

Abstract
Immunophilins are protein chaperones with peptidylprolyl isomerase activity that belong to one of two large families, the cyclosporin-binding cyclophilins (CyPs) and the FK506-binding proteins (FKBPs). Each family displays characteristic and conserved sequence features that differ between the two families. We report a novel group of dual-family immunophilins that contain both CyP and FKBP domains for which we propose the name FCBP (FK506- and cyclosporin-binding protein). The FCBP of Toxoplasma gondii, a protozoan parasite, contained N-terminal FKBP and C-terminal CyP domains joined by tetratricopeptide repeats. Structure-function analysis revealed that both domains were functional and exhibited family-specific drug sensitivity. The individual domains of FCBP inhibited calcineurin (protein phosphatase 2B) in the presence of the appropriate drugs. In binding studies, FCBP recruited calcineurin in the presence of FK506 and a putative target of rapamycin homolog in the presence of rapamycin. Two additional FCBP sequences in Flavobacterium and one in Treponema (spirochete) were also identified in which the CyP and FKBP domains were in the reverse order. T. gondii growth was inhibited by cyclosporin and FK506 in a moderately synergistic manner. The knockdown of FCBP by RNA interference revealed its essentiality for T. gondii growth. Clearly, the FCBPs are novel chaperones and potential targets of multiple immunosuppressant drugs.
The immunophilin superfamily consists of highly conserved proteins with rotamase or peptidylprolyl cis-trans-isomerase (PPIase)1 activity that act as chaperones by accelerating the isomerization of X-Pro-peptide bonds (1, 2). They belong to two major families that bind specific immunosuppressant molecules of fungal origin (3). The cyclophilins (CyPs) bind cyclosporin A (CsA), a cyclic undecapeptide (4, 5), and the FKBPs (FK506-binding proteins) bind macrolides such as FK506 (tarcolimus) (6) and rapamycin (sirolimus) that are structurally unrelated to CsA. In both families, the drugs bind to and inhibit the respective PPIase domains that are roughly 100 amino acids long. Thus, the PPIase domain is essentially synonymous with the drug-binding domain. For instance, the FKBP PPIase domain is often called FK506-binding domain (FKBD) (Figs. 1 and 2). Despite the common PPIase activity, there is no significant sequence homology between these two families (1). Furthermore, no immunophilin has been characterized to date that contains both CyP and FKBP domains (1).
Fig 1. Representative domain arrangements of FKBPs, Cyps, and the dual-family FCBPs.
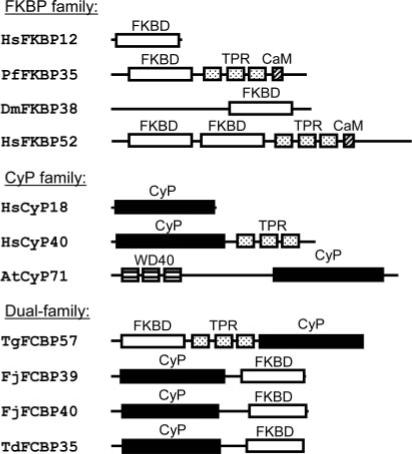
The “single-family” enzymes (FKBP, CyP) are shown for comparison only. The proteins are named by the initials of the organism (Dm, Drosophila melanogaster; Hs, Homo sapiens; Td, T. denticola; At, Arabidopsis thaliana) followed by the domain name(s) and the theoretical molecular mass in kDa. The accessory domains, i.e. TPR, WD40, and the putative CaM-binding domains, are also indicated. CyP domains are black rectangles, and FKBDs are white.
Fig. 2. Sequence homology of the dual-family immunophilins of Tg, Fj, and T. denticola (Td).

The nomenclature has been described in Fig. 1. Highlighted residues are important for various functions, including PPIase activity, substrate binding, and interaction with immunosuppressant drugs (CsA for the CyP domain and FK506 for the FKBP domain). A, homology of the FKBP domains. The prototype human FKBP (hFKBP12) and the P. falciparum FKBP35 (21, 22) are included for comparison. B, homology of the CyP domains. The prototype human CyP (hCyP18) is included for comparison. Residues important for CN binding are underlined. The W (for Trp), marked with overhead circle, is especially important for CsA binding but is replaced by H (for His) in Toxoplasma, which explains the relative CsA resistance of the latter (Fig. 4). Note how the residue numbers differ between the Toxoplasma and the bacterial FCBPs because of the reversal of the domain arrangement.
In the generally accepted nomenclature, the immunophilins are named with a prefix of one or two letters indicating the species followed by a number for the calculated molecular mass in kDa. Thus, the archetypal 12-kDa human FKBP is hFKBP12 or Homo sapien FKBP12. As a rule, the small immunophilins (e.g. hFKBP12) contain a single PPIase domain, whereas the larger ones may contain additional domains important for protein-protein interaction (1) such as WD40, calmodulin (CaM)-binding domain, and tetratricopeptide repeat (TPR) (Fig. 1). The TPR motif is a degenerate 34 amino acid sequence that folds into a pair of anti-parallel α-helices (7). In immunophilins containing three TPR motifs as in FKBP52 and CyP40 (Fig. 1), TPR1 and TPR2 are separated by 15−16 amino acid residues, whereas TPR2 and TPR3 are closer and separated by zero to one residue. Interestingly, the sequences of the TPR are more conserved within the family, i.e. a FKBP TPR is more similar in sequence to another FKBP TPR than to a CyP TPR (1).
The immunosuppressive activity of CsA and FK506 does not correlate with PPIase inhibition but involves a mechanism in which the CsA-CyP or the FK506-FKBP complex binds to and inhibits cytosolic calcineurin (CN), a Ca2+-CaM-dependent protein phosphatase, also known as protein phosphatase 2B or protein phosphatase 3 (3, 5, 8, 9). The rapamycin-FKBP complex, in contrast, inhibits a protein named TOR (target of rapamycin) (10).
