

Abstract
Because NMDA complex and mitochondrial function are related, we hypothesized memantine would influence mitochondrial function. We addressed this in vitro by studying the effects of chronic and acute memantine exposures on mitochondrial function. For acute exposure experiments, mitochondria were isolated from NT2 cells and assayed for electron transport chain (ETC) enzyme function and peroxide production in buffers containing up to 60 uM memantine. For chronic exposure experiments, NT2 cells were maintained for at least two weeks in medium containing up to 60 uM memantine, following which we assayed cells or their mitochondria for ETC enzyme activities, cytochrome oxidase protein levels, oxidative stress, calcium levels, and mitochondrial DNA levels. The ability of the NMDA receptor antagonist aminophosphonovaleric acid (APV) to modify memantine's mitochondrial effects was evaluated. Acute and chronic memantine similarly affected complex I (increased at high concentrations) and IV (decreased at high concentrations) Vmax activities. APV did not alter the effects of chronic memantine exposure on citrate synthase and complex IV. We detected a lower mitochondrial peroxide production rate with acute exposure, and an increased mitochondrial peroxide production rate with chronic exposure. Micromolar memantine concentrations affect mitochondria, some of these effects are directly mediated, and acute and chronic effects may differ.
1. Introduction
Activation of NMDA receptor complexes elevates cytosolic calcium concentration [1, 2]. Cells can address increased cytosolic calcium in part by transferring these cations into negatively charged mitochondrial matrices [3]. Mitochondrial calcium content may in turn influence mitochondrial function. Alterations in oxidative phosphorylation status, electron transport chain (ETC) function, and mitochondrial reactive oxygen species (ROS) production are potential consequences [4, 5].
Less functionally obvious mitochondria-NMDA receptor complex relationships exist. For example, a mitochondrial DNA (mtDNA) encoded protein, ND2, actually serves to anchor NMDA receptor complexes to a regulatory tyrosine kinase [6]. Thus, in addition to playing an essential role in excitotoxic cascades, mitochondria and NMDA receptor complexes structurally overlap.
Memantine is a moderate-affinity NMDA receptor antagonist [7]. It can reside within NMDA receptor complex channels, and impedes calcium influx that might otherwise occur through these channels. It is widely used for the treatment of Alzheimer's disease (AD), and clinical trials show over a six month period AD patients randomized to memantine show less symptom progression than placebo-randomized AD patients [8, 9]. While it is postulated memantine's clinical effects arise from NMDA channel antagonism, this hypothesis has been challenged [10]. Other mechanisms that could potentially mediate memantine's clinical effects therefore require consideration.
AD is associated with numerous histologic and biochemical abnormalities. Mitochondrial dysfunction is observed in both degenerating and non-degenerating tissues of AD subjects [11, 12, 13]. Because of recognized inter-relationships between NMDA receptor complexes, cell calcium homeostasis, and mitochondria, we evaluated whether memantine affects mitochondrial function. We studied this under in vitro conditions using the NT2 teratocarcinoma cell line, a neuron-like tumor cell line that expresses critical parts of the NMDA receptor complex [14, 15]. We found memantine can influence mitochondrial function, and that at least part (if not all) of this occurs independent of NMDA channel antagonism.
2. Materials and Methods
2.1 Cell culture
Aside from addition of memantine or DL-2 amino-5-phosphono-valeric acid lithium salt (APV) to cell medium, human teratocarcinoma Ntera/D1 (NT2) neuronal precursor cells were maintained as previously described [16]. To accomplish memantine exposures, memantine-HCl powder (molecular weight 215.76) obtained from Forest Research Institute (Jersey City, NJ) was dissolved in sterile water to generate 1000× stock solutions. These 1000× stock solutions were then diluted in Optimem (Gibco BRL, Gaithersburg, MD) to create media containing 5−60 uM memantine. This concentration range exceeds serum levels of memantine obtained with human usage (0.5−1.0 uM), but is in accordance with the concentration spectrum typically used for in vitro studies [17, 18, 19]. To accomplish APV exposures, APV (formula weight 203.1; Sigma, St. Louis) was dissolved in sterile water to generate a 1000×, 50 mM stock solution. This stock solution was then diluted in Optimem to create media containing 50 uM APV.
For chronic exposure experiments, NT2 cells were maintained in media containing 0−60 uM memantine, with or without concomitant 50 uM APV. Cells were maintained in their designated medium for at least two weeks prior to any assays. Cells were harvested when flasks reached 90% confluency. We also routinely changed the culture medium one day prior to harvesting. Adherent cells were harvested and washed as previously described [20]. All experiments were independently repeated (at least 10 times) to ensure reproducibility.
2.2 Mitochondrial Enrichment
Suspended cells were disrupted in a prechilled, 45 ml nitrogen cavitation chamber (Parr Instrument Company, Moline, Ill) as previously described [20].
