyR (26%) can be taken up by the mitochondria. It remains to be determined whether the caffeine-insensitive component of the SR/ER Ca2+ store (20-30%) displays local Ca2+ communication with mitochondria. Discharge of the caffeine-insensitive store with Tg failed to support mitochondrial Ca2+ uptake in the present experiments but Tg has also been shown to mobilize RyR/IP3R-sensitive stores with little mitochondrial Ca2+ accumulation in permeabilized RBL-2H3 cells, suggesting that coordinated activation of the release sites is required for optimal activation of mitochondrial Ca2+ uptake sites (Csordás et al. 1999).
In naive RBL-2H3 mast cells IP3+ Tg-induced Ca2+ release to the cytosolic buffer was 1.43 ± 0.14 nmol (n = 8; 4.2 nmol (mg protein)−1) whereas in uncoupler-pretreated cells 1.8 ± 0.2 nmol (n = 11) was liberated. The size of the SR/ER store is smaller in RBL-2H3 cells than in H9c2 myotubes (4.2 vs. 20 nmol (mg protein)−1), consistent with the relatively low density of ER shown by the electron micrographs of RBL-2H3 cells (Fig. 2). It is noteworthy that IP3 releases the entire Tg-sensitive Ca2+ store in RBL-2H3 mast cells (e.g. Csordás et al. 1999). Similar to H9c2 myotubes, in uncoupler-pretreated RBL-2H3 mast cells, the augmented IP3+ Tg-induced Ca2+ release was followed by a small ionomycin-induced Ca2+ release (Fig. 5B; 1.23 ± 0.17 vs. 0.33 ± 0.16 nmol Ca2+ in naive and uncoupler-pretreated cells, n = 11 and 8, P < 0.01). Thus the mitochondrial Ca2+ uptake calculated from the difference in magnitude of the IP3+ Tg-induced [Ca2+]c responses recorded in naive and uncoupler-pretreated cells was 20% of the total release, whereas it was estimated to be 50% using the uncoupler-dependent change in ionomycin-induced release responses in RBL-2H3 mast cells. In mast cells secretory granules represent an uncoupler-sensitive and ionomycin-insensitive Ca2+ store. If Ca2+ is released from secretory granules during IP3-induced Ca2+ mobilization as reported (e.g. Nguyen et al. 1998), calculations based on the uncoupler-dependent change in the effect of IP3+ Tg may lead to underestimation of mitochondrial Ca2+ uptake. Our observation that most of the contacts between ER and mitochondria are very close in RBL-2H3 cells (Fig. 2) is also in support of a particularly effective mitochondrial Ca2+ load during IP3-induced Ca2+ release.
DISCUSSION
In the present study we have described a fluorescence imaging method that can be used for quantitative measurement of RyR- or IP3R-mediated SR/ER Ca2+ release and mitochondrial Ca2+ uptake in permeabilized cells. In essence, we established measurements of Ca2+ mobilized from the intracellular stores of permeabilized adherent cells to the cytosolic buffer. Although cell permeabilization would not necessarily be expected to leave unaffected Ca2+ delivery to the mitochondria, evidence is emerging that calcium signal transmission to the mitochondria is controlled by a local [Ca2+]c regulation between RyR/IP3R and mitochondrial Ca2+ uptake sites and that this local [Ca2+]c regulation is preserved in carefully permeabilized adherent cells (Rizzuto et al. 1993, 1998; Csordás et al. 1999; Hajnoczky et al. 1999). This idea is further supported by the present electron microscopy data demonstrating preservation of the morphology of the SR/ER-mitochondrial junctions in permeabilized cells and also by the imaging studies showing large RyR/IP3R-driven mitochondrial matrix [Ca2+] and NAD(P)H signals in individual permeabilized cells. Importantly, the experimental protocol used for cell permeabilization and calcium imaging was also designed to avoid changes in the loading state of Ca2+ stores. Thus, the calcium imaging approach we used can provide quantitative information relevant for the physiological redistribution of Ca2+ between SR/ER and mitochondria during RyR/IP3R-mediated Ca2+ mobilization.