In this communication, we report a novel class of immunophilins, which contain both CyP and FKBP domains. We suggest that they be called FCBPs (FK506- and cyclosporin-binding protein), in keeping with the existing single family names such as FKBP and CyP. We found the first FCBP in T. gondii, a primitive protozoan parasite of the Apicomplexan family, to which the lethal malaria parasite, Plasmodium falciparum, also belongs. A sequence search subsequently led to the characterization of three additional FCBPs, two in Flavobacterium johnsonii, a bacterium with gliding motility, and one in Treponema denticola, an oral spirochete bacterium that causes periodontal disease.
EXPERIMENTAL PROCEDURES
Materials
Ethyl-FK506 (ascomycin) was purchased from Calbiochem, cyclosporin A and bovine cyclophilin were from Sigma, and monoclonal His-tag antibody was from Novagen. Geldanamycin was a kind gift from NCI, National Institutes of Health (11). The peptide N-succinyl-Ala-Leu-Pro-Phe-p-nitroanilide was from Bachem. Synthetic peptides 18CGSGSDKVDVDPASIVD34 and 500TDKSNDRPKQDVQIVDC516 corresponded to the N- and C-terminal ends of TgFCBP57. Mice antibodies against these peptides were made as described previously (12) and named antibodies A and B, respectively. Similar antibody was also made against a peptide corresponding to the catalytic subunit of T. gondii CN (234DESKDDVGQSPEDSFTPND252) (GenBank™ accession number AAM97278). Immunoblot was performed using Super Signal Ultra chemiluminescence (Pierce). Sequence alignments were done through the ClustalW program.
T. gondii Calcineurin
T. gondii tachyzoites were grown on human foreskin fibroblast cells and purified as recommended (13). T. gondii CN holoenzyme was purified from the parasite pellet essentially as described for the P. falciparum enzyme with the exception that the Complete protein inhibitor mixture (Roche Applied Science) was added in the lysis buffer to minimize proteolysis (12). The final preparation of CN, eluted from the calmodulin-Sepharose column, contained two major polypeptides of 54 and 22 kDa (data not shown), corresponding to the catalytic (CnA) and regulatory (CnB) subunits (GenBank™ accession numbers AAM97278 and AAM97279), respectively.
Identification and Recombinant Expression of the FCBP cDNAs
A BLAST search of the T. gondii TigrScan predicted protein sequences in ToxoDB was performed. To our surprise, the same entry (TgTigr-Scan_2443) was identified using either hFKBP12 or hCyP40 as query. The cDNA sequence was confirmed by reverse transcription-PCR and sequencing. The full-length open reading frame was PCR-amplified using primers containing NdeI and BclI sites and cloned into the NdeI and BamHI sites of pET15b. Essentially, similar searches of the Gen-Bank™ later led to the F. johnsonii and T. denticola sequences. The F. johnsonii genes were previously called ppiA and ppiB in recognition of their PPIase homology sequences (14), and their clones were kindly provided by Dr. Mark J. McBride (University of Wisconsin, Milwaukee, WI). The open reading frames were cloned into the XhoI and BamHI sites of pET15b. All of the pET15b clones were introduced into Escherichia coli BL21(DE3) pLysS and induced by isopropyl 1-thio-β-d-galactopyranoside. The soluble N-terminally His-tagged recombinant proteins were purified by Ni2+-affinity chromatography using the His-Bind™ kit (Novagen) (15). The CyP and FKBP domains of TgFCBP57 were similarly expressed and purified. The internal TPR deletion mutants were generated by the PCR-based megaprimer method (16) and similarly expressed and purified.
Enzyme Assays
PPIase assays were carried out as described previously (17, 18) with minor modifications. 1.5-ml reactions contained 20 nm purified wild type or mutant His-tagged enzymes, 250 μg of 1-chloro-3-tosylamido-7-amino-2-heptanone-treated chymotrypsin (Sigma), 35 mm HEPES (pH 7.9), 0.014% Triton X-100, and 20 μm substrate peptide N-succinyl-Ala-Leu-Pro-Phe-p-nitroanilide. When used, the inhibitor was preincubated with the enzyme (60 min). The reaction was initiated by the simultaneous addition of chymotrypsin and substrate peptide. Reactions were performed at 10 °C, and the rise in A400 was recorded every 15 s for 4 min.
The chaperone assay involved refolding of RNase T1 (18). In this procedure, RNase T1 (50 μm) was first unfolded by incubating at 25 °C for 3 h in buffer A (50 mm Tris-Cl (pH 8.0), 1 mm EDTA) containing 6 m guanidine hydrochloride. Refolding was initiated by diluting 50 μl of this solution with 2.2 ml of buffer A containing recombinant FCBP or its domains (final concentration 20 nm) and was followed at 10 °C by measuring the increase in Trp fluorescence at 320 nm with excitation at 268 nm in a Hitachi UV-visible spectrophotometer.
Phosphatase assay for calcineurin was done in a 96-well plate format using the RII phosphopeptide as substrate, and the liberated phosphate was measured by malachite green assay according to the instructions of the manufacturer (Calbiochem). Phosphatase reactions were routinely initiated with the addition of substrate. Where indicated, the following additions were made at the indicated concentrations 10 min prior to initiating the reaction: Ca2+; Mg2+; Mn2+ (2 mm each); calmodulin (1 μm); purified recombinant FCBP or its deletion mutants (1 μm); and the indicated amounts of CsA or ethyl-FK506 (19). Reactions were followed with time to ensure linearity, and the results were corrected by subtraction of the corresponding values from an enzyme-free reaction.