2.3 Cytochrome oxidase, citrate synthase, and complex I Vmax assays
For cells maintained for at least two weeks in medium containing memantine, APV, neither, or both (“chronic” exposure experiments), cytochrome oxidase and citrate synthase Vmax activities were determined on the whole cell pellets as previously described [20]. Cytochrome oxidase, citrate synthase, and complex I Vmax activities were also determined on enriched mitochondria as previously described [20].
“Acute” exposure experiments were also performed in which enriched mitochondria from cells maintained in memantine-free and APV-free medium were assayed for cytochrome oxidase, citrate synthase, and complex I activity in buffers supplemented with the desired amount of memantine (10 uM and 60 uM memantine).
2.4 Cell oxygen consumption
Cell suspensions were placed in individual wells of a BD Oxygen Biosensor System plate (BD Biosciences, Franklin Lakes, NJ) as previously described [20]. Each suspension was run in triplicate. BD Oxygen Biosensor System plates contain a dye that fluoresces with exposure to dissolved oxygen. Plates were scanned using a Wallac Victor2 plate reader (PerkinElmer Life and Analytical Sciences Inc., Wellesly, MA) with an excitation wavelength of 485 nm and an emission wavelength of 630 nm, and fluorescence data were converted to normalized relative fluorescence (NRF) units as previously described [20]. Over the two hour reading period there is a linear fluorescence increase, and the mean NRF unit slopes for the independently analyzed cell suspensions of a particular condition were calculated.
2.5 Oxidative stress determinations
Cytoplasmic peroxide levels were estimated using 2',7'-dichlorodihydrofluorescein diacetate (DCF) (Molecular Probes, Inc., Eugene, OR) as previously described [20]. Fluorescence increase slopes were calculated, which reflect increasing DCF fluorescence over time. Results for each condition are expressed as a fluorescence increase slope relative to that of untreated cells.
Mitochondrial peroxide production by enriched mitochondria was assessed using the amplex red (10-acetyl-3,7-dihydroxyphenoxazine) reagent as previously described. Amplex red reacts with H2O2 to produce highly fluorescent resorufin. The slope of the resorufin fluorescence increase was calculated for each sample. The slopes for the suspensions containing mitochondria from memantine-treated cells were compared to the slopes of suspensions containing mitochondria from untreated cells.
The glutathione peroxidase (GPx) reaction follows the oxidation of NADPH to NADP+, which manifests as an absorbance decrease at 340 nm. We prepared post-nuclear fractions from memantine treated and untreated NT2 cells, and assayed the post-nuclear fractions using a DU series spectrophotometer (Beckman Coulter) as previously described [20]. We used post-nuclear fractions because GPx is found in both cytoplasm and mitochondria. GPx activities from the post-nuclear fractions of memantine-treated cells were compared to those of post-nuclear fractions from untreated cells. Data are reported as relative activities.
2.6 Protein electrophoresis and Western blotting
Immunochemical determinations of cytochrome oxidase subunit 4 (CO4) protein levels were performed using whole cell protein preparations as previously described [20], and the mean densities for each condition were calculated. CO4 protein levels provide a surrogate estimate of mitochondrial mass. Mean densities for the different memantine-treated cell groups were compared to that of the untreated cell group.
2.7 Real time PCR
We used real time PCR to compare relative amounts of mtDNA and nuclear DNA copy numbers within memantine-treated and untreated cells. Amplicons from the mtDNA D-loop and the ß-actin nuclear DNA region were generated for this analysis as previously described [20]. For these determinations we used an iCycler iQ Real Time PCR Detection System thermocycler (BioRad) and iQ SYBR Green Supermix (BioRad). Mitochondrial to nuclear DNA ratios were calculated by dividing the mtDNA quantity for each sample by its corresponding ß-actin quantity. The mtDNA : nuclear DNA ratios from memantine-treated cells were compared to that of untreated cells.
2.8 Calcium measurements
Calcium measurements were performed using the Fluo-4 NW Calcium Assay Kit (Molecular Probes, Eugene, Oregon). Cells were harvested as described above, and assayed according to the manufacturer's protocol for non-adherent cells. Briefly, after washing cells in PBS cell pellets were re-suspended in Component C Assay Buffer (1× HBSS + 20 mM Hepes) at a concentration of 2.5 × 106 cells/ml. 50 ul of this suspension were placed in flat bottom wells of a 96 well tissue culture plate, yielding ∼125,000 cells/well/50ul. Cells from each memantine treatment were used to prepare six wells. “Blank” wells received 100ul of Component C Assay Buffer only. Plates were incubated at 37°C to let cells settle while the dye mixture was being prepared.
The dye mixture was prepared by adding 5ml of Component C Buffer +100ul of 250 mM Stock B (probenecid acid, prepared according to manufacturer's protocol) to reagent bottle A (which contained the Fluo-4 dye component). This bottle was mixed vigorously by hand for 2 minutes to ensure the dye was completely dissolved. 50 ul of this dye solution was added to each well (except the blank wells). Plates were then incubated at 37°C for 30 minutes in a 5% CO2-containing incubator. No significant difference was noted between reading the plates immediately following this 30 minute, 37°C incubation versus reading the plates after an additional 30 minute incubation at room temperature (we determined this since our plate reader was not temperature controlled and calcium measurements were therefore performed at room temperature).