We have used a mode of stimulation in which synchronized activation of the release sites occurs in all permeabilized cells present in the incubation chamber. Also, we used relatively weak Ca2+ buffering in the cytosolic medium. These conditions have allowed us to monitor SR/ER and mitochondrial Ca2+ transport by measuring [Ca2+]c in the bulk bathing medium. Importantly, it has been demonstrated previously that propagation of RyR/IP3R-mediated Ca2+ signals to the mitochondria is relatively insensitive to changes in global [Ca2+]c buffering (Rizzuto et al. 1993; Csordás et al. 1999; Sharma et al. 2000; Szalai et al. 2000). This suggests that dilution of the cytosol fails to affect the Ca2+ buffering component which is important in the local Ca2+ transfer between SR/ER release sites and mitochondrial Ca2+ uptake sites. Furthermore, a fundamental feature of intracellular calcium signalling is that synchronized activation of IP3R and RyR establishes [Ca2+]c oscillations, the frequency of which is controlled by the intensity of stimulation. Mitochondrial Ca2+ uptake seems to be associated with the rapid rise of [Ca2+]c responses and so loading of mitochondria by Ca2+ released during calcium spikes in intact cells may be as large as we calculated using permeabilized cells exposed to maximal doses of caffeine or IP3.
The most important finding of this study is that a large fraction of the Ca2+ released via RyR or IP3R can be taken up by the mitochondria. In carefully permeabilized adherent H9c2 myotubes, 26% of Ca2+ released via RyR was delivered to the mitochondria, whereas in RBL-2H3 cells 50% of Ca2+ released through IP3R was taken up by the mitochondria. As the mitochondrial matrix volume represents a relatively small fraction of the total cytosolic volume, mitochondrial accumulation of 25-50% of Ca2+ released to the cytosol allows elevation of [Ca2+]m well above the [Ca2+]c level. Elevation of [Ca2+]m controls the function of mitochondrial effectors such as Ca2+-sensitive dehydrogenases and the permeability transition pore. Another important conclusion of our study is concerned with the spatial distribution of Ca2+ release through the SR/ER membrane. Since calcium signal transmission from SR/ER to the mitochondria depends on a local control between RyR/IP3 and mitochondrial Ca2+ uptake sites and only subregions of the SR/ER surface are close to the mitochondia (e.g. Rizzuto et al. 1998; see also Fig. 2), delivery of 25-50% of released Ca2+ to the mitochondria may occur only if Ca2+ release is concentrated at SR/ER subdomains facing mitochondria. Remarkably, this functional organization allows mitochondria to contribute to the control over activation and deactivation of RyR/IP3R, to the shaping of global [Ca2+]c signals and also to the recharging of reticular Ca2+ stores. Taken together, the local transfer of substantial amounts of Ca2+ to the mitochondria demonstrated in the present study is important for the multiple physiological roles of mitochondrial calcium signalling in the regulation of extra- and intramitochondrial Ca2+-dependent effector mechanisms.
Acknowledgments
We would like to thank Drs Suresh K. Joseph and Theodore F. Taraschi for comments on this manuscript. This work was supported by a Grant-In-Aid (to G.H.) from the American Heart Association and DK51526 (to G.H.) from NIH. G.H. is a recipient of a Burroughs Wellcome Fund Career Award in the Biomedical Sciences. P.P. is a recipient of a Juvenile Diabetes Foundation Postdoctoral Fellowship.
