

Abstract
Aims/hypothesis
Mutations in the GCK and HNF1A genes are the most common cause of the monogenic forms of diabetes known as ‘maturity-onset diabetes of the young’. GCK encodes the glucokinase enzyme, which acts as the pancreatic glucose sensor, and mutations result in stable, mild fasting hyperglycaemia. A progressive insulin secretory defect is seen in patients with mutations in the HNF1A and HNF4A genes encoding the transcription factors hepatocyte nuclear factor-1 alpha and -4 alpha. A molecular genetic diagnosis often changes management, since patients with GCK mutations rarely require pharmacological treatment and HNF1A/4A mutation carriers are sensitive to sulfonylureas. These monogenic forms of diabetes are often misdiagnosed as type 1 or 2 diabetes. Best practice guidelines for genetic testing were developed to guide testing and reporting of results.
Methods
A workshop was held to discuss clinical criteria for testing and the interpretation of molecular genetic test results. The participants included 22 clinicians and scientists from 13 countries. Draft best practice guidelines were formulated and edited using an online tool (http://www.coventi.com).
Results
An agreed set of clinical criteria were defined for the testing of babies, children and adults for GCK, HNF1A and HNF4A mutations. Reporting scenarios were discussed and consensus statements produced.
Conclusions/interpretation
Best practice guidelines have been established for monogenic forms of diabetes caused by mutations in the GCK, HNF1A and HNF4A genes. The guidelines include both diagnostic and predictive genetic tests and interpretation of the results.
For members of the EMQN MODY group see the Appendix. For details of their affiliations, see the Electronic supplementary material which is available to authorised users via the online version of this article (doi:10.1007/s00125-008-0942-y).
Keywords: Best practice, GCK, HNF1A, HNF4A, Maturity-onset diabetes of the young, MODY, Monogenic diabetes
Introduction
Maturity-onset diabetes of the young (MODY) describes the dominantly inherited disorder of non-insulin-dependent diabetes typically diagnosed before 25 years that was first recognised by Tattersall [1, 2]. MODY is the most common form of monogenic diabetes, accounting for an estimated 1–2% of diabetes in Europe [3, 4], but is often misdiagnosed as type 1 or type 2 diabetes.
The term MODY is used to describe a group of clinically heterogeneous, often non-insulin-dependent forms of diabetes that are defined at the molecular genetics level by mutations in different genes. All show dominant inheritance and are disorders of beta cell dysfunction, but variable features include the age at onset, severity of the hyperglycaemia (and hence risk of complications) and associated clinical features. The most recent classification of diabetes by the American Diabetes Association and the World Health Organization recognises these discrete subtypes of MODY [5].
Mutations in the GCK and HNF1A genes are the most frequent cause of MODY in all populations studied. They account for approximately 70% of cases (see Table 1). The ratio of GCK to HNF1A mutations varies between countries because of different recruitment strategies for genetic testing; blood glucose screening in young, asymptomatic individuals will identify a higher proportion of GCK mutations.
Table 1.