RNA Interference Analysis of TgFCBP57
RNAi studies were carried out essentially as described previously (20). Using the appropriate primers, a 500-bp segment of the TgFCBP57-coding sequence (nucleotides 384−883) was amplified by PCR and cloned into HindIII and SacI sites of pGEM-4 (Promega). The resultant clone was transcribed by a mixture of T7 and SP6 polymerases, and the double-stranded RNA (dsRNA) was treated with calf-intestinal phosphatase. dsRNA was electroporated into 5 × 106 T. gondii RH parasites in 400 μl of electroporation buffer containing 120 mm KCl, 0.15 mm CaCl2, 10 mm K2HPO4/KH2PO4 (pH 7.6), 25 mm HEPES (pH 7.6), 2 mm EDTA, 5 mm MgCl2, 2 mm ATP, and 5 mm glutathione. Electroporation conditions were as follows: 1.5 kEV pulse; 50 ohms; 50 microfarads; and 1 ms with a 0.2-cm electrode gap in the Bio-Rad Gene Pulser. ∼5 × 104 electroporated parasites were added to confluent human foreskin fibroblast cell monolayer in each well of a 24-well plate, and the media were replaced at 4 h to remove unattached parasites. To measure parasite growth, 2.5 μCi of [3H]uracil in 1 ml of medium was added/well at 18 and 24 h post-infection. Incorporation was allowed to occur for 4 h, following which 1 ml of 0.6 n cold trichloroacetic acid was added per well and the plate was left on ice for 1 h and then submerged in a running water bath overnight. The contents of each well were then dissolved in 500 μl of 0.1 n NaOH, and 250 μl was used for liquid scintillation counting.
RESULTS
Characterization of the Dual-Family Immunophilins
We and others (21, 22) recently identified a FKBP sequence in P. falciparum that was 35 kDa in size and contained three TPRs. This FKBP ortholog showed the unique ability to inhibit the plasmodial calcineurin in the absence of drugs. Because the identification of parasitic drug targets is a major interest in our laboratory, these results prompted us to search for FKBP orthologs in another clinically significant member of the Apicomplexan family, namely T. gondii. As described under “Experimental Procedures,” homology query with the prototype human FKBP12 as well as CyP40 identified the 521 amino acid long putative T. gondii sequence in which the FKBP and CyP homology domains were at the N and C termini, respectively (Figs. 1 and 2), and a TPR domain containing three TPR motifs linked the two. We named the dual-family immunophilin FCBP. Because of its theoretical molecular mass of 57 kDa, the T. gondii protein was named TgFCBP57. Subsequent Gen-Bank™ searches led us to two F. johnsonii (Fj) and one T. denticola ortholog mentioned before, and using the same nomenclature, we named them FjFCBP39, FjFCBP40, and TdFCBP35. Interestingly, the similarity of the three putative bacterial proteins with CyP and FKBP domains was also noted recently (14), although the proteins were neither expressed nor studied. The identity of TgFCBP57 was ascertained by its reaction with peptide antibodies A and B (“Experimental Procedures”). The His-tagged recombinant additionally reacted with anti-His antibody and was ∼2 kDa larger due to the tag (data not shown).
In Fig. 2, we compared the deduced primary structures of these FCBPs with representative FKBPs and CyPs, including the only FKBP in P. falciparum (PfFKBP35). Whereas the high sequence conservation is obvious in each domain, there are a number of differences between the eukaryotic and the prokaryotic FCBPs. The prokaryotic FKBP domains have variable insertions after the invariant FD-dipeptide (Fig. 2). In the CyP domains, the eukaryotic ones are more similar to each other than to their prokaryotic counterparts as evidenced by the gaps. The SIY-tripeptide, for example, is present in the human and T. gondii domains but not in the bacterial ones. Most importantly, the relative order of the two immunophilin domains is reversed. In the prokaryotic FCBPs, the CyP domain is N-terminal, whereas in the eukaryotic FCBP, the FKBP domain is N-terminal (Figs. 1 and 2). The prokaryotic FCBPs have a much shorter linker with no TPR between CyP and FKBP domains. Thus the N-terminal half of TgFCBP57, containing the FKBD followed by the three TPRs, appears very similar to the same domains in PfFKBP35 (Fig. 1), perhaps indicating a close evolutionary relationship.
PPIase and Chaperone Activities of the FCBPs
Recombinantly expressed TgFCBP57 (Fig. 3A) and both the flavobacterial enzymes (Fig. 5) exhibited potent PPIase activities. The FKBP and the CyP domains, when expressed individually, had approximately half of the activity of the full-length FCBP on a molar basis (Fig. 3A). Because the bacterial FCBPs lacked the TPRs but still possessed PPIase activity, we reasoned that the exact sequence of the linker may not be important for catalytic function in vitro. In fact, recombinant TgFCBP57 mutants in which one, two, or all three TPR motifs were deleted retained nearly full activity, confirming that the TPRs were not essential for PPIase activity.
Fig. 3. PPIase (A) and chaperone activity (B) of recombinant full-length T. gondii FCBP and its domains.
