

Abstract
Carnitine palmitoyltransferase-1 (CPT-1) catalyzes the rate-limiting step of mitochondrial β-oxidation of long chain fatty acids (LCFA), the most abundant fatty acids in mammalian membranes and in energy metabolism. Human deficiency of the muscle isoform CPT-1b is poorly understood. In the current study, embryos with a homozygous knockout of Cpt-1b were lost before embryonic day 9.5 − 11.5. Also, while there were normal percentages of CPT-1b+/−pups born from both male and female CPT-1b+/− mice crossed with wild-type mates, the number of CPT-1b+/− pups from CPT-1b+/− breeding pairs was under-represented (63% of the expected number). Northern blot analysis demonstrated ∼50% Cpt-1b mRNA expression in brown adipose tissue (BAT), heart and skeletal muscles in the CPT-1b+/− male mice. Consistent with tissue-specific expression of Cpt-1b mRNA in muscle but not liver, CPT-1+/− mice had ∼60% CPT-1 activity in skeletal muscle and no change in total liver CPT-1 activity. CPT-1b+/− mice had normal fasting blood glucose concentration. Consistent with expression of CPT-1b in BAT and muscle, ∼7% CPT-1b+/− mice (n=30) developed fatal hypothermia following a 3 hr cold challenge, while none of the CPT-1b+/+ mice (n=30) did. With a prolonged cold challenge (6 hr), significantly more CPT-1b+/− mice developed fatal hypothermia (52% CPT-1b+/− mice vs. 21% CPT-1b+/+ mice), with increased frequency in females of both genotypes (67% female vs. 38% male CPT-1b+/− mice, and 33% female vs. 8% male CPT-1b+/+ mice). Therefore, lethality of homozygous CPT-1b deficiency in the mice is consistent with paucity of human cases.
Keywords: carnitine palmitoyltransferase-1b, cold intolerance, gestational lethality
Introduction
Long chain fatty acids (LCFAs) are a major energy source for muscle and liver [1]. During prolonged fasting, most tissues depend on fatty acid β-oxidation for cellular energy [2]. Infants rely heavily on LCFA oxidation for energy due to their limited glycogen stores [3]. Carnitine palmitoyltransferase 1 (CPT 1), an important enzyme for mitochondrial fatty acid oxidation (FAO) found to exist in tissue-specific isoforms [4], regulates inward translocation of LCFAs into mitochondrial matrix, and is rate-limiting for β-oxidation of LCFAs inside mitochondria [5].
The activity and expression levels of each CPT-1 isoform, which are encoded on separate chromosomes [6,7], are not only tissue-specific, but are also affected by nutritional and hormonal regulation, by species, and by developmental stage [8,9]. CPT-1a, or the liver isoform, is expressed in liver, kidney, white adipose tissue (WAT, in male but not female mice [9,10]), testis, ovary, pancreatic islet, lung, spleen, brain, intestine [8,9], and at much lower levels in heart [10]. In contrast, the muscle isoform, or CPT-1b, is expressed in brown adipose tissue (BAT), heart, skeletal muscle, testis, and WAT (in male rats [11,12], in female but not male mice, and in humans [9]). CPT-1c is expressed predominantly in brain [7], and regulates energy homeostasis [13], but the substrates of this enzyme remain unknown.
Presently, CPT-1b deficiency in humans has been rarely reported and incompletely understood [14-17], because some earlier cases of muscle CPT deficiency were described before the discovery that CPT-1 and CPT-2 are two separate enzymes [14-18]. The clinical manifestations include recurrent rhabdomyolysis precipitated by fasting or exercise [14-18], and in one patient, by ibuprofen [17]. Therefore, it is still not clear whether the scarcity of human cases with CPT-1b deficiency is due to embryonic lethality or asymptomatic existence.
Methods
Mice
The mutant mouse line had been generated previously using a targeted mutagenesis strategy by replacing a segment of 1468 bp (exons 1−3) in mouse Cpt-1b with a 3 kb neo-tk cassette in the C57BL/6J × 129X1/SvJ ES cells [19]. Mice in the current study were the second generation from 3 male founders, which were offspring from a male chimera and C57BL/6J (B6J) females. Mice were fasted for ∼18 h and euthanized with CO2 before collecting blood for biochemical markers. The mice were also fasted for ∼18 h prior to cold tolerance testing. Alternatively, the mice used to measure mRNA expression and for collecting tissue for activity assays were not fasted before being euthanized with CO2 inhalation. Also, two different mating pair arrangements were setup to obtain fetal tissue for genotyping and to isolate the corresponding placenta for RNA preparation. One strategy included male CPT-1b+/+ mice mated with female CPT-1b+/− mice; the other included male CPT-1b+/− mice mated with female CPT-1b+/+ mice. At embryonic day 12−14, pregnant females were sacrificed. Blood was collected from the retro-orbital venous plexus with the exception of the mice used to measure mRNA expression where blood was collected by cardiac puncture. Blood samples were allowed to briefly coagulate at room temperature, and then placed on ice and centrifuged for serum collection. All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham. Mice were fed a standard rodent diet (Purina RMH2500).
Genotyping
Mice were genotyped by standard PCR analysis of tail DNA using PCR Master Mix (Roche, Indianapolis). The wild type allele was detected by the primer pair cpt1b-genoF1 (CTACTGAAGATTGGGCTCCT) and cpt-1bgenoR2 (CAGCAATGGTGCAGGAATCT), which produced a 625 bp product. The mutant allele was detected by the primer pair cpt1b-genoF1 and Neo-pR2 (ACC GCT TCC TCG TGC TTT ACG GTA), which produced a 440 bp product. The annealing temperature was 58 °C.
Gestation studies
After finding no live-born CPT-1b−/− mice, meetings timed were used to investigate the suspected gestational lethality and the fertility of that genotype. CPT-1b+/− breeding pairs were checked for plugs for several days to determine the time of conception. The day on which the female was found plugged was considered day 0.5, and embryos and fetuses were collected from day 9.5 − 11.5. The mice were euthanized with CO2, fetuses were excised, and DNA was isolated followed by PCR analysis for genotyping.
Inheritance pattern investigation
To investigate possible parental sex preference of transmitting the mutant allele, breeders were set up with either the male or the female transmitting the mutant allele and paired with wild-type mates. Live-born pups of both crosses were genotyped by PCR.
