

Abstract
Neuronal activity evokes localized changes in blood flow. Although this response, termed neurovascular coupling, is widely used to monitor human brain function and diagnose pathology, the cellular mechanisms that mediate the response remain unclear. We investigated the contribution of glial cells to neurovascular coupling in the acutely isolated mammalian retina. We found that light stimulation and glial cell stimulation can both evoke dilation or constriction of arterioles. Light-evoked and glial-evoked vasodilations were blocked by inhibitors of cytochrome P450 epoxygenase, the synthetic enzyme for epoxyeicosatrienoic acids. Vasoconstrictions, in contrast, were blocked by an inhibitor of ω-hydroxylase, which synthesizes 20-hydroxyeicosatetraenoic acid. Nitric oxide influenced whether vasodilations or vasoconstrictions were produced in response to light and glial stimulation. Light-evoked vasoactivity was blocked when neuron-to-glia signaling was interrupted by a purinergic antagonist. These results indicate that glial cells contribute to neurovascular coupling and suggest that regulation of blood flow may involve both vasodilating and vasoconstricting components.
Keywords: blood flow, glia, arachidonic acid metabolites, calcium, retina, ATP
Introduction
Activity within a localized brain region elicits increased blood flow to that region, supplying oxygen and nutrients to active neurons. This response, termed neurovascular coupling, was first described by Roy and Sherrington (1890). The response, which can be detected with functional magnetic resonance imaging (fMRI), positron emission tomography, and other techniques, is commonly used to monitor brain function. Despite its importance in basic and clinical studies, the cellular mechanisms underlying neurovascular coupling remain unclear (Iadecola, 2004; Koehler et al., 2006).
In recent years, it has been suggested that glial cells contribute to neurovascular coupling in the brain (Paulson and Newman, 1987; Harder et al., 1998; Zonta et al., 2003; Filosa et al., 2004; Mulligan and MacVicar, 2004; Koehler et al., 2006). Glia are well suited to mediate this response because of their close contacts with both neurons and blood vessels (Harder et al., 1998; Anderson and Nedergaard, 2003). In addition, glia generate increases in intracellular Ca2+ in response to neuronal activity (Schipke and Kettenmann, 2004; Newman, 2005) and can produce vasoactive agents after such calcium increases. These agents include the arachidonic acid metabolites epoxyeicosatrienoic acids (EETs), which are vasodilating, 20-hydroxyeicosatetraenoic acid (20-HETE), which is vasoconstricting, and prostaglandins, which can be either vasodilating or constricting (Amruthesh et al., 1993; Alkayed et al., 1996).
Recent studies have confirmed that glial cells can induce vasomotor responses. Unfortunately, the studies reached opposite conclusions. Zonta et al. (2003) and Takano et al. (2006) showed that Ca2+ increases in glial cells result in vasodilation mediated by production of cyclooxygenase (COX) metabolites. Mulligan and MacVicar (2004), in contrast, demonstrated that glial Ca2+ increases result in vasoconstriction mediated by production of the ω-hydroxylase metabolite 20-HETE.
We reexamined the role of glia in generating vasomotor responses to address this important conflict. Experiments were conducted on the acutely isolated mammalian retina. As in the brain, neuronal activity in the retina evokes increases in blood flow (Riva et al., 2005). We found that both light stimulation and glial cell stimulation evoked vasomotor responses in retinal arterioles. These responses were mediated by production of EETs and 20-HETE. Nitric oxide (NO) influenced whether vasodilating or vasoconstricting responses were produced, possibly by modulating the production of these arachidonic acid metabolites. In addition, light-evoked responses were blocked when neuron-to-glia signaling was interrupted, suggesting that glial cells are necessary mediators of neurovascular coupling.
Materials and Methods
Whole-mount retina
Experiments were performed on acutely isolated whole-mount retinas of male Long–Evans rats (175–300 g; Harlan, Indianapolis, IN). Animals were killed by overdose of sodium pentobarbital and cardiac perfused with saline, and the eyes were enucleated. Retinas were gently peeled from the back of the eye so that many retinal pigment epithelium (RPE) cells adhered to the retina as described previously (Newman and Zahs, 1998). These attached RPE cells permitted the isolated retina to remain responsive to light for several hours. The vitreous humor was removed from retinal pieces by gentle peeling with forceps. Retinas were held in place in a chamber with nylon threads attached to a platinum ring and superfused at 2–3 ml/min with bicarbonate-buffered Ringer’s solution at ∼37°C. The animals used in this study were treated in accordance with the guidelines of the Institutional Animal Care and Use Committee of the University of Minnesota.
In some experiments, retinas were preincubated in tetanus toxin (TeNT) to block neurotransmitter release (45 min in 100 μg/ml TeNT). The absence of light-evoked neuronal activity was confirmed with recordings from ganglion cells, which were unresponsive to light after incubation. In the absence of vessel tone, arterioles were preconstricted with the thromboxane analog 9,11-dideoxy-9α,11α methanoepoxy-prosta5Z, 13E-dien-1-oic acid (U-46619; 100 nm) (Filosa et al., 2004). After U-46619 treatment, arteriole diameter was often smaller that it would be in vivo, because the vessels were not experiencing normal intraluminal pressure. The reduction in diameter did not abolish responsiveness.
Retinal imaging
Retinas were imaged with a 40× water immersion objective, infrared-differential interference contrast optics (IR-DIC), and a cooled CCD camera (CoolSnap ES; Roper Scientific, Duluth, GA) to measure arteriole diameter. Arterioles were easily distinguished from venules by their morphology under IR-DIC optics. A vessel was always traced from its emergence at the optic disc to the site of imaging. The following characteristics were considered when identifying a vessel: branching pattern, the appearance of the vascular wall, and its response to mechanical pressure. At the vitreal surface of the retina, arteries branch into second- and third-order arterioles before capillaries emerge, whereas in veins, capillaries emerge directly from the main vessel. Arteries have thicker vascular walls with different smooth muscle appearance than veins, maintain tone ex vivo, and respond with stronger contractions to mechanical pressure applied with a pipette. The branching pattern of arteries and veins also differs beneath the surface of the retina and can be easily identified.
Images were captured and analyzed using MetaMorph image processing software (Molecular Devices, Downingtown, PA). The intraluminal diameter of arterioles, which was clearly demarcated in IR-DIC images, was measured manually from magnified, enhanced retinal images. Some arterioles displayed small luminal oscillations before stimulation. In these cases, the prestimulus baseline diameter was taken as the average diameter of the vessel.