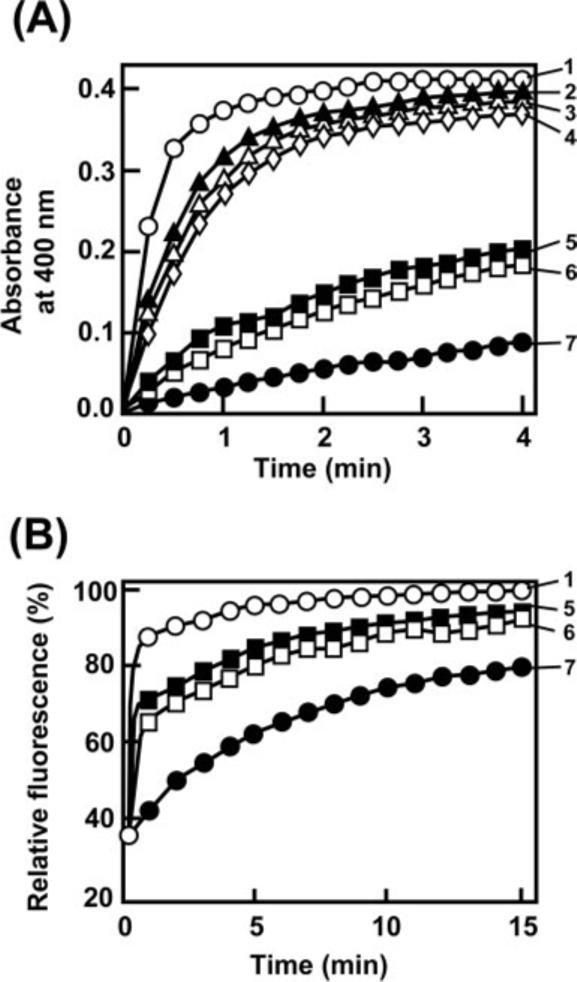
Assays were carried out as described under “Experimental Procedures.” The following His-tagged recombinants were assayed: 1, full-length TgFCBP57; 2, Δ1TPR (the most N-terminal TPR deleted); 3, Δ2TPR (two N-terminal TPRs deleted); 4, Δ3TPR (all three TPRs deleted); 5, FKBP domain (residues 25−154) only; 6, CyP domain (residue 338−521) only; and 7, no enzyme control. Note that both domains have independent and roughly equal PPIase and folding activity.
Fig. 5. Sensitivity of FCBPs to immunosuppressant drugs.

PPI-ase activity of the recombinant enzymes from T. gondii and F. johnsonii was determined in the presence of the indicated concentrations of CsA (gray bars) or ethyl-FK506 (black bars) or a mixture of the two (striped bars). The uninhibited activity of the corresponding enzymes (white bars) was taken as 100. Results represent an average of three experiments with mean ± S.E.
It is known that the PPIase and chaperone activities do not always correlate. For example, both CyP40 and FKBP52 display chaperone function independent of PPIase activity (23, 24). However, direct measurement of refolding of RNase T1, a traditional substrate for folding assay, revealed that the full-length TgFCBP57 as well as its FKBP and CyP domains had protein-folding activity (Fig. 3B). When compared on a molar basis, the individual domains contained approximately half of the activity of the full-length FCBP, matching the PPIase results above. Together, these results establish the modular nature of the FCBP and suggest that the individual FKBP and CyP domains, although quite diverse in sequence, both contribute to the activity of the full-length FCBP.
Drug Sensitivity of the FCBPs
As mentioned before, the PPIase activities of the CyPs and FKBPs are inhibited by cyclosporins and FK506, respectively. A number of amino acids known to be important for interactions with these drugs are in fact conserved in the corresponding domains of the FCBPs (Fig. 2). One important exception is His-474 of TgFCBP35, which is Trp in the other homologs (Fig. 2). Available evidence has shown that this change leads to CsA resistance. In three CyPs, namely E. coli Cyp20, human Cyp40, and a CyP from Brugia malayi (a causative agent of human filaria), the equivalent residue is His and changing it to Trp greatly increased the CsA affinity (25–27). For hCyp40, the values for the His → Trp mutant in fact approached the corresponding values for hCyP18 (25), which naturally contains a Trp in this position (Fig. 2). Therefore, it was important to investigate the drug sensitivity of a FCBP and its two PPIase domains. We first set out to determine the IC50 of CsA and FK506 on the full-length TgFCBP57; however, as high as a concentration of 10 μm of either inhibitor failed to reduce the PPIase activity by half (Fig. 4A). A simple explanation is that each drug only inhibited its cognate PPIase domain. For example, FK506 inhibited the FKBP domain only and not the CyP domain (5). To test this directly, we expressed each PPIase domain of TgFCBP57 recombinantly and determined their drug sensitivity. Indeed, the FKBP domain was highly sensitive to FK506 with an IC50 of 70 nm while being fully resistant to CsA (Fig. 4B). Similarly, the CyP domain was sensitive to CsA, although its IC50 was considerably higher at 750 nm (Fig. 4C). Based on the foregoing, we mutated the His-474 of the CyP domain to Trp, which resulted in a dramatic lowering of IC50 of CsA to 40 nm. As expected, the CyP domain (the wild type as well as the H474W mutant) was fully resistant to FK506. Thus each PPIase domain of TgFCBP57 individually exhibited sensitivity to the family-specific drug, further authenticating their membership in the respective families.
Fig. 4. Drug sensitivity of the individual domains of TgFCBP57.
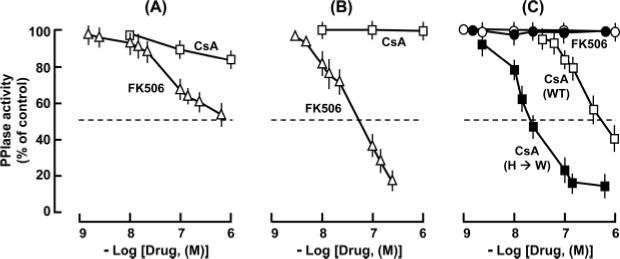
A, full-length enzyme. B, FKBP domain only (residues 25−154). C, CyP domain only (residues 338−521). Open symbols, WT; closed symbols, His-474 → Trp mutant (see Fig. 2) in which full CsA sensitivity was restored. Results represent the average of three experiments with mean ± S.E. bars.
We next tested the dual drug sensitivity of the FCBPs. The results (Fig. 5) show that CsA and FK506 additively inhibited all of the FCBPs. Thus the relative positioning of the CyP and FKBP domains did not affect drug-PPIase interaction in each domain, further confirming the modular nature of the FCBPs. The higher sensitivity of the bacterial enzymes toward CsA is easily explained by the conserved Trp corresponding to the His-474 in the T. gondii enzyme (Fig. 2).