During the 30 minute incubation period, a 100 uM solution of carbonyl cyanide p (trifluoromethoxy) phenylhydrazone (FCCP) (Sigma, St. Louis) was prepared in ethanol; this working solution was itself prepared by diluting a 100 mM FCCP-ethanol stock. Immediately prior to reading the plate, 11 ul of 100 uM FCCP solution was added to three wells from each treatment group. Thus, for each memantine exposure condition three wells were used to determine baseline cytoplasmic calcium levels, and three wells were used to determine the change in cytoplasmic calcium induced by mitochondrial respiratory chain uncoupling. As soon as the 30 minute incubation period was completed the plate was mixed quickly by hand and the fluorescence from each well was determined using a Perkin Elmer Wallac Victor2 1420 Multilabel Counter (excitation= 485 nm; emission=535 nm). Measurements were recorded over a 1.5 minute time course. Data were exported to an Excel spreadsheet and plotted by treatment group +/− FCCP. For baseline (no FCCP added) determinations, the initial readings from the memantine treatment conditions were normalized to those of the memantine-untreated group. The post-FCCP reading for each sample was compared to the baseline reading for that sample, and the difference in these two readings was used to calculate an FCCP-dependent percent change in each sample's calcium level. Baseline cytoplasmic calcium levels for each sample are reported as relative (to the memantine-untreated sample) fluorescence values. The cytoplasmic calcium levels of FCCP-treated cells are reported as percent changes to the non-FCCP baseline measurements.
2.9 Statistical analysis
Data are expressed as mean values ± standard error of the mean (SEM) for all independent assays of a particular condition. Two-tailed Student's t-tests were used to compare group means. P values of less than 0.05 were considered significant.
3. Results
3.1 Effects of chronic memantine exposure on cell respiration and Vmax activities
We used an assay of cell oxygen consumption to assess mitochondrial respiratory rates in memantine-treated cells. Assays were performed using oxygen biosensor plates, which facilitate quantitative assessments of cell oxygen consumption. Data were analyzed in terms of oxygen consumption rates and absolute oxygen consumption at 1 hour (Figure 1). For cells maintained in medium containing 10 uM or 60 uM memantine, there was an approximate 20−30% increase in both cell oxygen consumption rates and absolute oxygen consumption.
Figure 1.
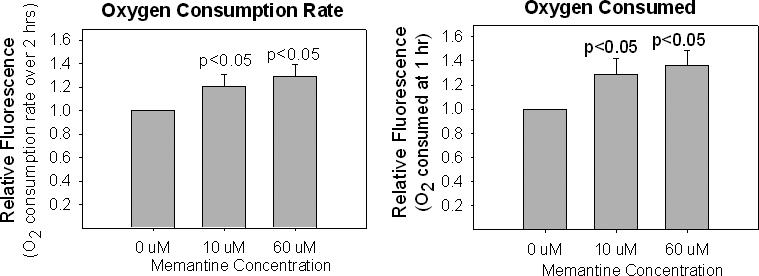
Chronic memantine exposure increases NT2 cell oxygen consumption. Data are reported two different ways. In (A), the oxygen consumption slopes from memantine treated cells are shown relative to the slope of untreated cells. In (B), the relative amounts of oxygen consumed over 1 hour are shown.
We next correlated our oxygen consumption data with electron transport chain (ETC) Vmax activites in memantine-treated cells. We measured the activity of complex IV (cytochrome oxidase) of the ETC chain in both chronically exposed whole cells and mitochondrial fractions prepared from chronically exposed cells. Whole cell cytochrome oxidase activities were determined across a broad range of memantine concentrations. The cytochrome oxidase activity for isolated mitochondria was obtained from mitochondrial fractions prepared from cells chronically exposed to 10 uM and 60 uM memantine. For both parameters, the Vmax activity was standardized to the total protein content of the assayed fraction. Activities from memantine-treated cells were normalized to the activity of mitochondria in or from cells that were not exposed to memantine. The results of our cytochrome oxidase Vmax determinations are shown in Figure 2.
Figure 2.

Relative to untreated cells, higher chronic memantine exposures reduce cytochrome oxidase Vmax activities. (A) Results of whole-cell cytochrome oxidase Vmax assays. (B) Results of enriched mitochondria cytochrome oxidase Vmax assays.
We were unable to detect a significant change in cytochrome oxidase activity with memantine concentrations of 20 uM or less. At the highest concentrations of memantine exposure studied (40 uM and 60 uM in whole cells, 60 uM for experiments using isolated mitochondria), cytochrome oxidase activity was diminished. At very high memantine concentrations (at least a 40-fold increase over clinically relevant serum levels), memantine reduces cytochrome oxidase activity.
In addition to standardizing our cytochrome oxidase data to total protein (in milligrams), we also standardized our cytochrome oxidase activity data using the activity of citrate synthase. Using this correction, cytochrome oxidase activity actually appeared increased in cells chronically exposed to 10 uM memantine (Figure 3).
