

Abstract
Phyloproteomics is a novel analytical tool that solves the issue of comparability between proteomic analyses, utilizes a total spectrum-parsing algorithm, and produces biologically meaningful classification of specimens. Phyloproteomics employs two algorithms: a new parsing algorithm (UNIPAL) and a phylogenetic algorithm (MIX). By outgroup comparison, the parsing algorithm identifies novel or vanished MS peaks and peaks signifying up or down regulated proteins and scores them as derived or ancestral. The phylogenetic algorithm uses the latter scores to produce a biologically meaningful classification of the specimens.
Keywords: Cancer, dichotomous development, mass spectrometry, phylogenetics, phyloproteomics, proteomics, serum, transitional clades
Introduction
The utilization of the serum proteome to accurately diagnose cancer has been challenging, and its future continues to be surrounded by uncertainties.1 Although statistical analysis of mass spectrometry (MS) profiles of serum proteins has gained enormous popularity and credibility,2-6 algorithmic analysis that produces biologically meaningful results with possible clinical diagnosis is still lacking. It now seems very simplistic to attempt to define cancer on the basis of statistical patterns, since cancer is a multifaceted evolving and adapting cellular condition with multiple proteomic profiles; some of these profiles cannot always be separated from noncancerous ones by narrowly defined statistical proteomic patterns on the basis of a limited number of spectral peaks. Cancer's incipience is marked by mutations that cause the malfunction of the apoptotic apparatus of the cell, and its promotion is characterized by different phases with each having its distinct proteomic profile.7,8 Advanced progression of cancer is marked by cellular dedifferentiation, loss of apoptosis, and metamorphosis into a primordial status where survival, and not function, becomes the cell's primary mission.8 In this latter stage, many proteins responsible for differentiation are not produced, and therefore missing MS peaks are as significant in defining the proteomic profiles of cancer.
The multiphasic nature of cancer progression combined with possible multiple developmental pathways8-11 entail the presence of a large number of proteomic changes for each type of cancer and its phases. These factors suggest that the proteomic profile of a cancer type is a hierarchical and continuous accumulation of proteomic change over time rather than one or a few simple distinct proteomic patterns. For an analytical tool to be successful in producing a clinical diagnosis, it has to uncover the hierarchical profile of cancer and be able to place a specimen within this profile.
In the present study, we propose that cancer can be promptly diagnosed, even at early stages, by phylogenetic analysis of the serum proteome. Since cancer is an evolutionary condition that involves genetic modifications and clonal production, it therefore requires an evolutionary method of analysis. Such an analysis is possible if an algorithm for sorting out the polarity (derived vs ancestral) of the MS values is available. We are demonstrating here through our polarity assessment algorithm (UNIPAL) that this task can be performed, and MS data can be analyzed with an evolutionary algorithm (Figure 1). Phyloproteomics is an evolutionary analytical tool that sorts out mass-to-charge (m/z) values into derived (apomorphic) or ancestral (plesiomorphic) and then classifies specimens according to the distribution pattern of their apomorphies into clades (a group composed of all the specimens sharing the same apomorphies). Phyloproteomics also illustrates the multiphasic nature of cancer by assigning cancer specimens to a hierarchical classification with each hierarchy defined by the apomorphic protein changes that are present in its specimens. The classification is presented in a graphical display termed cladogram or tree. The assumption that all cancerous specimens fit into well-defined proteomic models (patterns based on a few peaks) that distinguish them from noncancerous ones12-16 is replaced here by phylogenetically distinct clades of specimens with each clade sharing unique protein changes (synapomorphies) among its specimens.
Figure 1.

Schematic representation of phyloproteomic analysis. The process involves two steps. The first is the algorithmic sorting of the m/z values into derived (exists in some but not all specimens) and ancestral (in all specimens); the derived values are those signifying either novel, vanished, or up and down regulated peaks. The second step is a parsimony phylogenetic analysis that groups the specimens on the basis of the shared derived values.
Methods
Proteomic Data
We used mass spectrometry (MS) data of serum proteins generated by surface-enhanced laser desorption–ionization time-of-flight (SELDI-TOF) of 460 specimens from three types of cancer: ovarian (143), pancreatic (70), and prostate (36), as well as from noncancerous specimens (211). All sets of data used here are available from the NCI–FDA Clinical Proteomics Program (http://home.ccr.cancer.gov/ncifdaproteomics/ppatterns.asp) and are described and referred to in a few publications.12,13,15,17,18 From the prostate cancer data set, we included only the confirmed cancerous specimens.