Inhibition of Calcineurin by Individual FCBP Domains
Because the biochemical dissection experiments revealed the modular nature of the FCBPs, it was logical to ask whether each domain also maintained the ability to inhibit calcineurin in the presence of the cognate drug (4–6). We took purified T. gondii calcineurin and measured its phosphatase activity in the presence of the two recombinant domains of TgFCBP57 and either CsA or FK506. The CyP and FKBP domains clearly inhibited calcineurin in the presence of CsA and FK506, respectively, but not in the reciprocal combination (Fig. 6). Therefore, it is obvious that the modular arrangement of the FCBP extends to calcineurin inhibition as well.
Fig. 6. Inhibition of T. gondii CN activity by recombinant FKBP domain (residues 25−154) or CyP domain (residues 338−521) of TgFCBP57 in the presence of 10 μm CsA (gray bars) or ethyl-FK506 (black bars).

Uninhibited CN activity (white bar) was taken as 100. Results are average of three experiments with mean ± S.E.
Inhibition of T. gondii by CsA and FK506
The exact physiological target of CsA or FK506 in T. gondii is not known. However, because the immunophilins are potential pharmaceutical targets, an obvious question was whether these drugs would have anti-parasitic activity. Using an established [3H]-uridine incorporation assay that is specific for the salvage pathway of the parasite and absent in the host, both drugs were found to inhibit T. gondii growth with IC50 values at 0.7−0.8 μm extracellular drug concentration (Fig. 7A). When tested together, the two drugs showed a slightly synergistic inhibition (Fig. 7B).
Fig. 7. T. gondii drug sensitivity.
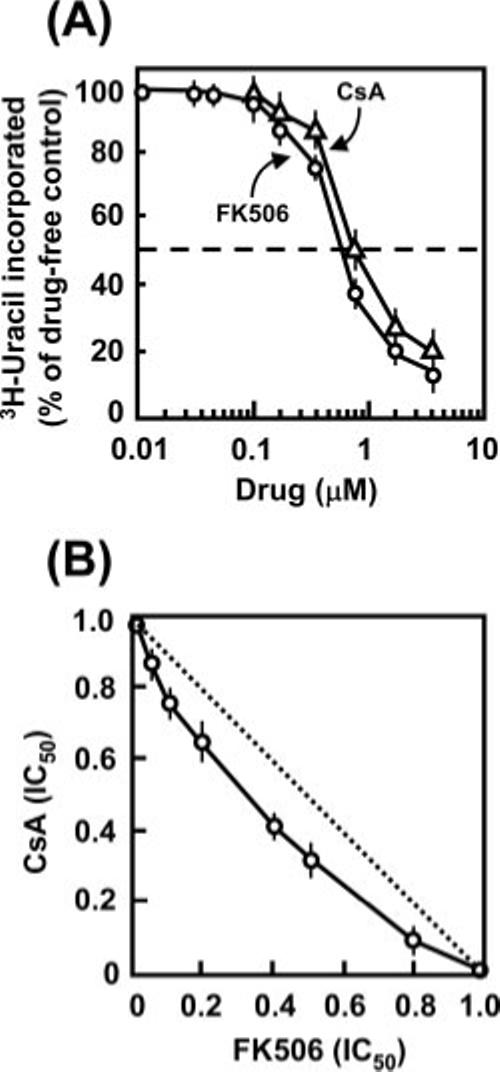
A, anti-toxoplasma activity of ethyl-FK506 and CsA. The IC50 of the two drugs are 0.65 and 0.76 μm, respectively. B, anti-toxoplasma activity of a combination of ethyl-FK506 and CsA. Note that the experimental line is slightly concave, indicating modest synergism, in contrast to the hypothetical straight line (dotted) that would have been obtained if there were no interaction between the drugs. The numbers on both axes are drug concentrations expressed as fractions of their individual IC50 values. Results represent average of three experimental sets with mean ± S.E.
Knockdown of TgFCBP57 by RNAi Inhibits T. gondii Growth
RNAi has emerged as an important tool to knock down specific gene expression (28). Recently, double-stranded RNA and short interfering RNA have been shown to trigger an RNAi-like effect in the Apicomplexa as well, especially in T. gondii (15, 20, 29, 30). We adopted a similar approach and targeted the TgFCBP57 mRNA by transfecting a dsRNA corresponding to an internal 500-bp region of the coding sequence, which led to successful silencing of TgFCBP57 (Fig. 8). Concomitantly, parasitic gene expression was significantly reduced, as measured by the incorporation of [3H]uracil.
Fig. 8. RNA interference phenotype of TgFCBP57 knockdown.
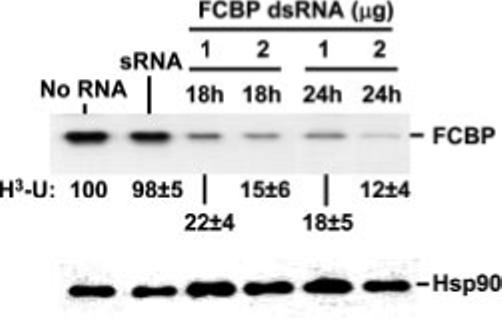
RNAi knockdown was done with dsRNA as described under “Experimental Procedures.” T. gondii cells, transfected with 1 or 2 μg of dsRNA, were used to infect human foreskin fibroblast cells, and at 18 and 24 h later, the cells were analyzed for TgFCBP57 by immunoblot. Results using antibody A (“Experimental Procedures”) are shown, although antibody B produced essentially the same result. [3H]Uracil incorporation of identical parallel cultures was measured and expressed as percentage of untreated (No RNA) value. Single-stranded sense RNA (sRNA,4 μg, 24 h) was used as another negative control. The numbers are average of three experiments with the mean ± S.E. as shown. Control Hsp90, detected by immunoblot (11), was unaffected (bottom panel).