Figure 3.
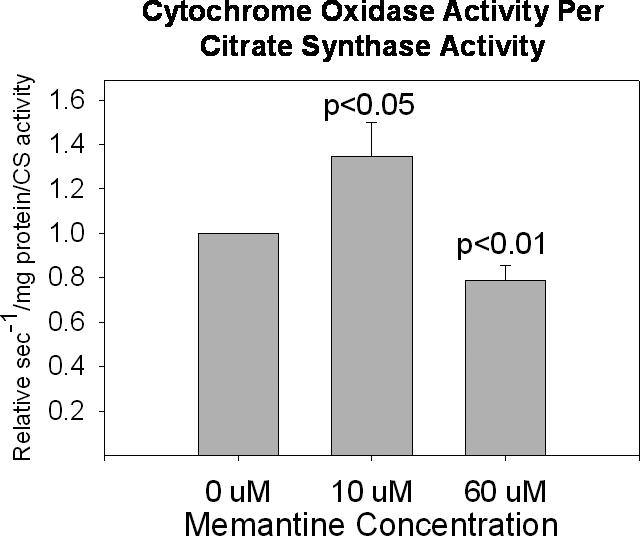
When citrate synthase activity is used to standardize cytochrome oxidase activities, cytochrome oxidase activity is increased with 10 uM chronic memantine exposure and decreased with 60 uM chronic memantine exposure.
Determinations of complex I activity are typically performed only on isolated mitochondrial fractions, as it is difficult to measure the Vmax of this enzyme using whole cells. We found 10 uM chronic memantine exposure had no effect on complex I activity, while 60 uM chronic memantine exposure resulted in a non-significant trend towards increased complex I activity (Figure 4). This trend became significant when complex I activities were standardized to citrate synthase activity.
Figure 4.
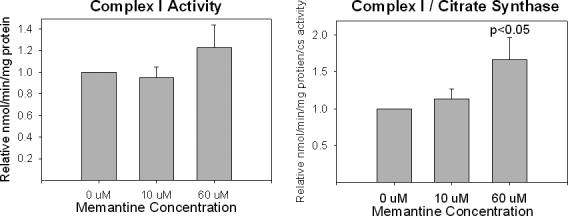
Effects of chronic memantine exposure on NT2 cell complex I activity. (A) No significant changes when corrected for total protein content. (B) When corrected for citrate synthase activity, the complex I Vmax activity is relatively increased by 60 uM memantine exposure.
We measured citrate synthase Vmax activities in both whole cells and in isolated mitochondria. The results of these assays are shown in Figure 5. Whole cell citrate activity was stable through a 40 uM concentration of memantine. With isolated mitochondria, however, relatively low memantine concentrations (10 uM) were associated with reduction of the citrate synthase Vmax. We evaluated the ability of an NMDA receptor antagonist, APV, to either potentiate or mitigate the effects of chronic memantine exposure on whole-cell cytochrome oxidase and citrate synthase activity. The results of these experiments are shown in Figure 6. APV did not alter the effects of chronic memantine exposure on citrate synthase and cytochrome oxidase. APV itself had no effect on citrate synthase and cytochrome oxidase activities.
Figure 5.
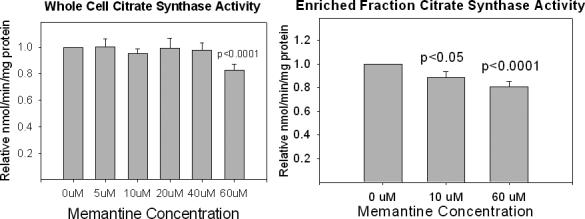
Effects of chronic memantine exposure on NT2 cell citrate synthase activity. (A) Assays performed on whole cells. (B) Assays performed on enriched mitochondrial fractions.
Figure 6.
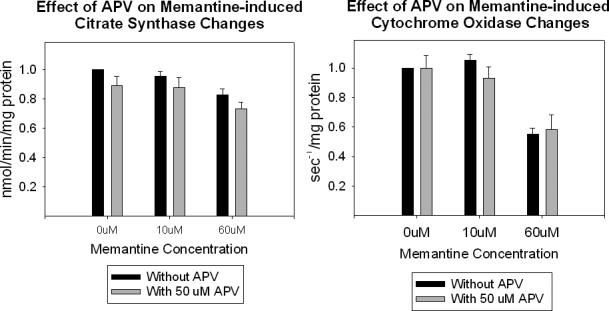
Effects of APV on chronic memantine-induced changes in (A) citrate synthase activity, and (B) cytochrome oxidase activity.
3.2 Effects of acute memantine exposure on Vmax activities
To definitively assess whether memantine could affect mitochondrial function independent of NMDA receptor complexes, we harvested mitochondria from cells not exposed to memantine at all and used them for Vmax assays in which the assay buffer contained 0 uM, 10 uM, or 60 uM memantine. Using this acute, direct exposure approach we found cytochrome oxidase activity was reduced in mitochondria exposed to 10 and 60 uM memantine (Figure 7). Interestingly, using this experimental paradigm the trend towards increased complex I activity seen when chronic 60 uM memantine exposure was used was again evident, and indeed statistically significant (p<0.05) (Figure 7). No change in citrate synthase activity was seen with even 60 uM memantine (data not shown). Memantine, therefore, does not directly affect citrate synthase activity.