Polarity Assessment and Phylogenetic Analysis
We employed the continuous range of mass-to-charge ratio (m/z) values of all specimens for the analysis. For polarity assessment (apomorphic [or derived] vs plesiomorphic [or ancestral]), data were polarized with a customized algorithm (UNIPAL) written by the authors that recognized novel and vanished MS peaks, as well as peaks signifying upregulated and downregulated proteins for each specimen. Each of these events was coded as equal; however, no standardization, normalization, or smoothing of the data was applied before or after polarity assessment—UNIPAL does not require any of these processes to carry out the polarization. Outgroups used to carry out polarity for each cancer type were selected from the noncancerous specimens; each outgroup encompassed the total variability within the noncancerous specimens.
UNIPAL requires a set of noncancerous specimens to be included in every separate data set in order to be used as an outgroup. It determines the polarity for every m/z value among the noncancerous specimens and then scores each value of the study group as derived or ancestral. The outgroup should be large enough to encompass all possible variations that exist within noncancerous specimens.
For phylogenetic analysis, we used MIX, the parsimony program of PHYLIP version 3.57c,19 to carry out separate phylogenetic parsimony analysis for each cancer type and then pooled all the specimens of the three cancer types plus the noncancerous in a larger analysis that included all 460 specimens. Processing with MIX was carried out in randomized and nonrandomized inputs; however, no significant differences were observed between the two. Phylogenetic trees were drawn using TreeView.20
Results and Discussion
The results of a phylogenetic analysis are best illustrated by a phylogenetic tree termed cladogram that shows the hierarchical classification in a graphical format. Parsimony analysis produced one most parsimonious cladogram (requiring the least number of steps in constructing a classification of specimens) for each of the pancreatic and prostate specimens (Figure 2a,b), five equally parsimonious cladograms for ovarian specimens (Figure 2c shows only one), and about 100 equally parsimonious cladograms for the inclusive analysis (Figure 3 summarizes only one). We examined all multiple equally parsimonious cladograms and found them to be fundamentally very similar in topology. They differed only in the internal arrangement of some minor branches where one or two specimens had equally plausible locations within their immediate clade.
Figure 2.
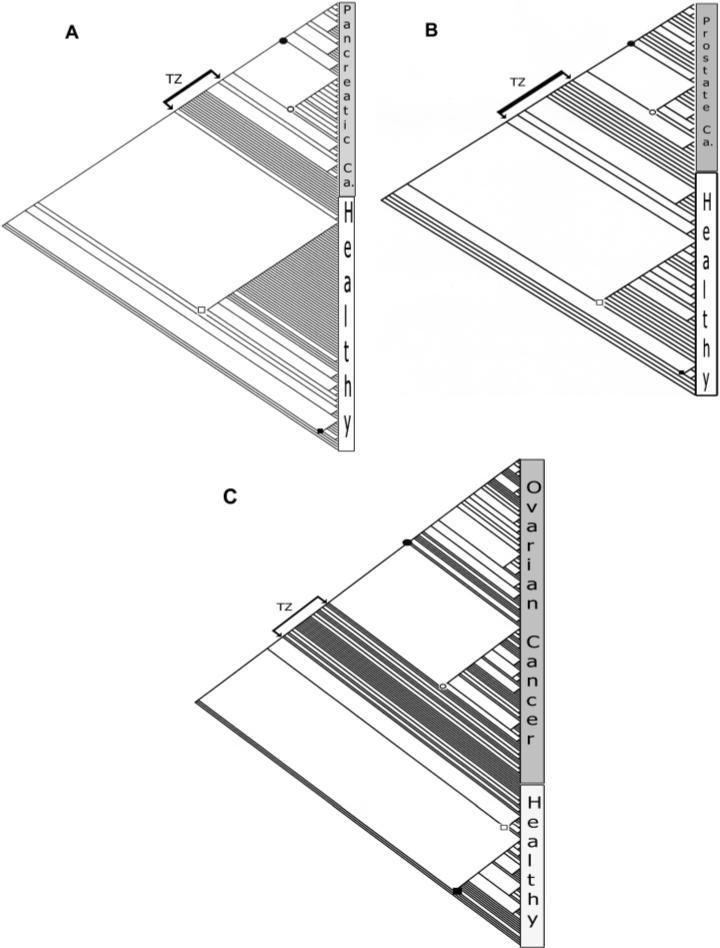
Phyloproteomic cladograms of three cancers: (A) pancreatic, (B) prostate, and (C) ovarian. The nodes of major clades are marked as follows: •, terminal cancer clade; ○, middle cancer clade; □, middle healthy clade; and ■, basal healthy clade. Transitional zones (TZ) are marked by bracketed arrows.