Northern blots analysis of mRNA levels and size
Northern blots were used to evaluate Cpt-1a and Cpt-1b tissue expression in both wild-type and mutant mice. Liver, BAT, heart, and skeletal muscle were collected aseptically for RNA analysis. A 100 mg sample of tissue was placed in 1 ml TRIzol reagent (Invitrogen), homogenized, and processed for RNA isolation. Gel electrophoresis, blotting, probe synthesis (Strip EZ DNA Ambion), and hybridization were carried out as described previously [20].
Real time quantitative RT-PCR analysis
First strand cDNA was generated from 1 μg of RNA in 20 μl volume reaction containing both oligo-(dT) and random hexamers (Invitrogen First Strand Synthesis Kit) according to the manufacturer's instructions. Real time quantitative RT-PCR was carried out in a 20 μl reaction volume containing 10 μl Supermix UDG (Invitrogen Life Technologies, Inc.), 2 μl cDNA, 0.3 μM LUX Cpt-1a and Cpt-1b primer pairs (Cpt-1a forward primer: CACTGCCCTTGGACCCAAATTGCAG[FAM]G, Cpt-1a reverse primer: AATTTGTGGCCCACCAGGAT; Cpt-1b forward primer: GACCATAGAGGCACTTCTCAGCATGG[FAM]C, Cpt-1b reverse primer: GCAGCAGCTTCAGGGTTTGT, [Invitrogen, Life Technologies]), 0.05 μM 18S RNA forward and reverse primers (JOE labeled, Invitrogen, Life Technologies). Cycling conditions included incubation at 50 °C for 2 min, a 2 min 95 °C denaturing step, followed by 49 cycles of 95 °C denaturation (manual ramp rate of 2 °C per second), incubation at 60 °C for 2 min (manual ramp rate of 2 °C per second). A final extension was achieved by denaturing at 95 °C for 1 sec (manual ramp rate of 2 °C per second), when melting curve was read from 55 to 95 °C every 0.5 °C (hold 1 sec), followed by incubation at 25 °C for 30 min.
Evaluation of carnitine palmitoyltransferase 1 activity
Frozen liver specimens were homogenized to yield a 10% (w/v) homogenate in 10 mM potassium phosphate/150 mM NaCl, pH 7.4, supplemented with protease inhibitor cocktail, and protein phosphatase inhibitor cocktail I and II (Sigma) at a 1/100 dilution. Carnitine palmitoyltransferase 1 (CPT-1) activity was determined in duplicate using the modified radiochemical forward assay by measuring the formation of 14C-labeled palmitoylcarnitine from [14C]-carnitine and palmitoyl-CoA at 37 °C, and is defined as the activity in nmoles/min/g wet weight inhibited by 200 μM malonyl-CoA [21,22]. Briefly, 10 μl aliquots of the 10% (w/v) liver homogenate were preincubated for 2 min with 50 μM palmitoyl-CoA at fixed palmitoyl-CoA/BSA ratio (0.85) alone or plus 200 μM malonyl-CoA in 250 μl reaction mixture containing 50 mM MOPS, 80 mM KCl, 1.0 mM EGTA, and 2.0 mM KCN. The reaction was initiated with [14C]carnitine (2.0 mM final concentration), specific radioactivity 1823 dpm/nmole, and stopped after one and a half min with 1.0 ml of 1 N HCl. Following extraction of [14C]-palmitoylcarnitine with water-saturated butanol and re-extraction of the butanol phase with butanol-saturated water an aliquot of the organic layer was used for scintillation counting.
Biochemical analysis
Total non-esterified fatty acids (NEFA) were measured by an enzymatic, colorimetric method (“NEFA-C” reagents, Wako Diagnostics, Richmond, Virginia). The assay was modified to accommodate a reduced sample size (10 μl), and use of a microplate reader for measurement of optical density at 550 nm. Serum acylcarnitine analysis was performed by using electrospray tandem mass spectrometry [23-25]. Urinary organic acid analysis was performed as described previously [26]. Glucose concentration was measured in 10 μl serum using an Ektachem DT II system (Johnson and Johnson Clinical Diagnostics).
Cold tolerance testing
Routine cold tolerance of male and female CPT-1b +/− and CPT-1b +/+ mice was assessed by exposure to 4 °C room temperature for 3 h with measurements at 1 h intervals, as we described previously [27]. Rectal temperatures were measured prior to cold exposure and repeated hourly using Barnant Thermocouple thermometer. A prolonged cold tolerance was assessed by exposure to 4 °C temperatures for 6 h. Following our standard protocol, cold challenge experiments were terminated when rectal temperatures dropped to 25 °C and the mice were deemed to have reached “fatal hypothermia”, or after a total of 6 h of exposure.
Histologic characterization
Male and female mice, age 6 − 8 weeks, were examined after cold-challenge. At necropsy, the mice were examined for internal and external abnormalities. Heart, liver, BAT, WAT, and skeletal muscle were fixed by immersion in formalin. Fixed tissues were processed routinely, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin (H&E).
Statistical analysis
Biochemical measurements were reported as mean values with standard deviations and p-values from Student's t tests. The χ2 tests of variance and Fisher's exact test were used to test whether the pattern of inheritance and fertility, respectively, were significantly different. Survival curves were generated by the Kaplan-Meier product-limit method and were compared using the log-rank test. Survival times were calculated from the study onset to death or termination due to study protocol (e.g., lowest tolerable temperature was reached and mouse was euthanized). Survival rates were reported as percentage surviving +/− standard errors. Results were considered significantly different with a p < 0.05.
Results
Initial characterization of Cpt-1b mutant mice
The CPT-1b+/− mice appear normal, with no differences in bodyweight in both male and female CPT-1b+/− mice, as measured in 6- to 7-week-old mice when fed, following an 18-hr-fast, or a 6-hr-cold challenge (data not shown). Also, the heart weight and the ratio of heart to bodyweight were not different across genotypes (data not shown). Moreover, there were no significant changes in the histology of liver, WAT, BAT and heart (data not shown).