Ca2+ imaging
After removal of the vitreous humor, retinas were incubated in the Ca2+-indicator dye fluo-4 AM (31 μg/ml) and pluronic acid (2.6 mg/ml) for 30 min at room temperature as described previously (Newman, 2001b). Retinas were viewed with epifluorescence optics (FITC filter set) and a Roper CoolSnap ES camera. In additional experiments, retinas were viewed with confocal microscopy (Odyssey scanner; Noran, Middleton, WI) using 488 nm excitation and a 500 nm long-pass barrier filter. To simultaneously image Ca2+ and vessel diameter, vessels were filled with fluorescein (50 μm), which was introduced through cardiac perfusion before enucleation.
Glial stimulation
Stimulation by photolysis of caged Ca2+.
Retinas were preincubated for 30 min in a caged-Ca2+ probe (o-nitrophenyl EGTA AM, 10 μm), pluronic acid (2.6 mg/ml), and fluo-4 (to monitor glial Ca2+). Four nanosecond flashes of 337 nm UV light focused to a 5 μm spot were used to photo release Ca2+ (VSL-337ND photolysis unit; Prairie Technologies, Middleton, WI). UV spots were focused onto individual astrocytes or Müller cells.
Glial stimulation by inositol 1,4,5-trisphosphate.
Inositol 1,4,5 trisphosphate (IP3) or caged-IP3 was introduced into individual glial cells through patch pipettes. Retinas were preincubated in collagenase/dispase (2 mg/ml) and DNase (0.1 mg/ml) for 15 min to digest the basal lamina at the vitreal surface of the retina. Whole-cell patch recordings were made from cells at the retinal surface. Patched cells were filled with Lucifer yellow contained in the pipette solution and identified by their morphology. Cell membrane potential and input resistance were monitored using an Axoprobe-1A microelectrode amplifier (Molecular Devices) and custom LabView software. Only cells that had stable membrane potentials more negative than −75 mV were used in experiments.
For IP3-stimulation experiments, the patch pipette solution contained 250 μm IP3. For caged-IP3-stimulation experiments, the patch pipette solution contained 250 μm caged-IP3 (NPE-caged Ins 1,4,5-P3). Vessels were allowed to come to a stable baseline diameter before photolysis. UV flashes focused onto the patched glial cell were used to photo liberate the caged IP3, as described above.
Glial stimulation by focal ATP ejection.
ATP (200 μm) was pressure-ejected from pipettes (1–4 s pulse duration) onto the vitreal surface of the retina.
Light stimulation of the retina
Diffuse flickering white light (100–250 ms flashes repeated every 250–1000 ms) was used to stimulate the retina. The light was focused onto the retina through the microscope objective lens.
Nitric oxide ramp
In some experiments, NO levels in the superfusate were controlled by addition of the NO donor 3-(2-hydroxy-2-nitroso-1-propylhydrazino)-1-propanamine (PAPA-NONOate) to the Ringer’s solution. The nitric oxide synthase (NOS) inhibitor Nω-Nitro-l-arginine methyl ester hydrochloride (L-NAME) was present during a 45 min preincubation period and throughout the experiment to keep endogenous NO levels to a minimum. NO concentrations were monitored with a 30 μm NO-sensitive electrode and an Apollo 4000 Free Radical Analyzer (World Precision Instruments, Sarasota, FL). Electrodes were calibrated before and after each experiment with solutions of known NO concentration.
For each experiment, endogenous NO was initially lowered to near zero by addition of 100 μm L-NAME. PAPA-NONOate was then added incrementally to gradually raise NO levels. NO levels in the superfusate were measured continuously in a small, closed fluid reservoir immediately upstream from the recording chamber. Uric acid (30 μm) was added to the superfusate to reduce the levels of NO2 resulting from NO oxidation (Keynes et al., 2003).
Statistics
Numerical values are given as mean ± SEM. Statistical significance was determined by the single-tailed Student’s t test.
Solutions and drugs
The bicarbonate-buffered Ringer’s solution contained the following (in mm): 117.0 NaCl, 3.0 KCl, 2.0 CaCl2, 1.0 MgSO4, 0.5 NaH2PO4, 15.0 dextrose, and 26 NaHCO3, bubbled with 5% CO2 in O2. The intracellular pipette solution contained the following (in mm): 5.0 Na-methanesulfonate, 136.0 K-methanesulfonate, 2.0 MgCl2, 2.0 MgATP, 0.2 NaGTP, 5.0 HEPES, pH 7.4. In some experiments, 0.1 mg/ml Lucifer yellow CH was added to the pipette solution to visualize glial cell contacts with blood vessels. Miconazole [1-(2,4-Dichloro-β-[(2,4-dichlorobenzyl)oxy]phenethyl)imidazole], aspirin (O-Acetylsalicylic acid), indomethacin [1-(4-Chlorobenzoyl)-5-methoxy-2-methyl-3-indoleacetic acid], L-NAME, fluorescein (free acid), diphenyleneiodonium chloride (DPI), suramin (sodium salt), Lucifer yellow (6-Amino-2,3-dihydro-1,3-dioxo-2-hydrazinocarbonylamino-1H-benz[d,e]isoquinoline-5,8-disulfonic acid Dilithium salt), IP3, PAPA-NONOate, and tetanus toxin from Clostridium tetani were purchased from Sigma-Aldrich (St. Louis, MO). U-46619, 2-(2-propynyloxy)-benzenhexanoic acid (PPOH), (±)5(6)-epoxy-8Z,11Z,14Z-eicosatrienoic acid (5,6-EET), (±)14(15)-epoxy-5Z,8Z,11Z-eicosatrienoic acid (14,15-EET), (±)11(12)-epoxy-5Z,8Z,14Z-eicosatrienoic (11,12-EET), and 17(18)-EpETE (17,18-EET) were purchased from Cayman Chemicals (Ann Arbor, MI). (S)-Nitroso-N-acetylpenicillamine (SNAP) was purchased from Tocris Cookson (Bristol, UK). N-(4-Butyl-2-methylphenyl)-N′-hydroxyformamidine (HET0016) was donated by Taisho Pharmaceutical (Tokyo, Japan). SKF525A (proadifen) was purchased from Calbiochem (La Jolla, CA). Caged IP3 [d-myo-inositol 1,4,5-triphosphate, P4(5)-(1-(2-nitrophenyl)ethyl) ester, tris(triethylammonium) salt (NPE-caged Ins 1,4,5-P3)] and caged calcium (o-nitrophenyl EGTA, AM) were purchased from Invitrogen (San Diego, CA).