Drug-dependent Binding of TgFCBP57 to Specific Targets
As the previous results implicated TgFCBP57 in the anti-parasitic activity of the immunosuppressant drugs, we initiated studies of the downstream targets to gain some insights into the mechanism. His-tagged recombinant TgFCBP57 was immobilized on the Ni2+ column, and soluble T. gondii lysate was passed through in the presence or absence of the drugs. Analysis of the bound proteins (Fig. 9) clearly revealed that TgFCBP57 associated with CN in the presence of FK506 and CsA, although the affinity with CsA was considerably lower. In contrast, rapamycin led to the recruitment of a protein of roughly 290 kDa, close in size to the mammalian homolog of TOR, also known as FKBP-rapamycin-associated protein. A homology search of the predicted proteins of T. gondii indeed revealed a prospective TOR sequence (TgTigrScan_2443 in ToxoDB), which is being characterized further (Fig. 10). Henceforth referred as TgTOR, the sequence showed striking similarity to the FKBP-rapamycin-binding domain (FRBD) as well as the phosphatidylinositol 3-kinase-like domain of human TOR. A synthetic peptide, TFRETLFLQKYGRELENAYT, corresponding to the central region of the putative FRBD of Tg-TOR significantly competed with the binding of TgTOR to the FCBP-rapamycin complex (Fig. 9, last lane), suggesting the authenticity of the sequence. An irrelevant peptide corresponding to the C-terminal 15 amino acids of respiratory syncytial viral P protein (GenBank™ accession number P12579) had no effect (data not shown), demonstrating specificity. Thus, the TOR pathway may represent an important downstream target of the FCBP-rapamycin complex in T. gondii.
Fig. 9. Drug-specific association of FCBP with downstream proteins.
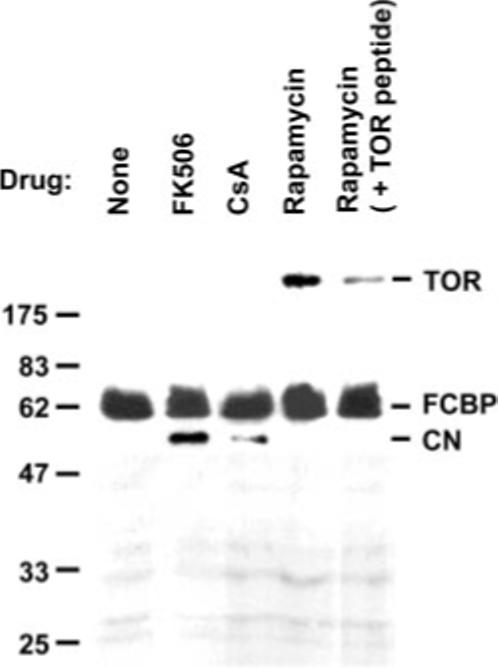
The experiment was done essentially as described previously (38). Purified His-tagged recombinant TgFCBP57 (10 μg) was immobilized on Ni2+-agarose column (60 μl), and 400 μg of T. gondii cell extract was passed through it. Where shown, the extract was pre-mixed with 10 μm drug. Where indicated (last lane), 0.2 μg of competing TOR peptide (FRBD region) was added to the extract. The column was then washed with 1 ml of wash buffer (Novagen) containing 0.2 mm imidazole. The bound proteins were stripped by boiling in 2× SDS sample buffer and analyzed by SDS-PAGE and silver staining. The catalytic subunit of T. gondii CN was further confirmed by immunoblot (data not shown). See “Results” for details.
Fig. 10. A putative T. gondii TOR homolog.

The C-terminal end of the deduced primary structures of human (P42345; hTOR) and T. gondii TOR (TgTOR) proteins are aligned. The TgTOR sequence is based on TgTigrScan_2443 in ToxoDB but was conceptually corrected and partially sequenced. The following domains are marked. Part of the FAT (FKBP-rapamycin-associated protein, ATM, and transformation/transcription domain-associated protein (TRRAP)) domain (smaller box), FRBD (larger box), and phosphatidylinositol 3-kinase-like domain (underlined) is shown. Identical residues are denoted by asterisk, and similar ones are represented by dots. We tentatively estimate the full-length TgTOR to be 2845 amino acids long.
DISCUSSION
Results presented here constitute the first report of dual-family immunophilins in which one PPIase domain was derived from the CyP family and the other was derived from the FKBP family. Therefore, the enzymes were tentatively named FCBP (Figs. 1 and 2). It is possible that more FCBPs will come to light as new genomes are sequenced and annotated; however, it is already apparent that FCBPs are not as ubiquitous or numerous as CyPs or FKBPs. For instance, we did not find any FCBP in another Treponema sp., namely Treponema pallidum (the causative agent of syphilis), although it contained putative CyP and FKBP orthologs with single family-type PPIase domains (GenBank™ accession numbers NP_219383 and NP_219298). Other spirochetes are also clinically highly significant and include Borrelia (Lyme disease) and Leptospira (leptospirosis); however, a homology search of their complete genome sequence did not reveal any dual-family FCBP. In contrast, CyPs and FKBPs are not only ubiquitous but often have multiple paralogs within the same organism. Humans, for example, have 15 FKBPs and at least 16 CyP-like sequences, whereas the lower eukaryote, Saccharomyces cerevisiae, encodes eight CyPs and four FKBPs (1).