Genes in which mutations cause MODY
| Gene symbol (other symbol) | ||||||
|---|---|---|---|---|---|---|
| GCK | HNF1A (TCF1) | HNF4A | PDX1 (IPF1) | NEUROD1 | HNF1B (TCF2) | |
| Protein | Glucokinase | Hepatocyte nuclear factor-1 alpha | Hepatocyte nuclear factor-4 alpha | Insulin promoter factor-1 | Neurogenic differentiation 1 | Hepatocyte nuclear factor-1 beta |
| Chromosome locus | 7p13 | 12q24.31 | 20q13.12 | 13q12.2 | 2q31.3 | 17q12 |
| Gene Accession no. | NM_000162.2 | NM_000545.4 | NM_000457.3a | NM_000209.2 | NM_002500.2 | NM_000458.1 |
| OMIM * (Gene) | 138079 | 142410 | 600281 | 600733 | 601724 | 189907 |
| OMIM # (Phenotype) | 125851 | 600496 | 125850 | 606392 | 606394 | 137920 |
| Mutation frequency (%) (not known in ~20% of cases) | 20–50 | 20–50 | ~5 | <1 | <1 | ~5 |
The Genbank reference sequence NM_000457.3 refers to the full length P1 transcript, which uses exons 1a and 1b, and not 1d. For the full length P2 transcript, which includes exon 1d, there is a human mRNA sequence AY680697 but no refseq. The convention is to use NM_000457.3 for exons 1a/1b and 2–10, with AY680697 for exon 1d only. HNF4A mutation descriptions in the literature use the translation start codon reported by Chartier et al. in 1994 [45]. However, an alternative start codon nine amino acids upstream was proposed in 1996 [46] and this alternative start codon is used in NM_000457.3. Hence the convention is to report HNF4A mutations using the amino acid methionine at codon 10 in NM_000457.3 as the start codon, with the A of this codon as the first nucleotide
aNomenclature for the HNF4A gene is complicated because the gene encodes nine isoforms expressed from two promoters. The liver-specific P1 promoter drives the expression of transcripts 1–3, which include exons 1a and 2–10, and transcripts 4–6, which include exons 1a, 1b, 1c and 2–10. Transcripts 7–9 are expressed from the pancreatic (P2) promoter located approximately 46 kb upstream of the HNF4A transcription start site and exhibit splicing of the upstream exon 1d to exon 2, without the inclusion of sequences from either exons 1a, 1b or 1c
Heterozygous loss-of-function GCK mutations result in mild, stable hyperglycaemia from birth. Microvascular complications are rare, reflecting the fact that HbA1c is normally just above the upper limit of the normal range. Treatment with oral hypoglycaemic agents or insulin is not needed because it rarely changes HbA1c [6]. A genetic diagnosis is important for the small number of children misdiagnosed with type 1 diabetes and treated with insulin [7]. The identification of GCK mutations in women with gestational diabetes can be useful for obstetric management, since their babies who do not inherit the mutation are at risk of macrosomia [8], and it can guide follow-up in the mothers.
Transcription factor mutations in the HNF1A or HNF4A genes cause a similar progressive diabetic phenotype although the penetrance of HNF4A mutations is lower (S. Ellard and A. T. Hattersley, unpublished data). Sensitivity to sulfonylureas means that some patients can transfer from insulin to oral agents [9, 10]. A low renal threshold for glucose is a feature of HNF1A mutations [11] and may provide a useful method of screening at-risk family members during childhood [12].
Mutations identified in the GCK, HNF1A and HNF4A genes include missense, nonsense, splicing, small deletions/insertions/duplications, and splice site and promoter region mutations [13, 14]. Partial and whole deletions have recently been reported in HNF1A and GCK [15]. The location of mutations within the HNF1A gene influences the age at diagnosis; the average age at diagnosis for patients with exon 1–6 mutations that affect all three HNF1A isoforms is younger than for those with mutations in exons 8–10 that affect only isoform HNF1A(A) [16, 17].
Rarer forms of MODY include heterozygous mutations in PDX1 (also known as IPF1; [18, 19]) and NEUROD1 [20, 21], but analysis of these genes is not usually included in routine molecular genetic testing for MODY. Dominantly inherited syndromic forms of diabetes may also be described as MODY subtypes. The renal cysts and diabetes syndrome results from HNF1B mutations, and other features include renal abnormalities, female genital malformations, hyperuricaemia, pancreatic atrophy and abnormal liver function tests [22–24]. Mutations in the CEL variable number tandem repeat cause a syndrome of diabetes and pancreatic exocrine dysfunction [25]. These syndromes and maternally inherited diabetes and deafness caused by the mitochondrial m.3243A→G mutation are not included in these guidelines since testing is guided by the non-endocrine pancreatic or extra-pancreatic clinical features.
A molecular genetic diagnosis of a GCK, HNF1A or HNF4A mutation is important because it confirms a diagnosis of MODY, classifies the subtype, predicts the likely clinical course and may change the patient’s treatment. First-degree relatives will be at 50% risk of inheriting the mutation and asymptomatic individuals may be offered predictive genetic testing (after appropriate genetic counselling) in order to provide reassurance (for those shown not to carry the mutation) or regular blood glucose monitoring with early diagnosis and appropriate treatment (for mutation carriers).