Early embryonic lethality, placental Cpt-1 expression, and skewed pattern of inheritance
Although an expected ratio among genotypes is 1:2:1 for CPT-1b+/+: CPT-1b+/−: CPT-1b−/−, after PCR analysis of 53 offspring from CPT-1b+/− breeders (Table 1A, first row), not a single CPT-1b−/− pup was found. Also, the expected numbers of pups with CPT-1b+/+ and CPT-1b+/− genotypes are indicated and were compared with the observed numbers (Table 1A, second row), suggesting an under-representation of CPT-1b+/− pups. Subsequently, we set up timed-mating experiments with CPT-1b+/− breeders and genotyped 30 fetuses at embryonic day 9.5−11.5 (Table 1B). After finding no CPT-1b−/− embryos, and 15 CPT-1b+/− and 15 CPT-1b+/+ embryos, we concluded that homozygous CPT-1b deficiency was lethal at earlier embryonic stages and that there was an under-representation of CPT-1b +/− fetuses.
Table 1.
| A. CPT-1b +/−(M) × CPT-1b +/− (F), 9 litters of live born pups | ||||
|---|---|---|---|---|
| pups | CPT-1b+/+ | CPT-1b+/− | CPT-1b−/− | Total |
| Observed (Expected)* | 31 (13.25) | 22 (26.5) | 0 (13.25) | 53 |
| Observed (Expected)* | 31 (17.7) | 22(35.3) | 0 (0) | 53 |
| B. CPT-1b +/−(M) × CPT-1b +/− (F), 12 breeding pairs, 4 pregnant females sacrificed | ||||
|---|---|---|---|---|
| fetuses | CPT-1b+/+ | CPT-1b+/− | CPT-1b−/− | Total |
| Observed (Expected) | 15 (7.5) | 15 (15) | 0 | 30 |
| Expected | 7.5 | 15 | 7.5 | 30 |
p<0.001 by chi-square test of variance
p<0.001 by chi-square test of variance
To investigate the potential role of CPT-1b expression in placenta, we used real time quantitative RT-PCR analysis of placental mRNA. We found that Cpt-1b mRNA levels in CPT-1b+/+ placenta (n=5) derived from CPT-1b+/+ female mice mated with CPT-1b+/− males were about ∼0.3% of that detected in BAT of 6-week-old non-fasted CPT-1b+/+ male mice (n=4, data not shown). In contrast, Cpt-1a mRNA levels in such placenta (n=5) were ∼15.1% of that detected in livers of the CPT-1b+/+ mice (n=4, data not shown). Therefore, Cpt-1a mRNA was found to be the predominant isoform expressed in placenta.
Transmission of the mutant allele from both the female and male parent of origin followed the Mendelian pattern (Tables 2A and 2B). Female CPT-1b +/− and male CPT-1b+/+ breeding pairs had 49% heterozygous pups and 51% wild-type pups (Table 2A), while male CPT-1b +/− and a B6J female CPT-1b +/+ breeding pairs produced 51% CPT-1b +/− and 49% wild-type offspring (Table 2B).
Table 2.
| A. Transmission via female CPT-1b +/− mice (mated with male CPT-1b +/+ mice) | |||
|---|---|---|---|
| Pups (10 litters) | CPT-1b+/+ | CPT-1b+/− | Total |
| Observed | 39 (51%) | 37 (49%) | 76 |
| Expected | 38 (50%) | 38 (50%) | 76 |
| B. Transmission via males CPT-1b +/− mice (mated with female CPT-1b +/+ mice) | |||
|---|---|---|---|
| Pups (26 litters) | CPT-1b+/+ | CPT-1b+/− | Total |
| Observed | 84 (49%) | 87 (51%) | 171 |
| Expected | 85.5 (50%) | 85.5 (50%) | 171 |
p>0.1 by chi-square test of variance
p>0.1 by chi-square test of variance
Reduced fertility
Deficiency in FAO may compromise reproduction, which was compared between the two genotypes. Two sets of breeders, CPT-1b+/− × CPT-1b+/−, and CPT-1b+/+ × CPT-1b+/+, were set up using 8-week-old littermates born from a founder CPT-1b +/− male and a C57BL/6J female. Among the 12 plugged CPT-1b+/− females sacrificed at embryonic day 9.5 − 11.5, 8 of them were found to have no fetuses at all, whereas 11 of 12 CPT-1b+/+ females were pregnant with a litter size similar to the B6J mice (n=6−9) (Table 3). Therefore, the CPT-1b+/− female mice in a heterozygous breeding pair are either less prone to conceive or more prone to undergo spontaneous abortion of fetuses.
Table 3.
Reproductive function study in plugged female mice at embryonic day 9.5−11.5
| Breeding pairs | With Fetuses | Without Fetuses | Plugged |
|---|---|---|---|
| CPT-1b+/− × CPT-1b+/− | 4 | 8 | 12 |
| CPT-1b+/+ × CPT-1b+/+ | 11 | 1 | 12 |
p<0.05 by Fisher's exact test
Analysis of Cpt-1a and Cpt-1b gene expression
The current mouse model with CPT-1b deficiency was developed with the 3 kb Neo-tk double cassette replacing the 1.4 kb exons 1−3 of Cpt-1b gene in the anti-sense orientation which, according to a previous study, is often related to rapid degradation of the “recombinant” RNA [28]. Indeed, in our northern blot analysis of the CPT-1b+/− samples of heart, BAT, and skeletal muscles, there was only one Cpt-1b band detected at 47 − 50% of the “integrated intensity” of that in the CPT-1b +/+ samples (Fig. 1A and 1B), suggesting the following: first, the mutant mRNA with a larger molecular weight is not detectable; second, there was no up-regulation of gene expression from the wild-type Cpt-1b allele; third, the normal absence of the Cpt-1a mRNA band in both wild-type and CPT-1b+/− samples of heart, BAT, and skeletal muscles suggested that there was no compensatory up-regulation.
Figure 1.

Levels of Cpt-1a and Cpt-1b mRNA. A) Northern blot analysis of total RNA in 6- to 8- week-old non-fasted CPT-1b+/− male mice (born from a founder CPT-1b+/− male and a C56BL/6J female): Cpt-1a and Cpt-1b mRNA expression in heart (H), brown adipose tissue (BAT), and skeletal muscles including quadricep (Q) and gastrocnemus muscles (G). B) Densitometry quantification of Cpt-1b mRNA expression. The integrated density reading of each band of Cpt-1b mRNA were first standardized with the amount of total RNA shown in (A), and then the average levels of which in the CPT-1b+/+ hearts (n=3) were arbitrarily set to 1. Mean+/− SE, *p<0.05, **p<0.005.