References
- Altschuld RA, Wenger WC, Lamka KG, Kindig OR, Capen CC, Mizuhira V, Vander Heide RS, Brierley GP. Structural and functional properties of adult rat heart myocytes lysed with digitonin. Journal of Biological Chemistry. 1985;260:14325–14334. [PubMed] [Google Scholar]
- Babcock DF, Herrington J, Goodwin PC, Park YB, Hille B. Mitochondrial participation in the intracellular Ca2+ network. Journal of Cell Biology. 1997;136:833–844. doi: 10.1083/jcb.136.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock DF, Hille B. Mitochondrial oversight of cellular Ca2+ signaling. Current Opinion in Neurobiology. 1998;8:398–404. doi: 10.1016/s0959-4388(98)80067-6. [DOI] [PubMed] [Google Scholar]
- Biden TJ, Wollheim CB, Schlegel W. Inositol 1,4,5-trisphosphate and intracellular Ca2+ homeostasis in clonal pituitary cells (GH3). Translocation of Ca2+ into mitochondria from a functionally discrete portion of the nonmitochondrial store. Journal of Biological Chemistry. 1986;261:7223–7229. [PubMed] [Google Scholar]
- Boitier E, Rea R, Duchen MR. Mitochondria exert a negative feedback on the propagation of intracellular Ca2+ waves in rat cortical astrocytes. Journal of Cell Biology. 1999;145:795–808. doi: 10.1083/jcb.145.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. Journal of Neurochemistry. 1996;67:2282–2291. doi: 10.1046/j.1471-4159.1996.67062282.x. [DOI] [PubMed] [Google Scholar]
- Chacon E, Ohata H, Harper IS, Trollinger DR, Herman B, Lemasters JJ. Mitochondrial free calcium transients during excitation-contraction coupling in rabbit cardiac myocytes. FEBS Letters. 1996;382:31–36. doi: 10.1016/0014-5793(96)00138-x. [DOI] [PubMed] [Google Scholar]
- Csordás G, Thomas AP, Hajnóczky G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO Journal. 1999;18:96–108. doi: 10.1093/emboj/18.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton RM, McCormack JG. On the role of the calcium transport cycle in heart and other mammalian mitochondria. FEBS Letters. 1980;119:1–8. doi: 10.1016/0014-5793(80)80986-0. [DOI] [PubMed] [Google Scholar]
- Drummond RM, Tuft RA. Release of Ca2+ from the sarcoplasmic reticulum increases mitochondrial [Ca2+] in rat pulmonary artery smooth muscle cells. Journal of Physiology. 1999;516:139–147. doi: 10.1111/j.1469-7793.1999.139aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Ca2+-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochemical Journal. 1992;283:41–50. doi: 10.1042/bj2830041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. Journal of Physiology. 1999;516:1–17. doi: 10.1111/j.1469-7793.1999.001aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnóczky G, Csordás G, Krishnamurthy R, Szalai G. Mitochondrial calcium signaling driven by the IP3 receptor. Journal of Bioenergetics and Biomembranes. 2000;32:15–25. doi: 10.1023/a:1005504210587. [DOI] [PubMed] [Google Scholar]
- Hajnóczky G, Hager R, Thomas AP. Mitochondria suppress local feedback activation of inositol 1,4,5-trisphosphate receptors by Ca2+ Journal of Biological Chemistry. 1999;274:14157–14162. doi: 10.1074/jbc.274.20.14157. [DOI] [PubMed] [Google Scholar]
- Hajnóczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Hajnóczky G, Thomas AP. Minimal requirements for calcium oscillations driven by the IP3 receptor. EMBO Journal. 1997;16:3533–3543. doi: 10.1093/emboj/16.12.3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansford RG. Effect of micromolar concentrations of free Ca2+ ions on pyruvate dehydrogenase interconversion in intact rat heart mitochondria. Biochemical Journal. 1981;194:721–732. doi: 10.1042/bj1940721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek JB, Walajtys-Rode E, Wang X. Hormonal stimulation, mitochondrial Ca2+ accumulation, and the control of the mitochondrial permeability transition in intact hepatocytes. Molecular and Cellular Biochemistry. 1997;174:173–179. [PubMed] [Google Scholar]
- Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. Journal of Cell Biology. 1997;137:633–648. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huser J, Blatter LA, Sheu SS. Mitochondrial calcium in heart cells: beat-to-beat oscillations or slow integration of cytosolic transients? Journal of Bioenergetics and Biomembranes. 2000;32:27–33. doi: 10.1023/a:1005556227425. [DOI] [PubMed] [Google Scholar]