Figure 7.
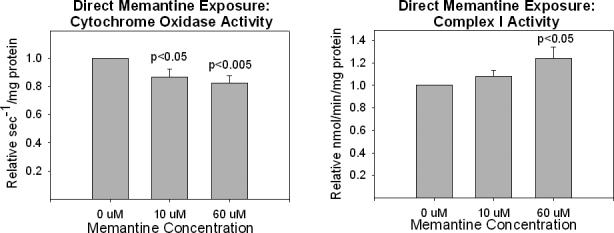
Effects of acute, direct memantine exposures on isolated mitochondria (A) cytochrome oxidase activity, and (B) complex I activity.
3.3 Effects of chronic and acute memantine on oxidative stress and calcium homeostasis
The mitochondrial respiratory chain contributes to cell oxidative stress levels, and through retrograde signal transduction can influence adaptive stress responses and antioxidant enzyme expression [21]. To assess the effects of memantine on cell and mitochondrial oxidative stress, we utilized three different approaches to measure peroxide production. First, we used a peroxide-sensitive fluorescent dye, DCF. DCF is localized to cell cytoplasm, and provides sensitive measurements of cytoplasmic peroxide levels. We further assayed the contribution of mitochondria to cell oxidative stress levels using another fluorescent dye, amplex red, to measure peroxide production from isolated mitochondria. Finally, cells that overproduce peroxide can compensate to some extent by upregulating antioxidant enzyme activities. We therefore measured glutathione peroxidase activities in our chronic memantine-exposed NT2 cells.
Figure 8 summarizes the results of assays performed on cells chronically maintained in memantine or on mitochondria isolated from cells chronically exposed to memantine. The DCF assay did not demonstrate any differences in cytoplasmic peroxide levels with either low (10 uM) or high (60 uM) chronic memantine exposures. However, the amplex red assay revealed both low and high dose chronic memantine exposures increased levels of mitochondrial peroxide production. Glutathione peroxidase, an antioxidant enzyme found in both cytoplasmic and mitochondrial compartments, was also increased with both memantine concentrations.
Figure 8.
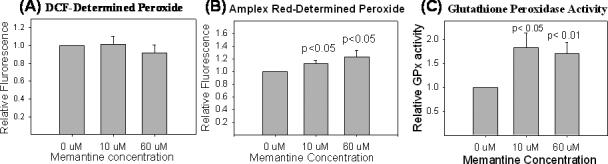
Cell peroxide production and oxidative stress levels in cells chronically exposed to memantine. (A) DCF fluorescence is comparable in cells exposed and not exposed to memantine. (B) Using amplex red fluorescence as a measure of peroxide, peroxide levels are higher in mitochondria isolated from treated cells. (C) Relative to untreated cells, memantine treated cells show increased glutathione peroxidase activity.
As we did with ETC activities, we assessed whether acute memantine exposures directly influenced mitochondrial peroxide production. We harvested mitochondria from cells not exposed to memantine at all, and used them in our amplex red assay in which the assay buffer contained 0 uM, 10 uM, or 60 uM memantine. Under these conditions, mitochondrial peroxide levels were actually reduced (Figure 9). It is worth noting additional experiments did not show evidence that memantine acts as a peroxide scavenger (data not shown).
Figure 9.
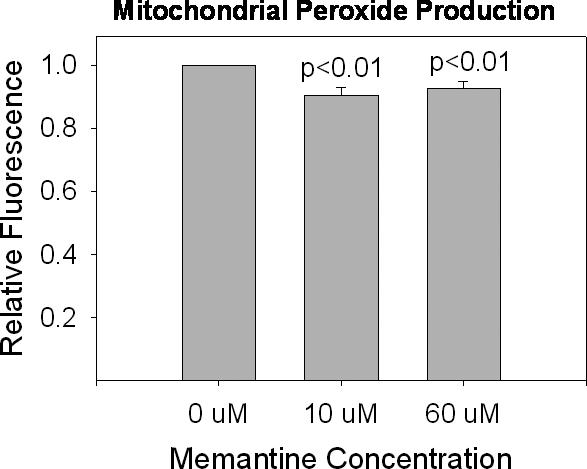
Mitochondrial peroxide production in mitochondria directly exposed to memantine. Using amplex red fluorescence as a measure of peroxide, peroxide levels are lower in mitochondria isolated from acutely treated cells as compared to mitochondria not acutely exposed to memantine.
We determined the effects of chronic memantine exposure on cytoplasmic and mitochondrial calcium levels using the dye fluo-4 AM. Fluo-4 AM enters cells and is retained within their cytoplasm. It fluoresces on reaction with calcium, with higher calcium concentrations resulting in higher fluorescence. We found that neither low nor high dose chronic memantine exposure altered fluo-4 AM fluorescence, suggesting that in our NT2 cell model, concentrations of up to 60 uM memantine do not change cytoplasmic calcium levels (Figure 10).