Methods
A group of European clinicians and scientists met on 22 May 2007 at a workshop to formulate best practice guidelines for molecular genetic testing in MODY. Discussions focused on clinical criteria for selection of patients for testing, methodologies, interpretation of results and reporting those results to the referring clinicians.
A draft document was posted on 24 August 2007 and an online editing tool was used by participants to produce consensus guidelines.
Results
Clinical criteria for testing
Mild fasting hyperglycaemia: testing for GCK mutations The finding of raised fasting blood glucose in the range of 5.5–8 mmol/l is unusual in children and young adults. This always raises concern that they may be about to develop type 1 diabetes or the patient has type 2 diabetes. However, a considerable proportion of young, non-obese patients with persistent mild fasting hyperglycaemia will have a heterozygous mutation in the GCK gene. In a cohort of 82 children with incidental hyperglycaemia, 43% had GCK mutations [26]. The phenotype associated with GCK mutations is remarkably similar for all mutations. The following features suggest a diagnosis of a GCK mutation:
The fasting hyperglycaemia is ≥5.5 mmol/l (98% patients), persistent (at least three separate occasions) and stable over a period of months or years [27].
HbA1c is typically just above the upper limit of normal and rarely exceeds 7.5%.
In an OGTT the increment [(2 h glucose) − (fasting glucose)] is small (71% of patients in the large European study reported by Stride et al. [27] had an increment <3 mmol/l). An increment of 4.6 mmol/l is often used to prioritise testing and corresponds to the 90th centile (S. Ellard and A. T. Hattersley, unpublished data).
Parents may have ‘type 2 diabetes’ with no complications or may not be diabetic. On testing, one parent will usually have a mildly raised fasting blood glucose (range of 5.5–8 mmol/l) unless the mutation has arisen de novo. Testing of apparently unaffected parents’ fasting glucose is important when considering a diagnosis of a glucokinase mutation.
Gestational diabetes: testing for GCK mutations Mutations of GCK cause mild fasting hyperglycaemia throughout life and this is often diagnosed during pregnancy when routine testing is performed. Since these patients have consistently raised fasting blood glucose levels, their babies who do not inherit the mutation may be macrosomic [28]. The diagnosis of a GCK mutation is important, not only as the child may subsequently be picked up as having a raised fasting blood glucose and this may lead to concern about type 1 diabetes, but also because the guidelines given to the mother are different from the normal ‘pre-type 2’ diabetic phenotype as they will not deteriorate with time. The following criteria identify when GCK testing is appropriate [29]:
Persistently raised fasting blood glucose in the range of 5.5–8 mmol/l before, during and after pregnancy.
An increment of <4.6 mmol/l on at least one OGTT (either during or after pregnancy).
A parent may have mild type 2 diabetes but often this has not been detected and so the absence of family history should not exclude the diagnosis.
Children and young adults with diabetes and a strong family history of diabetes: testing for HNF1A mutations The possibility of monogenic diabetes should be considered whenever a parent has diabetes even if they are thought to have type 1 or type 2 diabetes. The most common form of MODY is caused by HNF1A mutations. The clinical characteristics of patients with HNF1A mutations include:
Young-onset diabetes (typically before 25 years old in at least one family member).
Non-insulin-dependent outside the normal honeymoon period (3 years), e.g. not developing ketoacidosis in the absence of insulin, good glycaemic control on less than the usual replacement dose of insulin, or detectable C-peptide measured when on insulin with glucose >8 mmol/l.
Family history of diabetes (at least two generations). This may be insulin treated and considered to be ‘type 1’ diabetes or ‘type 2’ diabetes. At least two individuals within the family would typically be diagnosed in their 20s or 30s. There may also be an affected grandparent, although often these are diagnosed after 45 years. OGTTs in early stages tend to show a very large glucose increment, usually >5 mmol/l [27]. Some individuals may have a normal fasting level but a value within the diabetic range at 2 h.