CPT-1 enzyme activities
To investigate if the reduction of Cpt-1b mRNA to ∼50% in non-fasted male and female CPT-1b+/− mice resulted in reduced CPT-1 activity (Fig. 2), we determined that male CPT-1b+/−mice had ∼60% and ∼92% CPT-1 activity in muscle and liver, respectively, as compared to the male CPT-1b+/+ mice. Similarly, female CPT-1b+/− mice had ∼55% and ∼89% CPT-1 activity in muscle and liver, respectively, as compared to the female CPT-1b+/+ mice. This is consistent with the predominate expression of the CPT-1b and CPT-1a isoforms in skeletal muscle and liver, respectively, as well as the about ∼50% Cpt-1b mRNA levels and no up-regulation of Cpt-1a mRNA (Fig. 1A and B). Therefore, about 60% of total CPT-1 enzyme activity in skeletal muscle of non-fasted CPT-1b+/− mice appears to be sufficient for an unchallenged normal phenotype.
Figure 2. Enzyme activity of CPT-1 in skeletal muscle (gastroc) and liver.

Male CPT-1b+/− mice (n=5) had ∼60% and ∼92% CPT-1 activity in gastroc muscles and liver, respectively as compared to the male CPT-1b+/+ mice (n=4, **p<0.001). Results were also significantly different in female CPT-1b+/− mice (n=5) and CPT-1b+/+ mice (n=2, *p<0.05).
Cold challenge
Cold challenge is a useful metabolic test for mouse models with deficiency of FAO enzymes. Wild-type 129/S6 mice are completely “cold tolerant”, with less than 3 °C drop following a 3 hr cold-challenge [10]. In the present study, 65% and 30% of the CPT-1b+/+ male (n=17) and female (n=10) mice, respectively, on a mixed genetic background (third generation offspring from a B6J×129X1/SvJ chimera backcrossed to B6J females) were “cold tolerant” (as defined above), suggesting that the B6J genetic background of CPT-1b+/+ mice might predispose to increased susceptibility to cold. We found no significant difference in the percentage of mice demonstrating cold tolerance and fatal hypothermia between the two Cpt-1b genotypes within each sex (p>0.1, Fisher's exact test) (Fig. 3). Also, when data from male and female mice were combined, there was still no significant difference in the percentage of mice demonstrating cold tolerance and fatal hypothermia between the two Cpt-1b genotypes, although 6% male (n=17) and 7% female (n=15) CPT-1b+/− mice, respectively, developed fatal hypothermia (rectal temperature below 25 °C) over 3 hours, while both male (n=17) and female (n=13) CPT-1b+/+ mice did not.
Figure 3. Survival curves of 6 hr cold challenge.
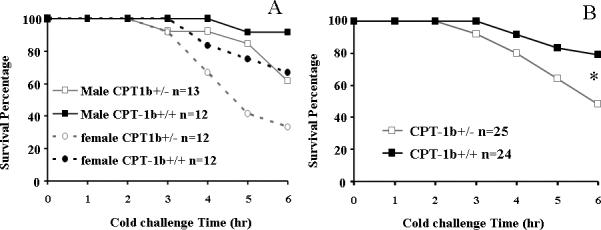
A) Among male mice, there were 5 CPT-1b+/− mice and 1 CPT-1b+/+ mouse with fatal hypothermia; the survival rates at 6 hr for the CPT-1b+/− mice and the CPT-1b+/+ mice were 62% and 92%, respectively. Among the female mice, there were 8 CPT-1b+/− mice and 4 CPT-1b+/+ mice with fatal hypothermia; the survival rates at 6 hr for the CPT-1b+/− mice and the CPT-1b+/+ mice were 33% and 67%, respectively. In both sexes, there was no significant difference between survival of the CPT-1b+/− and CPT-1b+/+ groups (p=0.160 in male, p=0.198 in female). B) When combining the data from both sexes, there were 13 CPT-1b+/− mice and 4 CPT-1b+/+ mice that developed fatal hypothermia. The survival rates at 6h for the CPT-1b+/− mice and the CPT-1b+/+ were 48% and 79%, respectively (*p<0.05 by Kaplan-Meier survival analysis: Gehan-Breslow test).
Among those taking a 6-hr cold challenge, 38% CPT-1b+/− male mice verses 8% CPT-1b+/+ control male mice developed fatal hypothermia (Fig. 3A). In females, 67% CPT-1b+/− mice compared to 33% CPT-1b+/+ littermates developed fatal hypothermia, suggesting an additive effect between female and Cpt-1b+/− genotype. When male and female data were combined (Fig. 3B), the survival curves were significantly different between genotypes.
Biochemical analysis
Because CPT-1b+/− mice had higher frequency of fatal hypothermia, we proceeded to evaluate biochemical measurements that might be altered by reduced CPT-1 enzyme activity. However, no consistent or significant differences in urine ketone body levels or other urine organic acids were noted when comparing these samples (data not shown). In contrast, consistent with CPT-1 being the rate-limiting enzyme for converting acyl-CoA to acylcarnitine, the levels of C18- and C16-acylcarnitines were mildly but significantly lowered in serum of CPT-1b+/− female mice as compared to those of CPT-1b +/+ female mice (Fig. 4), but differences were found in the males.
Figure 4. Serum acylcarnitines.

Following an 18 h fast, in CPT-1b+/− female mice (7−10 weeks old), there were small reductions in serum C16 and C18 acylcarnitine levels (n=6 for each genotype, mean+/−S.E, *p<0.05); however, the males (n=6 for each genotype, mean+/−S.E) showed no differences.
Because deficiencies in FAO are often related to (fasting) hypoglycemia, and increased plasma free fatty acid levels, these metabolites were examined. In both male and female mice aged 6−8 weeks, there was no difference in fasting free fatty acid concentrations in plasma between CPT-1b+/− and CPT-1b+/+ mice (data not shown). In both male and female mice aged 6−8 weeks, fasting blood glucose following 18-hr food removal did not differ between genotypes (Fig. 5). Interestingly, all mice had increased blood glucose levels following 3- but not 6-hr cold challenge; also, the blood glucose levels following 6-hr-cold are significantly less in female CPT-1b+/− mice compared to female CPT-1b+/+ mice.