Results
Light-evoked vasomotor responses
We first examined the effect of light stimulation on blood vessels in the retina. The diameter of arterioles at the vitreal surface of the retina was monitored with IR-DIC microscopy as retinas were stimulated with a flickering white light, which raises the activity level of neurons in the inner retina (Miller, 2001).
Light stimulation evoked both vasodilation and vasoconstriction. In most preparations, either dilating or constricting responses were observed in most of the arterioles tested. Both vasodilations (Fig. 1A,B) (movie S1, available at www.jneurosci.org as supplemental material) and vasoconstrictions (Fig. 1C,D) (movie S2, available at www.jneurosci.org as supplemental material) occurred over the entire length of vessels within the field of view (∼150 μm). Occasionally, transient (2–20 s) sphincter-like contractions were observed near vessel branch points (Fig. 1E,F) (movie S3, available at www.jneurosci.org as supplemental material). These localized constrictions sometimes occurred in vessels that otherwise showed little responsiveness or displayed widespread dilation. Similar activity-dependent sphincter-like contractions have been reported previously (Cauli et al., 2004).
Figure 1.

Light evokes vasodilation and vasoconstriction. A–F, IR-DIC images of arterioles at the vitreal surface of the retina. Shown are vessels before (A) and during (B) light-evoked vasodilation, before (C) and during (D) light-evoked constriction, and before (E) and during (F) light-evoked sphincter-like constriction. Scale bars, 10 μm. Arrowheads in all figures indicate the diameter of the vessel lumen. G, Time course of light-evoked vasodilation in six trials. H, Time course of vasodilation in a rapidly responding vessel (latency, <500 ms). I, Time course of vasoconstriction in five trials.
Response latencies of the light-evoked vasodilations and constrictions were short, with most responses beginning within 1–3 s after the onset of stimulation (Fig. 1G,I). In some trials, vasodilation latencies were as brief as 500 ms (Fig. 1H).
The type of light-evoked vasomotor response observed did not depend on stimulus intensity. Above a threshold brightness that evoked a response, brighter stimuli increased the magnitude of the vasomotor response in some cases but did not transform dilation to constriction or vice versa. Nor did the pre-existing tone of the arteriole change the nature of the light-evoked response. Addition of the thromboxane analog U-46619 (100 nm) to preconstrict vessels did not change the type of light-evoked response observed, suggesting that the initial diameter of a vessel was not a significant factor in determining the type of response evoked. When a preparation was stimulated repetitively at 5 min intervals, similar vascular responses were observed to each stimulus.
The magnitude of light-evoked vasodilations, compared with maximal vessel diameter, was also assessed. L-NAME pretreatment (100 μm; used to suppress vasoconstrictions; see below) constricted vessels an average of 25 ± 7% (n = 9). In the presence of L-NAME, retinas were excited with light flashes, evoking vasodilations. Preparations were then superfused with a 0 Ca2+, 5 mm EGTA solution to maximally dilate vessels. Maximal light-evoked dilations averaged 64 ± 6% (n = 9) of the dilations produced by the 0 Ca2+ solution. Before L-NAME perfusion, vessels showed some tone (73 ± 8%; n = 9 of maximum diameter measured in 0 Ca2+ solution).
Nitric oxide and vasomotor responses
NO plays a complex role in vasomotor activity. A principal effect of NO is to dilate vessels by raising cGMP levels in vascular smooth muscle cells (Ignarro et al., 1999). However, NO can also affect vasomotor responses by inhibiting cytochrome P450 epoxygenase or ω-hydroxylase, the synthetic enzymes for vasodilating EETs and the vasoconstrictor 20-HETE (Fig. 2E) (Fleming, 2001; Roman, 2002).
Figure 2.
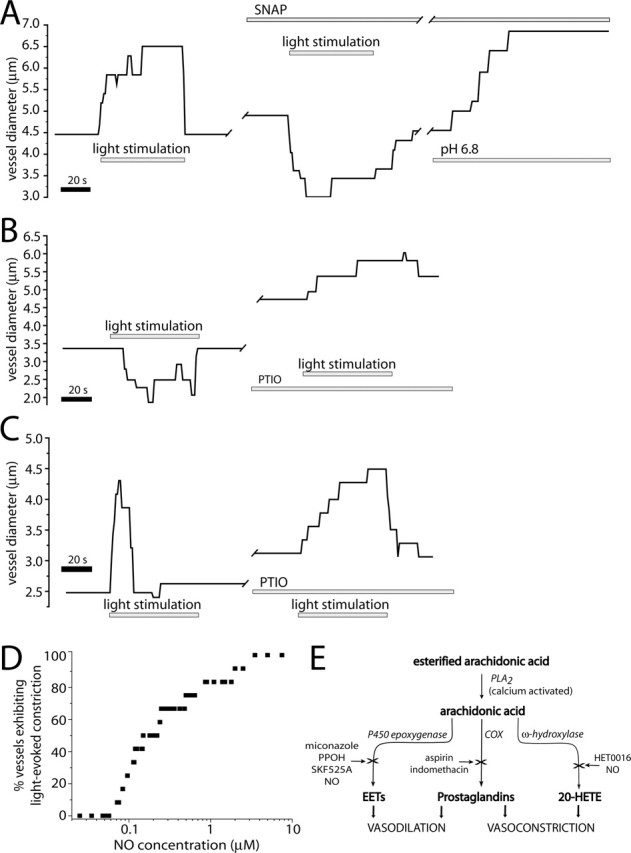
NO modulates light-evoked vasomotor responses. A, Light-evoked vasodilation is transformed into a vasoconstriction by addition of the NO donor SNAP (100 μm). The vessel dilates in the presence of SNAP and responds to a decrease in pH of the superfusate with an additional dilation. B, Light-evoked vasoconstriction is transformed into a vasodilation by addition of the NO scavenger PTIO (100 μm). The vessel dilates in the presence of PTIO. C, PTIO increases the duration of a light-evoked vasodilation. D, Raising NO levels increases the percentage of vessels that constrict to light stimulation. At <70 nm NO, light stimulation evoked vasodilation in all vessels (n = 12). As NO was raised, a greater percentage of vessels constricted. E, Schematic of arachidonic acid metabolism, showing synthesis of vasoactive EETs, prostaglandins, and 20-HETE. Synthetic enzymes are italicized and enzyme inhibition is indicated by an X.