The absence of pancreatic islet autoantibodies.
Glycosuria at blood glucose levels <10 mmol/l is often seen, as these patients have a low renal threshold [11].
Marked sensitivity to sulfonylureas resulting in hypoglycaemia despite poor glycaemic control before starting sulfonylureas [9, 30].
Several features suggesting monogenic diabetes rather than a diagnosis of young-onset type 2 diabetes should be considered: no marked obesity or evidence of insulin resistance in diabetic family members, absence of acanthosis nigricans and whether the family is from an ethnic background with a low prevalence of type 2 diabetes (e.g. of European descent).
Children and young adults with diabetes and a strong family history of diabetes: testing for HNF4A mutations Diabetes caused by mutations in the HNF4A gene is considerably less common (Table 1) than HNF1A mutations. The clinical characteristics are similar, except there is not a low renal threshold and the age of diagnosis may be later [31]. HNF4A mutations should be considered when HNF1A analysis does not detect a mutation but the clinical features are strongly suggestive of HNF1A. Patients are often sensitive to sulfonylureas [32]. HNF4A mutations are associated with macrosomia (approximately 56% of mutation carriers) and transient neonatal hypoglycaemia (approximately 15% of mutation carriers) [33, 34]. The possibility of HNF4A mutations should be considered when diabetic family members have marked macrosomia (>4.4 kg at term) or if diazoxide-responsive neonatal hyperinsulinism has been diagnosed in the context of familial diabetes.
Babies with diazoxide-responsive neonatal hyperinsulinaemic hypoglycaemia and a strong family history of diabetes: testing for HNF4A mutations Mutations of HNF4A are a cause of neonatal hypoglycaemia that remits during infancy or early childhood, with diabetes developing later in life [34]. Macrosomic babies with diazoxide-responsive hyperinsulinism and a strong family history of diabetes (see characteristic 3 in the section above entitled Children and young adults with diabetes and a strong family history of diabetes: testing for HNF1A mutations) should be considered for HNF4A mutation screening.
Testing methodology
The mutation screening methodology should be described in the report [e.g. sequencing, denaturing high-performance liquid chromatography (dHPLC), conformation-sensitive capillary electrophoresis (CSCE)] together with the sensitivity. PCR primers should be checked for primer binding site single nucleotide polymorphisms (SNPs; a useful tool is available at http://ngrl.man.ac.uk/SNPCheck/index.html). Gene dosage analysis may be useful if a diagnosis of MODY is strongly suspected and no mutation is found on mutation screening.
Interpretation of results
The textbox includes recommended interpretations for the most common reporting scenarios.
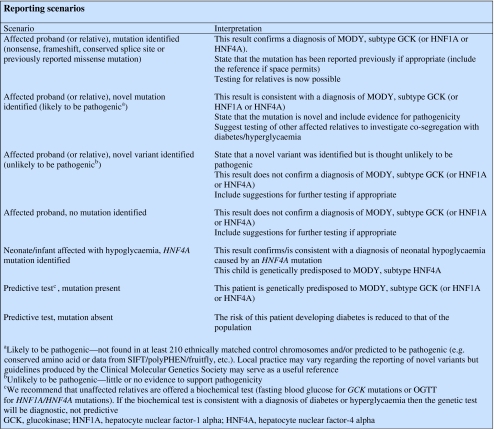
Reporting
Each laboratory has its own reporting format and general guidance on reporting is available from the European Molecular Genetics Quality Network (http://www.emqn.org), the UK Clinical Molecular Genetics Society (http://www.cmgs.org) and the Swiss Society of Medical Genetics (http://www.ssgm.ch). A one page report is the preferred format.
The report should state the methodology and specify the gene, exons and/or mutations tested for. If promoter sequences are examined then the report should specify the nucleotides analysed. An estimation of the assay sensitivity is particularly useful for pre-screening techniques such as dHPLC, CSCE etc. The use of mutation nomenclature approved by the Human Genome Variation Society (http://www.hgvs.org/mutnomen) is strongly recommended. The gene accession number (with version) is required in order to describe mutations unambiguously (see Table 1). The A nucleotide of the ATG start codon is numbered +1.