The mechanism of anti-toxoplasma activity of the immunosuppressants calls for further work. Because CsA is a weak inhibitor of TgFCBP57, a more probable target for CsA is CyP. A few CyPs are predicted in the T. gondii genome data base such as TgTwinScan_5726 and TgTwinScan_3965, but none has been characterized. These sequences have Trp corresponding to His-474 of TgFCBP57 and thus are likely to be sensitive to CsA. However, for FK506 and rapamycin, TgFCBP57 appears to be the sole proximal target because no other FKBP domain is to be found in the T. gondii genome.2 As mentioned before, P. falciparum also has only one FKBP sequence, viz. PfFKBP35 (Fig. 1). Further characterization of TgTOR will provide an important direction in rapamycin action. In an analogy to its mammalian homolog of TOR orthologs (10), TgTOR may act as a nutrient sensor, regulating parasitic cell growth and cell cycle progression, probably by regulating translation through the phosphorylation of translation factors. The phosphatidylinositol 3-kinase-like domain of TgTOR (Fig. 10) may well play a role in this process.
In all of the TPR-containing immunophilins (CyP and FKBP) characterized to date, the TPR domain is found downstream (C-terminal) of the PPIase domain (Fig. 1) (1). Thus, we speculate that TgFCBP57 evolved as a chimeric protein generated by the fusion of a FKBP-(TPR)3 sequence, such as PfFKBP35, with a CyP. However, there is an important difference between the FKBP domains of these two parasites. Whereas PfFKBP35 can inhibit calcineurin in the absence of any drug (21, 22), the FKBP domain of TgFCBP57 requires FK506 for this effect (Fig. 6).
Examples of duplicate enzymatic units in the same polypeptide are relatively rare in nature. Recently, a double protein phosphatase 2C was identified in P. falciparum in which the full-length protein as well as each half, expressed recombinantly, showed protein phosphatase 2C-like phosphatase activity (31). A few members of the chitinase family have two or more catalytic domains (32–34). Quite a few FKBPs are known that contain multiple FKBD (1); however, in most cases, it has not been tested whether each domain has PPIase activity. Human FKBP52 has two FKBDs (Fig. 1), but only the first one has PPIase activity (35). In all of the repeat-domain proteins, in general, the functionally duplicate segments show significant sequence homology but are not identical, thus ruling out simple gene duplication. It is also interesting to note that to our knowledge duplication of the PPIase domain has not yet been found in any CyPs (1). Currently, we do not know whether the FCBPs perform a unique function that cannot be preformed by the individual immunophilins. The exact physiological role(s) of FCBPs remain to be elucidated, although RNAi analysis suggested an essential role in T. gondii. Given their PPIase and chaperone activities, it is tempting to speculate that they play a role in protein folding and the formation of multiprotein signaling complexes such as the glucocorticoid receptor complex in human (36, 37). Perhaps the relative positioning of the CyP and FKBP domains in the FCBP allows them to simultaneously interact with two client proteins in such a multiprotein complex. The flexibility of the linker sequence may allow fine-tuning of the orientation, and the TPR domain may promote additional protein-protein interactions (7). Clearly, the determination of the tertiary structure of the FCBPs and identification of the interacting partners will shed light on the function of this remarkable new family of immunophilins.
Acknowledgments
We thank Dr. Mark J. McBride for the generous gifts of the F. johnsonii ppiA/B clones and Dr. Andrzej Galat for expert opinion. Preliminary genomic and cDNA sequence data was accessed via ToxoDB.org and/or www.tigr.org/tdb/t_gondii/. Genomic data were provided by The Institute for Genomic Research, and EST sequences were generated by Washington University. We thank these agencies for making the databases publicly accessible.
Footnotes
This work was supported by NIAID, National Institutes of Health Grant AI045803 (to S. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: PPIase, peptidylprolyl cis-trans-isomerase; CyP, cyclophilin; FKBP, FK506-binding protein; FKBD, FK506-binding domain; FCBP, FK506- and cyclosporin-binding protein; TPR, tetratricopeptide repeat; CsA, cyclosporin A; CN, calcineurin; ds, double-stranded; h, human; RNAi, RNA interference; TOR, target of rapamycin; Tg, T. gondii; Fj, F. johnsonii.
B. Adams, A. Musiyenko, R. Kumar, and S. Barik, unpublished observation.
REFERENCES
- 1.Galat A. Curr. Top. Med. Chem. 2003;3:1315–1347. doi: 10.2174/1568026033451862. [DOI] [PubMed] [Google Scholar]
- 2.Pratt WB, Galigniana MD, Harrell JM, DeFranco DB. Cell. Signal. 2004;16:857–872. doi: 10.1016/j.cellsig.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 3.Blankenship JR, Steinbach WJ, Perfect JR, Heitman J. Curr. Opin. Investig. Drugs. 2003;4:192–199. [PubMed] [Google Scholar]