We hypothesized that NO, possibly by selectively inhibiting either epoxygenase or ω-hydroxylase, determines whether vasodilations or vasoconstrictions are evoked by light stimulation. We tested whether NO affects light-evoked vasomotor responses by addition of NOS inhibitors. In the presence of L-NAME (100 μm), which blocks all forms of NOS, or DPI (3 μm), which blocks eNOS, light stimulation evoked only vasodilations; vasoconstrictions were completely eliminated. D-NAME (100 μm), the inactive analog of L-NAME, had no effect on light-evoked vasomotor activity.
We also manipulated NO levels with NO donors and scavengers. The primary effect of the NO donor SNAP (100 μm) was to block light-evoked vasodilations or to transform vasodilations into vasoconstrictions (Fig. 2A). SNAP also caused some vessels to relax. The SNAP block of light-evoked vasodilation was not attributable to a maximal relaxation of the vessels. In the presence of SNAP, vessels preserved sufficient tone so that superfusion with low-pH saline (which produces vasodilation through an alternate mechanism) resulted in additional dilation (Fig. 2A). Light-evoked vasoconstriction was unaffected by SNAP (data not shown).
The NO scavenger 2-pyenyl-4,4,5,5-tetramethylimidazoline-3-oxide-1-oxyl (PTIO) had the opposite effect on retinal arterioles. Vessels that displayed light-evoked vasoconstrictions generated dilations in the presence of PTIO (Fig. 2B). This was observed despite the fact that PTIO superfusion, in some cases, caused vessel relaxation (Fig. 2B,C). For vessels that initially displayed shorter light-evoked vasodilations, PTIO lengthened the duration of these vasodilations (Fig. 2C). The unexpected PTIO-induced increase in vessel diameter may be attributable to a disinhibition of epoxygenase produced by lowered NO levels (Fig. 2E). Although both SNAP and PTIO produced arteriole relaxation in some cases, their effect was variable and increases in vessel tone were also observed.
We determined the effect of NO levels on light-evoked vasomotor responses by monitoring the responses of single arterioles as superfusate NO levels were ramped from near 0 to several μm (Fig. 2D). Endogenous NO production was suppressed by the addition of L-NAME. Of the 12 arterioles studied, all dilated in response to light when NO was <70 nm. As NO was increased, light-evoked responses switched from dilation to constriction. For most vessels, this transition occurred for NO levels between 80 nm and 1 μm (Fig. 2D).
These results suggest that NO plays a pivotal role in determining the type of vascular response evoked by neuronal activity, with increased NO levels favoring vasoconstriction and decreased levels favoring vasodilation. Because NOS inhibition of NO production resulted in an increase in vasodilatory responses in the retina, we used NOS inhibitors to isolate and characterize vasodilation. The eNOS inhibitor DPI was used in most experiments, because it was less likely to interfere with NO signaling in neurons, which is mediated primarily through nNOS production of NO. Retinas were treated with DPI or L-NAME for at least 45 min before experimentation and for the duration of the experiment.
Arachidonic acid metabolites and light-evoked vasomotor responses
Previous studies (Harder et al., 1998; Zonta et al., 2003; Mulligan and MacVicar, 2004) have suggested that arachidonic acid metabolites mediate activity-dependent vasomotor responses in the CNS. To test this hypothesis, we examined whether production of the vasoconstrictor 20-HETE mediates light-evoked vasoconstriction in the retina. In control experiments, light stimulation evoked vasodilation in 40.5% of trials, whereas vasoconstriction was evoked in 28.5% of trials. (In the remaining trials, no vasomotor responses were evoked.) Addition of HET0016, which blocks 20-HETE synthesis by ω-hydroxylase, decreased the percentage of light-evoked vasoconstrictions to 6.3% and increased vasodilations to 87.5% (Fig. 3A), indicating that 20-HETE is a mediator of light-evoked vasoconstriction. We also examined whether HET0016 can change light-evoked vasoconstriction to vasodilation in individual vessels. In eight arterioles, which initially displayed light-evoked vasoconstriction (in the absence of NOS inhibitors), light-evoked vasodilations were observed in all eight after addition of 100 nm HET0016.
Figure 3.

Arachidonic acid metabolites mediate light-evoked vasomotor responses. A, The ω-hydroxylase inhibitor HET0016 (100 nm) decreases the percentage of light-evoked vasoconstrictions and increases the percentage of vasodilations. B, Pharmacology of light-evoked vasodilation. In the presence of the NOS inhibitors DPI (3 μm) or L-NAME (100 μm), the epoxygenase inhibitors PPOH (20 μm), SKF525A (20 μm), and miconazole (10 μm) reduce light-evoked vasodilation. The COX inhibitors aspirin (50 μm) and indomethacin (5 μm), in contrast, do not reduce vasodilation. In all figures, asterisks indicate a significant difference from controls (p < 0.05; Student’s t test). Error bars indicate SEM. The number of samples is given in parentheses. C, Superfusion of 5,6-EET (1 μm) increases the diameter of an arteriole.
The role of EETs and prostaglandins in mediating light-evoked vasodilation was also tested. Inhibition of EET synthesis by PPOH reduced the magnitude of light-evoked vasodilations to 3.8 and 23.7% of control in the presence of DPI and L-NAME, respectively (Fig. 3B). SKF525A and miconazole, two other inhibitors of epoxygenase with different mechanisms of action, similarly reduced the amplitude of light-evoked vasodilations (Fig. 3B). Inhibition of prostaglandin synthesis by aspirin or indomethacin, in contrast, did not reduce light-evoked vasodilation (Fig. 3B). We also examined whether PPOH can change light-evoked vasodilation to vasoconstriction in individual vessels. Of 11 arterioles, which initially displayed light-evoked vasodilation (in the absence of NOS inhibitors), light-evoked vasoconstrictions were observed in seven of them, after inhibition of EET production with 20 μm PPOH. The remaining four vessels became unresponsive to light with PPOH.
These results indicate that production of EETs, but not prostaglandins, contribute to light-evoked vasodilation in the retina. This conclusion is supported by the finding that superfusion of EET isomers 5,6-EET, 11,12-EET, and 17,18-EET evoked dilation of retinal arterioles, even in the presence of PPOH (Fig. 3C).
Glial-evoked vasomotor responses
We also investigated whether glial cell stimulation results in vasomotor responses in the retina. Glial cells were stimulated by increasing intracellular Ca2+ levels. Glial Ca2+ was raised by photolysis of caged Ca2+, by introduction of IP3 into cells, by photolysis of caged IP3, and by evoked intercellular glial Ca2+ waves. Both Müller cells (the principal retinal glial cells) and astrocytes [which are localized primarily within the nerve fiber layer at the vitreal surface of the retina (Newman, 2001a)] were tested.