Reports describing novel variants should state that the variant is novel and include the evidence in support of pathogenicity. This might include the absence from a large series of ethnically matched controls or MODY patients (testing of 210 normal chromosomes is necessary to achieve at least 80% power to detect a polymorphism present in 1% of the population [35]). Testing of other affected relatives is recommended in order to check for co-segregation and to calculate the LOD score in suitable sized pedigrees (LOD scores of ≥1 or ≥3 are suggestive or conclusive of linkage, respectively).
For missense variants the evidence for pathogenicity might include conservation across species and a significant amino acid substitution. Several programs are available that predict the pathogenicity of a missense variant based upon amino acid conservation (SIFT; http://www.blocks.fhcrc.org/sift/SIFT.html) or the structure and function of the protein (PolyPHEN; http://www.genetics.bwh.harvard.edu/pph) but they should be used to supplement other pieces of evidence rather than in isolation.
Both missense and silent variants can affect splicing if the mutation is within an exon splicing enhancer or exon splicing silencer. Splice predictor software programs (http://www.fruitfly.org or http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home) may aid interpretation. Base substitutions affecting the conserved splice donor (GT) site, splice acceptor (AG) site or the conserved A nucleotide within the branch site are highly likely to be pathogenic but splice predictor software may be useful in the interpretation of other intronic variants. Analysis of patient mRNA is often informative but lymphoblastoid cell lines are usually required because of the low levels of expression of the MODY genes in blood. Sequence analysis of RT-PCR products amplified from lymphoblastoid cell mRNA has demonstrated exon skipping, retention and the use of cryptic splice sites for a variety of intronic mutations in the GCK, HNF1A and HNF1B genes [36–38].
Novel promoter variants may be investigated by examination of known transcription binding sites or by in vitro transfection experiments [39–42]. They may also alter mRNA expression levels, which may be measured by allele-specific real-time PCR [43].
Polymorphisms
Some laboratories include details of polymorphisms detected in the report. The reasons for doing this include: (1) making all data available to the requesting clinician based on the rationale that a polymorphism may later be reclassified as a mutation; and (2) identifying heterozygous SNPs excludes a gene deletion involving the exon(s) in question.
However, this information can cause confusion or even misinterpretation of the result (A. T. Hattersley, unpublished data) and it certainly adds to the length of the report. While in some cases there may be reports in the literature of an association with type 2 diabetes or reduced insulin secretion, these polymorphisms do not cause MODY and we recommend that they should be excluded from the report.
Treatment
Individual treatment recommendations are outside the jurisdiction of a molecular genetics report since this is the referring clinician’s responsibility. It is useful to include an appropriate reference if there is evidence in the literature for a particular treatment (e.g. low-dose sulfonylureas in HNF1A/4A MODY) associated with the genetic diagnosis.
Other issues
Genetic counselling should be provided for all asymptomatic individuals requesting predictive testing. We recommend that unaffected relatives are offered a biochemical test first (fasting blood glucose for GCK mutations or OGTT for HNF1A/HNF4A mutations). If the biochemical test is consistent with a diagnosis of diabetes or hyperglycaemia then the genetic test will be diagnostic, not predictive.
For families requesting predictive testing for children too young to provide informed consent, referral to a specialist clinical genetics unit (or equivalent) is strongly recommended. Reasons for testing children include (1) to remove the uncertainty around the child’s status, and (2) to assist with management, as a negative test would mean that monitoring of blood glucose/glycosuria would not be necessary [44].
Conclusions
Molecular genetic testing is useful in patients with MODY because it confirms a diagnosis of monogenic diabetes, predicts likely clinical course, defines risk for relatives and determines treatment.