Figure 10.
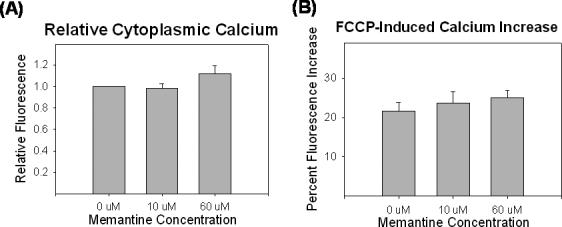
Effects of chronic memantine exposure on cytoplasmic and mitochondrial calcium levels. (A) Cytoplasmic calcium levels are unaffected by the memantine exposures. (B) Cytoplasmic calcium levels increase comparably following FCCP, suggesting mitochondria sequester equal amounts of calcium in memantine treated and untreated cells.
Because mitochondria play a critical role in buffering cell calcium levels, we also assayed cytoplasmic calcium levels immediately following addition of FCCP to NT2 cells. FCCP is a respiratory chain uncoupler that causes rapid depolarization of the mitochondrial membrane potential. This depolarization is accompanied by efflux of mitochondrial calcium stores. Mitochondrial calcium efflux, in turn, is detectable as an acute rise in cytoplasmic calcium levels. Indeed, FCCP caused an approximately 25% increase in cytoplasmic calcium levels. This increase was consistent for cells exposed to 0, 10, or 60 uM memantine (Figure 10). Because following FCCP exposure cytoplasmic calcium levels rose comparably between memantine conditions, mitochondria from cells maintained in each condition likely had equivalent amounts of calcium stored within them.
3.4 Effects of chronic memantine exposure on cell mitochondrial content
We evaluated the effects of chronic memantine exposure on cell mitochondrial content, as pharmacologic induction of mitochondrial biogenesis or mitochondrial autophagy is a potential consequence of drug exposure. We used two different approaches. We performed real time PCR determinations of cell mitochondrial DNA (mtDNA) copy number in cells chronically exposed to memantine, and also immunochemically quantified levels of a nuclear-encoded mitochondrial protein.
The real time PCR data are shown in Figure 11. We analyzed mtDNA copy number using primers to the D-loop. At both low (10 uM) and high (60 uM) memantine concentrations, mtDNA copy number was decreased. On the other hand, levels of the cytochrome oxidase subunit 4 (CO4) protein were not decreased (Figure 11).
Figure 11.
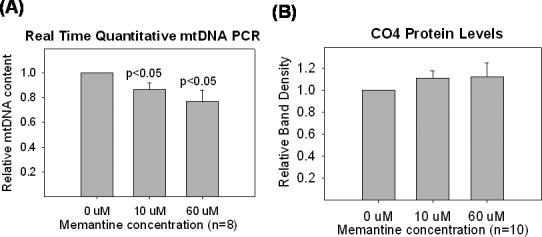
Effects of chronic memantine treatment on mtDNA and CO4 protein levels. (A) Chronic memantine treatment decreased NT2 cell mtDNA content. Data reflect the relative mtDNA content of memantine-treated cells versus that of cells not treated with memantine. (B) Chronic memantine treatment did not change levels of CO4, a nuclear-encoded ETC subunit. For the CO4 analysis, tubulin was used as an internal loading control, and tubulin levels were equivalent between memantine treated and untreated cells.
4. Discussion
Memantine is known to affect NMDA complex function. Because structural and functional relationships between NMDA receptor complexes and mitochondria exist, we hypothesized memantine would also influence mitochondrial function. Our studies did show memantine influences mitochondrial function. However, unlike what we predicted, mitochondrial effects are mostly or entirely mediated independent of NMDA channel modulation.
We reach this conclusion for several reasons. First, concurrent NMDA receptor antagonism with high concentrations of APV, which acts independent of the actual NMDA channel, should make NMDA receptor complexes exhibit similar minimum levels of function regardless of whether memantine resides within the complex's cation channel. APV, however, neither exacerbated nor mitigated memantine's mitochondrial effects. Second, the NMDA cation channel permits calcium influx, which could elevate cytoplasmic or mitochondrial calcium levels and thereby alter mitochondrial function. However, memantine concentrations up to 60 uM did not obviously alter cytoplasmic or mitochondrial calcium levels. Third (and most importantly), when mitochondria previously unexposed to memantine were separated from NMDA receptor complexes and exposed to memantine, complex I and IV activities changed similar to how they did when assayed in or isolated from cells chronically exposed to memantine. This strongly suggests memantine has a direct effect on ETC function. Still, it is important to note NMDA receptor complex function in undifferentiated NT2 cells is not as robust as in differentiated cells [15]. Our studies therefore do not rule out the possibility that under in vivo conditions or in different in vitro models, memantine might influence mitochondrial function via NMDA channel-mediated mechanisms.