Caged Ca2+ stimulation of glia
Glial cells were stimulated by flash photolysis of caged Ca2+ after retinal incubation with a membrane-permeant form of the compound. Photolysis of caged Ca2+ resulted in a Ca2+ increase within the stimulated glial cell and, in most trials, initiated a Ca2+ wave that propagated into neighboring glial cells (Fig. 4A–C) (movie S4, available at www.jneurosci.org as supplemental material). Glial stimulation evoked the same types of vasomotor responses observed with light stimulation. Vasodilations (Fig. 4D–F), vasoconstrictions (Fig. 4G,H), and sphincter-like constrictions (Fig. 4I,J) of neighboring arterioles were observed after glial stimulation. Response latencies were as brief as 1 s after photolysis. Glial-evoked vasodilations, constrictions, and sphincter-like constrictions were always preceded by an increase in glial Ca2+ in astrocytes and Müller cells (Fig. 4K–M). There was no correlation between the amplitude, duration, or spatial extent of the glial Ca2+ increases and the type of vasomotor response observed. Small, localized glial Ca2+ increases could evoke either dilations or constrictions. Large, widespread Ca2+ increases could also be associated with either vasodilations or vasoconstrictions.
Figure 4.

Photolysis of caged Ca2+ in glial cells evokes vasodilation and vasoconstriction. A–C, Fluorescence images showing a Ca2+ increase that propagates through several glial cells after an uncaging flash. D–F, Glial-evoked vasodilation. IR-DIC images of the region indicated in A are shown, each acquired 0.5 s after the corresponding image above. G, H, Glial-evoked vasoconstriction. Fluorescence images of a fluorescein-filled vessel before and after photolysis. Note the photolysis-evoked increase in glial Ca2+. I, J, Glial-evoked sphincter-like vasoconstriction. Fluorescence images of a fluorescein-filled vessel before and after photolysis. Yellow dots in A, G, and I indicate sites of uncaging flash. Scale bars: C, 20 μm; F, 5 μm; G, 10 μm; I, 10 μm. K–M, Time course of glial Ca2+ change and vessel dilation (K), constriction (L), and sphincter-like constriction (M).
We next asked whether the same arachidonic acid metabolites that mediate light-evoked vasodilation and constriction also mediate glial-evoked dilation and constriction. In control experiments, glial stimulation evoked vasodilation in 35.0% of trials, vasoconstriction in 30.0% of trials, and sphincter-like constriction in 35.0% of trials. Addition of the ω-hydroxylase inhibitor HET0016 to block 20-HETE synthesis decreased the percentage of vasoconstrictions and sphincter-like constrictions to 15.4 and 15.4%, respectively, while increasing the occurrence of vasodilations to 69.2% (Fig. 5A).
Figure 5.

Arachidonic acid metabolites mediate glial-evoked vasomotor responses. A, The ω-hydroxylase inhibitor HET0016 (100 nm) decreases the percentage of photolysis-evoked vasoconstrictions and increases the percentage of vasodilations. B, In the presence of DPI (3 μm), photolysis of caged Ca2+ evokes vasodilation that is blocked by the epoxygenase inhibitor PPOH (20 μm) but not by preincubation with TeNT.
PPOH inhibition of EET synthesis decreased the magnitude of glial-evoked vasodilation to 10% of control (Fig. 5B). In contrast, preincubation of retinas in tetanus toxin to block transmitter release from neurons did not reduce glial-evoked vasodilation (Fig. 5B, TeNT), demonstrating that dilation is mediated by direct glia to arteriole signaling and that activated glia do not signal back to vasoactive neurons. This was important to determine, because it has been demonstrated that in the retina, glial cells can modulate neuronal activity through release of gliotransmitters (Newman and Zahs, 1998; Newman, 2003). These experiments investigating glial-evoked vasodilation were conducted in the presence of DPI, similar to the conditions used to investigate light-evoked vasodilation. The results suggest that glial-evoked vasodilation is mediated by EET production, whereas vasoconstriction is mediated by 20-HETE production, as are light-evoked dilations and constrictions, respectively.
IP3 stimulation of glia
We also stimulated glial cells by raising intracellular IP3 levels, known to release Ca2+ from internal stores. IP3 was introduced into individual astrocytes or Müller cells through patch pipettes. Lucifer yellow was added to the pipette solution to visualize glial contacts with arterioles. Astrocytes send out multiple thin processes that envelop blood vessels (Fig. 6A), whereas Müller cells send out one or a few processes that contact vessels (Fig. 6B).
Figure 6.

IP3-stimulated glial cells evoke vasodilation. A, Lucifer yellow-filled astrocytes are shown in a fluorescence image. Multiple astrocyte processes envelop an arteriole. B, A Lucifer yellow-filled Müller cell contacting an arteriole is shown in a superimposed fluorescence and IR-DIC image. C, D, Arteriole dilation evoked by introduction of IP3 into a Müller cell. A vessel is shown before (C) and during (D) whole-cell recording with a pipette containing IP3 (250 μm). Scale bars: A, B, D, 20 μm. E, Photolysis of caged IP3 (250 μm) in Müller cells and astrocytes evokes vasodilation. F, Introduction of IP3 into glial cells evokes vasodilation. Photolysis of caged IP3 evokes vasodilation that is reduced by PPOH (20 μm) but not indomethacin (5 μm).
Neighboring arterioles dilated within 2–5 s after achieving whole-cell recordings, as IP3 diffused into the cell (Fig. 6C,D). Arteriole dilation averaged 38% (Fig. 6F). Leakage of solution from the pipette onto vessels before patching onto glial cells was not responsible for the observed vasodilation as direct ejection of the pipette contents, which included IP3, ATP, and high K+, onto arterioles caused vasoconstriction.
Glial cells were also stimulated by photolysis of caged IP3, introduced through patch pipettes. Flash photolysis was performed several minutes after achieving whole-cell patch recordings to allow vessels to stabilize before stimulating the glial cells. Photolysis evoked Ca2+ increases in the patched glial cells (monitored with a Ca2+ indicator dye) and produced rapid dilation of adjacent arterioles with latencies as brief as 1 s (Fig. 6E,F). In control experiments, photolysis flashes outside the margin of the caged IP3-filled cell or flashes onto patched cells not containing caged IP3 did not cause vasodilation. For reasons not presently clear, vasoconstriction was rarely seen after IP3 stimulation of glial cells.