Figure 3.

A phyloproteomic analysis showing dichotomous distribution of cancers into two clades. A schematic cladogram of a comprehensive phyloproteomic analysis composed of 460 specimens representing ovarian, pancreatic, and prostate cancers as well as noncancerous specimens. Specimens of every cancer type are classified into two clades: a terminal and middle, as well as transitional clades. Healthy specimens are classified into a major healthy clade and transitional clades.
A complete separation of the cancer specimens from noncancerous ones depended on the size of the noncancerous outgroup used to carry out polarity assessment. Polarizing the m/z values with the largest size outgroups (ones encompassing the largest amount of variation) available for each cancer type produced cladograms with separate groupings of cancerous and noncancerous specimens, that is, no cancer specimens grouped with the healthy and vice versa (100% sensitivity and specificity). However, with the use of randomly selected smaller outgroups, sensitivity dropped to 96% and below; this illustrates the significance of using the largest number possible for outgroup polarity assessment.
Each of the cladograms (Figure 2a–c) showed an upper bifurcation composed of cancerous specimens, while the lower end of the cladogram was occupied by a number of basal clades composed of noncancerous specimens and a central assemblage of noncancerous clades adjacent to cancerous ones. The latter assembly formed a distinct order of well-resolved and mostly single-specimen clades in the middle of the cladogram nested between the cancer and healthy clades (bracketed arrows in Figure 2a–c); we termed them transitional clades (TC). The transitional clades bordered their respective types (cancer or noncancer) in a tandem arrangement that formed a transitional zone (TZ) between the noncancer and cancer clades.
When data of all specimens of the three cancer types were pooled together with noncancerous ones and processed, each of the three cancers formed two large clades (the terminal and middle) and numerous small transitional clades adjacent to the noncancerous ones (Figure 3). The pancreatic and prostate clades formed sister groups in their terminal and middle clades, and their terminal clades were nested within the ovarian clades. The ovarian specimens formed two distinct clades at the upper part of the cladogram.
The cladograms revealed greater similarities in topology among cancer types. For each of the three cancer types, there were two large recognizable clades (the terminal and the middle) forming a major dichotomy that encompassed the majority of the specimens of each type (Figure 2a–c). This dichotomy persisted in the inclusive cladogram as well (Figure 3), with each of the cancers having two clades.
The use of mass spectrometry (MS) of serum proteins to produce clinically useful profiles has proved to be challenging and has generated some controversy.21-23 Although several methods have been published thus far,13-16 they all either had cancer type-specific sorting algorithms that produced below 95% specificity and did not apply well across other cancer types, did not utilize all potentially useful variability within the data, or were not widely tested.16,24 Furthermore, their relative success has been limited to diagnosis without any of the predictive conclusions potentially offered by phyloproteomics. Since cancer is an evolutionary condition produced by a set of mutations,7 its study should include evolutionary sound methods of analysis. Phylogenetics reveals both relatedness and diversity through a hypothesis of relationships among the specimens on the basis of the parsimonious distribution of novel m/z values of their proteomes.
This is the first report on the application of a phylogenetic algorithm to MS serum proteomic data for cancer analysis. By developing and applying an algorithm for polarity assessment and then using a parsimony phylogenetic algorithm for classifying specimens of three cancer types (ovarian, pancreatic, and prostate), we demonstrated that phylogenetics can successfully be applied to MS serum proteomic data for cancer analysis, diagnosis, typing, and possibly susceptibility assessment. Additionally, phyloproteomics points out the presence of distinct trends within cancer proteomic profiles.
Despite the good number of algorithms used for MS serum analysis,13-16 reproducibility and comparability of proteomic analyses are unattainable because of the lack of broadly acceptable universal methods of analysis. Phyloproteomics is composed of two algorithms that are applicable to MS data of any cancer (Figure 1). The first algorithm, UNIPAL, is a new polarity assessment program that we designed to work with MS data to produce a listing of novel derived values in a coded format, and the second algorithm is a popular phylogenetic parsimony program, MIX of the PHYLIP package,19 that uses the values generated by the first algorithm to classify the specimens. MIX is a robust analytical package that has been tested by scientists for the past 16 years, and is probably the most cited in phylogenetic studies. An added benefit to this approach is that it makes possible the comparison among results from different data sets and the evaluation of competing analytical tools.