Chronic memantine exposure enhanced some mitochondrial parameters and diminished others. Oxygen consumption was increased, and at the highest concentration studied complex I activity was also increased. At higher concentrations, though, cytochrome oxidase and citrate synthase activity were decreased. Our study did not address an underlying mechanistic explanation to explain these data. It does seem likely, though, that memantine acts as a mitochondrial irritant. While increased oxygen consumption could reflect increased oxidative phosphorylation capacity, it could also reflect decreased oxidative phosphorylation efficiency. Decreased complex IV activity is certainly consistent with a toxic effect. While we did observe an increase in citrate synthase-corrected complex IV activity in enriched mitochondria (at 10 uM), this may reflect reduction of the citrate synthase denominator rather than an actual increase in complex IV activity. Similarly, a reduced citrate synthase denominator could account for the increase in citrate synthase-corrected complex I activity we saw with 60 uM memantine. This would not, however, account for the apparent complex I activity increase seen with direct memantine exposure. Our finding of increased complex I activity with acute, direct 60 uM memantine exposure remains difficult to explain.
The Krebs cycle enzyme citrate synthase is encoded by a nuclear gene, translated in the cytoplasm, and imported to the mitochondrial matrix. It is often used to standardize Vmax measurements of ETC enzymes, as it is assumed citrate synthase activity is typically stable under different experimental conditions. Citrate synthase activity was reduced in whole cells or mitochondria isolated from cells chronically exposed to memantine, but not in isolated mitochondria exposed to memantine only during the actual assay. This could happen if memantine reduced mitochondrial citrate synthase import or retention. With whole cell assays, citrate synthase contained in the cytoplasm could still contribute to the total Vmax activity. With isolated mitochondria, only citrate synthase successfully imported into and retained within mitochondrial matrices is available for assay. Measuring citrate synthase activity in post-mitochondrial cytoplasmic fractions of memantine-exposed cells could perhaps address this possibility. Regardless, if irritated or otherwise abnormal mitochondria have difficulty importing or retaining citrate synthase, then citrate synthase may not constitute a universal marker of mitochondrial mass. If true, using citrate synthase activity to normalize ETC enzyme activity calculations could bias investigators from detecting activity reductions that do in fact exist, or introduce increased activity artifacts.
Mitochondrial DNA content was reduced, while CO4 protein levels were not. Different potential explanations for this are possible. Mitochondrial autophagy could account for these data. Mitochondrial degradation involves lysosomal digestion of mitochondria. During this process, the loss of intact, PCR-amplifiable intact mtDNA may exceed the loss of extractable, immunochemically detectable mitochondrial protein [22]. Alternatively, the number of mtDNA molecules per mitochondrion but not the total number of cell mitochondria may be influenced by memantine. This situation could occur if memantine affects mitochondrial import or retention of proteins participating in the mtDNA replication process. In any case, published data from others indicate a variety of mitochondrial assays are likely influenced by mitochondrial integrity and fragility [23].
Another one of this study's discrepant findings was that mitochondrial peroxide production was increased in cells chronically exposed to memantine, but not in mitochondria acutely exposed to memantine. As with our ETC enzyme activity data, we did not address potential underlying mechanisms for this. We can safely say, though, that chronically exposed cells have an opportunity to mount potentially compensatory physiologic changes. The change in peroxide production that occurred with prolonged exposure may represent a compensatory event. The increase in glutathione peroxidase activity we observed may also represent a compensatory event. We cannot say whether glutathione peroxidase activity was increased in the cytoplasm, mitochondria, or both. It does seem, though, that chronic memantine exposure did induce enough oxidative stress to trigger an increase in glutathione peroxidase activity, and that this increase was sufficient to abrogate any increase in cytoplasmic peroxide that chronic memantine exposure may have caused. Regardless, our data argue that when screening the effects of drugs on mitochondria, acute exposure paradigms may yield different results than chronic exposure paradigms.
It is worth considering whether mitochondrial effects mediate any of memantine's clinical effects. We saw changes in mitochondria and mitochondrial function at 10 uM memantine, a concentration that exceeds typically achieved serum levels ten-fold [19]. Although we did not observe an effect on cytochrome oxidase activity with 5 uM memantine, we did not comprehensively evaluate whether concentrations of less than 10 uM subtly affect mitochondria in other ways. If so, it is important to keep in mind what is ultimately toxic at higher concentrations may actually prove beneficial at lower concentrations, especially if robust compensatory mechanisms are triggered. For example, if complex IV activity was truly increased at the 10 uM concentration, and not an artifact of impaired citrate synthase import or retention, then memantine's mitochondrial effects might actually prove useful for the treatment of AD.