At the present time, molecular genetic testing for MODY is relatively expensive and phenotypic selection prior to testing is normal practice. With the development of new technologies it is likely that these costs will decrease in time and that the analysis of genes associated with monogenic diabetes may become routine for all newly diagnosed patients. In the meantime we hope that these guidelines will be useful in determining which patients should be offered testing, and in the interpretation and reporting of the test results.
Below is the link to the electronic supplementary material.
(PDF 12.9 kb)
Acknowledgements
Funding for the best practice meeting was provided by the EuroGentest Network of Excellence Project 2005-EU Contract No FP6-512148 (www.eurogentest.org) and the European Molecular Genetics Quality Network (http://www.emqn.org). A. T. Hattersley is a Wellcome Trust Research Leave Clinical Fellow.
Duality of interest The authors declare that there is no duality of interest associated with this manuscript.
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- CSCE
conformation-sensitive capillary electrophoresis
- dHPLC
denaturing high-performance liquid chromatography
- SNP
single nucleotide polymorphism
Appendix
Members of the EMQN MODY group are: C. Carette, L. Castano Gonzalez, G. de Nanclares Leal, R. Elles, G. Gaspar, D. Gasperikova, T. Hansen, M. Herr, O. Kamarainen, C. Kannengiesser, I. Klimes, G. Lacape, M. Losekoot, M. Malecki, P. Meyer, P. Njolstad, T. Predragovic, S. Pruhova and W. Wuyts.
Footnotes
For members of the EMQN MODY group see the Appendix. For details of their affiliations, see the Electronic supplementary material which is available to authorised users via the online version of this article (doi:10.1007/s00125-008-0942-y).
References
- 1.Tattersall RB (1974) Mild familial diabetes with dominant inheritance. Q J Med 43:339–357 [PubMed]
- 2.Tattersall RB, Fajans SS (1975) A difference between the inheritance of classical juvenile-onset and maturity-onset type diabetes of young people. Diabetes 24:44–53 [DOI] [PubMed]
- 3.Frayling TM, Evans JC, Bulman MP et al (2001) Beta-cell genes and diabetes: molecular and clinical characterization of mutations in transcription factors. Diabetes 50(Suppl 1):S94–S100 [DOI] [PubMed]
- 4.Lenderman H (1995) Is maturity-onset diabetes at young age (MODY) more common in Europe than previously assumed? Lancet 345:648 [DOI] [PubMed]
- 5.American Diabetes Association (2007) Diagnosis and classification of diabetes mellitus. Diabetes Care 30(Suppl 1):S42–S47 [DOI] [PubMed]
- 6.Gill-Carey O, Shields B, Colclough K, Ellard S, Hattersley AT (2007) Finding a glucokinase mutation alters patient treatment. Diabet Med 24(Suppl 1):6 (Abstract)
- 7.Schnyder S, Mullis P, Ellard S, Hattersley A, Fluck C (2005) Genetic testing for glucokinase mutations in clinically selected patients with MODY: a worthwhile investment. Swiss Med Wkly 135:352–356 [DOI] [PubMed]
- 8.Spyer G, Hattersley AT, Sykes JE, Sturley RH, MacLeod KM (2001) Influence of maternal and fetal glucokinase mutations in gestational diabetes. Am J Obstet Gynecol 185:240–241 [DOI] [PubMed]
- 9.Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT (2003) Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 362:1275–1281 [DOI] [PubMed]
- 10.Shepherd M, Pearson ER, Houghton J, Salt G, Ellard S, Hattersley AT (2003) No deterioration in glycemic control in HNF-1alpha maturity-onset diabetes of the young following transfer from long-term insulin to sulphonylureas. Diabetes Care 26:3191–3192 [DOI] [PubMed]