Figure 5. Blood glucose levels.
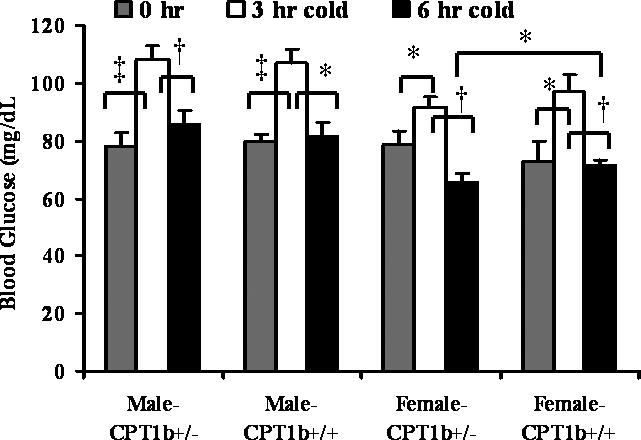
In both sexes (6−8 weeks old), there was no significant difference in blood glucose concentrations following either an 18 h fast or a 6h cold challenge between the two genotypes. Also, blood glucose levels in all 4 groups increased significantly following a 3 h but not a 6h cold challenge (n=12−14 for each group, mean+/−S.E, *p<0.05, †p<0.005, ‡p<0.001). Moreover, following a 6h cold challenge in female CPT-1b+/− mice (n=13), blood glucose concentrations were significantly lower than that in CPT-1b+/+ controls (n=12, mean+/−S.E, *p<0.05).
Histology
There were no significant lesions in BAT, heart, and liver in both male and female CPT-1b+/−mice for (n=6 male and 1 for females, respectively) compared to CPT-1b+/+ mice at 6 weeks of age (n=3 and 2, respectively).
Discussion
Inherited enzyme deficiencies of mitochondrial fatty acid β-oxidation are diseases with variable presentations from asymptomatic existence to life-threatening acute disease episodes. This is because deficiency in FAO not only reduces fat-derived energy supply but also leads to accumulation of fatty acid substrates or metabolites in the blood and other extra-cellular fluid compartment, in cytoplasm, and in mitochondria. Because CPT-1 is the rate-limiting enzyme for mitochondrial LCFA β-oxidation important in gametogenesis and embryogenesis [29-31], it is not surprising that, among available mouse models with enzyme deficiencies of FAO, a severe phenotype, or homozygous lethality is found in mice deficient in either CPT-1a [10] or CPT-1b (this study), in contrast to those with homozygous deficiency of any one of the four fatty acyl-CoA dehydrogenases (ACADs), which include very-long-, long-, medium-, and short-chain acyl-CoA dehydrogenases (VLCAD [32]; LCAD [33], MCAD [34]; and SCAD [35,36]). Also, among the four mouse models with deficiency in one of the ACADs, LCAD−/− mice have the most severe phenotype including fatty liver, fasting and cold intolerance and gestational loss [33]. Moreover, no live mice with a double homozygous LCAD(−/−)/VLCAD(−/−) genotype were detected from double heterozygous matings [32]. A potential explanation for embryonic lethality of CPT-1b−/− mice is that CPT-1b activity might be required during mouse pre-implantation development, because Cpt-1b mRNA is first and temporally detected at the 2-cell stage and reappears at the morula and blastocyst stage [30]. The importance of LCFA oxidation at this stage is also supported by our previous findings that the LCAD−/− genotype is associated with frequent gestational loss at the morula-to-blastocyst conversion [31].
In humans, loss of function mutations have been identified in genes encoding SCAD (ACADS [37]), MCAD (ACADM [38].), VLCAD (ACADVL [39]), and CPT-1A [40], but not CPT-1B or LCAD [41]. While homozygous CPT-1a deficiency is rare and potentially fatal in humans [40], heterozygous CPT-1a deficiency has not been reported with abnormalities. Similarly, the scarcity of reported CPT-1b deficient human cases is possibly due to embryonic lethality, as implied by our current mouse study. Also, heterozygous CPT-1b deficiency in humans would likely be free of symptoms, unless metabolically challenged with extreme exercise or offending drugs.
In our study, northern blot analysis suggested no up-regulation of Cpt-1b mRNA expression from the only functional copy of Cpt-1b gene in the CPT-1b+/− mice. Also, in contrast to the up-regulation of Cpt-1b mRNA expression in livers of CPT-1a+/− mice [10], there was no compensatory up-regulation of Cpt-1a mRNA in heart and BAT of CPT-1b+/− mice. Therefore, ∼50% of the wild-type levels of Cpt-1b mRNA, as well as ∼60% of the CPT-1 enzyme activities in CPT-1b predominant tissues were sufficient for a normal gross appearance, histology, and general physiologic functions in the CPT-1b+/− mice.
Interestingly, some animal models of FAO enzyme deficiency are associated with skewed patterns of inheritance [10]. Whereas LCAD+/− genotype is under-represented in offspring [31], probably due to frequent gestational loss, as similar to the LCAD−/− pups, it is not clear why both the VLCAD+/− [32] and CPT-1a+/− [10] genotypes are over-represented. In the current study, the mutant Cpt-1b (-) allele was under-represented during germ-line transmission in heterozygous matings.
Because Cpt-1b mRNA was found to be expressed at a very low level in wild-type placenta, as compare to Cpt-1a mRNA, we concluded that CPT-1a is the major isoform expressed. Furthermore, it is unlikely that CPT-1b deficiency is an important factor in the lethality of CPT-1b−/− embryos. The timing of Cpt-1b mRNA expression suggests that it is involved in spermatogenesis [8]; while CPT-1a is the sole isoform detected in immature testis, adult testis is much richer in Cpt-1b mRNA, which predominates in spermatids [9], and in meiotic and post-meiotic germ cells [8]. However, given the equal percentage of CPT-1b+/+ and CPT-1b+/− offspring from breeders consisting of either a CPT-1b+/− male mice with a B6/J female or a CPT-1b+/− female and a 129S6 male, it is unlikely that the percentage of sperm and oocyte with the mutant allele of Cpt-1b is reduced. Also, gestational loss of CPT-1b+/− pups is not a likely explanation. That is, the CPT-1b+/− breeders produced CPT-1b+/− pups and CPT-1b+/+ pups at a 1:1 ratio and that led us to postulate that perhaps the CPT-1b (+) sperm survive predominately before fertilization in heterozygous mating pairs. This is another possible explanation of the absence of CPT-1b−/− fetuses at embryonic day 9.5−11.5. Further study is required to investigate the mechanisms for under-representation of CPT-1b+/− pups from heterozygous mating.