- Ichas F, Jouaville LS, Mazat JP. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89:1145–1153. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Jouaville LS, Ichas F, Holmuhamedov EL, Camacho P, Lechleiter JD. Synchronization of calcium waves by mitochondrial substrates in Xenopus laevis oocytes. Nature. 1995;377:438–441. doi: 10.1038/377438a0. [DOI] [PubMed] [Google Scholar]
- Jouaville LS, Ichas F, Mazat JP. Modulation of cell calcium signals by mitochondria. Molecular and Cellular Biochemistry. 1998;184:371–376. [PubMed] [Google Scholar]
- Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proceedings of the National Academy of Sciences of the USA. 1999;96:13807–13812. doi: 10.1073/pnas.96.24.13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindman LA, Meyer T. Use of intracellular Ca2+ stores from rat basophilic leukemia cells to study the molecular mechanism leading to quantal Ca2+ release by inositol 1,4,5-trisphosphate. Biochemistry. 1993;32:1270–1277. doi: 10.1021/bi00056a011. [DOI] [PubMed] [Google Scholar]
- Landolfi B, Curci S, Debellis L, Pozzan T, Hofer AM. Ca2+ homeostasis in the agonist-sensitive internal store: functional interactions between mitochondria and the ER measured in situ in intact cells. Journal of Cell Biology. 1998;142:1235–1243. doi: 10.1083/jcb.142.5.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. The use of fura-2 for estimating Ca buffers and Ca fluxes. Neuropharmacology. 1995;34:1423–1442. doi: 10.1016/0028-3908(95)00144-u. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Chin WC, Verdugo P. Role of Ca2+/K+ ion exchange in intracellular storage and release of Ca2+ Nature. 1998;395:908–912. doi: 10.1038/27686. [DOI] [PubMed] [Google Scholar]
- Pralong WF, Spät A, Wollheim CB. Dynamic pacing of cell metabolism by intracellular Ca2+ transients. Journal of Biological Chemistry. 1994;269:27310–27314. [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Brini M, Chiesa A, Filippin L, Pozzan T. Mitochondria as biosensors of calcium microdomains. Cell Calcium. 1999;26:193–199. doi: 10.1054/ceca.1999.0076. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Renard-Rooney DC, Hajnóczky G, Seitz MB, Schneider TG, Thomas AP. Imaging of inositol 1,4,5-trisphosphate-induced Ca2+ fluxes in single permeabilized hepatocytes. Demonstration of both quantal and nonquantal patterns of Ca2+ release. Journal of Biological Chemistry. 1993;268:23601–23610. [PubMed] [Google Scholar]
- Robb-Gaspers LD, Burnett P, Rutter GA, Denton RM, Rizzuto R, Thomas AP. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO Journal. 1998;17:4987–5000. doi: 10.1093/emboj/17.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma VK, Ramesh V, Franzini-Armstrong C, Sheu SS. Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. Journal of Bioenergetics and Biomembranes. 2000;32:97–104. doi: 10.1023/a:1005520714221. [DOI] [PubMed] [Google Scholar]
- Simpson PB, Mehotra S, Lange GD, Russell JT. High density distribution of endoplasmic reticulum proteins and mitochondria at specialized Ca2+ release sites in oligodendrocyte processes. Journal of Biological Chemistry. 1997;272:22654–22666. doi: 10.1074/jbc.272.36.22654. [DOI] [PubMed] [Google Scholar]
- Simpson PB, Russell JT. Role of mitochondrial Ca2+ regulation in neuronal and glial cell signalling. Brain Research. 1998;26:72–81. doi: 10.1016/s0165-0173(97)00056-8. [DOI] [PubMed] [Google Scholar]
- Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nature Neuroscience. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- Szalai G, Csordás G, Hantash BM, Thomas AP, Hajnóczky G. Calcium signal transmission between ryanodine receptors and mitochondria. Journal of Biological Chemistry. 2000;275:15305–15313. doi: 10.1074/jbc.275.20.15305. [DOI] [PubMed] [Google Scholar]
- Szalai G, Krishnamurthy R, Hajnóczky G. Apoptosis driven by IP3-linked mitochondrial calcium signals. EMBO Journal. 1999;18:6349–6361. doi: 10.1093/emboj/18.22.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt-Gallitelli MF, Isenberg G. X-ray microanalysis of single cardiac myocytes frozen under voltage-clamp conditions. American Journal of Physiology. 1989;256:574–583. doi: 10.1152/ajpheart.1989.256.2.H574. [DOI] [PubMed] [Google Scholar]
- Wilson BS, Pfeiffer JR, Smith AJ, Oliver JM, Oberdorf JA, Wojcikiewicz RJ. Calcium-dependent clustering of inositol 1,4,5-trisphosphate receptors. Molecular Biology of the Cell. 1998;9:1465–1478. doi: 10.1091/mbc.9.6.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]