Phylogenetics has the intrinsic ability to reveal meaningful biological patterns by grouping together truly related specimens better than any other known methods (Table 1). Proteomic variability encompasses ancestral and derived variations, and only derived m/z intensity values are useful in classifying cancer types and subtypes into a meaningful hierarchy that reflects the phylogeny and ontogeny of their proteomic profiles. While clustering techniques use the presence of common peaks (without resolving their polarity) in order to create distinct patterns and then fit a specimen within a pattern,12,14,16 phylogenetics requires polarity assessment to sort out m/z intensities into derived and ancestral at first and then uses the distribution pattern of derived values among the specimens to produce their classification (i.e., the cladogram). Using only common intensity peaks without polarity assessment for pattern modelling has not been the most reliable means of classification.12,14 This is because clustering usually involves ancestral values and does not resolve multiple origins of a character (parallelisms), and both result in polyphyletic grouping (having unrelated specimens). Furthermore, phylogenetics can resolve the position of a novel specimen with new variations by placing it in a group that comprises its closest relatives on the basis of the number of apomorphic mutations it shares with them (Table 1).
Table 1.
The Advantages of Phylogenetic Analysis over Statistical Cluster Analysis
| phylogenetic analysis | cluster analysis |
|---|---|
| ■ produces a classification based on shared derived similarities and reflects phyletic relationships | ■ produces a classification based on overall similarity and may not reflect phyletic relationship |
| ■ uses one algorithm for the analysis of all types of cancers | ■ may require a specific algorithm for each cancer type |
| ■ discriminates between ancestral and derived states; uses only derived character states (apomorphies) | ■ does not discriminate between ancestral and derived character states; uses both |
| ■ resolves issues of parallelism (multiple independent origins) by parsimony or maximum likelihood | ■ does not resolve issues of parallelism |
| ■ offers predictivity | ■ does not offer predictivity. |
Phyloproteomics has a potential for cancer predictivity. Predictivity here is defined as the capacity of the classification to predict the characteristics of a specimen by determining the specimen's location within a cladogram. By using an ample number of well-characterized cancer specimens in an analysis, the unknown characters of a new specimen will be forecasted when it assembles within a clade in the cladogram. The specimen's location in a cladogram is always based on the type of mutations it carries and shares with the clade members, which will determine the diagnosis, cancer type, or possibly the susceptibility to developing cancer. Cladogram topology shows a hierarchical accumulation of novel serum protein changes across a continuum spanning from the transitional noncancerous specimens to the cancerous ones, with the latter having the highest number of apomorphic mutations.
Cladograms also revealed that the three types of cancer have fundamentally similar topologies; they all have one major dichotomy that indicates two lineages within each type (represented on the cladograms by the terminal clade and the middle clade [Figures 2–3]). If this typification holds true for additional cancer types, then it is possible that ontogenetically all types of cancers undergo two major common pathways in their development. There are only a few recent reports that support a dichotomous pattern of development8 in colorectal cancer,9 glioblastomas,10 and pancreatic carcinoma.11 Dichotomies may arise in cancer because of the selective advantages of cells harboring various mutations; the surviving mutations can be genetic or chromosomal,8,9 point mutation or amplification,10 or differential expression of alleles.11
Noncancerous transitional clades, present in all cladograms and mostly composed of individual specimens, are the closest sister groups to cancer clades. Because of their proximity to cancer clades, we hypothesize that these specimens, assumed to be from cancer-free individuals, represent the early stages of cancer development that cannot yet be morphologically or microscopically diagnosed as cancerous. For diagnostic purposes, cancerous and noncancerous transitional specimens will always be challenging to classify by other techniques. Occasionally, these specimens are distinct from one another by only very few apomorphies. The mostly single specimen composition of the transitional clades attests to their uniqueness.
Current diagnosis of cancer is not based on the number of mutations or synapomorphies; therefore, the determination of the status of a transitional specimen is still subjective unless a clear definition that is based on derived mutations is established by pathologists. Till then we suggest that the position of a transitional specimen within the transitional zone determines its diagnosis; if a specimen is on the upper end of the transitional zone (i.e., bordering cancer clades), then it is a cancerous specimen, and those occurring in the middle and lower end of the transitional zone are to be called high risk specimens.