Mice with impaired FAO often have reduced cold tolerance. Exposure to cold is detected by the brain, leading to activation of the sympathetic nervous system, which heavily innervates thermogenic targets such as BAT and skeletal muscle. Fatal hypothermia is 100% in homozygous knockout mice of VLCAD, LCAD, MCAD, and SCAD, whereas “single heterozygous” enzyme-deficient mice are usually cold-tolerant [42]. There was ∼33% fatal hypothermia in double heterozygous with VLCAD/LCAD, LCAD/SCAD, VLCAD/SCAD combinations on a mixed genetic background [42]. In the current study, it took an extended challenge (4 − 6 hrs), yet significantly more CPT-1b+/− mice developed fatal hypothermia following a 6-hr cold challenge (Fig. 3B). This and the finding that 6−7% CPT-1b+/− mice verses none of the control mice had fatal hypothermia following a 3-hr cold challenge are consistent with tissue-specific expression of this enzyme in BAT and skeletal muscle, which mediates nonshivering and shivering thermogenesis, respectively. Also, it is interesting that there was a trend for an increase in frequency of fatal hypothermia in female verses male mice with both CPT-1b+/+ and CPT-1b+/− genotypes. While we cannot rule out the lower body weight of female littermates might contribute to increased susceptibility to cold, as compared to the males, we found no correlation between lower body weight and fatal hypothermia within each genotype and each sex (data not shown). In fact, those that developed fatal hypothermia tended to be slightly heavier. Moreover, the additive effects of being female with Cpt-1b+/− genotype is consistent with cold susceptibility as an autosomal dominant trait of the mutant Cpt-1b gene with variable penetrance.
Glucose metabolism can be altered in some mouse models deficient in a FAO enzyme. For example, CPT-1a+/− mice have lower fasting blood glucose and higher fasting free fatty acid in plasma [10]. In the current study, young CPT-1b+/− mice at 6−9 weeks of age had normal indexes of glucose homeostasis: random and fasting blood glucose and fasting insulin. Also, while it has not been observed previously in mice of other genetic backgrounds such as the 129S6 mice (data not shown), blood glucose concentrations significantly increased following a 3-hr cold challenge in both Cpt-1b genotypes and sexes (Fig. 5). Cold challenge increases FAO, which might inhibit glucose utilization in these mice. Therefore, simply reducing Cpt-1b mRNA levels by half is not sufficient to alter glucose metabolism enough to cause any changes in plasma insulin and blood glucose levels.
In conclusion, we have developed a mouse model of CPT-1b deficiency that resulted in no homozygous fetuses found at any stages examined. We did find a normal mode of allelic transmission from the CPT-1b+/− male and female mice in breeding pairs with wild-type mates. In contrast, the number of CPT-1b+/− pups produced from CPT-1b+/− breeding pairs was under-represented. Heterozygous CPT-1b mice had virtually a normal phenotype, including normal body weight, normal fasting blood glucose levels. Also, increased blood glucose following a 3-hr cold challenge might reflect decreased glucose utilization during cold challenge. Moreover, CPT-1b+/− mice tended to have decreased cold tolerance as compared to CPT-1b+/+ mice at >3 hrs of cold challenge, although the difference was not significant. Following an extended time of cold challenge, there was a significant increase in the percentage of CPT-1b+/− mice developing lethal hypothermia compared to CPT-1b+/+ mice. Thus since there have been few human patients described with CPT-1b deficiency, this may also represent a high rate of gestational lethality in humans.
Acknowledgements
We thank Drs. Greg Cox and Roger Sher for providing some of the founder mice, Drs. Ada Elgavish and Jianfang Hu for helpful discussion and valuable assistance in biostatistics. This publication was made possible by Grant Number R01-RR02599 from the National Center for Research Resource (NCRR), a component of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH. This work was also supported by the Office of Biological and Environmental Research, U.S. Department of Energy (Y.Y.), under contract DE-AC05-00OR22725 with UT-Battelle, LLC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Neely JR, Rovetto MJ, Oram JF. Myocardial utilization of carbohydrate and lipids. Prog. Cardiovasc. Dis. 1972;15:289–329. doi: 10.1016/0033-0620(72)90029-1. [DOI] [PubMed] [Google Scholar]
- 2.Mitchell GA, Kassovska-Bratinova S, Boukaftane Y, Robert MF, Wang SP, Ashmarina L, Lambert M, Lapierre P, Potier E. Medical aspects of ketone body metabolism. Clin. Invest. Med. 1995;18:193–216. [PubMed] [Google Scholar]
- 3.Wolf H, Stave U, Novak M, Monkus EF. Recent investigations on neonatal fat metabolism. J. Perinat. Med. 1974;2:75–87. doi: 10.1515/jpme.1974.2.2.75. [DOI] [PubMed] [Google Scholar]
- 4.Britton CH, Schultz RA, Zhang B, Esser V, Foster DW, McGarry JD. Human liver mitochondrial carnitine palmitoyltransferase I: characterization of its cDNA and chromosomal localization and partial analysis of the gene. Proc. Natl. Acad. Sci. USA. 1995;92:1984–1988. doi: 10.1073/pnas.92.6.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGarry JD, Brown NF. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997;244:1–14. doi: 10.1111/j.1432-1033.1997.00001.x. [DOI] [PubMed] [Google Scholar]