- 11.Menzel R, Kaisaki PJ, Rjasanowski I, Heinke P, Kerner W, Menzel S (1998) A low renal threshold for glucose in diabetic patients with a mutation in the hepatocyte nuclear factor-1alpha (HNF-1alpha) gene. Diabet Med 15:816–820 [DOI] [PubMed]
- 12.Stride A, Ellard S, Clark P et al (2005) Beta-cell dysfunction, insulin sensitivity, and glycosuria precede diabetes in hepatocyte nuclear factor-1alpha mutation carriers. Diabetes Care 28:1751–1756 [DOI] [PubMed]
- 13.Ellard S, Colclough K (2006) Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha (HNF1A) and 4 alpha (HNF4A) in maturity-onset diabetes of the young. Hum Mutat 27:854–869 [DOI] [PubMed]
- 14.Gloyn AL (2003) Glucokinase (GCK) mutations in hyper- and hypoglycemia: maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemia of infancy. Hum Mutat 22:353–362 [DOI] [PubMed]
- 15.Ellard S, Thomas K, Edghill EL et al (2007) Partial and whole gene deletion mutations of the GCK and HNF1A genes in maturity-onset diabetes of the young. Diabetologia 50:2313–2317 [DOI] [PubMed]
- 16.Bellanné-Chantelot C, Carette C, Riveline J et al (2008) The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3 Diabetes 57:503–508 [DOI] [PubMed]
- 17.Harries LW, Ellard S, Stride A, Morgan NG, Hattersley AT (2006) Isomers of the TCF1 gene encoding hepatocyte nuclear factor-1 alpha show differential expression in the pancreas and define the relationship between mutation position and clinical phenotype in monogenic diabetes. Hum Mol Genet 15:2216–2224 [DOI] [PubMed]
- 18.Cockburn BN, Bermano G, Boodram LL et al (2004) Insulin promoter factor-1 mutations and diabetes in Trinidad: identification of a novel diabetes-associated mutation (E224K) in an Indo-Trinidadian family. J Clin Endocrinol Metab 89:971–978 [DOI] [PubMed]
- 19.Stoffers D, Ferrer J, Clarke W, Habener J (1997) Early-onset diabetes mellitus (MODY4) linked to IPF1. Nat Genet 17:138–139 [DOI] [PubMed]
- 20.Kristinsson SY, Thorolfsdottir ET, Talseth B et al (2001) MODY in Iceland is associated with mutations in HNF-1alpha and a novel mutation in NeuroD1. Diabetologia 44:2098–2103 [DOI] [PubMed]
- 21.Malecki MT, Jhala U, Antonellis A et al (1999) Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet 23:323–328 [DOI] [PubMed]
- 22.Bellanne-Chantelot C, Clauin S, Chauveau D et al (2005) Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes 54:3126–3132 [DOI] [PubMed]
- 23.Edghill EL, Bingham C, Ellard S, Hattersley AT (2006) Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J Med Genet 43:84–90 [DOI] [PMC free article] [PubMed]
- 24.Horikawa Y, Iwasaki N, Hara M et al (1997) Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet 17:384–385 [DOI] [PubMed]
- 25.Raeder H, Johansson S, Holm PI et al (2006) Mutations in the CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat Genet 38:54–62 [DOI] [PubMed]
- 26.Feigerlova E, Pruhova S, Dittertova L et al (2006) Aetiological heterogeneity of asymptomatic hyperglycaemia in children and adolescents. Eur J Pediatr 165:446–452 [DOI] [PubMed]
- 27.Stride A, Vaxillaire M, Tuomi T et al (2002) The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia 45:427–435 [DOI] [PubMed]
- 28.Hattersley AT, Beards F, Ballantyne E, Appleton M, Harvey R, Ellard S (1998) Mutations in the glucokinase gene of the fetus result in reduced birth weight. Nat Genet 19:268–270 [DOI] [PubMed]
- 29.Ellard S, Beards F, Allen LIS et al (2000) A high prevalence of glucokinase mutations in gestational diabetic subjects selected by clinical criteria. Diabetologia 43:250–253 [DOI] [PubMed]