So far, we have not yet carried out any correlations between specimens on the cladograms and patients' survival. Therefore, it is uncertain at this stage of the analysis if the terminal clades of cancers represent the advanced stages of cancer progression or if the two major clades have any prediction on prognosis.
Searching for biomarkers is a challenging process in biomedical research, and phyloproteomics offers the capacity to uncover many possible ones. The phylogenetic program, MIX, lists the shared derived m/z intensity values (synapomorphies) of each clade it produces, and each synapomorphy is a possible biomarker. In other words, the cladogram serves as a map showing the apomorphic m/z values of all potential biomarkers and their effective levels of groupings. A synapomorphy may represent a novel protein, a disappeared protein, or an up/down regulated protein; thus, these proteins corresponding to the apomorphic m/z values need to be identified if they are to be explored as biomarkers. Since the cladograms have hierarchical arrangement (i.e., presenting various levels of groupings) one can look for biomarkers at various levels of the cladogram. An apomorphic protein (we would like to call it apotein) that defines a clade will serve as a potential biomarker for the clade, while another apotein defining a nested subclade within the clade will be restricted as biomarker to the subgroup within the clade.
Conclusion
Phyloproteomics offers a new paradigm in cancer analysis that reveals relatedness and diversity of cancer specimens in a phylogenetic sense; its predictive power is a useful tool for diagnosis, characterizing cancer types, discovering biomarkers, and identifying universal characteristics that transcend several types of cancer. The implications of the new paradigm are of valuable clinical, academic, and scientific value.
References
- 1.Hede K. $104 million proteomics initiative gets green light. J. Natl. Cancer Inst. 2005;97(18):1324–1325. doi: 10.1093/jnci/dji325. [DOI] [PubMed] [Google Scholar]
- 2.Issaq HJ, Conrads TP, Prieto DA, Tirumalai R, Veenstra TD. SELDI-TOF MS for diagnostic proteomics. Anal. Chem. 2003;75(7):148A–155A. [PubMed] [Google Scholar]
- 3.Marvin LF, Roberts MA, Fay LB. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry in clinical chemistry. Clin. Chim. Acta. 2003;337(1−2):11–21. doi: 10.1016/j.cccn.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 4.Merchant M, Weinberger SR. Recent advancements in surface-enhanced laser desorption/ionization–time-of-flight-mass spectrometry. Electrophoresis. 2000;21(6):1164–1177. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1164::AID-ELPS1164>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 5.Pusch W, Flocco MT, Leung SM, Thiele H, Kostrzewa M. Mass spectrometry-based clinical proteomics. Pharmacogenomics. 2003;4(4):463–476. doi: 10.1517/phgs.4.4.463.22753. [DOI] [PubMed] [Google Scholar]
- 6.Srinivas PR, Srivastava S, Hanash S, Wright GL., Jr. Proteomics in early detection of cancer. Clin. Chem. 2001;47(10):1901–1911. [PubMed] [Google Scholar]
- 7.Wyllie AH, Bellamy CO, Bubb VJ, Clarke AR, Corbet S, Curtis L, Harrison DJ, Hooper ML, Toft N, Webb S, Bird CC. Apoptosis and carcinogenesis. Br. J. Cancer. 1999;80(Suppl 1):34–37. [PubMed] [Google Scholar]
- 8.Loeb KR, Loeb LA. Significance of multiple mutations in cancer. Carcinogenesis. 2000;21(3):379–385. doi: 10.1093/carcin/21.3.379. [DOI] [PubMed] [Google Scholar]
- 9.Chung DC. The genetic basis of colorectal cancer: insights into critical pathways of tumorigenesis. Gastroenterology. 2000;119(3):854–865. doi: 10.1053/gast.2000.16507. [DOI] [PubMed] [Google Scholar]