PPOH block of EET production dramatically reduced IP3-evoked arteriole vasodilation, both in the absence and presence of NOS inhibitors. In the absence of NOS inhibition, PPOH reduced the amplitude of glial-evoked arteriole vasodilation to 15% of control (Fig. 6F). In the presence of DPI, PPOH reduced the IP3-evoked vasodilation to 0% of control. Addition of the COX inhibitor indomethacin, in contrast, did not reduce vasodilation. These results confirm that, in the retina, glial-evoked vasodilation is mediated by production of EETs but not by COX products.
Glia Ca2+ waves and vasomotor responses
Glial cells were also stimulated by focal ejections of ATP, which evoked intercellular Ca2+ waves that propagated through the network of glial cells at the retinal surface (Newman and Zahs, 1997). When a propagated glial Ca2+ wave reached an arteriole, a vasomotor response was often initiated. Both vasodilations (Fig. 7A–D (movie S5, available at www.jneurosci.org as supplemental material), and vasoconstrictions (Fig. 7G,H) were observed. These vasomotor responses occurred even when the initial site of glial cell stimulation was >100 μm from a vessel. The onset of arteriole vasodilations (Fig. 7E) and vasoconstrictions (Fig. 7I) was preceded by an increase in Ca2+ within adjacent glial cells. We observed no correlation between the amplitude or duration of glial Ca2+ waves and the type of vasomotor response observed. Small, brief Ca2+ increases in glial cells near vessels could evoke either dilations or constrictions. Large, prolonged Ca2+ increases in perivascular glial cells could also evoke either type of vasomotor response.
Figure 7.

Propagated glial Ca2+ waves evoke vasodilation and vasoconstriction in distant arterioles. A–D, Fluorescence images showing an ATP-evoked Ca2+ wave propagating through glial cells at the vitreal surface of the retina. Dilation occurs when the Ca2+ wave reaches the vessel. The site of focal ATP ejection, used to initiate the Ca2+ wave, was just beyond the top right corner of the images. Scale bar, 20 μm. E, Time course of Ca2+ increase and vessel dilation in A–D, measured at the arrowheads in the images. Arrows in E indicate the time of acquisition of images A–D. F, Vessel dilation after arrival of glial Ca2+ waves is reduced by the epoxygenase inhibitor PPOH (20 μm) but not by preincubation in TeNT. L-NAME (100 μm) or DPI (3 μm) is present in superfusate. G, H, Fluorescence images before and after the arrival of a glial Ca2+ wave. Vasoconstriction occurs after the arrival of the Ca2+ wave. The vessel is filled with fluorescein. Scale bar, 20 μm. I, Time course of Ca2+ increase and vessel constriction after initiation (arrow) of a Ca2+ wave.
The mean amplitude of vasodilations initiated by the arrival of a glial Ca2+ wave was 23%. These vasodilations were reduced to 30% of control by PPOH (Fig. 7F), confirming that vessel dilation is mediated by EET production. Preincubation of retinas in tetanus toxin to block neurotransmitter release did not reduce vasodilations evoked by glial Ca2+ waves (Fig. 7F), demonstrating that glia are capable of directly producing vessel dilation and do not require neuronal release of transmitters onto vessels.
Neuron-to-glia signaling
In the retina, neuron-to-glia signaling is mediated by neuronal release of ATP and activation of glial purinergic receptors, which results in increased glial Ca2+ (Newman, 2005). Neuron-to-glia signaling is blocked by the purinergic antagonist suramin. If light-evoked vasomotor responses are mediated by glial cells, then interruption of neuron-to-glia signaling should block the vasomotor response. This proved to be the case (Fig. 8). In the presence of DPI, 100 μm suramin reduced the amplitude of light-evoked vasodilations from 26 to 1%. Similarly, in the presence of L-NAME, vasodilations were reduced from 38 to 1%. Light-evoked vasoconstrictions were also blocked by suramin. The amplitude of vasoconstrictions was reduced from 17 to 0% after suramin addition.
Figure 8.

Suramin blocks light-evoked but not glial-evoked vasomotor responses. In the presence of DPI (3 μm) or L-NAME (100 μm), light-evoked vasodilations are almost completely blocked by 100 μm suramin. Light-evoked vasoconstrictions are completely abolished by suramin. In contrast, glial-evoked vasodilations are not reduced by suramin. Glial cells were stimulated by photolysis of caged IP3.
It could be argued that suramin blocks light-evoked vasodilation and vasoconstriction, not because glia mediate neurovascular coupling but because suramin directly interferes with vasomotor responses or, alternately, because suramin reduces light-evoked neuronal activity. These are both unlikely. Glia-evoked vasodilation, as opposed to light-evoked vasodilation, was not reduced by suramin (Fig. 8). In the presence of DPI, the amplitude of vasodilations evoked by glial stimulation (photolysis of caged-IP3) was not reduced, demonstrating that the purinergic antagonist does not block the ability of the vessel to dilate. In addition, we have shown previously that suramin does not reduce light-evoked neuronal activity in the mammalian retina (Newman, 2005). Together, these results support the hypothesis that light-evoked responses in the retina are mediated through glial cell signaling.
Discussion
Our findings support the hypothesis that glial cells mediate neurovascular coupling in the CNS. In accordance with this hypothesis, our data show the following. First, selective stimulation of glial cells results in large, short-latency vasodilations and vasoconstrictions. These glial-evoked vasomotor responses are a result of direct glia-to-vessel signaling and do not involve neurons as intermediaries. Second, light-evoked and glial-evoked vasomotor responses are mediated by the same arachidonic acid metabolites. Both light- and glial-evoked dilation is mediated by epoxygenase production of EETs, whereas light- and glial-evoked constriction is mediated by ω-hydroxylase production of 20-HETE. Third, light-evoked vasodilations and vasoconstrictions are blocked when neuron-to-glia signaling is interrupted. Together, these results indicate that light-evoked vasomotor responses are mediated, in large part, by signaling through glial cells.