- 4.Jin L, Harrison SC. Proc. Natl. Acad. Sci. U. S. A. 2002;99:13522–13526. doi: 10.1073/pnas.212504399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huai Q, Kim HY, Liu Y, Zhao Y, Mondragon A, Liu JO, Ke H. Proc. Natl. Acad. Sci. U. S. A. 2002;99:12037–12042. doi: 10.1073/pnas.192206699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kissinger CR, Parge HE, Knighton DR, Lewis CT, Pelletier LA, Tempczyk A, Kalish VJ, Tucker KD, Showalter RE, Moomaw EW, Gastinel LN, Habuka N, Chen X, Maldonado F, Barker JE, Bacquet R, Villafranca JE. Nature. 1995;378:641–644. doi: 10.1038/378641a0. [DOI] [PubMed] [Google Scholar]
- 7.D'Andrea LD, Regan L. Trends Biochem. Sci. 2003;28:655–662. doi: 10.1016/j.tibs.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Feske S, Okamura H, Hogan PG, Rao A. Biochem. Biophys. Res. Commun. 2003;311:1117–1132. doi: 10.1016/j.bbrc.2003.09.174. [DOI] [PubMed] [Google Scholar]
- 9.Liu J, Farmer JD,, Jr., Lane WS, Friedman J, Weissman I, Schreiber SL. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 10.Hay N, Sonenberg N. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 11.Kumar R, Musiyenko A, Barik S. Malar. J. 2003;2:30. doi: 10.1186/1475-2875-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar R, Musiyenko A, Oldenburg A, Adams B, Barik S. BMC Mol. Biol. 2004;5:6. doi: 10.1186/1471-2199-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roos DS, Donald RG, Morrissette NS, Moulton AL. Methods Cell Biol. 1994;45:27–63. doi: 10.1016/s0091-679x(08)61845-2. [DOI] [PubMed] [Google Scholar]
- 14.McBride MJ, Braun TF. J. Bacteriol. 2004;186:2295–2302. doi: 10.1128/JB.186.8.2295-2302.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar R, Adams B, Oldenburg A, Musiyenko A, Barik S. Malar. J. 2002;1:5. doi: 10.1186/1475-2875-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burke E, Barik S. Methods Mol. Biol. 2003;226:525–532. doi: 10.1385/1-59259-384-4:525. [DOI] [PubMed] [Google Scholar]
- 17.Nair SC, Rimerman RA, Toran EJ, Chen S, Prapapanich V, Butts RN, Smith DF. Mol. Cell. Biol. 1997;17:594–603. doi: 10.1128/mcb.17.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki Y, Haruki M, Takano K, Morikawa M, Kanaya S. Eur. J. Biochem. 2004;271:1372–1381. doi: 10.1111/j.1432-1033.2004.04049.x. [DOI] [PubMed] [Google Scholar]
- 19.Sewell TJ, Lam E, Martin MM, Leszyk J, Weidner J, Calaycay J, Griffin P, Williams H, Hung S, Cryan J. J. Biol. Chem. 1994;269:21094–21102. [PubMed] [Google Scholar]
- 20.Al-Anouti F, Ananvoranich S. Antisense Nucleic Acid Drug Dev. 2002;12:275–281. doi: 10.1089/108729002320351593. [DOI] [PubMed] [Google Scholar]
- 21.Kumar R, Adams B, Musiyenko A, Shulyayeva O, Barik S. Mol. Biochem. Parasitol. 2005;141:163–173. doi: 10.1016/j.molbiopara.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 22.Monaghan P, Bell A. Mol. Biochem. Parasitol. 2005;139:185–195. doi: 10.1016/j.molbiopara.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 23.Freeman BC, Toft DO, Morimoto RI. Science. 1996;274:1718–1720. doi: 10.1126/science.274.5293.1718. [DOI] [PubMed] [Google Scholar]
- 24.Ward BK, Allan RK, Mok D, Temple SE, Taylor P, Dornan J, Mark PJ, Shaw DJ, Kumar P, Walkinshaw MD, Ratajczak T. J. Biol. Chem. 2002;277:40799–40809. doi: 10.1074/jbc.M207097200. [DOI] [PubMed] [Google Scholar]
- 25.Hoffmann K, Kakalis LT, Anderson KS, Armitage IM, Handschumacher RE. Eur. J. Biochem. 1995;229:188–193. doi: 10.1111/j.1432-1033.1995.tb20454.x. [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Chen CM, Walsh CT. Biochemistry. 1991;30:2306–2310. doi: 10.1021/bi00223a003. [DOI] [PubMed] [Google Scholar]
- 27.Page AP, Landry D, Wilson GG, Carlow CK. Biochemistry. 1995;34:11545–11550. doi: 10.1021/bi00036a030. [DOI] [PubMed] [Google Scholar]
- 28.Meister G, Tuschl T. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 29.Malhotra P, Dasaradhi PV, Kumar A, Mohmmed A, Agrawal N, Bhatnagar RK, Chauhan VS. Mol. Microbiol. 2002;45:1245–1254. doi: 10.1046/j.1365-2958.2002.03105.x. [DOI] [PubMed] [Google Scholar]
- 30.McRobert L, McConkey GA. Mol. Biochem. Parasitol. 2002;119:273–278. doi: 10.1016/s0166-6851(01)00429-7. [DOI] [PubMed] [Google Scholar]
- 31.Mamoun CB, Sullivan DJ,, Jr., Banerjee R, Goldberg DE. J. Biol. Chem. 1998;273:11241–11247. doi: 10.1074/jbc.273.18.11241. [DOI] [PubMed] [Google Scholar]
- 32.Howard MB, Ekborg NA, Taylor LE,, 2nd, Weiner RM, Hutcheson SW. J. Bacteriol. 2004;186:1297–1303. doi: 10.1128/JB.186.5.1297-1303.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Royer V, Fraichard S, Bouhin H. Biochem. J. 2002;366:921–928. doi: 10.1042/BJ20011764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka T, Fukui T, Imanaka T. J. Biol. Chem. 2001;276:35629–35635. doi: 10.1074/jbc.M105919200. [DOI] [PubMed] [Google Scholar]
- 35.Pirkl F, Fischer E, Modrow S, Buchner J. J. Biol. Chem. 2001;276:37034–37041. doi: 10.1074/jbc.M102595200. [DOI] [PubMed] [Google Scholar]
- 36.Wu B, Li P, Liu Y, Lou Z, Ding Y, Shu C, Ye S, Bartlam M, Shen B, Rao Z. Proc. Natl. Acad. Sci. U. S. A. 2004;101:8348–8353. doi: 10.1073/pnas.0305969101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ratajczak T, Ward BK, Minchin RF. Curr. Top. Med. Chem. 2003;3:1348–1357. doi: 10.2174/1568026033451934. [DOI] [PubMed] [Google Scholar]
- 38.Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, Abraham RT. J. Biol. Chem. 1995;270:815–822. doi: 10.1074/jbc.270.2.815. [DOI] [PubMed] [Google Scholar]