- 10.Hayashi Y, Yamashita J, Watanabe T. Molecular genetic analysis of deep-seated glioblastomas. Cancer Genet Cytogenet. 2004;153(1):64–68. doi: 10.1016/j.cancergencyto.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 11.Adsay NV, Merati K, Andea A, Sarkar F, Hruban RH, Wilentz RE, Goggins M, Iocobuzio-Donahue C, Longnecker DS, Klimstra DS. The dichotomy in the preinvasive neoplasia to invasive carcinoma sequence in the pancreas: differential expression of MUC1 and MUC2 supports the existence of two separate pathways of carcinogenesis. Mod. Pathol. 2002;15(10):1087–1095. doi: 10.1097/01.MP.0000028647.98725.8B. [DOI] [PubMed] [Google Scholar]
- 12.Petricoin EE, Paweletz CP, Liotta LA. Clinical applications of proteomics: proteomic pattern diagnostics. J. Mammary Gland Biol. Neoplasia. 2002;7(4):433–440. doi: 10.1023/a:1024042200521. [DOI] [PubMed] [Google Scholar]
- 13.Alexe G, Alexe S, Liotta LA, Petricoin E, Reiss M, Hammer PL. Ovarian cancer detection by logical analysis of proteomic data. Proteomics. 2004;4(3):766–783. doi: 10.1002/pmic.200300574. [DOI] [PubMed] [Google Scholar]
- 14.Conrads TP, Fusaro VA, Ross S, Johann D, Rajapakse V, Hitt BA, Steinberg SM, Kohn EC, Fishman DA, Whitely G, Barrett JC, Liotta LA, Petricoin EF, III, Veenstra TD. High-resolution serum proteomic features for ovarian cancer detection. Endocr.-Relat. Cancer. 2004;11(2):163–178. doi: 10.1677/erc.0.0110163. [DOI] [PubMed] [Google Scholar]
- 15.Zhu W, Wang X, Ma Y, Rao M, Glimm J, Kovach JS. Detection of cancer-specific markers amid massive mass spectral data. Proc. Natl. Acad. Sci. U.S.A. 2003;100(25):14666–14671. doi: 10.1073/pnas.2532248100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adam BL, Qu Y, Davis JW, Ward MD, Clements MA, Cazares LH, Semmes OJ, Schellhammer PF, Yasui Y, Feng Z, Wright GL., Jr. Serum protein fingerprinting coupled with a pattern-matching algorithm distinguishes prostate cancer from benign prostate hyperplasia and healthy men. Cancer Res. 2002;62(13):3609–3614. [PubMed] [Google Scholar]
- 17.Petricoin EF, Ornstein DK, Paweletz CP, Ardekani A, Hackett PS, Hitt BA, Velassco A, Trucco C, Wiegand L, Wood K, Simone CB, Levine PJ, Linehan WM, Emmert-Buck MR, Steinberg SM, Kohn EC, Liotta LA. Serum proteomic patterns for detection of prostate cancer. J. Natl. Cancer Inst. 2002;94(20):1576–1578. doi: 10.1093/jnci/94.20.1576. [DOI] [PubMed] [Google Scholar]
- 18.Petricoin EF, III, Ardekani AM, Hitt BA, Levine PJ, Fusaro VA, Steinberg SM, Mills GB, Simone C, Fishman DA, Kohn EC, Liotta LA. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 2002;359(9306):572–577. doi: 10.1016/S0140-6736(02)07746-2. [DOI] [PubMed] [Google Scholar]
- 19.Felsenstein J. PHYLIP: Phylogeny Inference Package, version 3.2. Cladistics. 1989. pp. 164–166.
- 20.Page RD. TreeView: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 1996;12(4):357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- 21.Baggerly KA, Morris JS, Coombes KR. Reproducibility of SELDI-TOF protein patterns in serum: comparing datasets from different experiments. Bioinformatics. 2004;20(5):777–785. doi: 10.1093/bioinformatics/btg484. [DOI] [PubMed] [Google Scholar]
- 22.Sorace JM, Zhan M. A data review and re-assessment of ovarian cancer serum proteomic profiling. BMC Bioinformatics. 2003;4(1):24. doi: 10.1186/1471-2105-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Check E. Proteomics and cancer: running before we can walk? Nature. 2004;429(6991):496–497. doi: 10.1038/429496a. [DOI] [PubMed] [Google Scholar]
- 24.Ornstein DK, Rayford W, Fusaro VA, Conrads TP, Ross SJ, Hitt BA, Wiggins WW, Veenstra TD, Liotta LA, Petricoin EF., III Serum proteomic profiling can discriminate prostate cancer from benign prostates in men with total prostate specific antigen levels between 2.5 and 15.0 ng/mL. J. Urol. 2004;172(4 Pt 1):1302–1305. doi: 10.1097/01.ju.0000139572.88463.39. [DOI] [PubMed] [Google Scholar]