The results reveal that glia can evoke both vasodilations and vasoconstrictions mediated by EET and 20-HETE production, respectively. As shown previously, glial cells can synthesize both arachidonic acid metabolites (Amruthesh et al., 1993; Alkayed et al., 1996; Harder et al., 1998). Glial production of EETs and 20-HETE could be initiated by increases in glial Ca2+, which would activate Ca2+-dependent phospholipase A2 and result in arachidonic acid production. Light-evoked neuronal activity has been shown previously to increase glial Ca2+ in the retina (Newman, 2005). However, our results do not exclude a downstream site of EET or 20-HETE production. For instance, synthesis of these arachidonic acid metabolites could occur in vascular smooth muscle cells after release of other signaling molecules from activated glial cells. Moreover, unlike COX metabolites, EETs and 20-HETE can also be stored in membranes for later release, adding to the complexity of this pathway. Release of these metabolites from the membrane could account for the residual vasomotor responses seen in the presence of epoxygenase and ω-hydroxylase inhibitors.
We observed no correlation between the amplitude, duration, or spatial extent of glial Ca2+ increases after glial stimulation and the type of vasomotor response observed. Indeed, no such correlation would be expected if the nature of the vasomotor response (dilation or constriction) is determined by NO levels, as we argue in the following paragraphs. We should note that our results do not exclude the possibility of glial-evoked vasomotor responses that are generated by a Ca2+-independent mechanism.
Our findings suggest that NO levels may regulate whether neuronal activity results in vasodilation or constriction in the retina. NO has multiple effects on vasomotor responses. NO can induce vasodilation by stimulating guanylyl cyclase, leading to the production of cGMP and activation of K+ channels in vascular smooth muscle. NO can also induce vasodilation by inhibiting ω-hydroxylase, resulting in a decrease in the vasoconstricting metabolite 20-HETE (Alonso-Galicia et al., 1998; Sun et al., 2000). Both of these mechanisms contribute to vasodilation in the brain (Alonso-Galicia et al., 1999). However, NO could also favor vasoconstriction. NO could inhibit cytochrome P450 epoxygenase, the enzyme that metabolizes arachidonic acid to vasodilating EETs (Udosen et al., 2003). By reducing EET production, NO would promote vasoconstriction.
Although NO has both vasodilating and vasoconstricting effects, it is generally considered to be a vasodilating agent. However, our results suggest that in the retina, NO promotes vasoconstriction. Activity-evoked vasodilations were generally observed in the presence of NOS inhibitors and NO scavengers, whereas activity-evoked vasoconstrictions were seen in the presence of NO donors. Arterioles that initially dilated in response to light in nominally zero NO conditions constricted as NO levels were raised. These findings suggest that EET production by epoxygenase is more sensitive to NO inhibition than is 20-HETE production by ω-hydroxylase. High NO levels would inhibit epoxygenase production of vasodilatory EETs, resulting in activity-dependent vasoconstriction mediated by 20-HETE production. Low NO levels would release epoxygenase from inhibition, resulting in greater activity-dependent vasodilation. Comparative studies to determine the relative sensitivity of epoxygenase and ω-hydroxylase to NO inhibition have not been conducted.
The net effect of NO on vasomotor responses is most likely complex and may depend on the precise site of its synthesis, which would determine which cells it acts on. In the proximal retina, nNOS is expressed in processes of amacrine cells lying close to capillaries and larger vessels (Roufail et al., 1995). In addition, Müller cells and astrocytes both express eNOS (Haverkamp et al., 1999). Precise control of NO synthesis within retinal neurons and glial cells could determine whether glial activation results in vasodilation or constriction and may control the spatial distribution of these responses. Further studies are needed to determine the relationship between the cellular sources of NO, regulation of its synthesis, and its effects on neurovascular coupling.
Our results are in agreement with previous studies demonstrating the involvement of glial cells in mediating neurovascular coupling. Zonta et al. (2003) found that in the hippocampal slice, glial stimulation results in arteriole dilation mediated by production of COX metabolites. Recently, Takano et al. (2006) reported that in somatosensory cortex in vivo, glial stimulation elicited vasodilation via production of COX-1 metabolites. In contrast, we found that in the retina, EET production, but not prostaglandin production, mediates vasodilation. This discrepancy could be a result of differences in the neurovascular coupling mechanisms operating in different CNS regions.
Mulligan and MacVicar (2004) found that in hippocampal slices, glial stimulation results in vasoconstriction mediated by 20-HETE production, in agreement with our own findings. However, they did not observe glial-evoked vasodilations. The lack of a vasodilation response could be caused by high levels of NO in their preparation, leading to inhibition of EET production, because they report that glial-evoked vasodilations were observed after NOS inhibition by L-NAME.
Filosa et al. (2004) reported that in cortical slices, evoked Ca2+ increases in astrocytes result in a decrease in Ca2+ oscillations in adjacent vascular smooth muscle cells and to changes in vasomotor activity, demonstrating functional glia to vascular signaling. Cauli et al. (2004) report that stimulation of interneurons in cortical slices can induce either vasodilation or vasoconstriction, in agreement with our own findings that both types of vasomotor responses can be evoked by neuronal activity.
Our finding that light stimulation and glial stimulation evoke both vasodilation and vasoconstriction suggests that regulation of blood flow in the CNS may also involve both vasomotor responses. For instance, neuronal activity in a localized CNS region could evoke vasodilation within that region and vasoconstriction in more distant vessels, resulting in an increased redistribution of blood flow to the vasodilating region. It is noteworthy that fMRI studies of activity-dependent hemodynamic changes indicate that a center-surround-like regulation of blood flow may occur in vivo (Harel et al., 2002; Shmuel et al., 2002; Devor et al., 2005). Active vasoconstriction may contribute to such an effect. It is difficult to predict what the spatial distribution of glial-evoked vasodilations and vasoconstrictions will be in vivo. The distribution will depend on a number of factors that could not be reproduced in our experiments, including the spatial distribution of neuronal activation, the resting level of NO, and the spatial distribution of NO production.
Regulation of blood flow in response to neuronal activity is a complex process and is undoubtedly mediated by several mechanisms. Our results suggest that glial cells are involved in generating activity-dependent vasomotor responses and can induce both vasodilation and vasoconstriction. NO, which plays a central role in neurovascular coupling, may exert its effect, in part, by modulating glial cell regulation of these vasomotor responses.
Footnotes
This work was supported by National Institutes of Health Grant EY004077 (E.A.N.) and a National Science Foundation predoctoral fellowship (M.R.M.). We thank Drs. K. R. Zahs and John Garthwaite for helpful discussions.