- 30.Pearson ER, Liddell WG, Shepherd M, Corrall RJ, Hattersley AT (2000) Sensitivity to sulphonylureas in patients with hepatocyte nuclear factor-1alpha gene mutations: evidence for pharmacogenetics in diabetes. Diabet Med 17:543–545 [DOI] [PubMed]
- 31.Pearson ER, Pruhova S, Tack CJ et al (2005) Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor 4alpha mutations in a large European collection. Diabetologia 48:878–885 [DOI] [PubMed]
- 32.Fajans SS, Brown MB (1993) Administration of sulfonylureas can increase glucose-induced insulin secretion for decades in patients with maturity-onset diabetes of the young. Diabetes Care 16:1254–1261 [DOI] [PubMed]
- 33.Fajans SS, Bell GI (2007) Macrosomia and neonatal hypoglycaemia in RW pedigree subjects with a mutation (Q268X) in the gene encoding hepatocyte nuclear factor 4alpha (HNF4A). Diabetologia 50:2600–2601 [DOI] [PubMed]
- 34.Pearson ER, Boj SF, Steele AM et al (2007) Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med 4:e118 [DOI] [PMC free article] [PubMed]
- 35.Collins JS, Schwartz CE (2002) Detecting polymorphisms and mutations in candidate genes. Am J Hum Genet 71:1251–1252 [DOI] [PMC free article] [PubMed]
- 36.Bulman MP, Harries LW, Hansen T et al (2002) Abnormal splicing of hepatocyte nuclear factor 1 alpha in maturity-onset diabetes of the young. Diabetologia 45:1463–1467 [DOI] [PubMed]
- 37.Harries LW, Ellard S, Jones RW, Hattersley AT, Bingham C (2004) Abnormal splicing of hepatocyte nuclear factor-1 beta in the renal cysts and diabetes syndrome. Diabetologia 47:937–942 [DOI] [PubMed]
- 38.Sun F, Knebelmann B, Pueyo ME et al (1993) Deletion of the donor splice site of intron 4 in the glucokinase gene causes maturity-onset diabetes of the young. J Clin Invest 92:1174–1180 [DOI] [PMC free article] [PubMed]
- 39.Gragnoli C, Lindner T, Cockburn BN et al (1997) Maturity-onset diabetes of the young due to a mutation in the hepatocyte nuclear factor-4 alpha binding site in the promoter of the hepatocyte nuclear factor-1 alpha gene. Diabetes 46:1648–1651 [DOI] [PubMed]
- 40.Hansen SK, Parrizas M, Jensen ML et al (2002) Genetic evidence that HNF-1alpha-dependent transcriptional control of HNF-4alpha is essential for human pancreatic beta cell function. J Clin Invest 110:827–833 [DOI] [PMC free article] [PubMed]
- 41.Raeder H, Bjorkhaug L, Johansson S et al (2006) A hepatocyte nuclear factor-4 alpha gene (HNF4A) P2 promoter haplotype linked with late-onset diabetes: studies of HNF4A variants in the Norwegian MODY registry. Diabetes 55:1899–1903 [DOI] [PubMed]
- 42.Thomas H, Jaschkowitz K, Bulman M et al (2001) A distant upstream promoter of the HNF-4alpha gene connects the transcription factors involved in maturity-onset diabetes of the young. Hum Mol Genet 10:2089–2097 [DOI] [PubMed]
- 43.Harries LW, Hattersley AT, Ellard S (2004) Messenger RNA transcripts of the hepatocyte nuclear factor-1alpha gene containing premature termination codons are subject to nonsense-mediated decay. Diabetes 53:500–504 [DOI] [PubMed]
- 44.Shepherd M, Ellis I, Ahmad AM et al (2001) Predictive genetic testing in maturity-onset diabetes of the young (MODY). Diabet Med 18:417–421 [DOI] [PubMed]
- 45.Chartier FL, Bossu JP, Laudet V, Fruchart JC, Laine B (1994) Cloning and sequencing of cDNAs encoding the human hepatocyte nuclear factor 4 indicate the presence of two isoforms in human liver. Gene 147:269–272 [DOI] [PubMed]
- 46.Drewes T, Senkel S, Holewa B, Ryffel GU (1996) Human hepatocyte nuclear factor 4 isoforms are encoded by distinct and differentially expressed genes. Mol Cell Biol 16:925–931 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Below is the link to the electronic supplementary material.
(PDF 12.9 kb)