- 6.Britton CH, Mackey DW, Esser V, Foster DW, Burns DK, Yarnall DP, Froguel P, McGarry JD. Fine chromosome mapping of the genes for human liver and muscle carnitine palmitoyltransferase I (CPT1A and CPT1B) Genomics. 1997;40:209–211. doi: 10.1006/geno.1996.4539. [DOI] [PubMed] [Google Scholar]
- 7.Price N, van der F, Leij V, Jackson C, Corstorphine R, Thomson A, Sorensen V, Zammit A novel brain-expressed protein related to carnitine palmitoyltransferase I. Genomics. 2002;80:433–442. doi: 10.1006/geno.2002.6845. [DOI] [PubMed] [Google Scholar]
- 8.Adams SH, Esser V, Brown NF, Ing NH, Johnson L, Foster DW, McGarry JD. Expression and possible role of muscle-type carnitine palmitoyltransferase I during sperm development in the rat. Biol. Reprod. 1998;59:1399–1405. doi: 10.1095/biolreprod59.6.1399. [DOI] [PubMed] [Google Scholar]
- 9.Brown NF, Hill JK, Esser V, Kirkland JL, Corkey BE, Foster DW, McGarry JD. Mouse white adipocytes and 3T3-L1 cells display an anomalous pattern of carnitine palmitoyltransferase (CPT) I isoform expression during differentiation. Inter-tissue and interspecies expression of CPT I and CPT II enzymes. Biochem. J. 1997;327:225–231. doi: 10.1042/bj3270225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nyman LR, Cox KB, Hoppel CL, Kerner J, Barnoski BL, Hamm DA, Tian L, Schoeb TR, Wood PA. Homozygous carnitine palmitoyltransferase 1a (liver isoform) deficiency is lethal in the mouse. Mol. Genet. Metab. 2005;86:179–187. doi: 10.1016/j.ymgme.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 11.Esser V, Brown NF, Cowan AT, Foster DW, McGarry JD. Expression of a cDNA isolated from rat brown adipose tissue and heart identifies the product as the muscle isoform of carnitine palmitoyltransferase I (M-CPT I). M-CPT I is the predominant CPT I isoform expressed in both white (epididymal) and brown adipocytes. J. Biol. Chem. 1996;271:6972–6977. doi: 10.1074/jbc.271.12.6972. [DOI] [PubMed] [Google Scholar]
- 12.Yamazaki N, Shinohara Y, Shima A, Terada H. High expression of a novel carnitine palmitoyltransferase I like protein in rat brown adipose tissue and heart: isolation and characterization of its cDNA clone. FEBS Lett. 1995;363:41–45. doi: 10.1016/0014-5793(95)00277-g. [DOI] [PubMed] [Google Scholar]
- 13.Wolfgang MJ, Kurama T, Dai Y, Suwa A, Asaumi M, Matsumoto S, Cha SH, Shimokawa T, Lane MD. The brain-specific carnitine palmitoyltransferase-1c regulates energy homeostasis. Proc. Natl. Acad. Sci. USA. 2006;103:7282–7287. doi: 10.1073/pnas.0602205103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DiMauro S, DiMauro PM. Muscle carnitine palmityltransferase deficiency and myoglobinuria. Science. 1973;182:929–931. doi: 10.1126/science.182.4115.929. [DOI] [PubMed] [Google Scholar]
- 15.Hostetler KY, Hoppel CL, Romine JS, Sipe JC, Gross SR, Higginbottom PA. Partial deficiency of muscle carnitine palmitoyltransferase with normal ketone production. N. Engl. J. Med. 1978;298:553–557. doi: 10.1056/NEJM197803092981007. [DOI] [PubMed] [Google Scholar]
- 16.Bertorini T, Yeh YY, Trevisan C, Stadlan E, Sabesin S, DiMauro S. Carnitine palmityl transferase deficiency: myoglobinuria and respiratory failure. Neurology. 1980;30:263–271. doi: 10.1212/wnl.30.3.263. [DOI] [PubMed] [Google Scholar]
- 17.Ross NS, Hoppel CL. Partial muscle carnitine palmitoyltransferase-A deficiency. Rhabdomyolysis associated with transiently decreased muscle carnitine content after ibuprofen therapy. J. Am. Med. Assoc. 1987;257:62–65. doi: 10.1001/jama.257.1.62. [DOI] [PubMed] [Google Scholar]
- 18.Pula TP, Max SR, Zielke HR, Chacon M, Baab P, Gumbinas M, Reed WD. Selective carnitine palmitoyltransferase deficiency in fibroblasts from a patient with muscle CPT deficiency. Ann. Neurol. 1981;10:196–198. doi: 10.1002/ana.410100211. [DOI] [PubMed] [Google Scholar]
- 19.Chick WS, Mentzer SE, Carpenter DA, Rinchik EM, Johnson D, You Y. X-rayinduced deletion complexes in embryonic stem cells on mouse chromosome 15. Mamm.Genome. 2005;16:661–671. doi: 10.1007/s00335-005-0011-5. [DOI] [PubMed] [Google Scholar]
- 20.Goetzman ES, Tian L, Wood PA. Differential induction of genes in liver and brown adipose tissue regulated by peroxisome proliferator-activated receptor-[alpha] during fasting and cold exposure in acyl-CoA dehydrogenase-deficient mice. Mol. Genet. Metab. 2005;84:39–47. doi: 10.1016/j.ymgme.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 21.Hoppel CL, Kerner J, Turkaly P, Turkaly J, Tandler B. The malonyl-CoA-sensitive form of carnitine palmitoyltransferase is not localized exclusively in the outer membrane of rat liver mitochondria. J. Biol. Chem. 1998;273:23495–23503. doi: 10.1074/jbc.273.36.23495. [DOI] [PubMed] [Google Scholar]
- 22.Kerner J, Distler AM, Minkler P, Parland W, Peterman SM, Hoppel CL. Phosphorylation of rat liver mitochondrial carnitine palmitoyltransferase-I: effect on the kinetic properties of the enzyme. J. Biol. Chem. 2004;279:41104–41113. doi: 10.1074/jbc.M406570200. [DOI] [PubMed] [Google Scholar]
- 23.Rashed MS, Ozand PT, Bucknall MP, Little D. Diagnosis of inborn errors of metabolism from blood spots by acylcarnitines and amino acids profiling using automated electrospray tandem mass spectrometry. Pediatr. Res. 1995;38:324–331. doi: 10.1203/00006450-199509000-00009. [DOI] [PubMed] [Google Scholar]