Acknowledgments
This research was funded by grants from the Forest Research Institute and the National Institute on Aging (AG022407).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Beal MF. Mechanisms of excitotoxicity in neurologic diseases. Faseb J. 1992;6:3338–44. [PubMed] [Google Scholar]
- 2.Albin RL. Greenamyre JT. Alternative excitotoxic hypotheses. Neurology. 1992;42:733–8. doi: 10.1212/wnl.42.4.733. [DOI] [PubMed] [Google Scholar]
- 3.Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–60. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- 4.Brookes PS, Yoon Y, Robotham JL, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–33. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 5.Hongpaisan J, Winters CA, Andrews SB. Strong calcium entry activates mitochondrial superoxide generation, upregulating kinas signaling in hippocampal neurons. J Neurosci. 2004;24:10878–87. doi: 10.1523/JNEUROSCI.3278-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gingrich JR, Pelkey KA, Fam SR, Huang Y, Petralia RS, Wenthold RJ, Salter MW. Unique domain anchoring of Src to synaptic NMDA receptors via the mitochondrial protein NADH dehydrogenase subunit 2. Proc Natl Acad Sci USA. 2004;101:6237–41. doi: 10.1073/pnas.0401413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Disc. 2006;5:160–70. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- 8.Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. Memantine Study Group. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med. 2003;348:1333–41. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- 9.Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I, Memantine Study Group Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291:317–24. doi: 10.1001/jama.291.3.317. [DOI] [PubMed] [Google Scholar]
- 10.Creeley C, Wozniak DF, Labruvere J, Taylor GT, Olney JW. Low doses of memantine disrupt memory in adult rats. J Neurosci. 2006;26:3923–32. doi: 10.1523/JNEUROSCI.4883-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parker WD, Jr, Filley CM, Parks JK. Cytochrome oxidase deficiency in Alzheimer's disease. Neurology. 1990;40:1302–3. doi: 10.1212/wnl.40.8.1302. [DOI] [PubMed] [Google Scholar]
- 12.Parker WD, Jr., Parks J, Filley CM, Kleinschmidt-DeMasters BK. Electron transport chain defects in Alzheimer's disease brain. Neurology. 1994;44:1090–6. doi: 10.1212/wnl.44.6.1090. [DOI] [PubMed] [Google Scholar]
- 13.Swerdlow RH, Kish SJ. Mitochondria in Alzheimer's disease. Int Rev Neurobiol. 2002;53:341–85. doi: 10.1016/s0074-7742(02)53013-0. [DOI] [PubMed] [Google Scholar]
- 14.Younkin DP, Tang CM, Hardy M, Reddy UR, Shi QY, Pleasure SJ, Lee VM, Pleasure D. Inducible expression of neuronal glutamate receptor channels in the NT2 human cell line. Proc Natl Acad Sci USA. 1993;90:2174–8. doi: 10.1073/pnas.90.6.2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardy M, Younkin D, Tang CM, Pleasure J, Shi QY, Williams M, Pleasure D. Expression of non-NMDA glutamate receptor channel genes by clonal human neurons. J Neurochem. 1994;63:482–9. doi: 10.1046/j.1471-4159.1994.63020482.x. [DOI] [PubMed] [Google Scholar]
- 16.Binder DR, Dunn WH, Jr, Swerdlow RH. Molecular characterization of mtDNA depleted and repleted NT2 cell lines. Mitochondrion. 2005;5:255–65. doi: 10.1016/j.mito.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 17.Parson CG, Quack G, Bresink I, Baran L, Przegalinski E, Kostowski W, Krzascik P, Hartmann S, Danysz W. Comparison of the potency, kinetics and voltage-dependency of a series of uncompetitive NMDA receptor antagonists in vitro with anticonvulsive and motor impairment activity in vivo. Neuropharmacology. 1995;34:1239–58. doi: 10.1016/0028-3908(95)00092-k. [DOI] [PubMed] [Google Scholar]
- 18.Chen HS, Lipton SA. Pharmacological Implications of Two Distinct Mechanisms of Interaction of Memantine with N-Methyl-D-aspartate-Gated Channels. J Pharmacol Exp Ther. 2005;314:961–71. doi: 10.1124/jpet.105.085142. [DOI] [PubMed] [Google Scholar]
- 19.Volbracht, van Beek J, Zhu C, Blomgren K, Leist M. Neuroprotective properties of memantine in different in vitro and in vivo models of excitotoxicity. European J Neurosci. 2006;23:2611–22. doi: 10.1111/j.1460-9568.2006.04787.x. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh S, Patel N, Rahn D, McAllister J, Sadeghi S, Horwitz G, Berry D, Wang KX, Swerdlow RH. The thiazolidinedione pioglitazone alters mitochondrial function in human neuronal-like cells. Mol Pharmacol. 2007;71:1695–1702. doi: 10.1124/mol.106.033845. [DOI] [PubMed] [Google Scholar]
- 21.Brookes PS, Levonen AL, Shiva S, Sarti P, Carley-Usmar VM. Mitochondria: regulators of signal transduction by reactive oxygen and nitrogen species. Free Radic Biol Med. 2002;33:755–64. doi: 10.1016/s0891-5849(02)00901-2. [DOI] [PubMed] [Google Scholar]
- 22.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, et al. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. 2001;21:3017–23. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Navarro A, Boveris A. Rat brain and liver mitochondria develop oxidative stress and lose enzymatic activities on aging. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1244–49. doi: 10.1152/ajpregu.00226.2004. [DOI] [PubMed] [Google Scholar]