References
- Alkayed NJ, Narayanan J, Gebremedhin D, Medhora M, Roman RJ, Harder DR (1996). Molecular characterization of an arachidonic acid epoxygenase in rat brain astrocytes. Stroke 27:971–979. [DOI] [PubMed] [Google Scholar]
- Alonso-Galicia M, Sun CW, Falck JR, Harder DR, Roman RJ (1998). Contribution of 20-HETE to the vasodilator actions of nitric oxide in renal arteries. Am J Physiol Renal Physiol 275:F370–F378. [DOI] [PubMed] [Google Scholar]
- Alonso-Galicia M, Falck JR, Reddy KM, Roman RJ (1999). 20-HETE agonists and antagonists in the renal circulation. Am J Physiol Renal Physiol 277:F790–F796. [DOI] [PubMed] [Google Scholar]
- Amruthesh SC, Boerschel MF, McKinney JS, Willoughby KA, Ellis EF (1993). Metabolism of arachidonic acid to epoxyeicosatrienoic acids, hydroxyeicosatetraenoic acids, and prostaglandins in cultured rat hippocampal astrocytes. J Neurochem 61:150–159. [DOI] [PubMed] [Google Scholar]
- Anderson CM, Nedergaard M (2003). Astrocyte-mediated control of cerebral microcirculation. Trends Neurosci 26:340–344. [DOI] [PubMed] [Google Scholar]
- Cauli B, Tong XK, Rancillac A, Serluca N, Lambolez B, Rossier J, Hamel E (2004). Cortical GABA interneurons in neurovascular coupling: relays for subcortical vasoactive pathways. J Neurosci 24:8940–8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor A, Ulbert I, Dunn AK, Narayanan SN, Jones SR, Andermann ML, Boas DA, Dale AM (2005). Coupling of the cortical hemodynamic response to cortical and thalamic neuronal activity. Proc Natl Acad Sci USA 102:3822–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filosa JA, Bonev AD, Nelson MT (2004). Calcium dynamics in cortical astrocytes and arterioles during neurovascular coupling. Circ Res 95:e73–e81. [DOI] [PubMed] [Google Scholar]
- Fleming I (2001). Cytochrome P450 and vascular homeostasis. Circ Res 89:753–762. [DOI] [PubMed] [Google Scholar]
- Harder DR, Alkayed NJ, Lange AR, Gebremedhin D, Roman RJ (1998). Functional hyperemia in the brain: hypothesis for astrocyte-derived vasodilator metabolites. Stroke 29:229–234. [DOI] [PubMed] [Google Scholar]
- Harel N, Lee SP, Nagaoka T, Kim DS, Kim SG (2002). Origin of negative blood oxygenation level-dependent fMRI signals. J Cereb Blood Flow Metab 22:908–917. [DOI] [PubMed] [Google Scholar]
- Haverkamp S, Kolb H, Cuenca N (1999). Endothelial nitric oxide synthase (eNOS) is localized to Muller cells in all vertebrate retinas. Vision Res 39:2299–2303. [DOI] [PubMed] [Google Scholar]
- Iadecola C (2004). Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci 5:347–360. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Cirino G, Casini A, Napoli C (1999). Nitric oxide as a signaling molecule in the vascular system: an overview. J Cardiovasc Pharmacol 6:879–886. [DOI] [PubMed] [Google Scholar]
- Keynes RG, Griffiths C, Garthwaite J (2003). Superoxide-dependent consumption of nitric oxide in biological media may confound in vitro experiments. Biochem J 369:339–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler RC, Gebremedhin D, Harder DR (2006). Role of astrocytes in cerebrovascular regulation. J Appl Physiol 100:307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RF (2001). The physiology and morphology of the vertebrate retina. In: Retina (Ryan SJ, ed.) pp. 138–170. St. Louis: Mosby.
- Mulligan SJ, MacVicar BA (2004). Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 431:195–199. [DOI] [PubMed] [Google Scholar]
- Newman EA (2001a). Glia of the retina. In: Retina (Ryan SJ, ed.) pp. 89–103. St. Louis: Mosby.
- Newman EA (2001b). Propagation of intercellular calcium waves in retinal astrocytes and Müller cells. J Neurosci 21:2215–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA (2003). Glial cell inhibition of neurons by release of ATP. J Neurosci 23:1659–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA (2005). Calcium increases in retinal glial cells evoked by light-induced neuronal activity. J Neurosci 25:5502–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA, Zahs KR (1997). Calcium waves in retinal glial cells. Science 275:844–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA, Zahs KR (1998). Modulation of neuronal activity by glial cells in the retina. J Neurosci 18:4022–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson OB, Newman EA (1987). Does the release of potassium from astrocyte endfeet regulate cerebral blood flow? Science 237:896–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva CE, Logean E, Falsini B (2005). Visually evoked hemodynamical response and assessment of neurovascular coupling in the optic nerve and retina. Prog Retin Eye Res 24:183–215. [DOI] [PubMed] [Google Scholar]
- Roman RJ (2002). P-450 Metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82:131–185. [DOI] [PubMed] [Google Scholar]
- Roufail E, Stringer M, Rees S (1995). Nitric oxide synthase immunoreactivity and NADPH diaphorase staining are co-localised in neurons closely associated with the vasculature in rat and human retina. Brain Res 684:36–46. [DOI] [PubMed] [Google Scholar]
- Roy C, Sherrington C (1890). On the regulation of blood supply in the brain. J Physiol (Lond) 11:85–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipke CG, Kettenmann H (2004). Astrocyte responses to neuronal activity. Glia 47:226–232. [DOI] [PubMed] [Google Scholar]
- Shmuel A, Yacoub E, Pfeuffer J, Van de Moortele PF, Adriany G, Hu X, Ugurbil K (2002). Sustained negative BOLD, blood flow and oxygen consumption response and its coupling to the positive response in the human brain. Neuron 36:1195–1210. [DOI] [PubMed] [Google Scholar]
- Sun CW, Falck JR, Okamoto H, Harder DR, Roman RJ (2000). Role of cGMP versus 20-HETE in the vasodilator response to nitric oxide in rat cerebral arteries. Am J Physiol Heart Circ Physiol 279:H339–H350. [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M (2006). Astrocyte-mediated control of cerebral blood flow. Nat Neurosci 9:260–267. [DOI] [PubMed] [Google Scholar]
- Udosen IT, Jiang H, Hercule HC, Oyekan AO (2003). Nitric oxide-epoxygenase interactions and arachidonate-induced dilation of rat renal microvessels. Am J Physiol Heart Circ Physiol 285:H2054–H2063. [DOI] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann K-A, Pozzan T, Carmignoto G (2003). Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci 6:43–50. [DOI] [PubMed] [Google Scholar]