- 24.Chace DH, Hillman SL, Van Hove JL, Naylor EW. Rapid diagnosis of MCAD deficiency: quantitative analysis of octanoylcarnitine and other acylcarnitines in newborn blood spots by tandem mass spectrometry. Clin. Chem. 1997;43:2106–2113. [PubMed] [Google Scholar]
- 25.Vianey-Saban C, Guffon N, Delolne F, Guibaud P, Mathieu M, Divry P. Diagnosis of inborn errors of metabolism by acylcarnitine profiling in blood using tandem mass spectrometry. J. Inherit. Metab. Dis. 1997;20:411–414. doi: 10.1023/a:1005306818025. [DOI] [PubMed] [Google Scholar]
- 26.Wood PA, Armstrong D, Sauls D, Davisson MT. Screening mutant mice for inborn errors of metabolism. Lab Anim. Sci. 1988;38:15–19. [PubMed] [Google Scholar]
- 27.Guerra C, Koza RA, Walsh K, Kurtz DM, Wood PA, Kozak LP. Abnormal nonshivering thermogenesis in mice with inherited defects of fatty acid oxidation. J. Clin. Invest. 1998;102:1724–1731. doi: 10.1172/JCI4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000;26:99–109. [PubMed] [Google Scholar]
- 29.Risley MS. Support of Xenopus laevis spermatogenesis in vitro by different energy substrates. Biol. Reprod. 1990;42:511–522. doi: 10.1095/biolreprod42.3.511. [DOI] [PubMed] [Google Scholar]
- 30.Gentile L, Monti M, Sebastiano V, Merico V, Nicolai R, Calvani M, Garagna S, Redi CA, Zuccotti M. Single-cell quantitative RT-PCR analysis of Cpt1b and Cpt2 gene expression in mouse antral oocytes and in preimplantation embryos. Cytogenet. Genome Res. 2004;105:215–221. doi: 10.1159/000078191. [DOI] [PubMed] [Google Scholar]
- 31.Berger PS, Wood PA. Disrupted blastocoele formation reveals a critical developmental role for long-chain acyl-CoA dehydrogenase. Mol. Genet. Metab. 2004;82:266–272. doi: 10.1016/j.ymgme.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 32.Cox KB, Hamm DA, Millington DS, Matern D, Vockley J, Rinaldo P, Pinkert CA, Rhead WJ, Lindsey JR, Wood PA. Gestational, pathologic and biochemical differences between very long-chain acyl-CoA dehydrogenase deficiency and long-chain acyl-CoA dehydrogenase deficiency in the mouse. Hum. Mol. Genet. 2001;10:2069–2077. doi: 10.1093/hmg/10.19.2069. [DOI] [PubMed] [Google Scholar]
- 33.Kurtz DM, Rinaldo P, Rhead WJ, Tian L, Millington DS, Vockley J, Hamm DA, Brix AE, Lindsey JR, Pinkert CA, O'Brien WE, Wood PA. Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc. Natl. Acad. Sci. USA. 1998;95:15592–15597. doi: 10.1073/pnas.95.26.15592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tolwani RJ, Hamm DA, Tian L, Sharer JD, Vockley J, Rinaldo P, Matern D, Schoeb TR, Wood PA. Medium-chain acyl-CoA dehydrogenase deficiency in gene-targeted mice. PLoS. Genet. 2005;1:e23. doi: 10.1371/journal.pgen.0010023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hinsdale ME, Kelly CL, Wood PA. Null allele at Bcd-1 locus in BALB/cByJ mice is due to a deletion in the short-chain acyl-CoA dehydrogenase gene and results in missplicing of mRNA. Genomics. 1993;16:605–611. doi: 10.1006/geno.1993.1237. [DOI] [PubMed] [Google Scholar]
- 36.Wood PA, Amendt BA, Rhead WJ, Millington DS, Inoue F, Armstrong D. Short-chain acyl-coenzyme A dehydrogenase deficiency in mice. Pediatr. Res. 1989;25:38–43. doi: 10.1203/00006450-198901000-00010. [DOI] [PubMed] [Google Scholar]
- 37.Naito E, Ozasa H, Ikeda Y, Tanaka K. Molecular cloning and nucleotide sequence of complementary DNAs encoding human short chain acyl-coenzyme A dehydrogenase and the study of the molecular basis of human short chain acyl-coenzyme A dehydrogenase deficiency. J. Clin. Invest. 1989;83:1605–1613. doi: 10.1172/JCI114058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yokota I, Indo Y, Coates PM, Tanaka K. Molecular basis of medium chain acylcoenzyme A dehydrogenase deficiency. An A to G transition at position 985 that causes a lysine-304 to glutamate substitution in the mature protein is the single prevalent mutation. J. Clin. Invest. 1990;86:1000–1003. doi: 10.1172/JCI114761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aoyama T, Souri M, Ueno I, Kamijo T, Yamaguchi S, Rhead WJ, Tanaka K, Hashimoto T. Cloning of human very-long-chain acyl-coenzyme A dehydrogenase and molecular characterization of its deficiency in two patients. Am. J. Hum. Genet. 1995;57:273–283. [PMC free article] [PubMed] [Google Scholar]
- 40.Bennett MJ, Boriack RL, Narayan S, Rutledge SL, Raff ML. Novel mutations in CPT 1A define molecular heterogeneity of hepatic carnitine palmitoyltransferase I deficiency. Mol. Genet. Metab. 2004;82:59–63. doi: 10.1016/j.ymgme.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 41.Yamaguchi S, Indo Y, Coates PM, Hashimoto T, Tanaka K. Identification of very-longchain acyl-CoA dehydrogenase deficiency in three patients previously diagnosed with longchain acyl-CoA dehydrogenase deficiency. Pediatr. Res. 1993;34:111–113. doi: 10.1203/00006450-199307000-00025. [DOI] [PubMed] [Google Scholar]
- 42.Schuler AM, Gower BA, Matern D, Rinaldo P, Vockley J, Wood PA. Synergistic heterozygosity in mice with inherited enzyme deficiencies of mitochondrial fatty acid beta-oxidation. Mol. Genet. Metab. 2005;85:7–11. doi: 10.1016/j.ymgme.2004.09.006. [DOI] [PubMed] [Google Scholar]